

Abstract
We describe the design and synthesis of novel bivalent ligands based on the conjugation of 4-anilidopiperidine derivatives with enkephalin analogues. The design of non-peptide analogues is explored with 5-amino substituted tetrahydronaphthalen-2yl)methyl containing 4-anilidopiperidine derivatives, while non-peptide-peptide ligands are explored by conjugating the C-terminus of enkephalin analogues (H-Xxx-DAla-Gly-Phe-OH) to the amino group of 4-anilidopiperidine small molecule derivatives with and without a linker. These novel bivalent ligands are evaluated for biological activities at μ and δ opioid receptors. They exhibit very good affinities at μ and δ opioid receptors, and potent agonist activities in MVD and GPI assays. Among these the lead bivalent ligand 17 showed excellent binding affinities (0.1 nM and 0.5 nM) at μ and δ opioid receptors respectively, and was found to have very potent agonist activities in MVD (56 +/- 5.9 nM) and GPI (4.6 +/− 1.9 nM) assays. In vivo the lead bivalent ligand 17 exhibited a short duration of action (< 15 min) comparable to 4-anilidopiperidine derivatives, and moderate analgesic activity. The ligand 17 has limited application against acute pain but may have utility in settings where a highly reversible analgesic is required.
Keywords: Opioids, Opioid receptors, bivalent ligands, enkephalins
Graphical abstract
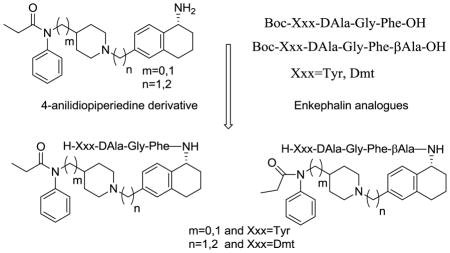
Opioid analgesics are widely used for the treatment of moderate to severe pain, and endogenous opioids exert their biological influences through three distinct opioid receptors μ, δ and κ. Even though opioids have been using as pain relievers for chronic and neuropathic pain, they also produce a number of adverse side effects that can limit their clinical utility, including nausea and vomiting, constipation and respiratory depression. During the last two decades the scientific community has explored possible approaches to address the side effects of opioids by making bivalent ligands at both μ and δ opioid receptors. There is a growing interest in developing bivalent/bifunctional ligands targeting a variety of G-protein coupled receptors including opioid, dopamine, serotonin and muscarinic receptors.1 Opioid ligands which interact with multiple receptors expand the therapeutic index of monovalent agonists and may limit side effects.2 The increasing interest in creating hybrid opioid molecules during the last decade resulted in an number of publications and patents from different research groups.3–5 There is convincible evidence from the literature that δ receptor agonists, as well as δ receptor antagonists, can provide beneficial modulation to the pharmacological effects of μ agonists. For example, δ agonists can enhance the analgesic potency and efficacy of μ agonists, and δ antagonists can prevent or diminish the development of tolerance and physical dependence by μ agonists.6–9 The emerging promising approach in the area of opioid based drug development is the opioid ligands possessing mixed μ agonist/ δ agonist profile and mixed μ agonist/δ antagonist profile.10,11 Since the discovery of fentanyl, medicinal chemistry efforts have yielded thousands of 4-anilidopiperidine analogs. Fentanyl is a well-known μ-selective synthetic analgesic (ED50 0.011 mg/kg) which is 50–100 times more potent than morphine, has a prominent place due to high potency, low cardiovascular toxicity, fast onset and short duration of action. Enkephalin analogues were derived from endogenous opioid peptides such as enkephalins occurring naturally in the brain, along with other endogenous opioid peptides. The latter function as both neuromodulators and hormones and are responsible for a broad spectrum of physiological effects. The enkephalin peptides, which carry an opioid message sequence showed good opioid affinity and bioactivity at μ and δ opioid receptors. Here we took an approach to conjugate these two different opioid ligands to achieve μ and δ opioid receptor interaction.
The opioid ligands which interact with multiple receptors expand the therapeutic index. Based on the above hypothesis we have designed and synthesized novel bivalent ligands for μ and δ receptors based on two distinct classes of opioid ligands, the peptide sequence derived from the endogenous opioid peptides and the non-peptide moiety from 4-anilidopiperidine derivatives. Portoghese et al12 incorporated enkephalin peptides into Fentanyl, and synthesized 1-and 2- substituted fentanyl analogues based on the structural analogy between the aromatic rings of fentanyl and the Tyr1 and Phe4 opioid peptides. The resulting analogues showed very weak or no opioid activity. In the last decade, our laboratory has worked on novel opioid ligands based on the 4-anilidopiperidine and modified enkephalin analogues and the conjugation of enkephalin analogues to different sites of the fentanyl molecule.13–15 In continuation of our efforts to develop novel bivalent opioid ligands, we report the design of hybrid opioid molecules which contain a peptide and a non-peptide moiety. The peptide moiety contains an enkephalin analogue like Xxx-DAla-Gly-Phe-OH (Xxx=Tyr, Dmt), and the non-peptide molecule contains a (tetrahydronaphthalen-2yl) methyl moiety with an amino substitution in the 5th position. The peptide moiety is attached to different 4-anilidopiperidine cores N-phenyl-N-(piperidin-4-yl)propionamide/N-phenyl-N-(piperidin-4-ylmethyl)propionamide.16 The rationale for choosing the 5-substituted tetrahydronaphthalen-2yl) methyl moiety is that the amine functionality can be conjugated to the enkephalin analogues, thus maintaining the functionality of 4-anilidopiperidine core unhindered, and also to introduce an additional hydrophobic moiety of cyclohexyl which can improve the lipophilicity of the molecule. The C-terminal of the enkephalin opioid peptide is attached to the amino group of the small molecule with and without a linker.
The method for synthesis of 4-anilidopiperidine analogues 1, 2 and 3 was reported in our earlier communication.17 The enkephalin analogues 4 (Boc-Tyr-DAla-Gly-Phe-OH) and 13 (Boc-Dmt-DAla-Gly-Phe-OH) were prepared by solution phase peptide synthesis following the Nα-Boc strategy by previously reported.15 The Boc protected enkephalin analogue 4 (Boc-Tyr-DAla-GlyPhe-OH) was coupled with 4-anilidopiperidine derivatives 1, 2 and 3 by using HATU/DIPEA in acetonitrile to yield crude hybrid analogues of small molecules and peptides 5, 6 and 7. Without further purification of hybrid analogues 5, 6 and 7, the deprotection of the N-terminal Boc-group was performed with 50% trifluoroacetic acid in dichloromethane, thus obtaining the final hybrid analogues 8, 9 and 10. Boc-β-Ala-OH was coupled with 4-anilidopiperidine analogue 3 by using HBTU/DIPEA in acetonitrile to yield the compound 11 followed by Boc-deprotection to give compound 12. The Boc-protected enkephalin analogues 4 (Boc-Tyr-DAla-Gly-Phe-OH) and 13 (Boc-Dmt-DAla-Gly-Phe-OH) were condensed with compound 12, followed by Boc-deprotection with 50% trifluoroacetic acid in dichloromethane, thus obtaining the crude final bivalent ligands 16 and 17. All the final crude ligands were washed with diethyl ether twice for partial purification and were purified by RP-HPLC (>95%) in 40–50% overall yields and characterized by analytical HPLC, 1H-NMR, HRMS, and TLC. The assignments of NMR resonances are available in the Supporting Information.
Opioid binding affinities (see Table 1) of the new bivalent analogues for the human δ-opioid receptor (hDOR) or the rat μ-opioid receptor (rMOR) were determined by radio ligand competition analysis using [3H] DPDPE to label the δ-opioid receptor and [3H] DAMGO to label the MOR opioid receptor in cell membrane preparations from transfected cells that stably express the respective receptor type.18 Detailed radioligand experimental procedures are given in the Supporting Information. The functional bioactivity profiles of selected ligands (see Table 2) were determined in MVD and GPI/LMMP smooth muscle preparations, as described previously.19 IC50 values, relative potency estimates and their associated errors were determined by fitting the data to the Hill equation by a computerized non-linear least-square method. Detailed GPI and MVD in vitro bioassays experimental procedures are given in the Supporting Information.
Table 1.
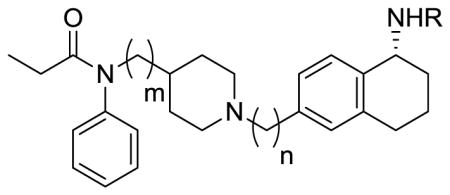
Binding Affinities of the Bivalent ligands at DOR and MOR
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R | m | n | log IC50 | Binding Kib (nM) | log IC50 | Ki ratio μ/δ | aLogP | |
| MORa (μ) | DORa (δ) | ||||||||
|
| |||||||||
| 1 | H | 1 | 1 | −5.88 ± 0.16 | 580 | 6000 | −9.56 ± 0.30 | 1/10 | 4.17 |
|
| |||||||||
| 2 | H | 0 | 2 | −5.76 ± 0.39 | 760 | >10000 | n.d. | 1/13 | 4.04 |
|
| |||||||||
| 3 | H | 0 | 1 | −7.02 ± 0.11 | 40 | 10000 | n.d. | 1/250 | 3.53 |
|
| |||||||||
| 8 | H-Tyr-DAla-Gly-Phe | 1 | 1 | −7.67 ± 0.10 | 10 | 320 | −6.16 ± 0.14 | 1/32 | 4.88 |
|
| |||||||||
| 9 | H-Tyr-DAla-Gly-Phe | 0 | 2 | −7.65 ± 0.12 | 10 | 720 | −5.80 ± 0.11 | 1/72 | 4.87 |
|
| |||||||||
| 10 | H-Tyr-DAla-Gly-Phe | 0 | 1 | −8.23 ± 0.03 | 3 | 370 | −6.10 ± 0.06 | 1/122 | 4.61 |
|
| |||||||||
| 16 | H-Tyr-DAla-Gly-Phe-βAla | 0 | 1 | −8.89 ± 0.03 | 1 | 34 | −7.14 ± 0.08 | 1/34 | 4.32 |
|
| |||||||||
| 17 | H-Dmt-DAla-Gly-Phe-βAla | 0 | 1 | −9.56 ± 0.30 | 0.1 | 0.5 | −8.92 ± 0.04 | 1/5 | 4.45 |
|
| |||||||||
| Fentanyl | 5.9 | 570 | |||||||
|
| |||||||||
| YD | 2.8 | 300 | |||||||
|
| |||||||||
| AGFNH2 | |||||||||
Competition assays were carried out using rat brain membranes.
Competition against radiolabeled ligand, data collected from at least 2 independent experiments
n.d.: not determined.
Table 2.
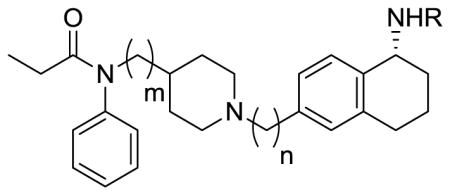
In vitro functional activity profiles of bivalent ligands
| ||||||
|---|---|---|---|---|---|---|
| Compound | R | m | n | MVD IC50a (nM) | GPI IC50a (nM) | Selectivity for MOR |
| 3 | H | 0 | 1 | 7.5% at 1uM | 1000 +/− 360 | |
| 8 | H-Tyr-DAla-Gly-Phe | 1 | 1 | 230+/−29 | 190+/− 33 | 1.2 |
| 9 | H-Tyr-DAla-Gly-Phe | 0 | 2 | 500+/−74 | 150+/− 23 | 3.2 |
| 10 | H-Tyr-DAla-Gly-Phe | 0 | 1 | 910+/−170 | 210+/− 39 | 4.2 |
| 16 | H-Tyr-DAla-Gly-Phe-βAla | 0 | 1 | 190 +/− 31 | 10+/−4.2 | 18 |
| 17 | H-Dmt-DAla-Gly-Phe-βAla | 0 | 1 | 56 +/− 5.9 | 4.6+/−1.9 | 12 |
| Fentanyl | 9.5 ± 4.0 | 3.4 ± 0.45 | ||||
| YDAGFNH2 | 0.72 ± 0.20 | 40.0 ± 13.0 | ||||
Concentration at 50% inhibition of muscle contraction at electrically stimulated isolated tissues; these values represent the mean of four tissues within 95% confidence limit.
A series of novel bivalent ligands was designed and synthesized by attaching C-terminal of enkephalin analogues to the amino group of small molecules 1, 2 and 3 and tested for biological activities at μ and δ opioid receptors. According to the alogP values we expect that these bivalent ligands maintain highly lipophilic character and that will increase the cell permeability and consequently their bioavailability.20 The designed bivalent ligands showed very good affinity towards both μ and δ opioid receptors. The 4-anilidopiperidine small molecules 1 and 2 showed moderate binding affinity of 580 nM and 760 nM respectively towards the μ opioid receptor, and the compound 3 showed good binding affinity 40 nM with 250 fold selectivity towards the μ opioid receptor. The small molecules 1, 2 and 3 showed weak or no binding affinity toward the δ opioid receptor with very weak or no opioid activity. The bivalent ligands 8 and 9 containing small molecule 1 and 2 attached to the C-terminus of enkephalin analogues ((H-Xxx-DAla-GlyPhe-OH). These analogues showed good binding affinity (10 nM) towards the μ opioid receptor and moderate binding affinity (320 nM and 723 nM) respectively at the δ opioid receptor. Similarly, enkephalin analogues attached to the small molecule 3 and the resultant bivalent ligand 10 showed a good binding affinity of 3 nM, a 3 fold increase at μ opioid receptor and similar binding affinity at the δ opioid receptor like compounds 8 and 9. In our further bivalent ligands 16 and 17, we introduced a linker β-alanine between the enkephalin analogue and 4-anilidopiperidine small molecule 3, and in the enkephalin peptide sequence replacement of Tyr with Dmt. The compound 16 showed good binding affinity towards both μ (1 nM) and δ opioid receptor (34 nM) and when compared with the bivalent ligand 10 absence of linker between enkephalin analogue and small molecule exhibits a 3 fold increase in μ receptor affinity and 10 fold increase in δ receptor affinity. Ligand 17 showed excellent binding affinity (0.1 and 0.5 nM) at both the μ and δ opioid receptor when Tyr is replaced by Dmt in the enkephalin peptide. The lead bivalent ligand 17 showed excellent binding affinity toward both the μ and δ opioid receptor, and did not show any activity for the κ opioid receptor.
The bivalent ligands (8, 9 and 10), which contain different 4-anilidopiperidine small molecules 1, 2 and 3 attached to the enkephalin analogue showed moderate agonist activity in both the MVD and GPI assay. The introduction of linker in between enkephalin analogues and small molecule 3 and the replacement of Tyr with Dmt (16 and 17) showed very good agonist activities at both mouse vas deferens MVD (IC50=190+/- 31 and 10+/-4.2) and guinea pig ileum GPI (IC50= 10+/−4.2, and 4.6+/−1.9) assays respectively. In the MVD assay, the bivalent ligand 16 displayed about 18-fold lower potency as compared to the potencies determined in the GPI assay. Similarly, a 12-fold lower potency was seen with compound 17 in this assay. The bivalent ligand 17 showed good binding affinity as well as good agonist activities at both the μ and δ opioid receptors.
The lead ligand 17 was chosen for in vivo assay studies and the ligand 17 (0.01, 0.1, 1, and 10 μg in 5 μL; n=5/treatment) was evaluated in rats using a radiant heat assay. Paw withdrawal latencies (PWLs) 5 min after spinal administration of ligand 17 at 1 μg and 10 μg in 5 μL were significantly higher than those vehicle-treated rats and baseline values (p < 0.01) with a short duration as shown in the time-effect curve (Figure 2 top). The area under the curve (AUC) was calculated to determine if increasing the dose increased the duration of effect. No significant differences in the AUC were observed (Figure 2 top). The % antinociception was calculated and a dose response curve constructed to determine if responses at the peak time of effect (5 min) were dose-dependent (Figure 2 bottom). Despite significant antinociception at 0.1, 1, and 10 μg compared to 5 μL of vehicle (p = 0.03), the efficacy of ligand 17 was not dose-related (Slope = 8.2 ± 7.4; R2= 0.06; Figure 2 bottom). The lead bivalent ligand 17 exhibited short duration of action (< 15 min) comparable to 4-anilidopiperidine derivatives and moderate in vivo activity. Detailed in vivo experimental procedures are given in the experimental section.
Figure 2.

Ligand 17 was evaluated in SD rats using a radiant heat assay; Ligand 17 dose-dependency was assessed by constructing a dose response curve (top); Ligand 17 antinociceptive dose-response curve (bottom).
In conclusion, we have achieved very good opioid bivalent ligands obtained by conjugating C-terminal of enkephalin analogues attached to the amino group of different 4-anilidopiperidine core which contained a 5-amino substituted tetrahydronapthalene methyl group. The bivalent ligands (8, 9 and 10) which contain the 4-anilidopiperidine core attached to the enkephalin analogues showed moderate binding affinity, as well as moderate agonist activity at both μ and δ opioid receptors. Introduction of the linker (β-alanine) in between the small molecule and enkephalin analogues produced the potent bivalent ligands 16 and 17. When Tyr1 was replaced with Dmt1, the bivalent ligand 17 showed very good binding affinity as well as potent agonist activities at both μ and δ opioid receptors. In in vivo the lead bivalent ligand 17 exhibited short duration of action (~ 15 min), comparable to 4-anilidopiperidine derivatives21 and moderate in vivo activity. The ligand 17 has limited application against acute pain due to short duration of action but may have utility in settings where a highly reversible analgesic is required.
All amino acid derivatives were purchased from Novabiochem and ChemImpex International. All reactions were performed under nitrogen, unless otherwise noted. All reactions were performed under nitrogen, unless otherwise noted. Analytical HPLC was performed on a Hewlett-Packard 1090 [C-18, Vydac, 4.6mm_250mm, 5 μm] and preparative RP-HPLC on Vydac 214 TP C-4 column. 1H NMR spectra were recorded on a Bruker DRX 500 spectrometer in DMSO-d6 solution. Chemical shifts are reported in δ units with respect to residual proton signal of DMSO at 2.5 ppm. HRMS and unit mass spectra were taken in the positive ion mode under ESI methods. All final compounds were obtained in >95% purity, as established by analytical HPLC.
In vivo Assay
Adult male Sprague-Dawley rats (225–300 g; Harlan, Indianapolis, IN) were kept in a temperature-controlled environment with lights on 07:00–19:00 with food and water available ad libitum. All animal procedures were performed in accordance with the policies and recommendations of the International Association for the Study of Pain, the National Institutes of Health, and with approval from the Animal Care and Use Committee of the University of Arizona for the handling and use of laboratory animals.
Surgical Methods
Rats were anesthetized (ketamine/xylazine anesthesia, 80/12 mg/kg i.p.; Sigma-Aldrich) and placed in a stereotaxic head holder. The cisterna magna was exposed and incised, and an 8-cm catheter (PE-10; Stoelting) was implanted as previously reported,22 terminating in the lumbar region of the spinal cord. Catheters were sutured (3-0 silk suture) into the deep muscle and externalized at the back of the neck. After a recover period (≥ 7 days) after implantation of the indwelling cannula, vehicle (10% DMSO: 90% MPH2O) or ligand 17 (0.01, 0.1, 1, 10 μg; n=5/treatment) were injected in a 5μL volume followed by a 9 μL saline flush. Catheter placement was verified at completion of experiments.
Behavioral Assay
Paw-flick latency23 was determined as follows. Rats were allowed to acclimate to the testing room for 30 minutes prior to testing. Basal paw withdrawal latencies (PWLs) to an infrared radiant heat source were measured (Intensity=40) and ranged between 16.0 and 20.0 seconds. A cutoff time of 33.0 seconds was used to prevent tissue damage. After a single, intrathecal injection (i.t.) of ligand 17 or vehicle, PWLs were re-assessed 8 times up to 4 hours or until they returned to baseline values. Maximal percent efficacy was calculated and expressed as:
Preparation of compound 5
To an ice-cold stirred solution of the Boc-protected enkephalin peptide 4 (Boc-Tyr-DAla-Gly-Phe-OH) (141 mg, 0.22 mmol, 1 eq) in dry acetonitrile (8 mL) was added DIPEA (0.16 mL, 0.9 mmol, 4 eq) and HATU (0.13 g, 0.36 mmol, 1.4 eq) followed by (R)-N-((1-((5-amino-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)methyl)-N-phenylpropionamide 1 (0.1 g, 0.22 mmol, 1 eq). The resulting reaction mixture was stirred for 4–6 h at room temperature. The solvent was stripped of under reduced pressure, and the resultant residue was diluted with dichloromethane (70 mL) and washed with 5% potassium hydrogen sulfate solution twice and followed by diluted sodium bicarbonate solution two times. The organic layer was washed with water followed by brine and dried over sodium sulfate. The organic phase was evaporated to dryness in vacuo. The resultant residue was washed with diethyl ether a couple of times and dried. This produced the enkephalin conjugated small molecule 5 (0.15 g, 65% of yield) as a light brown colored solid and used for the further reaction without purification.
Preparation of compound 6
Prepared as described for compound 5 from 2 (0.1 g, 0.226 mmol, 1eq) ((R)-N-(1-(2-(5-amino-5,6,7,8-tetrahydronaphthalen-2-yl)ethyl)piperidin-4-yl)-N-phenylpropionamide) and enkephalin peptide 4 (141 mg, 0.226 mmol, 1eq) (Boc-Tyr-DAla-GlyPhe-OH) afforded the compound 6 (0.14 g, 60% of yield) as a brown coloured solid and used for the further reaction without purification.
Preparation of compound 7
Prepared as described for compound 5 from 3 (0.1 g, 0.19 mmol, 1eq) (R)-N-(1-((5-amino-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)-N-phenylpropionamide and enkephalin peptide 4 (0.12 mg, 0.19 mmol, 1 eq) (Boc-Tyr-DAla-GlyPhe-OH) afforded the product 7 (0.13 g, 65% of yield) as a light brown coloured solid and used for the further reaction without purification.
Preparation of compound 8
To an ice-cold stirred solution of the enkephalin conjugated small molecule 5 (0.12 g, 0.12 mmoles, 1eq) in dry dichloromethane (4 mL) was added 4 mL of trifluoroacetic acid. The resulting mixture was stirred for 2 h, and the solvent was stripped of under reduced pressure. The resultant residue was washed with diethyl ether a couple of times and dried. This produced the final ligand 8 as a light brown colored solid. The crude final ligand 8 was isolated by preparative RP-HPLC (10–90% of acetonitrile containing 0.1% TFA in water within 40 min) to give pure ligand 8 (50 mg, 45% of yield) as a white powder. ESI MS m/z 844 (MH)+. HRMS [M + H]+ 844.47549 (theoretical 844.47561); 1H NMR (499 MHz, DMSO-d6) δ 9.74 – 9.31 (m, 2H), 8.61 (d, J = 7.4 Hz, 1H), 8.46 (d, J = 8.4 Hz, 1H), 8.27 (t, J = 5.9 Hz, 1H), 8.16 – 8.06 (m, 2H), 8.01 (t, J = 7.5 Hz, 1H), 7.45 (t, J = 7.5 Hz, 2H), 7.38 (q, J = 7.3, 6.6 Hz, 1H), 7.34 – 7.30 (m, 2H), 7.28 – 7.13 (m, 9H), 7.04 (dd, J = 8.6, 6.8 Hz, 2H), 6.73 – 6.68 (m, 2H), 4.92 – 4.83 (m, 1H), 4.56-4.50 (m, 1H), 4.35 – 4.30 (m, 1H), 4.17 (d, J = 4.9 Hz, 2H), 4.01 (q, J = 6.5 Hz, 1H), 3.80-3.73 (m, 1H), 3.66 (dd, J = 16.8, 5.7 Hz, 1H), 3.55 (d, J = 7.2 Hz, 2H), 3.34 – 3.25 (m, 2H), 3.12 (d, J = 8.4 Hz, 1H), 2.93 (m, 2H), 2.89 – 2.76 (m, 4H), 2.70 (m, 1H), 1.95 (q, J = 7.6, 7.2 Hz, 2H), 1.87 – 1.79 (m, 2H), 1.79 – 1.44 (m, 5H), 1.35 (q, J = 12.8, 12.1 Hz, 2H), 1.24 (d, J = 7.0 Hz, 1H), 1.08 (d, J = 7.0 Hz, 2H), 0.90 – 0.86 (m, 3H).
Preparation of compound 9
Prepared as described for compound 8 from 6 (0.1 g, 0.10 mmol, 1eq) afforded the crude product 9. The crude final ligand 9 was isolated by preparative RP-HPLC (10–90% of acetonitrile containing 0.1% TFA in water within 40 min) to give pure ligand 9 (40 mg, 40% of yield) as a white powder. ESI MS m/z 844 (MH)+. HRMS [M + H]+ 844.47507(theoretical 844.47561);1H NMR (499 MHz, DMSO-d6) δ 9.40 – 9.30 (bs, 1H), 9.28-9.18 (bs,1H), 8.61 (d, J = 7.5 Hz, 1H), 8.41 (d, J = 8.5 Hz, 1H), 8.27 (t, J = 5.9 Hz, 1H), 8.12 (bs, 2H), 7.97 (d, J = 8.2 Hz, 1H), 7.54 – 7.43 (m, 3H), 7.30 – 7.16 (m, 7H), 7.04 (dd, J = 9.0, 7.1 Hz, 3H), 7.00 – 6.93 (m, 2H), 6.73 – 6.67 (m, 2H), 4.88 – 4.79 (m, 1H), 4.77-4.70 (m, 1H), 4.55-4.50 (m, 1H), 4.35-4.28 (m, 1H), 4.05 – 3.92 (m, 1H), 3.81 – 3.69 (m, 1H), 3.64 (dd, J = 16.8, 5.7 Hz, 1H), 3.59 – 3.50 (m, 2H), 3.19 – 3.06 (m, 4H), 2.96 – 2.77 (m, 6H), 2.71 – 2.57 (m, 2H), 2.02 – 1.92 (m, 2H), 1.84 (q, J = 7.4 Hz, 2H), 1.77 – 1.57 (m, 3H), 1.56 – 1.43 (m, 3H), 1.24 (d, J = 6.9 Hz, 1H), 1.08 (d, J = 7.0 Hz, 2H), 0.89 (t, J = 7.4 Hz, 3H).
Preparation of Compound 10
Prepared as described for compound 8 from 7 (0.1 g, 0.107 mmol, 1eq) afforded the crude product 10. The crude final ligand 10 was isolated by preparative RP-HPLC (10–90% of acetonitrile containing 0.1% TFA in water within 40 min) to give pure ligand 10 (45 mg, 43% of yield) as a white powder. ESI MS m/z 830 (MH)+. HRMS [M + H]+ 830.45976 (theoretical 830.45996); 1H NMR (499 MHz, DMSO-d6) δ 8.62 – 8.57 (m, 1H), 8.44 (d, J = 8.5 Hz, 1H), 8.27 (t, J = 6.0 Hz, 1H), 8.21 – 8.06 (m, 2H), 8.01 (d, J = 8.2 Hz, 1H), 7.52 – 7.41 (m, 3H), 7.29 – 7.18 (m, 7H), 7.15 (d, J = 3.5 Hz, 3H), 7.04 (dd, J = 8.5, 6.6 Hz, 2H), 6.71 (m, 2H), 4.90-4.84 (m, 1H), 4.76 – 4.65 (m, 1H), 4.55-4.48 (m, 1H), 4.41 – 4.29 (m, 1H), 4.14 (d, J = 5.6 Hz, 2H), 4.04 – 3.93 (m, 1H), 3.84 – 3.70 (m, 1H), 3.65 (dd, J = 16.7, 5.4 Hz, 1H), 3.29 (d, J = 11.8 Hz, 2H), 3.10 (t, J = 12.2 Hz, 2H), 2.97 – 2.89 (m, 2H), 2.88-2.80 (m, 2H), 2.76 – 2.61 (m, 2H), 1.97 – 1.88 (m, 2H), 1.82 (q, J = 7.5 Hz, 2H), 1.76-1.60 (m,3H), 1.53 – 1.42 (m, 3H), 1.28 – 1.21 (m, 1H), 1.09 (d, J = 7.0 Hz, 2H), 0.87 (t, J = 7.4 Hz, 3H).
Preparation of compound 11
To an ice-cold stirred solution of the Boc-protected β-alanine (47 mg, 0.24 mmoles, 1.2 eq) in dry acetonitrile (5 mL) was added DIPEA (0.13 mL, 0.79 mmol, 4 eq) and HBTU (0.13 g, 0.345 mmol, 1.4 eq) followed by (R)-N-(1-((5-amino-5,6,7,8- tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)-N-phenylpropionamide 3 (0.1 g, 0.19 mmol, 1 eq). The resulting reaction mixture was stirred for 6 h at room temperature. The solvent was stripped of under reduced pressure, and the resultant residue was diluted with dichloromethane (50 mL) and washed with 5% potassium hydrogensulfate solution twice and followed by diluted sodium bicarbonate solution for two times. The organic layer was washed with water followed brine and dried over sodium sulfate. The organic phase was evaporated to dryness in vacuo. The resultant residue was washed with diethyl ether a couple of times and dried. This produced the β-alanine conjugated small molecule 11 (70 mg, 63% of yield) as a dark brown colored solid and used for the further reaction without purification.
Preparation of compound 12
To an ice-cold stirred solution of the β-alanine conjugated small molecule 11 (70 mg, 0.12 mmol, 1eq) in dry dichloromethane (3 mL) was added 3 mL of trifluoroacetic acid. The resulting mixture was stirred for 2 h, and the solvent was stripped of under reduced pressure. The resultant residue was washed with diethyl ether a couple of times and dried. This produced the compound 12 (70 mg, 100% quantitative yield) as a brown colored solid and used for the further reaction without purification.
Preparation of compound 14
To an ice-cold stirred solution of the Boc-protected enkephalin peptide 4 (Boc-Tyr-DAla-Gly-Phe-OH) (86 mg, 0.13 mmol, 1eq) in dry acetonitrile (5 mL) was added DIPEA (0.1 mL, 0.55 mmol, 4 eq) and HATU (84 mg, 0.22 mmol, 1.6eq) followed by β-alanine conjugated small molecule 12 (80 mg, 0.13 mmol, 1eq). The resulting reaction mixture was stirred for 6 h at room temperature. The solvent was stripped of under reduced pressure, and the resultant residue was diluted with dichloromethane (50 mL) and washed with 5% potassium hydrogensulfate solution twice and followed by diluted sodium bicarbonate solution for two times. The organic layer was washed with water followed by brine and dried over sodium sulfate. The organic phase was evaporated to dryness in vacuo. The resultant residue was washed with diethyl ether a couple of times and dried. This produced the enkephalin conjugated small molecule 14 (90 mg, 61% of yield) as a light brown colored solid and used for the further reaction without purification.
Preparation of compound 15
Prepared as described for compound 14 from β-alanine conjugated small molecule 12 (0.1 g, 0.226 mmol, 1eq) and enkephalin peptide 13 (141 mg, 0.226 mmol, 1 eq) (Boc-Dmt-DAla-Gly-Phe-OH) afforded the compound 15 (0.14 g, 60% of yield) as a brown coloured solid and used for the further reaction without purification.
Preparation of compound 16
To an ice-cold stirred solution of the enkephalin conjugated small molecule 14 (90 mg 0.084 mmoles, 1eq) in dry dichloromethane (3 mL) was added 3 mL of trifluoroacetic acid. The resulting mixture was stirred for 2 h, and the solvent was stripped of under reduced pressure. The resultant residue was washed with diethyl ether a couple of times and dried. This produced the crude final ligand 16 as a brown colored solid. The crude final ligand 16 was isolated by preparative RP-HPLC (10–90% of acetonitrile containing 0.1% TFA in water within 40 min) to give pure ligand 16 (35 mg, 41% of yield) as a white powder. ESI MS m/z 901 (MH)+. HRMS [M + H]+ 901.49597 (theoretical 901.49707); 1H NMR (499 MHz, DMSO-d6) δ 9.36 (bs, 1H) 8.56 (d, J = 7.4 Hz, 1H), 8.22 (m, 3H), 8.18 – 8.06 (m, 3H), 8.02 (d, J = 8.3 Hz, 1H), 7.46 (m, 3H), 7.28 – 7.07 (m, 10H), 7.07 – 6.98 (m, 2H), 6.74 – 6.65 (m, 2H), 4.92 (m, 1H), 4.70 (m, 1H), 4.57 (m, 1H), 4.39 – 4.23 (m, 2H), 4.13 (d, J = 4.4 Hz, 2H), 3.98 (m, 1H), 3.71 – 3.55 (m, 2H), 3.35 – 3.25 (m, 2H), 3.10 (q, J = 12.3 Hz, 2H), 3.04 – 2.83 (m, 3H), 2.83 – 2.61 (m, 3H), 1.95 – 1.78 (m, 6H), 1.77 – 1.61 (m, 2H), 1.53-1.42 (m, 2H), 1.18 – 1.14 (m, 3H), 1.06 (d, J = 7.0 Hz, 3H). 0.87 (t, J = 7.4 Hz, 3H).
Preparation of compound 17
Prepared as described for compound 16 from 15 (80 mg 0.077 mmol, 1eq) afforded the crude compound 17. The crude final ligand 17 was isolated by preparative RP-HPLC (10–90% of acetonitrile containing 0.1% TFA in water within 40 min) to give pure ligand 17 (32 mg, 40% of yield) as a white powder. ESI MS m/z 929 (MH)+. HRMS [M + H]+ 929.52844 (theoretical 929.52837); 1H NMR (499 MHz, DMSO-d6) δ 8.40 – 8.27 (m, 2H), 8.24 (d, J = 8.6 Hz, 1H), 8.17 (t, J = 5.7 Hz, 1H), 8.11 – 8.05 (m, 2H), 7.99 (d, J = 8.4 Hz, 1H), 7.52 – 7.40 (m, 4H), 7.26 – 7.15 (m, 9H), 7.14 – 7.10 (m, 2H), 6.41 (s, 2H), 5.00 – 4.92 (m, 1H), 4.75–4.65 (m, 1H), 4.48 – 4.40 (m, 1H), 4.30 – 4.21 (m, 1H), 4.17 – 4.08 (m, 2H), 3.73 – 3.64 (m, 1H), 3.63 – 3.55 (m, 1H), 3.37 – 3.20 (m, 4H), 3.14–3.04 (m, 2H), 3.03 – 2.90 (m, 2H), 2.85 (dd, J = 13.8, 4.6 Hz, 1H), 2.76 – 2.65 (m, 2H), 2.27 (t, J = 7.2 Hz, 2H), 2.17 (s, 6H), 1.96 – 1.78 (m, 6H), 1.75 – 1.60 (m, 2H), 1.52 – 1.40 (m, 2H), 0.91 – 0.83 (m, 7H).
Supplementary Material
Figure 1.

Design principle of bivalent ligands.
Scheme 1.

Preparation of enkephalin and 4-anilidopiperidine derivative bivalent ligands
Acknowledgments
The work was supported by grants from the U.S. Public Health Service NIDA (Grants 314450 NIDA 2P01 DA006284). We thank Christine Kasten for assistance with the manuscript.
Abbreviations
- ACN
Acetonitrile
- Boc
tert-butyloxycarbonyl
- CHO
Chinese hamster ovary
- DCM
Dichloromethane
- DIPEA
N,N-Diisopropylethylamine
- DALEA
[D-Ala2, Leu5] enkephalin amide
- DAMGO
[D-Ala2,NMePhe4,Gly5-ol]enkephalin
- Dmt
2,6-dimethyltyrosine
- DPDPE
c[D-Pen2,DPen5]enkephalin
- GPI
guinea pig isolated ileum
- HBTU
N,N,N′,N′-Tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium3-oxid hexafluorophosphate
- HRMS
High resolution mass spectrometry
- hDOR
human δ opioid receptor
- MVD
mouse vas deferens
- rMOR
rat μ opioid receptor
- RT
Room temperature
- RP-HPLC
reversed-phase high-performance liquid chromatography
- SAR
structure-activity relationship
- TFA
trifluoroacetic acid
- TLC
thin layer chromatography
- Tyr
Tyrosine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Messer WS., Jr Curr Pharm Design. 2004;10:2015. doi: 10.2174/1381612043384213. [DOI] [PubMed] [Google Scholar]
- 2.Porreca F, Takemori AE, Sultana M, Portoghese PS, Bowen WD, Mosberg HI. J Pharmacol Exp Ther. 1992;263:147. [PubMed] [Google Scholar]
- 3.Mosberg HI, Yeomans L, Harland AA, Bender AM, Sobczyk-Kojiro K, Anand JP, Clark JM, Jutkiewicz EM, Traynor JR. J Med Chem. 2013;56:2139. doi: 10.1021/jm400050y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mosberg HI, Yeomans L, Anand JP, Porter V, Sobczyk-Kojiro K, Traynor JR, Jutkiewicz EM. J Med Chem. 2014:3148. doi: 10.1021/jm5002088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ballet S, Betti C, Novoa A, Tömböly C, Nielsen CU, Helms HC, Lesniak A, Kleczkowska P, Chung NN, Lipkowski AW, Brodin B, Tourwé D, Schiller PW. ACS Med Chem Lett. 2014;5:352. doi: 10.1021/ml4004765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaught JL, Takemori AE. J Pharmacol Exp Ther. 1979;208:86. [PubMed] [Google Scholar]
- 7.Horan P, Tallarida RJ, Haaseth RC, Matsunaga TO, Hruby VJ, Porreca F. Life Sci. 1992;50:1535. doi: 10.1016/0024-3205(92)90144-e. [DOI] [PubMed] [Google Scholar]
- 8.He L, Lee NM. J Pharmacol Exp Ther. 1998;285:1181. [PubMed] [Google Scholar]
- 9.Horan PJ, Mattia A, Bilsky EJ, Weber S, Davis TP, Yamamura HI, Malatynska E, Appleyard SM, Slaninova J, Misicka A, Lipkowski AW, Hruby VJ, Porreca F. J Pharmacol Exp Ther. 1993;265:1446. [PubMed] [Google Scholar]
- 10.Ananthan S. AAPS J [electronic resource] 2006;8:E118. [Google Scholar]
- 11.Davis MP. Expert Opin Drug Discov. 2010;5:1007. doi: 10.1517/17460441.2010.511473. [DOI] [PubMed] [Google Scholar]
- 12.Essawi MYH, Portoghese PS. J Med Chem. 1983;26:348. doi: 10.1021/jm00357a007. [DOI] [PubMed] [Google Scholar]
- 13.Lee YS, Kulkarni V, Cowell SM, Ma SW, Davis P, Lai J, Porreca F, Vardanyan R, Hruby VJ. J Med Chem. 2011;54:382. doi: 10.1021/jm100982d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrov RR, Vardanyan R, Lee YS, Ma SW, Davis P, Begay LJ, Lai J, Porreca F, Hruby VJ. Bioorg Med Chem Lett. 2006;16:4946. doi: 10.1016/j.bmcl.2006.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee YS, Petrov RR, Park C, Ma SW, Davis P, Lai J, Porreca F, Hruby VJ. J Med Chem. 2007;50:5528. doi: 10.1021/jm061465o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dan P, Birgitte EL, Gordon M, Ostergaard NE, Paul RJ. WO 2007093603. 2007
- 17.Deekonda S, Wugalter L, Kulkarni V, Rankin D, Largent-Milnes TM, Davis P, Neemah MB, Lai J, Vanderah TW, Porreca F, Hurby VJ. Bioorg Med Chem. doi: 10.1016/j.bmc.2015.07.071. (Manuscript under revision) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Misicka A, Lipkowski AW, Horvath R, Davis P, Kramer TH, Yamamura HI, Hruby VJ. Life Sci. 1992;51:1025. doi: 10.1016/0024-3205(92)90501-f. [DOI] [PubMed] [Google Scholar]
- 19.Kramer TH, Davis P, Hruby VJ, Burks TF, Porreca F. J Pharmacol Exp Ther. 1993;266:577. [PubMed] [Google Scholar]
- 20.Lee K, Jung WH, Park CW, Hong CY, Kim IC, Kim S, Oh YS, Kwon OH, Lee SH, Park HD, Kim SW, Lee YH, Yoo YJ. Bioorg Med Chem Lett. 1998;18:2563. doi: 10.1016/s0960-894x(98)00456-9. [DOI] [PubMed] [Google Scholar]
- 21.Yaksh TL, Noueihed RY, Durant PA. Anesthesiology. 1986;64:54. doi: 10.1097/00000542-198601000-00009. [DOI] [PubMed] [Google Scholar]
- 22.Yaksh TL, Rudy TA. Physiol Behav. 1976;17:1031. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- 23.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. Pain. 1988:3277. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.