

Abstract
Metabolic disease is accompanied by a range of cellular defects (“comorbidities”) whose origin is uncertain. To investigate this pathophysiological phenomenon we used the Spontaneously Hypertensive Rat (SHR), which besides an elevated arterial blood pressure also has many other comorbidities, including a defective glucose and lipid metabolism. We have shown that this model of metabolic disease has elevated plasma matrix metalloproteinase (MMP) activity, which cleaves the extracellular domain of membrane receptors. We hypothesize here that the increased MMP activity also leads to abnormal cleavage of the scavenger receptor and fatty acid transporter CD36. To test this idea, chronic pharmaceutical MMP inhibition (CGS27023A) of the SHR and its normotensive control, the Wistar Kyoto Rat (WKY), was used to determine if inhibition of MMP activity serves to maintain CD36 receptor density and function. Surface density of CD36 on macrophages from the heart, spleen, and liver was determined in WKY, SHR, CGS-treated WKY (CGS WKY), and CGS-treated SHR (CGS SHR) by immunohistochemistry with an antibody against the CD36 ectodomain. The extracellular CD36 density was lower in SHR heart and spleen macrophages compared to that in the WKY. MMP inhibition by CGS served to restore the reduced CD36 density on SHR cardiac and splanchnic macrophages to levels of the WKY. To examine CD36 function, culture assays with murine macrophages (RAW 264.7) after incubation in fresh WKY or SHR plasma were used to test for adhesion of light-weight donor red blood cell (RBC) by CD36. This form of RBC adhesion to macrophages was reduced after incubation in SHR compared WKY plasma. Analysis of the supernatant macrophage media by Western blot shows a higher level of CD36 extracellular protein fragments following exposure to SHR plasma compared to WKY. MMP inhibition in the SHR plasma compared to untreated plasma, served to increase the RBC adhesion to macrophages and decrease the number of receptor fragments in the macrophage media. In conclusion, these studies bring to light that plasma in the SHR model of metabolic disease has an unchecked MMP degrading activity which causes cleavage of a variety of membrane receptors, including CD36, which attenuates several cellular functions typical for the metabolic disease, including RBC adhesion to the scavenger receptor CD36. In addition to other cell dysfunctions chronic MMP inhibition restores CD36 in the SHR.
Keywords: CD36, MMP, TIMP, Spontaneously Hypertensive Rat (SHR), MMP inhibitor CGS27023A
Introduction
CD36 is an 88 kDa Class B scavenger receptor expressed on macrophages and other types of cells, such as platelets, endothelial cells, and adipocytes (Febbraio et al., 2001; Brookes & Cooper, 2007). CD36 functions as a scavenger receptor involved in phagocytosis of apoptotic cells and recognition of diacyl lipids of bacteria during infection. Among its functions, CD36 is a receptor for the glycoprotein thrombospondin-1 on microvascular endothelial cells contributing to anti-angiogenic activity important in cancer therapy (Febbraio et al., 2001; Simantov & Silverstein, 2003). As a fatty acid transporter, CD36 can recognize and internalize oxidized-low density lipoprotein (ox-LDL) (Febbraio et al., 2001; Taylor et al., 2005) important in lipid metabolism and accumulation of cholesterol in foam cells of atherosclerotic plaques (Itabe et al., 2011).
The SHR has mutations in the CD36 gene and defective adipocyte transport of long chain fatty acids (LCFAs) associated with type 2 diabetes (Febbraio et al., 2001; Lauzier et al., 2011). The North American strain of SHR has a reduced expression of CD36 and is not a natural CD36 null model (Bonen et al., 2009). Specifically, the main 2.9 kb mRNA transcript of CD36 is not present in the North American strain of SHR while the 3.8 kb and 5.4 kb transcripts are present (Bonen et al., 2009). Given the presence of these transcripts, there may be other mechanisms responsible for the defective adipose transport in the SHR in addition to genetic mutation. Functionally, SHR hearts have a reduced capacity to use exogenous LCFAs for β-oxidation and triglyceride formation prior to developing left ventricular hypertrophy (Lauzier et al., 2011). This is possibly due to alterations in CD36 protein by post-translational modifications (Lauzier et al., 2011). In addition, a decrease in fatty acid and glucose metabolism was observed in stroke-prone SHR (SHRSP), and yet the quantitative trait loci on rat chromosome 4 with CD36 defects only accounted for 40% of insulin dysfunction (Collison et al., 2000).
Defective fatty acid metabolism in the SHR may be in part due to proteolytic cleavage of the CD36 scavenger receptor or fatty acid transporter (FAT) as a result of elevated matrix metalloproteinase (MMP) activity in SHR plasma and its microvascular endothelium. Compared to its normotensive control, the Wistar Kyoto Rat (WKY), MMP levels, specifically MMP-2 and MMP-9, in the SHR are elevated (Chen et al., 2012; DeLano & Schmid-Schönbein, 2008; Tran et al., 2010), leading to ectodomain receptor cleavage and cellular dysfunctions (Chen et al., 2010; Delano et al., 2011; DeLano & Schmid-Schönbein, 2008; Rodrigues et al., 2010; Tran et al., 2010) generating its co-morbidities, such as insulin resistance. Levels of the endogenous tissue inhibitors of metalloproteinases (TIMPs), such as TIMP-1 and TIMP-2 with the ability to inhibit MMP-9 and MMP-2, respectively (Bernardo & Fridman, 2003; Brumann et al., 2012) are undetermined and will be presented in this report.
MMP activities can be chronically attenuated in the SHR with a broad-spectrum MMP inhibitor (CGS27023A, CGS; Novartis, Basel, Switzerland) in the drinking water, an intervention that normalizes its blood pressure (Chen et al., 2012). The process of red blood cell removal via adhesion to scavenger receptors on macrophages in the SHR is impaired by MMP cleavage of the red blood cell glycocalyx (surface proteoglycans) (Pot et al., 2011). We will examine here whether CD36, a polyinosinic acid sensitive scavenger receptor on Kupffer cells (liver macrophages), important for clearance of oxidatively damaged red blood cells, is proteolytically cleaved by elevated MMPs in the SHR (Terpstra & van Berkel, 2000; Yesner et al., 1996). By immunolabeling of the CD36 extracellular domain, we quantify the extracellular CD36 density on the macrophage cell membranes of frozen heart, spleen, and liver tissue sections in WKY, SHR, CGS WKY, and CGS SHR. To investigate the effect of red blood cell (RBC) removal by spleen cell attachment we determine in-vitro red blood cell adhesion to cultured murine macrophages in the presence of WKY, SHR and CGS-inhibited SHR plasma. Cleavage of the CD36 receptor by MMP activity is determined by detection of CD36 ectodomain fragments in supernatants from murine macrophage cultures.
Material and methods
Animals
All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California, San Diego. Male Wistar Kyoto Rats (WKY) and Spontaneously Hypertensive Rats (SHR) (Harlan Laboratories, Inc., Indianapolis, IN) at 8 weeks of age were given standard diet ad libitum (n = 3 per group). A separate group of age-matched WKY (n = 3) and SHR (n = 3) was given 3 mg/kg/day of the broad-spectrum matrix metalloproteinase inhibitor CGS27023A (Novartis Pharmaceuticals, Inc., Summit, NJ) in their drinking water for a period of 24 weeks. These animals are denoted as CGS-treated WKY (CGS WKY) and CGS-treated SHR (CGS SHR). The dosage of the MMP inhibitor CGS27023A was based on the effective anti-metastatic dose (5–25 mg/kg) in a mouse melanoma model administered through an osmotic pump (Kasaoka et al., 2008) but was slightly decreased to adjust for an enhanced drinking volume of the water with the inhibitor. The treatment had no detectable side effects on the SHR or the WKY rats and significantly decreased the SHR’s plasma MMP activity and systolic blood pressure by 24 weeks (Chen et al., 2012).
Plasma collection and tissue harvest
Animals (WKY, SHR, CGS WKY, and CGS SHR) were anesthetized with 50 mg/kg of sodium pentobarbital (Abbott Laboratories, North Chicago, Illinois) through a catheter in the femoral artery. Whole blood was drawn from the arterial catheter and centrifuged at 1000 × g for 15 min. Plasma in the supernatant was collected and stored at −80 °C (later used for Western blot analysis).
After euthanasia (120 mg/kg, Beuthanasia, Intervet, Inc., Summit, NJ), a midline incision was made and the abdominal and chest cavity were exposed. The frontal half of the heart, distal edge of the right liver lobe, and distal edge of the spleen were dissected, further cut into a tissue of size within 1.5 cm3, placed in a mold filled with O.C.T. embedding medium (Sakura Tissue-Tek Optimal Cutting Temperature Compound), and flash-frozen with liquid nitrogen chilled 2-methylbutane (Fisher Scientific, Waltham, MA).
The heart, liver, and spleen were stored at −80 °C, and 5 μm-thick sections of each organ were cut with a cryostat (Bright Instrument Co. Ltd., Huntingdon, Cambridgeshire, England; OTF Microtome Cryostat 5030 Series) at chamber temperatures of −19 °C, −12 °C, and −16 °C, respectively. Each section was adhered to a single microscope slide. The heart and spleen were removed for analysis given the SHR’s defective cardiac fatty acid transport and red cell removal mechanism, respectively (Lauzier et al., 2011; Pot et al., 2011; Terpstra & van Berkel, 2000). In addition, the liver was investigated due to increased levels of plasma soluble CD36 in patients prone to develop fatty liver and increased fatty acid transport in the liver of the North American strain of SHR (Bonen et al., 2009; Garciarena et al., 2009; Lauzier et al., 2011; Pot et al., 2011; Terpstra & van Berkel, 2000).
Immunohistochemistry of frozen tissue sections
Two 5 μm-thick sections of each organ (heart, spleen, and liver) were used per animal (WKY, SHR, CGS WKY, and CGS SHR; n = 3) for immunolabeling of the extracellular domain of the CD36 scavenger receptor on macrophages. Individual tissue sections (frontal cross section of the heart, spleen, and liver) were placed on a microscope slide within a ~1.5 cm square, which was marked with a hydrophobic barrier (ImmEdge pen, #H-4000; Vector Laboratories, Burlingame, CA) around the tissue sections to prevent leakage of staining reagents. Tissue sections were fixed in acetone at −20 °C for 5 min. Endogenous peroxidase from the tissue was blocked with Peroxo-Block (#00-2015; Life Technologies, Grand Island, NY) for 45 s at room temperature, and non-specific binding was prevented by blocking with 2.5% normal horse serum for 1 h at room temperature (#MP-7405: ImmPRESS anti-goat Ig peroxidase kit; Vector Laboratories Inc., Burlingame, CA).
After blocking for peroxidase and non-specific binding, we washed with 1× PBS and apply primary antibody (#sc-5522; polyclonal goat IgG; final concentration 5.5 μg/ml; 3:100 dilution in 1× PBS from 200 μg/ml stock CD36 antibody solution; Santa Cruz Biotechnology Inc., Santa Cruz, CA) against the extracellular domain of CD36 on macrophage membranes of tissue sections for 1.5 h at room temperature in a closed and moisturized box lined with wet towels. As a negative control, the primary antibody was replaced with normal goat IgG (#sc-2028; final concentration 5.5 μg/ml; 1.5:100 dilution from 400 μg/ml stock solution; Santa Cruz Biotechnology Inc., Santa Cruz, CA) on a separate tissue section. 1× PBS was used as a rinse buffer to remove non-bound antibody. Subsequently, secondary antibody (#MP-7405; ImmPRESS anti-goat Ig peroxidase kit; Vector Laboratories, Inc., Burlingame, CA) was applied to tissue sections for 30 min at room temperature. After washes, ImmPACT DAB (#SK-4105; Vector Laboratories, Inc., Burlingame, CA) was applied and slides were developed for 15 s, 30 s, and 25 s for the heart, spleen, and liver sections, respectively. Finally, slides were placed through an ethanol gradient (50% ethanol for 1 min, 70% ethanol for 1 min, and twice in 100% ethanol for 1 min) as well as 30 s in xylene for dehydration of slides. Slides were dried in the hood for 3 h and mounted with coverslips.
A separate primary antibody against the extracellular domain (between amino acid #25–75; Protein Accession #P31996; UniProt Consortium) of CD68 (#sc-5474; polyclonal goat IgG; 3:100 dilution from stock 200 μg/ml solution; final concentration 5.5 μg/ml; Santa Cruz Biotechnology, Inc., Burlingame, CA) was used as a macrophage marker on separate tissue sections.
Digital image analysis of tissue sections
Brightfield micrographs of macrophages (with CD36 labeled by immunohistochemistry) in the heart, liver, and spleen frozen sections were captured (SM-LUX microscope, Leitz Wetzlar, Germany) using a 100× oil-immersion objective (numerical aperture 1.25) and digitized (VC500 One-Touch Video Capture USB 2.0, Diamond Multimedia, Chatsworth, CA). The morphology of macrophages in the heart, liver, and spleen was determined by comparisons with sections labeled with the CD68 macrophage marker.
Macrophage count
The number of macrophages in WKY (n = 3) and SHR (n = 3) per image (area = 0.0016 cm2) of heart, spleen, and liver sections under the 10× objective captured in a light microscope (SM-LUX microscope, Leitz Wetzlar, Germany) was counted and averaged over 5 images per tissue section per organ.
Measurements of CD36 density on macrophages in the heart, spleen, and liver
CD36 densities on macrophages in WKY, SHR, CGS WKY, and CGS SHR were determined by two digital methods (ImageJ, version 1.42q; 8-bit). First, a cellular contour for each macrophage was drawn with a freehand selection tool (ImageJ). The average pixel gray value was calculated within the cell area as a qualitative measurement. In order to confirm these measurements as a quantitative index for the CD36 density, a second method was used. To detect peak label intensities on the membrane, a straight-line segment across the macrophage cell membrane (from the cytoplasmic to the extracellular side of a cell) was selected at 5 different and random membrane locations for each cell. The peak intensity for each line was recorded and the 5 peak values (in each line) averaged for each cell. 30 macrophages per animal per group (WKY, SHR, CGS WKY, and CGS SHR) per organ (heart, liver, and spleen) were sampled and averaged.
Western blot detection of plasma TIMP-1 and TIMP-2 levels
Precast gels (Bio-Rad 4–20% Mini-PROTEAN TGX; Bio-Rad Laboratories, Inc., Hercules, CA; #456-1099) were used to resolve proteins in plasma samples of WKY, SHR, CGS WKY, and CGS SHR (n = 3 rats per group) via electrophoresis. Equal volumes of plasma (2.5 μl) were loaded into the gel. Gel electrophoresis was run at a constant voltage (160 V) and transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Inc., Hercules, CA; #162-0097) at constant current (300 mA). Membranes were then blocked with 5% milk in Tris-Buffered Saline and Tween 20 (TBST) followed by an overnight primary antibody incubation (TIMP-1 antibody #sc-6834 or TIMP-2 antibody #sc-9905; 1:500 dilution; final concentration 0.4 μg/ml; Santa Cruz Biotechnology Inc., Santa Cruz, CA) and a secondary anti-goat IgG polyclonal antibody (Sigma-Aldrich Co., LLC; #A5420; 1:5000 dilution). Antibody dilutions were prepared in 5% milk in Tris-Buffered Saline and Tween 20 (TBST). Supersignal West Pico Chemiluminescent substrate (Thermo Fisher Scientific, Waltham, MA; #34096) was used for imaging with digital analysis (ImageJ). The band density was normalized by a reference sample (the initial WKY plasma sample) that was added on each gel in a separate lane parallel to the experimental samples (WKY, SHR, CGS WKY, and CGS SHR).
Quantification of red blood cell attachment after plasma incubation
Plasma was obtained from WKY (n = 3) and SHR (n = 3) rats by centrifugation at 600 ×g for 10 min. One WKY rat was selected as the source of low-weight density red blood cells (LRBCs). These were isolated from WKY whole blood by centrifugation at 600 ×g for 10 min and collection of the top fraction of erythrocytes. SHR plasma (n = 3) was incubated for 20 min at room temperature with CGS at a final concentration of 7.6 μM. Plasma samples were diluted in a 1:10 ratio. Murine macrophages (Raw 264.7) were cultured in Dulbecco’s modified eagle medium (DMEM; ATCC, Manassas, Virginia) and 10% fetal bovine serum (FBS; ATCC, Manassas, Virginia) at 37 °C in an atmosphere of 5% CO2 and allowed to grow to about 100% confluency before being exposed to either DMEM, WKY, SHR, or CGS-SHR plasma. Immediately following the administration of plasma, 7 μl of WKY LRBCs was added to each well. Macrophages were incubated in the presence of a given plasma variant or DMEM and WKY LRBCs for 30 min at 37 °C. Cells were starved in serum free media for 12 h prior to experimentation. Cultures were then removed from the incubator and given 2× washes of PBS. After each wash cells were manually shaken for 10 s to remove unattached LRBCs. The shear stress applied was on the order of 10 dyn/cm2. Images were recorded at 40× and in each field of view the ratio of attached LRBCs to macrophages and number of macrophages was calculated to yield an average number of attached LRBCs per macrophage.
Soluble CD36 fragments in supernatant fluid after incubation with WKY and SHR plasma
Supernatant media fluid was removed from macrophages cultured in 24 well plates following 1 h of incubation in WKY (n = 3), SHR (n = 3), or CGS SHR (n = 3) plasma. CGS SHR plasma was prepared by adding CGS inhibitor to SHR plasma aliquots at 7.6 μM concentrations. WKY, SHR, and CGS SHR plasma samples were then diluted 1:5 in DMEM. After incubation, aliquots of 150 μl were centrifuged (Savant DNA 110 SpeedVac Concentrator, Thermo Fisher Scientific, Waltham, MA; # DNA110-220) for 65 min to concentrate samples. An equal volume of sample buffer (Bio-Rad Laboratories, Inc., Hercules, CA; # 161-0737) with beta-mercaptoethanol (Sigma-Aldrich Co., LLC, St. Louis, MO; # M6250) was added to samples. Equal volumes of samples were added to lanes of a 15% polyacrylamide gel.
For Western blotting, proteins were transferred to a 0.45 μm pore size nitrocellulose membrane (Bio-Rad Laboratories, Inc., Hercules, CA; #162-0097). Following blockade with 5% nonfat dry milk in TBST, primary antibodies against the extracellular domain of CD36 (Abcam, Cambridge, England; ab80978) were applied at a 1:500 dilution. Secondary antibodies (anti-rabbit IgG polyclonal antibody; Sigma-Aldrich Co., LLC; #A5420) were applied at a 1:5000 dilution. All antibody dilutions were prepared in 5% milk in Tris-Buffered Saline and Tween 20 (TBST). Supersignal West Pico Chemiluminescent substrate (Thermo Fisher Scientific, Waltham, MA; #34096) was used for imaging. DMEM was used as a negative control.
Statistical analysis
Results are presented as mean ± standard deviation. Single-factor analysis of variance (ANOVA) was used to evaluate statistical significance for groups greater than 2, and Student’s t-test was used to compare between two groups. For culture experiments, multiple comparison t-test with a post hoc Bonferroni correction was used. A value of p < 0.05 was considered statistically significant.
Results
Our previous measurements indicate that the gelatinase levels (MMP-2 and MMP-9) in SHR plasma are significantly higher than those in the normotensive WKY control (Chen et al., 2012; DeLano & Schmid-Schönbein, 2008; Timlin et al., 2005). Chronic MMP inhibition for 24 weeks by a broad-spectrum MMP inhibitor CGS restores SHR gelatinase levels to significantly lower levels found in the WKY (Chen et al., 2012). Furthermore the TIMP-1 levels (with molecular weight of 22 kD) in SHR plasma were on average significantly lower than those in the WKY (Fig. 1). TIMP-2 levels (molecular weight ~ 21 kD) in SHR plasma were significantly decreased compared to that in the WKY rats (Fig. 2). Chronic MMP inhibition by CGS reduced TIMP-1 levels further and reached a value that was not different compared to control groups (CGS WKY vs WKY; CGS SHR vs. SHR; Fig. 1). TIMP-2 levels in WKY plasma were not significantly increased by CGS treatment compared to that in the WKY without treatment, and neither was TIMP-2 levels in CGS SHR plasma significantly decreased compared to that in the SHR (Fig. 2).
Fig. 1.
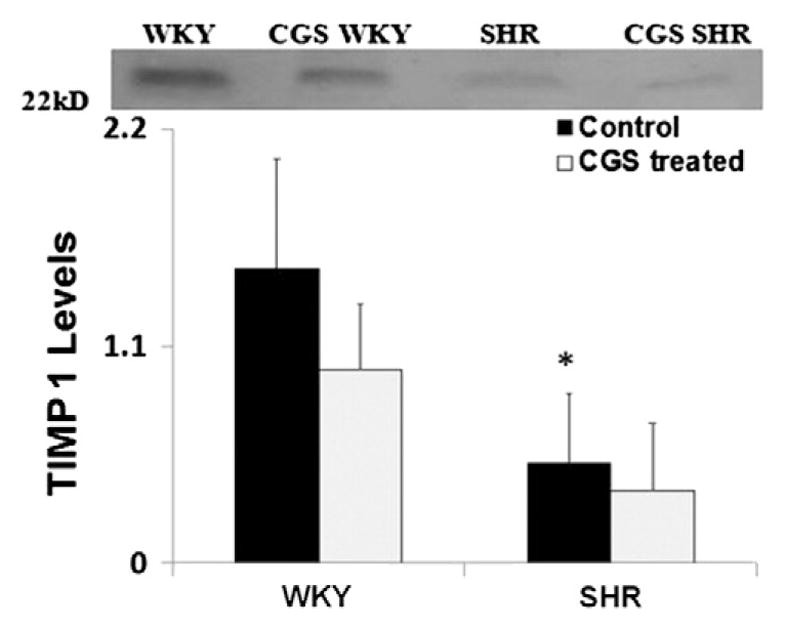
TIMP-1 levels in WKY, SHR, CGS WKY, and CGS SHR plasma. Representative TIMP-1 band at 22 kDa (the molecular weight of TIMP-1) for Western blots performed. Plasma from WKY, SHR, CGS WKY, and CGS SHR (n = 4 rats per group) were tested. One WKY plasma sample was used as normal reference between blots and included in every Western blot. Histogram shows average relative band density for TIMP-1 levels in plasma of WKY, SHR, CGS WKY, and CGS SHR. By Student’s t-test, *p < 0.05 WKY vs. SHR. By single-factor ANOVA, p < 0.01 among WKY, SHR, CGS WKY, and CGS SHR.
Fig. 2.
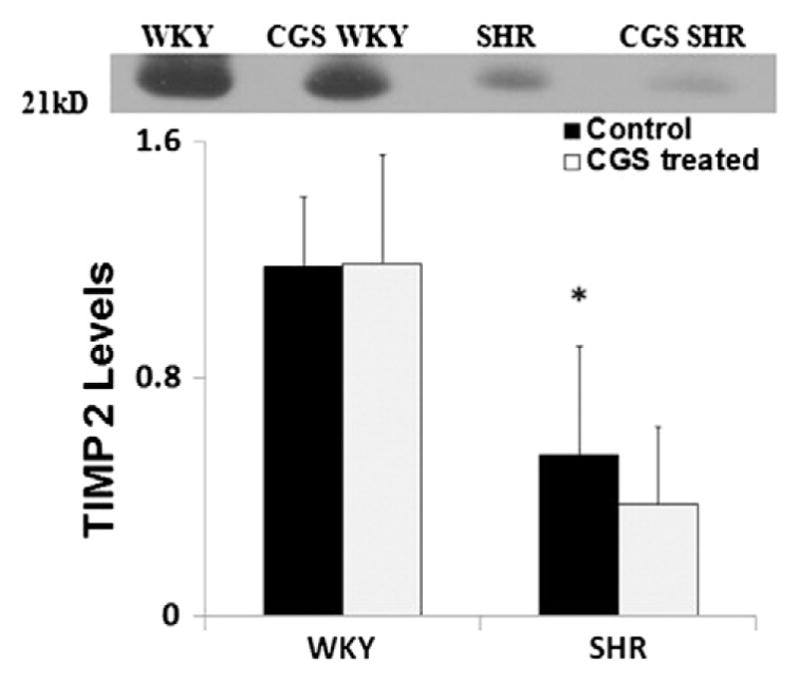
TIMP-2 levels in WKY, SHR, CGS WKY, and CGS SHR plasma by Western blot. Representative TIMP-2 band at 21 kDa (the molecular weight of TIMP-2) for Western blots performed. Plasma from WKY, SHR, CGS WKY, and CGS SHR (n = 4 rats per group) were tested. One WKY plasma sample was used as a normalizing reference between blots and included in every Western blot. The histogram shows the average relative band density for TIMP-2 levels in plasma of WKY, SHR, CGS WKY, and CGS SHR. By Student’s t-test, *p < 0.05 WKY vs. SHR. By single-factor ANOVA, p < 0.01 among WKY, SHR, CGS WKY, and CGS SHR.
Identification of CD68-positive macrophages in the heart, spleen, and liver of WKY and SHR
CD68-positive macrophages as determined by antibody labeling of its extracellular domain were found on both WKY and SHR heart, spleen, and liver (Fig. 3A). SHR macrophages in the heart and the spleen on average assume a rounder shape than the WKY (Fig. 3A). In contrast, the SHR macrophages in the liver tend to have a more extended shape (Fig. 3A). The average CD68 label density on WKY macrophage membrane was higher compared to those of the SHR (Fig. 3A; blue arrows (WKY) vs. black arrows (SHR)). The SHR macrophage density (cell count) in the heart and spleen was significantly higher than that in the WKY (Fig. 3B).
Fig. 3.

WKY and SHR macrophages labeled with CD68 macrophage marker by immunohistochemistry in frozen tissue sections. A. Representative micrographs of heart, spleen, and liver macrophages labeled with CD68 antibody are shown along with respective negative controls in which the primary CD68 antibody was replaced by an irrelevant goat IgG. Scale bar = 20 μm. Single cell macrophage boundaries are outlined by dashed lines. Blue arrows in micrographs of WKY macrophages point towards areas on the cell membrane with higher CD68 label density compared to those of the SHR. Black arrows in micrographs of SHR macrophages indicate areas of reduced CD68 label. B. Average macrophage count per 0.0016 cm2 area of heart, spleen, and liver sections (n = 3; average of 5 image frames per animal per tissue section) in WKY and SHR. *p < 0.05 WKY vs. SHR macrophage count in the heart. **p < 0.05 WKY vs. SHR macrophage count in the spleen.
Extracellular domain density of CD36 on macrophages in the SHR heart
Macrophages were found near blood vessels and between myocardial cells in the atria and ventricles of the heart. In heart sections, the SHR macrophages possess a rounder morphology than that of the WKY (Fig. 4A). The average extracellular CD36 density in SHR heart macrophages as measured by both light absorption (Fig. 4B) and peak intensity in the cell membrane was significantly lower than that in the WKY (Fig. 4A; black arrows (SHR) vs. blue arrows (WKY)). After chronic CGS treatment, the CD36 label density for CGS SHR was significantly higher than that of the SHR and reached similar levels as in the WKY (Figs. 4B and C). Although the CD36 label density in the CGS WKY group was on average higher than that of the WKY, it was not significantly different (Figs. 4B and C).
Fig. 4.

Immunolabeling of extracellular CD36 on macrophage membranes in the heart. A. Representative micrographs of WKY, SHR, CGS WKY, and CGS SHR macrophages with CD36 labeled (brown) in 5 μm frozen heart sections. Single cell macrophage boundaries are outlined by dashed lines. Negative control (normal goat IgG) has near undetectable intensity levels. Scale bar = 20 μm. Blue arrows shown on the micrograph of WKY macrophage indicate areas on the cell membrane that have higher and more evenly distributed CD36 density than that of the SHR. Black arrows point towards areas on the SHR cell membrane that have low CD36 ectodomain density. B. Histogram of average CD36 label density by light absorption where a contour of each cell was drawn. The log10 of {the mean intensity within the contour of a given cell was normalized to background intensity without cells}. The light absorption was measured in 30 cells per animal and averaged between 3 rats (n = 3) per following groups of animals: WKY, SHR, CGS WKY, and CGS SHR. By Student’s t-test, *p < 0.05 WKY vs. SHR. **p < 0.05 SHR vs. CGS SHR. By single-factor ANOVA, p < 0.01 among WKY, SHR, CGS WKY, and CGS SHR.
Extracellular CD36 label density on SHR macrophages in the spleen
Macrophages in the spleen were found in the interstitial space of the red pulp and at locations near blood vessels. The morphology of macrophages in the spleen sections varied across the SHRs and WKYs. However, the majority of SHR macrophages had rounder shapes than the WKY (Fig. 5A). We found that the average extracellular CD36 density in SHR spleen macrophages as measured by both light absorption (Fig. 5B) and peak intensity was significantly lower than that in the WKY (Fig. 5A; black arrows (SHR) vs. blue arrows (WKY)). After chronic MMP inhibition in the SHR, the CD36 label density was significantly higher than that in the untreated SHR group (Fig. 5B). Extracellular CD36 label density in the CGS SHR spleen macrophages was not significantly different from that in the WKY. There was also no significant increase in the extracellular CD36 label density on spleen macrophages of CGS WKY compared to that in the WKY (Fig. 5B).
Fig. 5.

Immunolabeling of extracellular CD36 on macrophage membranes in the spleen. A. Representative micrographs of WKY, SHR, CGS WKY, and CGS SHR macrophages with CD36 labeled (brown) in frozen spleen sections. Single cell macrophage boundaries are outlined by dashed lines. Negative control with replacement of the CD36 primary antibody with normal goat IgG is shown on the right panel. Scale bar = 20 μm. Blue arrows on the micrograph of WKY macrophage indicate areas on the WKY cell membrane with higher and more evenly distributed CD36 label density compared to that of the SHR. Black arrows point towards areas on the SHR cell membrane that have low CD36 ectodomain density. B. Histogram of average CD36 label density by light absorption where a contour of each cell was drawn. The log10 of {the mean intensity within the contour of a given cell was normalized to background intensity without cells}. The light absorption was measured in 30 cells per animal and averaged between rats (n = 3) in each of the following animal groups: WKY, SHR, CGS WKY, and CGS SHR. By Student’s t-test, *p < 0.05 WKY vs. SHR. **p < 0.05 SHR vs. CGS SHR. By single-factor ANOVA, p < 0.05 among WKY, SHR, CGS WKY, and CGS SHR.
Extracellular CD36 label density in SHR liver macrophages
Liver macrophages (Kupffer cells) were found near blood vessels in liver sections. The morphology of SHR Kupffer cells varied from cell to cell, but the majority of macrophages had an extended shape (Fig. 6A). The WKY had relatively few extended macrophages present in its liver compared to the SHR (Fig. 6A; rounder morphology of WKY macrophage). In contrast to the extracellular CD36 label density in the heart and spleen, the SHR liver macrophages (Fig. 6A; black arrows) showed an elevated average CD36 label density compared to that in the WKY (Fig. 6A; blue arrows) as measured by both light absorption (Fig. 6B) and peak intensity. In addition, the average extracellular CD36 label density in CGS SHR was not significantly different from that in the WKY (Fig. 6B).
Fig. 6.

Immunolabeling of extracellular CD36 on macrophage membranes in the liver. A. Representative micrographs of WKY, SHR, CGS WKY, and CGS SHR macrophages with CD36 labeled (brown) in frozen liver sections. In negative controls, the CD36 primary antibody was replaced with normal goat IgG. Scale bar = 20 μm. Blue arrows in the micrograph of WKY macrophage point towards areas on the cell membrane with sparse CD36 label. Black arrows in the micrograph of SHR macrophage point towards areas on the cell membrane with higher CD36 label density compared to the WKY. B. Histogram of average CD36 label density by light absorption where a contour of each cell was drawn. The log10 of {the mean intensity within the contour of a given cell was normalized to background intensity without cells}. The light absorption was measured in 30 cells per animal and averaged between 3 rats (n = 3) per following animals: WKY, SHR, CGS WKY, and CGS SHR. By Student’s t-test, *p < 0.05 WKY vs. SHR.
Attachment of LRBCs to macrophage cultures after incubation with WKY plasma as compared to SHR plasma
The level of LRBC attachment to macrophages was significantly reduced after incubation with SHR as compared to WKY plasma (Fig. 7). CGS SHR plasma incubation resulted in further reduction LRBC below the levels with WKY plasma and significantly reduced attachment compared to SHR plasma. The average number of LRBC per macrophage (attachment ratio) between CGS SHR and WKY plasma incubations was also significantly reduced.
Fig. 7.

Red blood cell attachment to macrophages after incubation in WKY, SHR, and CGS SHR plasma. A. Representative micrographs of cultured murine macrophages (black arrows) incubated in control incubation medium (DMEM) and diluted WKY, SHR and CGS-inhibited SHR plasma in the presence of light-weight red blood cells (red arrows). Panel inset shows the smaller red blood cell adhered to a larger macrophage (green arrow). Excess red blood cells were removed by mild fluid shear. Micrographs show the relative ratios of attached red blood cells to macrophages. B. Histogram of average ratio of light-weight red blood cell attachment to macrophages after DMEM, and diluted WKY, SHR and CGS-SHR plasma conditions. The ImageJ cell counter tool was used to generate the ratio of attached light-weight red blood cells over macrophages per field of view. Ratios from 10 fields of view were calculated per animal with n = 4 animals used WKY plasma group and n = 5 for the SHR and CGS SHR groups. p < 0.0001 among all groups by single-factor ANOVA ****p < 0.0001 WKY plasma vs. SHR plasma and **p < 0.01 CGS-SHR plasma vs. SHR plasma by Student’s t-test with Bonferroni correction.
Extracellular CD36 levels in supernatant from macrophages incubated in SHR and WKY plasma
Extracellular CD36 levels (molecular weight ~53kD) in supernatant incubated with SHR plasma were significantly higher than supernatant from cell cultures incubated with WKY or SHR plasma with CGS (Fig. 8).
Fig. 8.
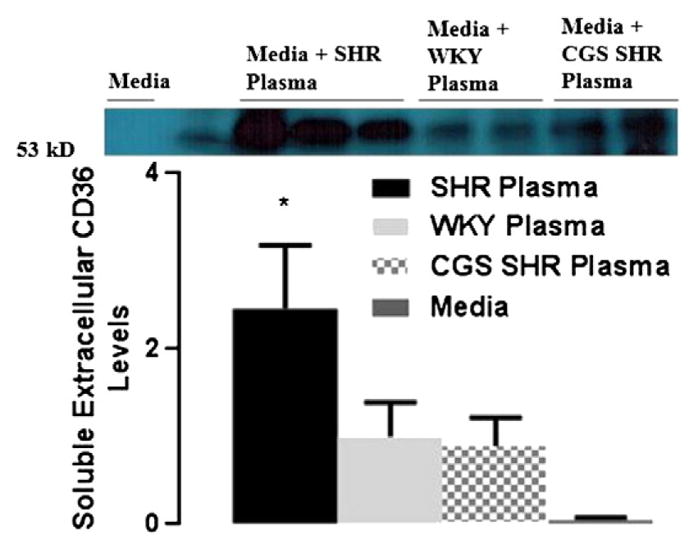
Soluble extracellular CD36 levels in macrophage culture supernatant after exposure to plasma of WKY, SHR, CGS WKY, and CGS SHR. A. Representative extracellular CD36 band at 53 kDa (the molecular weight of extracellular CD36) for Western blots performed. Culture supernatant from WKY, SHR, and CGS SHR (n = 3 rats per group) was tested. One WKY plasma sample was used as control reference for every Western blot. Histogram of normalized band density (relative to WKY plasma band density) for extracellular CD36 levels in culture supernatant of WKY, SHR, and CGS SHR. **p < 0.01 WKY and CGS SHR vs. SHR by Student’s t-test with Tukey post hoc correction. ***p < 0.001 among WKY, SHR, and CGS SHR by single-factor ANOVA.
Discussion
The current experiments provide evidence that the elevated MMP activity in the SHR is accompanied by reduced TIMP-1,2 levels. Compared to its WKY control, the enhanced proteolytic activity in the SHR is accompanied by increased cleavage of the CD36 ectodomain on macrophages in several tissues and reduced adhesion to a subpopulation of light-weight RBCs to which macrophages show a tendency to adhere. Macrophages exposed in-vitro to SHR plasma form higher concentrations of the extracellular domain fragments than the WKY and chronic treatment with a MMP inhibitor attenuates the ectodomain cleavage in the SHR.
The reduction in TIMP-1 and TIMP-2 levels (Figs. 1, 2) in the SHR is in line with the elevation in gelatinase (MMP-2 and MMP-9) activity levels (Chen et al., 2012; DeLano & Schmid-Schönbein, 2008; Tran et al., 2010)
CGS treatment did not significantly affect plasma TIMP-1 and TIMP-2 levels in control animals (WKY and SHR in Figs. 1, 2). Also, the inhibition mechanism of TIMPs is based on binding to the active sites or catalytic domains of MMPs (Nagase et al., 2006). Several genetic and post-translational modifications can lead to inactivation of TIMPs, including mutations of the amino acid threonine into glycine in TIMP-1, carbamylation of N-terminal amino groups in TIMPs, and additions of the amino acid alanine at the N-terminus in TIMPs (Nagase et al., 2006). Dissection of the regulation of TIMPs both in response to MMP levels and via other biochemical mechanisms is needed to understand the endogenous inhibition process as well as the real-time plasma TIMP levels in the SHR.
The SHR has elevated levels of tissue macrophages
In line with previously reported elevated macrophage counts in the SHR brain and kidney (Kobori et al., 2005; Liu et al., 1994), we also found higher macrophage density in the heart and spleen in SHR rats indicating higher rates of tissue infiltration. In contrast, the rounder shape of the SHR macrophages is in line with a potentially lower phagocytic activity of damaged or apoptotic cells.
The SHR heart and spleen displayed reduced CD36 density
The susceptibility of the SHR heart to apoptosis (Garciarena et al., 2009) may lead to higher number of apoptotic cells present and subject to clearance. This fact compounded with CD36 reduction may lead to reduced phagocytic clearance in this organ. Similarly, although the SHR may be in greater need for clearance of apoptotic cells in the spleen, the rounder shape of its macrophages may also be an indication of a lower level of phagocytosis in its spleen. Increased extracellular CD36 label density in the CGS SHR spleen and heart macrophages to the levels of the WKY indicates that CGS was able to restore the extracellular CD36 label density in the SHR to levels similar to that in the WKY (Figs. 5B, C).
In contrast to the spleen and heart, the liver of the SHR displayed a greater CD36 density than that of the treated and untreated WKY rats
This is in line with previous observations indicating an increase in fatty acid transport in the liver of the North American strain of SHR compared to controls (Bonen et al., 2009). It is associated with the thymus of the SHR that has enhanced atrophy at 15-weeks of age (Suzuki et al., 1999) which affects liver functions via the hypothalamus–pituitary–gonad axis (Li et al., 1996). Specifically, male thymectomized rats have significant reductions in liver microsome cytochrome P-450 and aminopyrine-N-demethylase activities (Li et al., 1996). Gonadal steroid hormones affect CD36 expression as well as blood pressure in deoxycorticosterone-salt hypertensive rats (Crofton & Share, 1997; Zeng et al., 2003). Hence the steroid hormone receptor may be part of the pathways for the increased CD36 membrane density in Kupffer cells of the SHR. The MMP blockade in the CGS SHR and CGS WKY lead to a non-significant reduction in extracellular CD36 label density compared to that in the SHR and WKY, respectively (Figs. 6B, C). The control by hormone receptor could be part of the CD36 density on Kupffer cells in the SHR and requires further investigation.
Adhesion of LRBCs to murine macrophages after incubation with SHR plasma
LRBCs constitute a population of red blood cells prone to removal from the circulation by the spleen (Pot et al., 2011). The capacity of macrophages to attach to and phagocytize LRBCs as a mechanism for removal in the spleen is reduced in the SHR due to factors extant in the plasma. Macrophage activation by plasma is needed for cell surface receptors to initiate attachment (Zhang et al., 2003), but the attachment may be reduced in SHR and partially restored during MMP inhibition by a mechanism that involves CD36 cleavage. Incubation of macrophages in SHR plasma with its enhanced MMP activity resulted in a greater amount of extracellular CD36 fragments in supernatant medium. Incubation of SHR plasma with CGS reduced the ectodomain levels of CD36 on macrophage supernatants. Thus, MMPs are a possible source of CD36 cleavage. The level of CD36 extracellular fragments in the supernatant of cultures incubated with WKY plasma may also stem from MMP activity. WKY MMP activity levels are also slightly elevated above the activity in plasma of the rat Wistar stain from which the SHR and WKY were bred (Tran et al., 2010).
Potential MMP cleavage sites on the CD36 scavenger receptor
The current evidence for enhanced CD36 receptor cleavage in the presence of unchecked MMP activity is in line with the presence of potential cleavage sites in its ectodomain. Screening the linear sequence of the CD36 receptor yields four potential cleavage sites for MMP-2 and MMP-9 at the amino acid scissile bond, serine and leucine (Tran et al., 2010). The actual cleavage site in-vivo depends on the conformation of the receptor in the physiological state as well as ligands specific for the CD36 receptor that may influence cell signaling. Proteolytic cleavage yields soluble fragments of the receptor that we can identify by ELISA. But there may also be additional cleavage sites on the ectodomain so that binding sites for antibodies may be cleaved and the receptor fragment levels determined in the current experiments may underestimate the actual degree of receptor cleavage.
Soluble CD36 levels in the plasma of patients with the metabolic syndrome
Levels of soluble CD36 in the plasma can serve as indirect evidence for proteolytic ectodomain cleavage, as we have shown previously for other receptors in the SHR, such as soluble P-selectin (Chen et al., 2012). Detected by ELISA, soluble CD36 in the plasma is higher in SHR (Fig. 8) and reduced after MMP inhibition (CGS-SHR). Soluble CD36 levels are also higher in plasma of patients with impaired glucose regulation, insulin resistance, and metabolic syndrome as well as patients who are prone to develop fatty livers (Handberg et al., 2012).
Transcriptional regulation of CD36
The expression of the CD36 scavenger receptor is controlled by transcription factors and receptor ligand levels. In normal physiological conditions, CD36 is transcriptionally regulated by nuclear receptors, including the pregnane X receptor (PXR), the peroxisome proliferator activated receptor-γ (PPAR-γ), and the liver X receptor (LXR) (He et al., 2011). LXR can bind to the liver X receptor response element (LXRE) in the CD36 gene promoter (He et al., 2011). The liver contributes to fatty acid metabolism by receiving and responding to fatty acids from diet, adipose stores, and fatty acids produced de novo (Terpstra & van Berkel, 2000). Low CD36 expression is associated with greater fatty acid flux to the liver leading to an increase in CD36 ligands such as ox-LDL with subsequent upregulation in CD36, accumulation of cholesterol in macrophages, dyslipidemia, and insulin resistance (Handberg et al., 2012). Upregulation of CD36 is due to the ability of ox-LDL to activate the transcription factor PPAR-γ(Feng et al., 2000). In monocytes, CD36 mRNA upregulation is triggered by exposure to interleukin-4 and monocyte-colony stimulating factor while lipopolysaccharide and dexamethasone exposure lead to its downregulation (Yamashita et al., 2007). Non-diabetic hypertensives have a higher incidence of fatty liver than healthy controls (Brookes & Cooper, 2007). In the case of the SHR, the PPAR-γ protein expression is significantly upregulated in the kidney, liver, heart, and brain compared to the WKY (Sun et al., 2008). Agonists and activators of PPAR-γ have also been shown to lower blood pressure for hypertensives without diabetes (Sun et al., 2008). Further investigation of the role of PPAR-γ in CD36 receptor signaling in the SHR is required.
Ligand uptake of the CD36 receptor
Ox-LDL, fibrillar amyloids, apoptotic cells, and infected red blood cells are key ligands to the CD36 scavenger receptor (Collins et al., 2009). Uptake of ox-LDL by the scavenger receptor CD36 is an actin-dependent process (Collins et al., 2009). Ligands to CD36 promote micropinocytosis and internalization of the ligand–receptor complexes (Collins et al., 2009) through actin-dependent endocytosis and involves activation of Src family kinases, June N-terminal Kinases (JNK), and Rho family Guanosine Triphosphate hydrolyzing enzyme (GTPase) (Collins et al., 2009). In line with reduced CD36 expression on SHR macrophages, these cells internalize significantly fewer non-opsonized Plasmodium falciparum parasite-infected red blood cells compared to WKY macrophages expressing high levels of CD36 (Patel et al., 2004). As a genetic strain the SHR has mutations in the CD36 gene (Lauzier et al., 2011). Comparisons with genetic knockouts of CD36 may provide insights into potential links between the CD36 knockouts and the SHR pathology.
Fatty acid metabolism in CD36 knockout mice, CD36 deficient patients, SHR rats, and normotensive WKY controls
As indicated by the CD36 knockout mice, CD36 is necessary in mitochondrial fatty acid oxidation, specifically palmitate oxidation during muscle contraction (Holloway et al., 2009). Not only is the Liver X Receptor (LXR) agonist no longer capable of increasing hepatic and circulating levels of triglycerides and free fatty acids in CD36 knockout mice (He et al., 2011), but cultured macrophages of CD36(−/−) also exhibit reduced uptake of oxidized-LDL (Collins et al., 2009). CD36 deficient patients have higher plasma glucose levels, serum triglyceride levels, and blood pressure as well as lower HDL-cholesterol levels (Turk et al., 2001). Higher baseline lipoprotein levels in the circulation could be an indicator for inadequate fatty acid metabolism. Transgenic mice that over-express CD36 have lower blood lipid levels, and hypertensive rats have impaired fatty acid and glucose metabolism due to CD36 deficiency compared to the WKY rat strain. This evidence is in line with the current results (Figs. 4 and 5) (Aitman et al., 1999). Fatty acid levels were not measured in the WKY, SHR, or treated groups, and so the effect of CGS on these factors remains to be investigated. Interestingly, lack of CD36 in CD36 knockout mice can be protective against atherosclerotic plaques with reduced pro-inflammatory migration and accumulation of macrophages to inflammatory sites (Cai et al., 2012; Kuchibhotla et al., 2008).
SHR with CD36 transgene
Compared to SHR with a CD36 variant gene and insulin resistance, congenic SHR-4 (SHR with a wildtype CD36 transgene) exhibit reduced levels of serum free fatty acids and insulin (Klevstig et al., 2011). Reduced insulin resistance in SHR-4 was confirmed to be associated with protein kinase Cs (PKCs) in insulin signaling pathways (Klevstig et al., 2011). Cultured macrophages exposed to PKC activators have up-regulated mRNA expression for CD36, and certain inhibitors of PKCs reduced expressions of CD36 time-dependently (Feng et al., 2000). The wild type CD36 gene and physiological levels of CD36 receptor expression is required for normal fatty acid and glucose metabolism. In fact, the wild type CD36 transgene introduced into the SHR improves metabolic disorders but has no effect on hypertension (Pravenec et al., 2003). This is in line with our basic hypothesis that receptor cleavage involved in ectodomain cleavage of the β2-adrenergic receptor leads to arteriolar constriction and consequently elevation of the central blood pressure (Rodrigues et al., 2010), cleavage of the insulin receptor causes insulin resistance (DeLano & Schmid-Schönbein, 2008), cleavage of the VEGF-R2 leads to endothelial apoptosis and capillary rarefaction (Tran et al., 2010), cleavage of the fMet-Leu-Phe receptor (FPR) on SHR leukocytes leads to a defective pseudopod formation (Chen et al., 2010), or cleavage of leukocyte adhesion molecules (CD18, P-selectin, PSGL-1) leads to a defective adhesion to the endothelium and reduced migration into tissue under acute inflammatory conditions (Chen et al., 2012; DeLano & Schmid-Schönbein, 2008; Fukuda & Schmid-Schönbein, 2003). Each of these functional defects in the SHR is attenuated by chronic treatment with an MMP inhibitor, which restores a normal ectodomain receptor density in the SHR. The evidence presented in the current study shows that proteolytic cleavage of the CD36 ectodomain on SHR macrophages in the heart and spleen could potentially contribute to impaired fatty acid and glucose metabolism.
Changes in systolic blood pressure with inhibitor CGS
Systolic blood pressure for all animals groups was measured in a prior study with results showing a decrease in blood pressure in the SHR group compared to the WKY rat control levels (Chen et al., 2012).
Use of Wistar-Kyoto rats as normotensive control compared to Wistar rats
The WKY rats are the low pressure “normotensive” control for the SHR both of them are derived from the WKY strain. There is variability in the WKY strain with regard to size and mean blood pressures depending on the breeder (Kurtz & Morris, 1987). While the blood pressure of the WKY strain is suitable as a normotensive control the SHR, the blood pressure and the MMP activity levels of the WKY strain is higher than that of the original Wister strain. As it pertains to CD36, Wistar rats also maintain a higher level of the receptor compared to the SHR (Purushothaman et al., 2011). This supports the utility of WKY as a low-pressure control for studies of receptor levels.
Conclusions
As a model of genetic hypertension and a rodent strain with several co-morbidities encountered in the metabolic syndrome, the SHR’s impaired fatty acid metabolism and impaired red blood cell removal may be associated with proteolytic cleavage of the CD36 scavenger receptor. Enhanced plasma MMP activity together with reduced plasma TIMP-1 and TIMP-2 levels, the SHR has elevated proteolytic activity in not only its plasma but also its microvascular endothelium that may lead to a reduced extracellular CD36 density on macrophages in the heart and the spleen. In contrast, SHR’s liver extracellular CD36 density is higher than the control WKY rat as detected by immunohistochemistry. Chronic MMP inhibition by CGS significantly restored the reduced extracellular CD36 density in the SHR heart and spleen. As a pharmaceutical intervention, the broad-spectrum MMP inhibitor CGS could serve as a potential candidate to prevent proteolytic receptor cleavage and cellular dysfunctions in the microcirculation and surrounding tissues.
Acknowledgments
The author’s would like to acknowledge Robert Dolan for his assistance with Westerns of the CD36 extracellular fragments in culture supernatant.
The research was supported by the National Heart Lung and Blood Institute (NHLBI) Grant HL-10881.
Abbreviations
- CGS
CGS27023A (broad spectrum MMP inhibitor)
- CGS SHR
CGS-treated Spontaneously Hypertensive Rat
- CGS WKY
CGS-treated Wistar Kyoto Rat
- FAT
Fatty Acid Transporter
- FPR
Formyl Peptide Receptor
- GTPase
Guanosine Triphosphate Hydrolyzing Enzyme
- IL-4
Interleukin-4
- JNK
June N-terminal Kinase
- LRBC
Light-weight red blood cell
- LXR
Liver X receptor
- LXRE
Liver X response element
- MMP
Matrix Metalloproteinase
- M-CSF
Monocyte-Colony Stimulating Factor
- Ox-LDL
Oxidized-Low Density Lipoprotein
- PKC
Protein Kinase C
- PXR
Pregnane X Receptor
- PPAR-γ
Peroxisome Proliferator Activated Receptor-γ RBC, Red blood cell
- SHR
Spontaneously Hypertensive Rat
- SHRSP
Stroke-Prone Spontaneously Hypertensive Rat
- sCD36
Soluble CD36
- TIMP-1
Tissue Inhibitor of the Metalloproteinase-1
- TIMP-2
Tissue Inhibitor of the Metalloproteinase-2
- VEGF-R2
Vascular Endothelial Growth Factor-Receptor 2
- WKY
Wistar Kyoto Rat
Footnotes
Competing interests
The authors declare no competing financial or non-financial conflict(s) of interest(s).
Authors’ contributions
MHS and AYC contributed to the study with conception, design, experimentation, analysis, interpretation, composition, and revision of the manuscript. DCM contributed to animal handling and facilitation of experimental design and execution, specifically cell cultures and the cell adhesion assay. JC contributed to the study by facilitation of experimental design and execution, specifically Western blots of TIMP-1 and TIMP-2. GWSS contributed to the study with overall conception, design, guidance, supervision, interpretation, composition and writing of the manuscript.
Contributor Information
Angela Y. Chen, Email: vistacove2009@gmail.com.
Jason Chow, Email: jcchow09@gmail.com.
Diana C. Muñoz, Email: dianamunoz07@yahoo.com.
Geert W. Schmid-Schönbein, Email: gwss@bioeng.ucsd.edu.
References
- Aitman TJ, Glazier AM, Wallace CA, Cooper LD, Norsworthy PJ, Wahid FN, Al-Majali KM, Trembling PM, Mann CJ, Shoulders CC, Graf D, St Lezin E, Kurtz TW, Kren V, Pravenec M, Ibrahimi A, Abumrad NA, Stanton LW, Scott J. Identification of Cd36 (Fat) as an insulin-resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat Genet. 1999;21(1):76–83. doi: 10.1038/5013. [DOI] [PubMed] [Google Scholar]
- Bernardo MM, Fridman R. TIMP-2 (tissue inhibitor of metalloproteinase-2) regulates MMP-2 (matrix metalloproteinase-2) activity in the extracellular environment after pro-MMP-2 activation by MT1 (membrane type 1)-MMP. Biochem J. 2003;374(Pt 3):739–745. doi: 10.1042/BJ20030557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonen A, Han XX, Tandon NN, Glatz JF, Lally J, Snook LA, Luiken JJ. FAT/CD36 expression is not ablated in spontaneously hypertensive rats. J Lipid Res. 2009;50(4):740–748. doi: 10.1194/jlr.M800237-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes MJ, Cooper BT. Hypertension and fatty liver: guilty by association? J Hum Hypertens. 2007;21(4):264–270. doi: 10.1038/sj.jhh.1002148. [DOI] [PubMed] [Google Scholar]
- Brumann M, Kusmenkov T, Ney L, Kanz KG, Leidel BA, Biberthaler P, Mutschler W, Bogner V. Concentration kinetics of serum MMP-9 and TIMP-1 after blunt multiple injuries in the early posttraumatic period. Mediat Inflamm. 2012;2012:435463. doi: 10.1155/2012/435463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Wang Z, Ji A, Meyer JM, van der Westhuyzen DR. Scavenger receptor CD36 expression contributes to adipose tissue inflammation and cell death in diet-induced obesity. PLoS One. 2012;7(5):e36785. doi: 10.1371/journal.pone.0036785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AY, DeLano FA, Valdez SR, Ha JN, Shin HY, Schmid-Schönbein GW. Receptor cleavage reduces the fluid shear response in neutrophils of the spontaneously hypertensive rat. Am J Physiol Cell Physiol. 2010;299(6):C1441–C1449. doi: 10.1152/ajpcell.00157.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AY, Ha JN, Delano FA, Schmid-Schönbein GW. Receptor cleavage and P-selectin-dependent reduction of leukocyte adhesion in the spontaneously hypertensive rat. J Leukoc Biol. 2012;92(1):183–194. doi: 10.1189/jlb.0112010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RF, Touret N, Kuwata H, Tandon NN, Grinstein S, Trimble WS. Uptake of oxidized low density lipoprotein by CD36 occurs by an actin-dependent pathway distinct from macropinocytosis. J Biol Chem. 2009;284(44):30288–30297. doi: 10.1074/jbc.M109.045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison M, Glazier AM, Graham D, Morton JJ, Dominiczak MH, Aitman TJ, Connell JM, Gould GW, Dominiczak AF. Cd36 and molecular mechanisms of insulin resistance in the stroke-prone spontaneously hypertensive rat. Diabetes. 2000;49(12):2222–2226. doi: 10.2337/diabetes.49.12.2222. [DOI] [PubMed] [Google Scholar]
- Crofton JT, Share L. Gonadal hormones modulate deoxycorticosterone-salt hypertension in male and female rats. Hypertension. 1997;29(1 Pt 2):494–499. doi: 10.1161/01.hyp.29.1.494. [DOI] [PubMed] [Google Scholar]
- DeLano FA, Schmid-Schönbein GW. Proteinase activity and receptor cleavage: mechanism for insulin resistance in the spontaneously hypertensive rat. Hypertension. 2008;52(2):415–423. doi: 10.1161/HYPERTENSIONAHA.107.104356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano FA, Chen AY, Wu KI, Tran ED, Rodrigues SF, Schmid-Schönbein GW. The autodigestion hypothesis and receptor cleavage in diabetes and hypertension. Dis Models. 2011;8(1):37–46. doi: 10.1016/j.ddmod.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001;108(6):785–791. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Han J, Pearce SF, Silverstein RL, Gotto AM, Jr, Hajjar DP, Nicholson AC. Induction of CD36 expression by oxidized LDL and IL-4 by a common signaling pathway dependent on protein kinase C and PPAR-gamma. J Lipid Res. 2000;41(5):688–696. [PubMed] [Google Scholar]
- Fukuda S, Schmid-Schönbein GW. Regulation of CD18 expression on neutrophils in response to fluid shear stress. Proc Natl Acad Sci U S A. 2003;100(23):13152–13157. doi: 10.1073/pnas.2336130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garciarena CD, Caldiz CI, Portiansky EL, Chiappe de Cingolani GE, Ennis IL. Chronic NHE-1 blockade induces an antiapoptotic effect in the hypertrophied heart. J Appl Physiol. 2009;106(4):1325–1331. doi: 10.1152/japplphysiol.91300.2008. [DOI] [PubMed] [Google Scholar]
- Handberg A, Hojlund K, Gastaldelli A, Flyvbjerg A, Dekker JM, Petrie J, Piatti P, Beck-Nielsen H, Investigators R. Plasma sCD36 is associated with markers of atherosclerosis, insulin resistance, and fatty liver in a nondiabetic healthy population. J Intern Med. 2012;271(3):294–304. doi: 10.1111/j.1365-2796.2011.02442.x. [DOI] [PubMed] [Google Scholar]
- He J, Lee JH, Febbraio M, Xie W. The emerging roles of fatty acid translocase/CD36 and the aryl hydrocarbon receptor in fatty liver disease. Exp Biol Med. 2011;236(10):1116–1121. doi: 10.1258/ebm.2011.011128. [DOI] [PubMed] [Google Scholar]
- Holloway GP, Jain SS, Bezaire V, Han XX, Glatz JF, Luiken JJ, Harper ME, Bonen A. FAT/CD36-null mice reveal that mitochondrial FAT/CD36 is required to upregulate mitochondrial fatty acid oxidation in contracting muscle. Am J Physiol Regul Integr Comp Physiol. 2009;297(4):R960–R967. doi: 10.1152/ajpregu.91021.2008. [DOI] [PubMed] [Google Scholar]
- Itabe H, Obama T, Kato R. The dynamics of oxidized LDL during atherogenesis. J Lipids. 2011;2011:418313. doi: 10.1155/2011/418313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasaoka T, Nishiyama H, Okada M, Nakajima M. Matrix metalloproteinase inhibitor, MMI270 (CGS27023A) inhibited hematogenic metastasis of B16 melanoma cells in both experimental and spontaneous metastasis models. Clin Exp Metastasis. 2008;25(7):827–834. doi: 10.1007/s10585-008-9198-7. [DOI] [PubMed] [Google Scholar]
- Klevstig MJ, Markova I, Burianova J, Kazdova L, Pravenec M, Novakova O, Novak F. Role of FAT/CD36 in novel PKC isoform activation in heart of spontaneously hypertensive rats. Mol Cell Biochem. 2011;357(1–2):163–169. doi: 10.1007/s11010-011-0886-2. [DOI] [PubMed] [Google Scholar]
- Kobori H, Ozawa Y, Suzaki Y, Nishiyama A. Enhanced intrarenal angiotensinogen contributes to early renal injury in spontaneously hypertensive rats. J Am Soc Nephrol. 2005;16(7):2073–2080. doi: 10.1681/ASN.2004080676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla S, Vanegas D, Kennedy DJ, Guy E, Nimako G, Morton RE, Febbraio M. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc Res. 2008;78(1):185–196. doi: 10.1093/cvr/cvm093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz TW, Morris RC., Jr Biological variability in Wistar-Kyoto rats. Implications for research with the spontaneously hypertensive rat. Hypertension. 1987;10:127–131. doi: 10.1161/01.hyp.10.1.127. [DOI] [PubMed] [Google Scholar]
- Lauzier B, Merlen C, Vaillant F, McDuff J, Bouchard B, Beguin PC, Dolinsky VW, Foisy S, Villeneuve LR, Labarthe F, Dyck JR, Allen BG, Charron G, Des Rosiers C. Post-translational modifications, a key process in CD36 function: lessons from the spontaneously hypertensive rat heart. J Mol Cell Cardiol. 2011;51(1):99–108. doi: 10.1016/j.yjmcc.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Li L, Zhou J, Xing S. Thymus influences liver functions through hypothalamus–pituitary–gonad axis in rats. Adv Neuroimmunol. 1996;6(3):289–293. doi: 10.1016/s0960-5428(96)00024-1. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jacobowitz DM, Barone F, McCarron R, Spatz M, Feuerstein G, Hallenbeck JM, Siren AL. Quantitation of perivascular monocytes and macrophages around cerebral blood vessels of hypertensive and aged rats. J Cereb Blood Flow Metab. 1994;14(2):348–352. doi: 10.1038/jcbfm.1994.43. [DOI] [PubMed] [Google Scholar]
- Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69(3):562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Patel SN, Serghides L, Smith TG, Febbraio M, Silverstein RL, Kurtz TW, Pravenec M, Kain KC. CD36 mediates the phagocytosis of Plasmodium falciparum-infected erythrocytes by rodent macrophages. J Infect Dis. 2004;189(2):204–213. doi: 10.1086/380764. [DOI] [PubMed] [Google Scholar]
- Pot C, Chen AY, Ha JN, Schmid-Schönbein GW. Proteolytic cleavage of the red blood cell glycocalyx in a genetic form of hypertension. Cell Mol Bioeng. 2011;4(4):678–692. doi: 10.1007/s12195-011-0180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pravenec M, Landa V, Zidek V, Musilova A, Kazdova L, Qi N, Wang J, St Lezin E, Kurtz TW. Transgenic expression of CD36 in the spontaneously hypertensive rat is associated with amelioration of metabolic disturbances but has no effect on hypertension. Physiol Res. 2003;52(6):681–688. [PubMed] [Google Scholar]
- Purushothaman S, Sathik MB, Nair R. Reactivation of peroxisome proliferator-activated receptor alpha in spontaneously hypertensive rat: age-associated paradoxical effect on the heart. J Cardiovasc Pharmacol. 2011;58:254–262. doi: 10.1097/FJC.0b013e31822368d7. [DOI] [PubMed] [Google Scholar]
- Rodrigues SF, Tran ED, Fortes ZB, Schmid-Schönbein GW. Matrix metalloproteinases cleave the beta2-adrenergic receptor in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2010;299(1):H25–H35. doi: 10.1152/ajpheart.00620.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simantov R, Silverstein RL. CD36: a critical antiangiogenic receptor. Front Biosci. 2003;8:s874–s882. doi: 10.2741/1168. [DOI] [PubMed] [Google Scholar]
- Sun L, Ke Y, Zhu CY, Tang N, Tian DK, Gao YH, Zheng JP, Bian K. Inflammatory reaction versus endogenous peroxisome proliferator-activated receptors expression, re-exploring secondary organ complications of spontaneously hypertensive rats. Chin Med J. 2008;121(22):2305–2311. [PubMed] [Google Scholar]
- Suzuki H, Delano FA, Jamshidi N, Katz D, Mori M, Kosaki K, Gottlieb RA, Ishii H, Schmid-Schonbein GW. Enhanced DNA fragmentation in the thymus of spontaneously hypertensive rats. Am J Physiol. 1999;276(6 Pt 2):H2135–H2140. doi: 10.1152/ajpheart.1999.276.6.H2135. [DOI] [PubMed] [Google Scholar]
- Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol. 2005;23:901–944. doi: 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- Terpstra V, van Berkel TJ. Scavenger receptors on liver Kupffer cells mediate the in-vivo uptake of oxidatively damaged red blood cells in mice. Blood. 2000;95(6):2157–2163. [PubMed] [Google Scholar]
- Timlin MT, Barrows BR, Parks EJ. Increased dietary substrate delivery alters hepatic fatty acid recycling in healthy men. Diabetes. 2005;54(9):2694–2701. doi: 10.2337/diabetes.54.9.2694. [DOI] [PubMed] [Google Scholar]
- Tran ED, DeLano FA, Schmid-Schönbein GW. Enhanced matrix metalloproteinase activity in the spontaneously hypertensive rat: VEGFR-2 cleavage, endothelial apoptosis, and capillary rarefaction. J Vasc Res. 2010;47(5):423–431. doi: 10.1159/000281582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk BE, Huang LL, Piro ET, Cantley LC. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat Biotechnol. 2001;19(7):661–667. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- Yamashita S, Hirano K, Kuwasako T, Janabi M, Toyama Y, Ishigami M, Sakai N. Physiological and pathological roles of a multi-ligand receptor CD36 in atherogenesis; insights from CD36-deficient patients. Mol Cell Biochem. 2007;299:19–22. doi: 10.1007/s11010-005-9031-4. [DOI] [PubMed] [Google Scholar]
- Yesner LM, Huh HY, Pearce SF, Silverstein RL. Regulation of monocyte CD36 and thrombospondin-1 expression by soluble mediators. Arterioscler Thromb Vasc Biol. 1996;16(8):1019–1025. doi: 10.1161/01.atv.16.8.1019. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Tao N, Chung KN, Heuser JE, Lublin DM. Endocytosis of oxidized low density lipoprotein through scavenger receptor CD36 utilizes a lipid raft pathway that does not require caveolin-1. J Biol Chem. 2003;278(46):45931–45936. doi: 10.1074/jbc.M307722200. [DOI] [PubMed] [Google Scholar]
- Zhang X, Fitzsimmons RL, Cleland LG, Ey PL, Zannettino AC, Farmer EA, Sincock P, Mayrhofer G. CD36/fatty acid translocase in rats: distribution, isolation from hepatocytes, and comparison with the scavenger receptor SR-B1. Lab Invest. 2003;83(3):317–332. doi: 10.1097/01.lab.0000059923.67198.ba. [DOI] [PubMed] [Google Scholar]