

Summary
Multi-organ failure contributes to mortality in bacterial sepsis. Platelet and immune cell activation contribute to organ injury during sepsis, but the mechanisms by which bacterial virulence factors initiate these responses remain poorly defined. We demonstrate that during lethal sepsis, Staphylococcus aureus α-toxin simultaneously alters platelet activation and promotes neutrophil inflammatory signaling through interactions with its cellular receptor ADAM10. Platelet intoxication prevents endothelial barrier repair and facilitates formation of injurious platelet-neutrophil aggregates, contributing to lung and liver injury that is mitigated by ADAM10 deletion on platelets and myeloid lineage cells. While plateletor myeloid-specific ADAM10 knockout does not alter sepsis mortality, double-knockout animals are highly protected. These results define a pathway by which a single bacterial toxin utilizes a widely expressed receptor to coordinate progressive, multi-organ disease in lethal sepsis. As an expression-enhancing ADAM10 polymorphism confers susceptibility to severe human sepsis, these studies highlight the importance of understanding molecular host-microbe interactions.
Graphical abstract
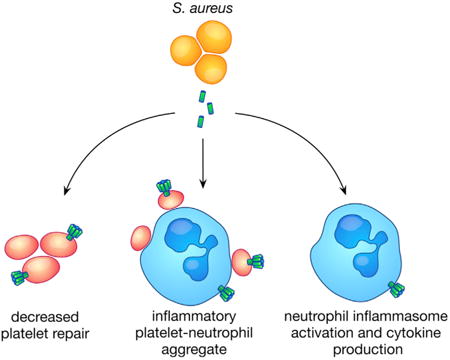
Introduction
Bacterial sepsis is a significant cause of infectious disease mortality worldwide (Angus and van der Poll, 2013). Among the pathogens associated with human sepsis, Staphylococcus aureus remains the leading etiologic agent, associated with the highest mortality (Ani et al., 2015). The success of S. aureus as an intra-vascular pathogen depends on virulence factors that contribute to immunoevasion and promote survival in the bloodstream (Powers and Bubeck Wardenburg, 2014). S. aureus disables circulating immune cells (Okumura and Nizet, 2014), inhibits complement (Spaan et al., 2013), alters coagulation (Bhakdi et al., 1988; McAdow et al., 2012), and attaches to the vascular endothelium (Edwards et al., 2010). To develop strategic approaches for the prevention of sepsis, detailed knowledge of the molecular mechanisms by which specific virulence factors perturb complex multi-cellular interactions in the vasculature is required.
S. aureus α-toxin (Hla) is a secreted, pore-forming cytotoxin expressed by most human disease isolates (Berube and Bubeck Wardenburg, 2013). Hla interacts with its receptor ADAM10 to facilitate cellular injury (Wilke and Bubeck Wardenburg, 2010). ADAM10 is a widely expressed zinc-dependent metalloprotease that contributes to tissue barrier regulation, cell migration and differentiation, and control of cellular activation through enzymatic cleavage of the extracellular domain of its substrates (Reiss and Saftig, 2009). The Hla-ADAM10 interaction leads to ADAM10 activation, resulting in pathologic cleavage of ADAM10 substrates E-cadherin on epithelial cells and endothelial VE-cadherin (Inoshima et al., 2011, 2012; Powers et al., 2012). These molecular events culminate in tissue barrier disruption, exacerbating S. aureus pneumonia and dermonecrotic skin injury and contributing to vascular leakage (Inoshima et al., 2011, 2012; Powers et al., 2012). Conditional ADAM10 knockout in the pulmonary epithelium and epidermal keratinocytes blunts disease, and an active-site inhibitor of ADAM10 improves the outcome of lethal invasive disease and severe skin infection (Inoshima et al., 2011, 2012; Powers et al., 2012; Sampedro et al., 2014). Illustrating the tissue specificity of Hla cellular action, loss of ADAM10 expression on myeloid lineage cells exacerbates skin infection, but mitigates pneumonia (Becker et al., 2014). Analysis of ADAM10 polymorphisms in human sepsis revealed that the C allele of the rs653765 ADAM10 promoter polymorphism confers susceptibility to the most severe forms of disease (Cui et al., 2015). The CC rs653765 genotype was associated with significantly higher ADAM10 expression and a concomitant increase in ADAM10 substrates CX3CL1, IL-6R, and TNFα, underscoring the importance of a detailed molecular understanding of the Hla-ADAM10 interaction in lethal S. aureus sepsis.
In contrast to single-organ system disease, sepsis is associated with multi-organ dysfunction. Diffuse endothelial injury is thought to underlie systemic injury in sepsis, contributing to organ-specific disease manifestations (Lee and Slutsky, 2010). Injury of the pulmonary endothelium triggers neutrophil infiltration, microvascular aggregation of activated platelets and neutrophils, hemorrhage, and extravasation of protein-rich fluid into the airspace, referred to as sepsis-associated acute lung injury (Matthay and Zemans, 2011). Similarly, injury of the liver sinusoidal endothelium facilitates platelet aggregation and activation of Kupffer cells and recruited neutrophils, leading to inflammatory cytokine release (Nesseler et al., 2012; Singer et al., 2006). Endothelial activation through inflammation or injury initiates the platelet repair response, marginalizing platelets and facilitating adhesion to sub-endothelial collagen (Lee and Slutsky, 2010). The platelet-collagen interaction delivers a potent platelet activation signal, resulting in release of prothrombotic, proinflammatory mediators from granule stores to further augment platelet activation and thrombus formation (Li et al., 2010). Platelet activation contributes to innate immunity through antimicrobial peptide release (Yeaman, 2010), bacterial trapping against liver sinusoidal macrophages in sepsis (Wong et al., 2013), and promotion of platelet-neutrophil aggregate (PNA) formation that facilitates neutrophil extracellular trap extrusion (Caudrillier et al., 2012). Mirroring circulating platelets, innate immune cells also interact with activated endothelial cells (Ley et al., 2007). Macrophages recruit neutrophils to sites of injury where they aid in host defense and augment inflammation by elaboration of cytokines and reactive oxygen species (Mantovani et al., 2011; McDonald et al., 2013). While platelet and immune cell activation contributes to organ-specific pathogenesis during sepsis, the role of specific bacterial virulence factors in these processes remains unknown.
The broad expression profile of ADAM10 suggests that α-toxin may contribute to progressive, multi-organ disease observed in lethal sepsis by simultaneously targeting multiple host cells in bloodstream, causing a synergistic injury. Platelet activation by Hla triggers granule release and the formation of PNAs in vitro (Bhakdi et al., 1988; Parimon et al., 2013), also stimulating IL-1β secretion by monocytes, macrophages, and neutrophils (Bhakdi et al., 1989; Nygaard et al., 2013). Our knowledge of Hla-mediated destruction of the endothelial barrier (Powers et al., 2012) led us to hypothesize that pleiotropic targeting of these distinct cell populations by Hla may underlie sepsis progression. We therefore employed a genetic strategy to understand the role of the Hla-ADAM10 complex on multiple hematopoietic lineages during lethal bloodstream infection.
Results
Hla Impairs Platelet Aggregation through Glycoprotein Modification
Vascular injury poses a threat of hemorrhage, necessitating a rapid, multi-step repair process initiated by platelet adherence to von Willebrand factor (vWF) extruded from endothelial cells. We reasoned that endothelial injury promoted by the Hla-ADAM10 complex should prompt a rapid platelet response to restore vascular integrity. We evaluated this process in human pulmonary artery endothelial cells (HPAECs) following exposure to Hla or its non-toxinogenic variant (HlaH35L) that fails to form a lytic pore or activate the metalloprotease (Inoshima et al., 2011; Wilke and Bubeck Wardenburg, 2010). vWF was extruded upon treatment with Hla or histamine, an agonist of vWF deposition (Figure 1A, red), whereas vWF was not observed following treatment with HlaH35L or the ADAM10 inhibitor GI254023X that blocks toxin action (Figure 1A). We next examined whether vWF release enhanced platelet-endothelial interactions in a flow chamber system where labeled human platelets were circulated over toxin-treated HPAECs. The addition of a sub-cytolytic concentration of Hla, but not HlaH35L, caused platelet-endothelial tethering within 30 min (Figure 1B).
Figure 1. Hla Alters Platelet Aggregation.
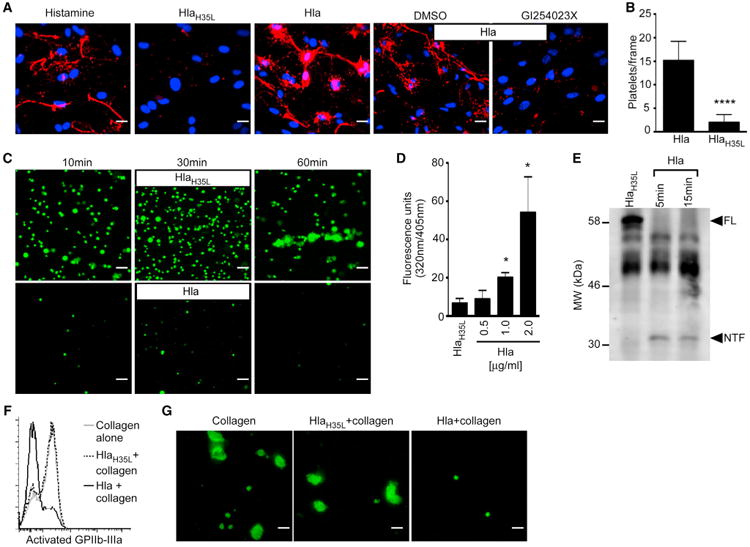
(A) von Willebrand factor (red) release from primary human pulmonary artery endothelial cells (HPAECs) was assessed following stimulation with histamine, HlaH35L, Hla, or Hla in the presence of the ADAM10 inhibitor GI254023X or vehicle control DMSO. Dapi (blue) denotes cell nuclei.
(B) Quantification of calcein AM-labeled human platelets that adhere to HlaH35L or Hla-treated HPAECs as in (A), ****p ≤ 0.0001.
(C) Adherence of Hla or HlaH35L-treated calcein AM-labeled platelets to collagen-coated slides.
(D) Toxin-induced metalloprotease activity in platelets stimulated with Hla in the presence of a fluorogenic ADAM10 substrate for 30 min, *p ≤ 0.05.
(E) GPVI immunoblot analysis of lysates prepared from human platelets treated with HlaH35L or Hla. FL, full-length GPVI; NTF, N-terminal GPVI fragment.
(F) Activated GPIIb-IIIa expression on human platelets pre-treated with Hla (black line) or HlaH35L (dotted line) followed by treatment with collagen compared to collagen-alone control (gray line).
(G) Aggregation of calcein AM-labeled platelets on plate-bound human fibrinogen following treatment as in (C). Scale bars, 10 μm. mean ± SD.
To further analyze Hla action on platelets, we examined downstream mechanisms of platelet-dependent endothelial repair. Platelet-vWF tethering promotes the interaction of platelet glycoprotein VI (GPVI) with exposed sub-endothelial collagen, augmenting platelet activation (Nieswandt et al., 2001). We utilized the flow-based system to examine whether Hla modulates platelet binding to type-1 fibrillar collagen. HlaH35L-treated platelets adhered to collagen and formed aggregates over 60 min, whereas Hla-treated platelets displayed limited collagen interaction (Figure 1C). This finding was reminiscent of the impaired platelet-collagen interaction observed in human GPVI deficiency (Moroi and Jung, 2004). As GPVI is a native ADAM10 substrate (Bender et al., 2010), we examined human platelet ADAM10 activity following toxin treatment. Hla led to increased ADAM10 activity (Figure 1D), triggering GPVI proteolysis detectable by release of the N-terminal extracellular fragment (Figure 1E, NTF). Collagen-bound GPVI engenders platelet morphologic changes and upregulates GPIIb-IIIa-dependent adhesion to fibrinogen to facilitate platelet-platelet interactions (Li et al., 2010). Collagen treatment of human platelets led to activated GPIIb-IIIa expression (Figure 1F, gray line), while Hla pre-treatment abrogated this response (black line), consistent with GPVI loss. HlaH35L pre-treatment did not impair GPIIb-IIIa activation (dotted line). Hla also impaired collagen-induced platelet aggregation on fibrinogen (Figure 1G).
ADAM10 Is Required for Modification of Platelet Adhesion by Hla
Multiple stimuli contribute to platelet activation via ligand engagement of platelet surface receptors (Li et al., 2010). In response to Hla, platelets secrete alpha- and dense-granules, which are effectors of activation (Bhakdi et al., 1988). To examine the necessity of ADAM10 in platelet activation by Hla, we generated mice harboring platelet-specific ADAM10 deletion utilizing platelet factor 4 (PF4) promoter-driven Cre recombinase expression (Figure S1A, ADAM10−), and confirmed loss of ADAM10 expression in knockout mice (Figure S1B, PF4 ADAM10−/−). Loss of platelet ADAM10 expression did not alter hematopoiesis, consistent with prior studies (Bender et al., 2010) (Figures S1C– S1F). ADAM10−/− platelets were Hla-insensitive measured by LDH (Figure S1G) and ATP release (Figure S1H); however, they remained responsive to collagen (Figure S1I). ADAM10−/− platelets displayed a marked reduction in cell-associated metalloprotease activity upon toxin treatment in contrast to control platelets (Figure S1J).
We hypothesized that loss of platelet ADAM10 expression would preserve the adhesive properties of Hla-treated platelets, maintaining platelet-collagen and platelet-platelet interactions. We thus examined physiologic adhesive events mediated by platelet GPVI and GPIIb-IIIa in control and ADAM10−/− platelets in response to Hla. While control platelets exhibited a loss of GPVI expression upon Hla exposure, ADAM10−/− platelets maintained GPVI (Figure 2A). Consistent with GPVI cleavage, toxin-treated control platelets did not bind fibrillar collagen underflow conditions, while ADAM10−/− platelets were readily adherent (Figure 2B). Soluble and plate-bound collagen are potent stimuli for human platelet activation ex vivo; however, mouse platelets are optimally stimulated by a peptide derived from collagen (collagen-related peptide, CRP). Hla pre-treated control platelets failed to display activated GPIIb-IIIa in response to CRP (Figure 2C, left panel, black line), in contrast to HlaH35L pre-treated platelets (dotted line) or CRP alone (gray line). ADAM10−/− platelets were Hla resistant, expressing activated GPIIb-IIIa following CRP treatment (Figure 2C) and preserving fibrinogen binding (Figure 2D).
Figure 2. ADAM10 Alters Platelet Adhesive Properties in Response to Hla and Contributes to Sepsis-Associated Lung Injury.
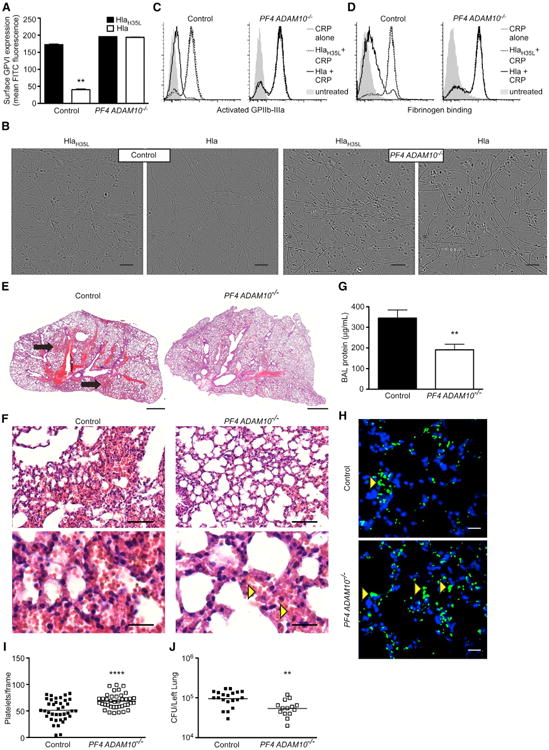
(A) GPVI expression on control or PF4 ADAM10−/− platelets following treatment with HlaH35L or Hla, **p ≤ 0.01. Data are represented as mean ± SD.
(B) Adherence of control or PF4 ADAM10−/− platelets flowed over plate-bound collagen following treatment with HlaH35L or Hla. Scale bars, 10 μm.
(C) Analysis of activated GPIIb-IIIa expression on control (left) or PF4 ADAM10−/− (right) platelets pre-treated with Hla (black line) or HlaH35L (dotted line) followed by treatment with collagen-related peptide (CRP). CRP alone (gray line), untreated (gray shading) platelets served as controls.
(D) Binding of FITC-labeled fibrinogen to control and PF4 ADAM10−/− platelets treated as in (C).
(E) H&E-stained lung sections from control or PF4 ADAM10−/− mice 4 hr after infection with S. aureus USA300. Scale bars, 1,000 μm, and images representative of greater than three mice per condition from two independent experiments.
(F) Higher-magnification images of lung sections in (E), where yellow arrows demonstrate red cells in the vasculature of PF4 ADAM10−/−-infected mice. Scale bars, 100 μm (upper) and 20 μm (lower).
(G) Protein content in bronchoalveolar lavage (BAL) fluid from control (n = 12) and PF4 ADAM10−/− (n = 9) mice infected as in (E), **p ≤ 0.01.
(H) Platelets and nuclei (blue, dapi) in control or PF4 ADAM10−/− lungs 4 hr post infection as in (E). Scale bars, 10 μm.
(I) Quantification of platelets per frame from control or PF4 ADAM10−/− lungs (n = 3) of the images (n = 20) in (D).
(J) S. aureus colony-forming unit (CFU) recovery from lung tissue 4 hr after infection of control (n = 8) and PF4 ADAM10−/− (n = 9) mice as in (E), **p ≤ 0.01. Data are represented as mean ± SD; see also Figures S1 and S2.
Platelet Intoxication Contributes to Sepsis-Associated Acute Lung Injury
Acute lung injury is a common complication of sepsis and other systemic injuries, in which disturbances of platelet function contribute to pathology (Looney et al., 2009; Zarbock et al., 2006). To assess whether alterations in platelet adhesion by Hla modulate S. aureus sepsis-associated ALI, we examined lung pathology in control and PF4 ADAM10−/− mice following lethal intravenous challenge with S. aureus USA300, an epidemic human isolate. Control mice demonstrated intrapulmonary hemorrhage 4 hr post-infection (Figure 2E, black arrows). While red cells are visualized in the capillaries of infected PF4 ADAM10−/− mice (Figure 2F, yellow arrows), these mice do not develop overt hemorrhage (Figure 2F). Consistent with decreased pathologic evidence of ALI, we also observed a reduction in bronchoalveolar protein extravasation in PF4 ADAM10−/− mice relative to controls (Figure 2G). Our observation that Hla perturbs platelet adhesion and promotes lung injury suggests that Hla renders platelets less adherent to sites of microvascular injury in sepsis. We therefore visualized platelets in the lungs 4 hr post-infection. Control mice display a reduction in platelet accumulation compared to knockout mice, suggesting a defect in adhesive function of Hla-sensitive platelets (Figures 2H and 2I). These early manifestations of ALI in control mice relative to PF4 ADAM10−/− mice are accompanied by greater recovery of S. aureus from saline-perfused lungs, a measure of bacteria that are firmly attached to the vasculature or lung tissue (Figure 2J). Consistent with toxin-mediated exacerbation of ALI, mice infected with Hla-deficient S. aureus exhibited decreased alveolar hemorrhage (Figures S2A and S2B), bronchoalveolar protein (Figure S2C), and S. aureus recovery (Figure S2D). The increased dissemination of wild-type bacteria into the lung does not merely reflect a differential bloodstream load (Figure S2E).
Hla Augments Host Inflammation through PNA Formation
The functional role of platelet adhesion is not limited to endothelial repair, as activated platelets also adhere to neutrophil PSGL-1 through surface display of P-selectin extruded from α-granules (Brown et al., 1998). We hypothesized that perturbation of platelet activation by Hla may broadly alter inter-cellular interactions and host inflammation. Treatment of control platelets with CRP and Hla led to P-selectin expression within 15 min, in contrast to HlaH35L treatment (Figure 3A, mouse; and Figure 3B, human). PF4 ADAM10−/− platelets failed to express P-selectin in response to Hla (Figure 3A), while this response was preserved to CRP. To examine the Hla-ADAM10 complex in PNA formation, we utilized a whole-blood flow-cytometric approach examining co-staining with neutrophil-specific markers (GR1, mouse; or CD11b, human) and platelet GPIbα. To detect PNAs, gated GR1+ or CD11b+ cells were examined for associated platelets (population gating, Figure S3A). In murine cells, the baseline detection of neutrophils (GR1 staining, Figure 3C, left) and PNA (GR1+/GPIbα+ population, untreated) was similar between control (upper panel) and PF4A DAM10−/− (middle panel) mice. Likewise, CRP treatment induced a GR1+/GPIbα+ PNA population in both groups of mice. HlaH35L treatment mirrored the untreated GR1+/GPIbα+ cell population, while Hla treatment of control blood enhanced PNA formation (upper panel). In contrast, Hla treatment of blood harvested from PF4 ADAM10−/− mice did not enhance the GR1+/GPIbα+ population (middle panel), indicative of the role of the platelet toxin-receptor complex in triggering PNA formation. In human cells, the presence of PNA formation indicated by a CD11b+/GPIbα+ population was similarly observed upon Hla treatment in the presence of neutrophils (Figure 3C, lower panel) (Parimon et al., 2013).
Figure 3. Hla Promotes PNA Formation.
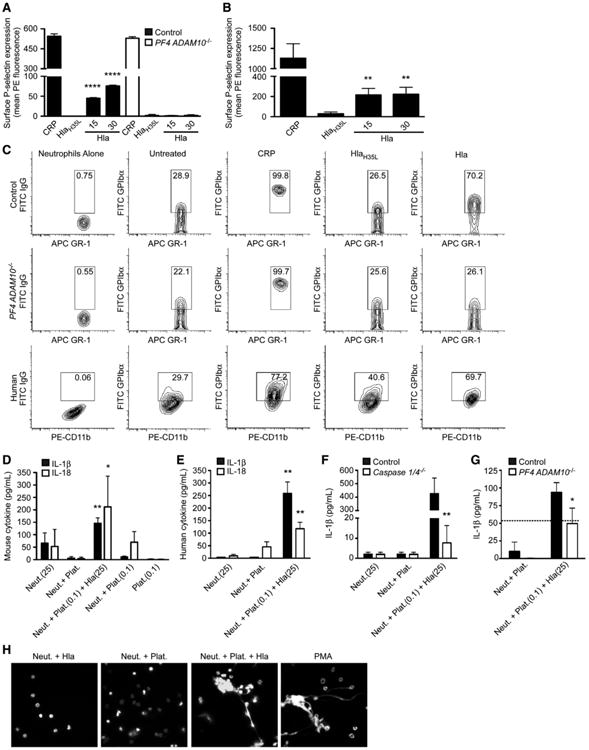
(A and B) P-selectin expression on mouse (A) and human (B) platelets following treatment with collagen-related peptide (CRP), HlaH35L, or Hla; **p ≤ 0.01, ****p ≤ 0.0001.
(C) PNA formation from control and PF4 ADAM10−/− mice (GR1+/GPIbα+) or humans (CD11b+/GPIbα+) either left untreated or treated with CRP, HlaH35L, or Hla.
(D–G) IL-1β (βλαχκ) and IL-18 (white) production following PNA formation in platelet-neutrophil suspensions from wild-type mice (D), human (E), control and Caspase 1/4−/− mice (F), or control and PF4 ADAM10−/− mice (G); (D) and (E) *p ≤ 0.05, **p ≤ 0.01 compared to Neut. or Neut.+Plat. (F) and (G) *p ≤ 0.05, **p ≤ 0.01 compared to control, where dotted line denotes the response of control neutrophils to Hla. Data are represented as mean ± SD. In (D)–(G), (0.1) and (25) denote treatment with toxin concentrations in μg/ml.
(H) NET release examined in mouse platelet-neutrophil suspensions treated as in (D)–(G) or with phorbol myristate acetate (PMA). See also Figure S3.
PNA formation stimulates chemokine and cytokine synthesis (Neumann et al., 1997; Weyrich et al., 1996). We have previously observed that murine neutrophils resist lytic intoxication, demonstrating that the Hla-ADAM10 interaction on neutrophils primarily triggers IL-1β release (Becker et al., 2014). Similarly, human neutrophils exhibit resistance to the lytic effects of Hla relative to human platelets (Figure S3B) (Nygaard et al., 2013; Valeva et al., 1997). We hypothesized that neutrophil stimulation by adherent platelets in concert with Hla may augment neutrophil cytokine production, leading to a state of enhanced inflammation. To assess this, neutrophils were incubated with untreated platelets or platelets stimulated for 4 hr with a low concentration of Hla (0.1 μg/ml) to promote PNA formation. Platelet-neutrophil suspensions were then left untreated or treated with a toxin concentration (25 μg/ml) that stimulates neutrophil IL-1β release (Becker et al., 2014). Both mouse and human neutrophils stimulated with toxin-pre-treated platelets produced significantly more IL-1β (Figure 3D, mouse; and Figure 3E, human, black) ανδ IΛ-18 (white) in response to Hla (Neut.+Plat.(0.01)+Hla(25)) compared to toxin-treated neutrophils alone (Neut.(25)). IL-1 β ανδ IΛ-18 production by unexposed cells (Neut.+Plat.) or neutrophils and platelets only exposed to low Hla concentrations (Neut.+Plat.(0.01)) confirmed the synergistic release of IL-1 β ανδ IΛ-18 elicited from toxin-exposed neutrophils engaged in PNA. Limited cytokine release was observed upon neutrophil exposure to pre-treated platelets alone (Neut.+Plat.(0.01), Figure 3D) or platelets exposed to Hla (Plat.(0.01), Figure 3D).
α-toxin contributes to activation of the NLRP3 inflammasome, leading to caspase-1 cleavage and production of mature IL-1 β ανδ IΛ-18 in macrophages and in the context of S. aureus infection of mice (Craven et al., 2009; Kebaier et al., 2012; Muñoz-Planillo et al., 2009). To examine the role of caspase-1 activation in PNA-mediated cytokine release downstream of the Hla-ADAM10 complex, we utilized mice harboring a caspase 1 deletion (Caspase 1/4−/−, de facto caspase 4 deletion) (Kuida et al., 1995). Neutrophils from Caspase 1/4−/− mice exposed to platelets and Hla failed to elicit a robust IL-1β response compared to controls (Figure 3F) demonstrating the vital role of the caspase-mediated pathway in toxin-stimulated inflammatory cytokine production by PNA. Stimulation of control or PF4 ADAM10−/− platelets with a low concentration of Hla in the presence of neutrophils, followed by neutrophil stimulation with Hla, revealed that only control platelets facilitated synergistic IL-1β production, confirming the necessity of host ADAM10 in this pathway (Figure 3G, where dotted line indicates neutrophils alone treated with Hla). Consistent with this pro-inflammatory effect of toxin-activated platelets, simultaneous exposure of neutrophils to platelets and Hla resulted in neutrophil extracellular trap release (NETs, Figure 3H) (Clark et al., 2007). In contrast, neutrophils exposed to an Hla concentration that facilitates PNA formation (1 μg/ml) or platelets alone did not release NETs (Figure 3H), while NET release was confirmed with PMA treatment.
The ADAM10-Hla Interaction Augments Liver IL-1β Production
These data indicate that Hla alters platelet activation, abrogating the beneficial function of platelets in vascular repair while inducing a potentially deleterious pro-inflammatory state by augmenting platelet-neutrophil interactions and enhancing the potency of Hla on neutrophils. Within the liver, immune cell activation and Kupffer cell-mediated neutrophil recruitment enhance inflammatory cytokine production and hepatocellular injury (Nesseler et al., 2012). As S. aureus sepsis initiates a rapid interaction of platelets and innate immune cells in the hepatic vasculature (Wong et al., 2013), we evaluated liver inflammation by examining tissue-specific IL-1β induction in septic PF4 ADAM10−/− mice or myeloid lineage-specific ADAM10−/− mice (LysM ADAM10−/−) (Becker et al., 2014) 72 hr post-infection. Both PF4 ADAM10−/− (Figure 4A) and LysM ADAM10−/− (Figure 4B) mice demonstrated a decrease in liver IL-1β compared to controls. Diminished IL-1β production in LysM ADAM10−/− mice was not attributable to altered platelet-neutrophil interactions (Figure S4A). These findings suggest that concomitant intoxication of both cell types in the septic liver may facilitate maximal IL-1β production during infection. To extend these studies, we generated mice harboring simultaneous deletion of ADAM10 in both lineages (PF4/LysM ADAM10−/−). Similar to single-knockout mice, liver IL-1β production was significantly decreased in septic PF4/LysM ADAM10−/− mice (Figure 4C). This response was IL-1β specific, as we did not detect alterations in TNF-α or IL-10 (Figures S4B and S4C).
Figure 4. Hla Targets Platelets and Myeloid Cells in the Liver to Stimulate IL-1β Production.
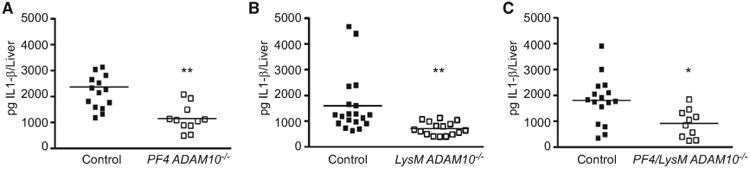
Liver IL-1β analysis 72 hr post infection of PF4 ADAM10−/− (A), LysM ADAM10−/− (B), and PF4/LysM ADAM10−/− (C) or corresponding controls, *p ≤ 0.05, **p ≤ 0.01. Data are represented as mean ± SD; see also Figures S4 and S5.
PF4/LysM ADAM10 Mice Are Protected from Multi-Organ Injury
We have demonstrated that Hla contributes to sepsis-associated lung and liver injury dependent on hematopoietic cell targeting. We hypothesized that PF4/LysM ADAM10−/− mice would exhibit reduced pathologic signs of multi-organ injury associated with lethal sepsis. Indeed, fewer areas of overt alveolar hemorrhage were observed in these mice compared to controls (Figure 5A), with a reduction in proteinaceous edema in double-knockout mice (392 ± 99 μg/ml in control versus 130 ± 22 μg/ml in PF4/LysM ADAM10−/−) mirroring PF4 ADAM10−/− mice. Lungs from LysM ADAM10−/− single-knockout mice remained susceptible to lung injury with BAL fluid protein (Figure S5A) and bacterial extravasation (Figure S5B) as controls, suggesting that platelet intoxication is a predominant contributor to ALI. Similarly, liver injury was reduced in PF4/LysM ADAM10−/− mice. While control livers displayed large areas of necrosis (Figure 5B, arrows), double-knockout mice experienced a lesser degree of liver injury following infection. Glycogen depletion, a marker of liver stress, was more prominent in control than PF4/LysM ADAM10−/− mice (Figure 5C, reduced PAS staining [magenta], noted by yellow arrows). Control mice demonstrated increased cellular apoptosis measured by active caspase-3 staining of liver sections (Figure 5D, brown) and elevated myeloperoxidase activity (MPO) in liver homogenates compared to PF4/LysM ADAM10−/− mice (Figure 5E). Assignment of pathology scores to quantify liver injury based on the markers described above in addition to hepatocyte vacuolation confirmed the protection conferred in double-knockout mice (Figure 5F). This reduction in liver injury was reflected as decreased serum alanine aminotransferase in PF4/LysM ADAM10−/− mice (Figure 5G).
Figure 5. The Hla-ADAM10 Interaction on Platelets and Myeloid Cells Contributes to Multi-Organ Injury.
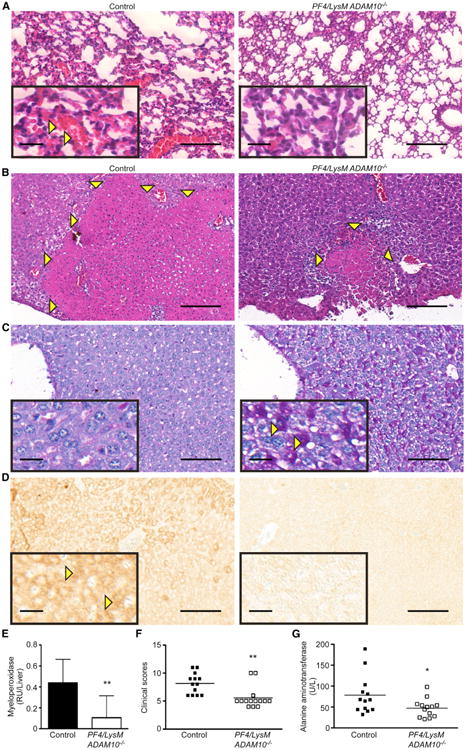
(A) H&E-stained lung sections from control or PF4/LysM ADAM10−/− mice 4 hr after intravenous S. aureus infection. Images representative of greater than three mice per condition from two independent experiments.
(B) H&E-stained liver sections from control or PF4/LysM ADAM10−/− mice 72 hr after infection.
(C) (C and D) Images of periodic-acid Schiff stain (PAS, C) or activated caspase-3 immunohistochemistry (D) of liver sections from mice as in (B). In (A)–(D) scale bars, 100 μm (larger image) and 20 μm (inset); images representative of greater than five mice per condition from two independent experiments.
(F) Liver myeloperoxidase activity in control (n = 16) or PF4/LysM ADAM10−/− (n = 10) mice as in (C). Data represent two independent pooled experiments, **p ≤ 0.01.
(G) Liver pathology scores from mice as in (B)–(D). Data represent two independent pooled experiments, **p ≤ 0.01.
(H) Serum alanine aminotransferase in infected control (n = 13) or PF4/LysM ADAM10−/− (n = 13) mice as in (C). Data represent two independent pooled experiments, *p ≤ 0.05. Data are represented as mean ± SD; see also Figure S6.
Hla Targets Platelets and Myeloid Cells to Cause Lethality
The contribution of Hla to multi-organ injury in sepsis led us to examine the role of simultaneous targeting of platelets and myeloid lineage cells in relation to mortality. Double-knockout mice were highly protected against lethal sepsis (Figure 6A). In contrast, PF4 ADAM10−/− and LysM ADAM10−/− single-knockout mice succumbed as control mice (Figures 6B and 6C). Survival of double-knockout mice is not simply a byproduct of decreased bacterial load as S. aureus recovery from multiple organs was comparable in control and PF4/LysM ADAM10−/− mice 3 days post-infection (Figures S6A–S6D). While these data suggest that Hla targeting of both platelets and myeloid lineage cells contributes to a synergistic increase in sepsis mortality, each of these cell populations remains vital for innate immunoprotection against S. aureus infection. Independent depletion of platelets, neutrophils, or macrophages renders C57Bl/6 mice fully susceptible to lethal sepsis (Figures S7A– S7C, respectively). Moreover, IL-1 receptor-deficient (IL-1R−/−) and Caspase 1/4−/− mice exhibit increased susceptibility to lethal infection (Figures S7D and S7E, respectively).
Figure 6. Hla Targets Platelets and Myeloid Lineage Cells to Contribute to Lethal Sepsis.
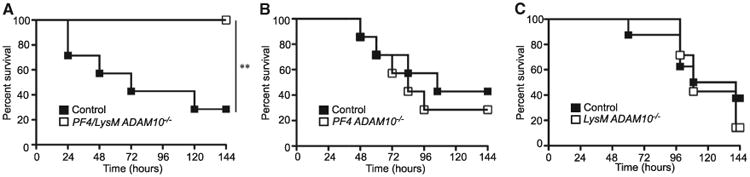
Survival following lethal S. aureus infection in PF4/LysM ADAM10−/− (A, n = 7), PF4ADAM10−/− (B, n = 7), LysMADAM10−/− (C, n = 7), or corresponding controls (n = 7, 7, and 8, respectively); **p ≤ 0.01. See also Figure S7.
Discussion
Severe bacterial sepsis remains a leading cause of infectious disease mortality. Modulation of the host response has been the primary focus of novel sepsis therapies, attempting to dampen the host pro-inflammatory response or mitigate endothelial dysfunction and coagulopathy (Angus and van der Poll, 2013). The inadequacy of these approaches is thought to reflect disease heterogeneity and gaps in our understanding of pathophysiology, especially related to pathogen-specific responses (Angus, 2011). Our studies of S. aureus α-toxin and its cognate receptor ADAM10 provide insight on the fundamental molecular mechanisms that underlie staphylococcal sepsis. The Hla-ADAM10 complex appears to contribute to intravascular injury through a three-pronged approach—initiation of endothelial injury (Powers et al., 2012), perturbation of platelet-mediated endothelial repair, and synergistic activation of pro-inflammatory pathways by infiltrating immune cells (Figure 7A). The demonstration that a single bacterial toxin can simultaneously alter discrete cellular processes on multiple cell types through a widely expressed receptor highlights a previously unrecognized role for bacterial toxins in coordination of disease within a tissue environment.
Figure 7. Model Depicting Multi-Cellular Targeting of S. aureus Hla in Sepsis.
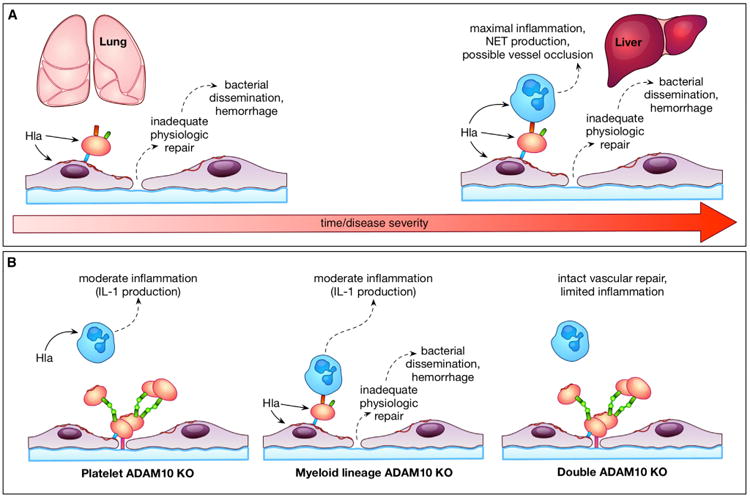
(A) S. aureus α-toxin contributes to temporospatial lung and liver injury by preventing adequate platelet repair and exacerbating the host inflammatory response.
(B) Consequences of selective deletion of ADAM10 on platelets or myeloid lineage cells.
This study extends our observations on the pathologic outcomes of toxin-dependent ADAM10 activation, identifying GPVI as a target of the toxin-receptor complex. Hla activates platelets at sub-cytolytic concentrations (Bhakdi et al., 1988) and augments formation of infective endocarditis thrombi (Bayer et al., 1997). While our findings in ADAM10 knockout platelets confirm toxin-induced activation, we observe a mechanism by which Hla blunts GPIIb-IIIa activation through GPVI proteolysis. Loss of GPIIb-IIIa expression precipitates a bleeding disorder in humans and experimental animals, as mutant platelets fail to establish fibrinogen-based platelet bridges required for aggregation and plugging of the injured endothelium (Tronik-Le Roux et al., 2000). Thus, while platelet activation should restore endothelial barrier function, early S. aureus infection is paradoxically associated with hemorrhage as toxin-induced platelet activation is uncoupled from platelet aggregation by the Hla-ADAM10 complex.
Platelets adhere to S. aureus via GPIIb-IIIa (Herrmann et al., 1993), and also utilize the activated integrin to rapidly trap bacteria in liver sinusoids through GPIb- and GPIIb-mediated interactions with resident macrophages or Kupffer cells (Wong et al., 2013). The loss of this interaction in GPIb- or GPIIb-deficient mice is associated with increased mortality, bacterial burden, and liver injury after B. cereus infection (Wong et al., 2013). Congruent with this observation, platelet depletion increases S. aureus infection mortality (Wong et al., 2013). These findings suggest that the improved outcomes we report in PF4/LysM ADAM10−/− mice may in part result from maintenance of activated glycoprotein expression that promotes bacterial clearance. While we do not observe altered tissue bacterial recovery following lethal infection, this outcome may be more readily evident in sub-lethal challenge.
In contrast to the beneficial role of platelets and myeloid lineage cells in bacterial clearance, recent evidence indicates that heterotypic PNAs exacerbate tissue injury (Caudrillier et al., 2012). Inhibition of aggregate formation in sepsis or acid-induced acute lung injury decreased immune cell infiltration and vascular permeability in the lung (Looney et al., 2009; Zarbock et al., 2006), and provided protection against pulmonary vascular permeability in a murine model of sickle cell disease (Polanowska-Grabowska et al., 2010). Platelet activation by Hla has previously been shown to promote the formation of PNAs (Parimon et al., 2013); however, our studies demonstrate that this interaction primes the neutrophil for caspase-dependent pro-inflammatory cytokine production and NET formation in response to Hla. Liver injury attributable to the toxin appears closely associated with this pro-inflammatory state. Hla activates the NLRP3 inflammasome to stimulate IL-1β secretion (Craven et al., 2009; Kebaier et al., 2012; Muñoz-Planillo et al., 2009). In S. aureus infection, myeloid lineage-specific ADAM10 knockout (Becker et al., 2014) or genetic disruption of Nlrp3 affords protection against disease, corresponding to a reduction in inflammatory cytokine generation (Craven et al., 2009; Kebaier et al., 2012). We demonstrate that increased liver IL-1β production and hepatic injury in sepsis correlate with platelet and myeloid lineage Hla susceptibility, a finding that is consistent with the recently described role of ADAM10 in meprin β-δριϖεν pro-inflammatory cytokine production (Li et al., 2014). While IL-1 receptor signaling is required for protection during bloodstream staphylococcal infection (Hultgren et al., 2002), hyper-inflammatory responses in the liver result in microvascular accumulation of neutrophils, which leads to acute liver injury (Ramaiah and Jaeschke, 2007). We do not observe differences in liver IL-1β production between PF4/LysM ADAM10−/− mice that survive sepsis and the corresponding PF4 ADAM10−/− or LysM ADAM10−/− single-knockout mice, suggesting that this marker of inflammation does not fully reflect the cumulative burden of tissue injury that results from bloodstream toxin action. While selective ADAM10−/− mice provide an opportunity to dissect the relative contribution of the Hla-ADAM10 interaction on discrete cell populations (Figure 7B), a complete understanding of pathogenesis will require knowledge of the concurrent microvascular effects of other S. aureus virulence factors, notably leukotoxins (Alonzo and Torres, 2014) and coagulopathy-inducing coagulase and vWF binding protein (vWBP) (McAdow et al., 2012; Thomer et al., 2013).
Our findings provide evidence that the progression of lethal staphylococcal sepsis results from two distinct insults that are separable on a cellular and molecular level. First, rapid severe disease ensues from poor initial control of bacterial infection by the innate immune system observed upon cell depletion or genetic ablation of essential protective inflammatory signaling pathways. Second, perturbation of host cellular function by Hla can dampen protective responses and enhance pro-inflammatory injury. This degree of complexity provides molecular insight on the observed failures of either antimicrobial therapies or targeted immunomodulation to prevent sepsis-associated mortality in humans, and establishes a temporal framework by which to consider bacterial insults in disease progression. Further resolution of these processes will require an appreciation of the spatiotemporal dynamics of toxin expression, cellular localization to the infection site, and toxin-mediated cellular injury. As Hla targets the endothelium, platelets, and myeloid lineage cells, a more detailed dissection of the independent contribution of these cell populations to toxin-mediated injury is necessary. It is predicted from our findings that Hla-induced endothelial damage may be the inciting insult that triggers platelet and immune cell marginalization within the vasculature, rendering these cells toxin-susceptible and potentially depleting these lineages from other areas of the body as observed in human sepsis. Recent evidence underscores the importance of endothelial targeting by Hla during bloodstream infection, as autophagy-deficient mice display increased ADAM10 on the endothelial cell surface and exhibit increased susceptibility to invasive staphylococcal infection (Maurer et al., 2015). In light of these studies, endothelial-specific deletion of ADAM10 may provide substantial protection against sepsis by limiting the downstream consequences of Hla on multiple cellular targets. While the ADAM10 rs653765 CC polymorphism confers susceptibility to severe sepsis caused by a broad array of pathogens, this clinical observation together with our mechanistic studies on the Hla-ADAM10 complex provides the rationale for clinical genotype analysis in S. aureus sepsis patients. Such focused studies may enable rapid genetic evaluation of patient-specific risk for disease progression, and thereby inform the strategic, early delivery of therapies demonstrated in pre-clinical sepsis models to neutralize Hla or inhibit ADAM10 activity (Powers et al., 2012).
The cellular complexity of the microvasculature, coupled with its dense distribution in every organ, may provide an optimized tissue microenvironment in which bacterial toxins rapidly amplify systemic injury. Recent studies of B. anthracis anthrax toxin lend strong support for the role of bacterial toxins as key mediators of multi-cellular and multi-organ pathology in sepsis. Utilizing conditional knockout mice harboring cell type-specific deletion of the toxin receptor CMG2 utilized by anthrax lethal toxin (LT) and edema toxin (ET), Liu et al. demonstrate that LT targeting of cardiomyocytes and vascular smooth muscle and ET targeting of hepatocytes contribute to lethality (Liu et al., 2013). In contrast to this two-toxin system, our findings on S. aureus Hla provide evidence that a single toxin-receptor interaction elicits a highly coordinated, systemic injury by activity on disparate cell populations in the bloodstream. Given the common clinical findings of endothelial injury, coagulopathy, and inflammation-induced injury in human sepsis, understanding pathogen-specific strategies for integration of these pathways may highlight fundamental, targetable features of disease.
Experimental Procedures
Ethics Statement
Animal and human studies were conducted in accord with protocols approved by the University of Chicago Institutional Animal Care and Use Committee and Institutional Review Board.
Animal Studies
PF4cre tranLd to Adam10loxP/loxP transgenic C57Bl/6 mice (Jackson Laboratories) to generate PF4cre Adam10loxP/loxP mice (PF4 ADAM10−/−). ADAM10 myeloid lineage knockout mice (LysM ADAM10−/−) (Becker et al., 2014) and caspase-1-deficient mice (Caspase 1/4−/−) (Kuida et al., 1995) have been previously described. IL-1 receptor knockout mice (IL-1R−/−) were purchased from Jackson Laboratories. S. aureus strain USA300 LAC or its isogenic Hla mutant (Inoshima et al., 2011) was prepared to deliver an inoculum of 5 × 107 bacteria/100 μl via retro-orbital intravenous injection to 6- to 8-week-old C57Bl/6 mice (Powers et al., 2012).
Tissue Analysis
BAL fluid from infected mice was collected by PBS instillation into the lungs and measured for protein content (Inoshima et al., 2011). Lungs perfused with 3 ml PBS or liver, heart, and spleen tissues were homogenized for CFU enumeration by serial dilution plating or ELISA for cytokine analysis. For histopathologic studies, H&E, periodic acid-Schiff, or caspase-3 staining was performed on formalin-fixed tissues, or frozen lung sections were stained with α-CD42 platelet antibody. Blinded scoring (scores ranging from 1 to 3, with 3 representing the most severe phenotype) was performed for liver necrosis, glycogen depletion, immune cell infiltration, and caspase-3 cleavage.
Cellular Analysis
Endothelial Studies
HPAECs (Lonza) were cultured in EBM-2 BulletKit media. Active Hla was prepared as described (Wilke and Bubeck Wardenburg, 2010). HPAECs exposed to Hla or HlaH35L (5 μg/ml) were stained with α-vWF antibody or exposed to platelets under shear stress of 150 s−1 after plating in IBIDI μ-slide VI flow chambers for platelet binding quantification. Cells were examined on an Olympus DSU Spinning Disk Confocal Microscope.
Platelet Studies
Platelets were exposed to Hla or HlaH35L (human, 1 μg/ml; or mouse, 0.25 μg/ml) to assess collagen or fibrinogen binding in IBIDI chambers. Platelet metalloprotease activity on 5 × 105 human or mouse platelets was quantified utilizing an ADAM10-specific fluorogenic peptide substrate in metalloprotease assay buffer. Platelet surface protein expression in response to collagen and toxin treatment was evaluated by immunoblot analysis and flow-cytometric analysis for GPVI or flow-cytometric analysis for activated GPIIbIIIa.
Platelet-Neutrophil Studies
Mouse or human whole blood was left untreated, treated with Hla or HlaH35L or CRP as above, then fixed prior to red cell lysis and staining for cell subset analysis. Gated GR-1+ or CD11b+ neutrophils were examined for GPIbα εξπρεσσι○ν to detect PNA. For PNA cytokine production, neutrophil-platelet suspensions were left untreated or treated with Hla (0.1 μg/ml) to facilitate PNA formation. Suspensions then exposed to Hla (25 μg/ml) were examined for supernatant IL-1β and IL-18 by ELISA. NET formation by PNAs on poly-L-lysine-coated slides was assessed by fluorescence microscopy following Hoechst staining.
Statistical Methods
Pairwise comparisons were performed using the Student's unpaired t test or Mantel-Cox log-rank test for comparison of survival following Kaplan-Meier estimation. p values of less than 0.05 were considered significant. Error bars represent ± SD.
Additional experimental details are contained within the Supplemental Information.
Supplementary Material
Highlights.
S. aureus α-toxin alters platelet adhesion and promotes neutrophil-platelet aggregation
Platelet intoxication exacerbates acute lung injury
Toxin-activated platelets and neutrophils contribute to liver injury
Protection of platelets and myeloid cells from α-toxin improves sepsis outcome
Acknowledgments
This work was supported by NIH award AI097434-01 and the Burroughs Wellcome Foundation Investigators in the Pathogenesis of Infectious Disease Fellowship (J.B.W.). The authors acknowledge membership in and support from the Region V “Great Lakes” RCE (NIH award 2-U54-AI-057153). M.E.P. was supported by NIH Grant T32 GM007183 and an American Heart Association pre-doctoral training fellowship (FP053181-01-PR). R.E.N.B. is a trainee of the NIH Medical Scientist Training Program at the University of Chicago (GM007281). We thank Dr. Steve P. Watson for guidance on platelet studies and the generous gift of CRP, and Dr. Tatyana Golovkina for providing Cas-pase 1/4−/− mice.
Footnotes
Supplemental Information: Supplemental Information includes seven figures and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi. org/10.1016/j.chom.2015.05.011.
Author Contributions: M.E.P. performed all experiments and contributed to data analysis and writing of the manuscript. R.E.N.B. contributed to animal and ex vivo experiments with PF4/LysM ADAM10−/− mice. A.S. performed caspase-3 analysis in liver tissues. J.R.T. developed the pathologic scoring system, performed all tissue pathology analysis, and contributed to the writing of the manuscript. J.B.W. contributed to data analysis and writing of the manuscript.
References
- Alonzo F, 3rd, Torres VJ. The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol Mol Biol Rev. 2014;78:199–230. doi: 10.1128/MMBR.00055-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus DC. The search for effective therapy for sepsis: back to the drawing board? JAMA. 2011;306:2614–2615. doi: 10.1001/jama.2011.1853. [DOI] [PubMed] [Google Scholar]
- Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:840–851. doi: 10.1056/NEJMra1208623. [DOI] [PubMed] [Google Scholar]
- Ani C, Farshidpanah S, Bellinghausen Stewart A, Nguyen HB. Variations in organism-specific severe sepsis mortality in the United States: 1999–2008. Crit Care Med. 2015;43:65–77. doi: 10.1097/CCM.0000000000000555. [DOI] [PubMed] [Google Scholar]
- Bayer AS, Ramos MD, Menzies BE, Yeaman MR, Shen AJ, Cheung AL. Hyperproduction of alpha-toxin by Staphylococcus aureus results in paradoxically reduced virulence in experimental endocarditis: a host defense role for platelet microbicidal proteins. Infect Immun. 1997;65:4652–4660. doi: 10.1128/iai.65.11.4652-4660.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker RE, Berube BJ, Sampedro GR, DeDent AC, Bubeck Wardenburg J. Tissue-specific patterning of host innate immune responses by Staphylococcus aureus α-toxin. J Innate Immun. 2014;6:619–631. doi: 10.1159/000360006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender M, Hofmann S, Stegner D, Chalaris A, Bosl M, Braun A, Scheller J, Rose-John S, Nieswandt B. Differentially regulated GPVI ectodomain shedding by multiple platelet-expressed proteinases. Blood. 2010;116:3347–3355. doi: 10.1182/blood-2010-06-289108. [DOI] [PubMed] [Google Scholar]
- Berube BJ, Bubeck Wardenburg J. Staphylococcus aureus α-toxin: nearly a century of intrigue. Toxins (Basel) 2013;5:1140–1166. doi: 10.3390/toxins5061140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakdi S, Muhly M, Mannhardt U, Hugo F, Klapettek K, Mueller-Eckhardt C, Roka L. Staphylococcal alpha toxin promotes blood coagulation via attack on human platelets. J Exp Med. 1988;168:527–542. doi: 10.1084/jem.168.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakdi S, Muhly M, Korom S, Hugo F. Release of interleukin-1 beta associated with potent cytocidal action of staphylococcal alpha-toxin on human monocytes. Infect Immun. 1989;57:3512–3519. doi: 10.1128/iai.57.11.3512-3519.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KK, Henson PM, Maclouf J, Moyle M, Ely JA, Worthen GS. Neutrophil-platelet adhesion: relative roles of platelet P-selectin and neutrophil beta2 (DC18) integrins. Am J Respir Cell Mol Biol. 1998;18:100–110. doi: 10.1165/ajrcmb.18.1.2314. [DOI] [PubMed] [Google Scholar]
- Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, Toy P, Werb Z, Looney MR. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122:2661–2671. doi: 10.1172/JCI61303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, Ting JP, Duncan JA. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE. 2009;4:e7446. doi: 10.1371/journal.pone.0007446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Gao Y, Xie Y, Wang Y, Cai Y, Shao X, Ma X, Li Y, Ma G, Liu G, et al. An ADAM10 promoter polymorphism is a functional variant in severe sepsis patients and confers susceptibility to the development of sepsis. Crit Care. 2015;19:73. doi: 10.1186/s13054-015-0796-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AM, Potts JR, Josefsson E, Massey RC. Staphylococcus aureus host cell invasion and virulence in sepsis is facilitated by the multiple repeats within FnBPA. PLoS Pathog. 2010;6:e1000964. doi: 10.1371/journal.ppat.1000964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann M, Lai QJ, Albrecht RM, Mosher DF, Proctor RA. Adhesion of Staphylococcus aureus to surface-bound platelets: role of fibrinogen/fibrin and platelet integrins. J Infect Dis. 1993;167:312–322. doi: 10.1093/infdis/167.2.312. [DOI] [PubMed] [Google Scholar]
- Hultgren OH, Svensson L, Tarkowski A. Critical role of signaling through IL-1 receptor for development of arthritis and sepsis during Staphylococcus aureus infection. J Immunol. 2002;168:5207–5212. doi: 10.4049/jimmunol.168.10.5207. [DOI] [PubMed] [Google Scholar]
- Inoshima I, Inoshima N, Wilke GA, Powers ME, Frank KM, Wang Y, Bubeck Wardenburg J. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat Med. 2011;17:1310–1314. doi: 10.1038/nm.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoshima N, Wang Y, Bubeck Wardenburg J. Genetic requirement for ADAM10 in severe Staphylococcus aureus skin infection. J Invest Dermatol. 2012;132:1513–1516. doi: 10.1038/jid.2011.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebaier C, Chamberland RR, Allen IC, Gao X, Broglie PM, Hall JD, Jania C, Doerschuk CM, Tilley SL, Duncan JA. Staphylococcus aureus α-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J Infect Dis. 2012;205:807–817. doi: 10.1093/infdis/jir846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MSS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Lee WL, Slutsky AS. Sepsis and endothelial permeability. N Engl J Med. 2010;363:689–691. doi: 10.1056/NEJMcibr1007320. [DOI] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- Li Z, Delaney MK, O'Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30:2341–2349. doi: 10.1161/ATVBAHA.110.207522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YJ, Fan YH, Tang J, Li JB, Yu CH. Meprin-β regulates production of pro-inflammatory factors via a disintegrin and metalloproteinase-10 (ADAM-10) dependent pathway in macrophages. Int Immunopharmacol. 2014;18:77–84. doi: 10.1016/j.intimp.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Liu S, Zhang Y, Moayeri M, Liu J, Crown D, Fattah RJ, Wein AN, Yu ZX, Finkel T, Leppla SH. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature. 2013;501:63–68. doi: 10.1038/nature12510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looney MR, Nguyen JX, Hu Y, Van Ziffle JA, Lowell CA, Matthay MA. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Invest. 2009;119:3450–3461. doi: 10.1172/JCI38432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11:519–531. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. 2011;6:147–163. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer K, Reyes-Robles T, Alonzo F, 3rd, Durbin J, Torres VJ, Cadwell K. Autophagy mediates tolerance to Staphylococcus aureus alpha-toxin. Cell Host Microbe. 2015;17:429–440. doi: 10.1016/j.chom.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAdow M, Missiakas DM, Schneewind O. Staphylococcus aureussecretes coagulase and von Willebrand factor binding protein tomodify the coagulation cascade and establish host infections. J Innate Immun. 2012;4:141–148. doi: 10.1159/000333447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald B, Jenne CN, Zhuo L, Kimata K, Kubes P. Kupffer cells and activation of endothelial TLR4 coordinate neutrophil adhesion within liver sinusoids during endotoxemia. Am J Physiol Gastrointest Liver Physiol. 2013;305:G797–G806. doi: 10.1152/ajpgi.00058.2013. [DOI] [PubMed] [Google Scholar]
- Moroi M, Jung SM. Platelet glycoprotein VI: its structure and function. Thromb Res. 2004;114:221–233. doi: 10.1016/j.thromres.2004.06.046. [DOI] [PubMed] [Google Scholar]
- Muñoz-Planillo R, Franchi L, Miller LS, Núñ ez G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol. 2009;183:3942–3948. doi: 10.4049/jimmunol.0900729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesseler N, Launey Y, Aninat C, Morel F, Mallé dant Y, Seguin P. Clinical review: The liver in sepsis. Crit Care. 2012;16:235. doi: 10.1186/cc11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann FJ, Marx N, Gawaz M, Brand K, Ott I, Rokitta C, Sticherling C, Meinl C, May A, Schö mig A. Induction of cytokine expression in leukocytes by binding of thrombin-stimulated platelets. Circulation. 1997;95:2387–2394. doi: 10.1161/01.cir.95.10.2387. [DOI] [PubMed] [Google Scholar]
- Nieswandt B, Brakebusch C, Bergmeier W, Schulte V, Bouvard D, Mokhtari-Nejad R, Lindhout T, Heemskerk JW, Zirngibl H, Fässler R. Glycoprotein VI but not alpha2beta1 integrin is essential for platelet interaction with collagen. EMBO J. 2001;20:2120–2130. doi: 10.1093/emboj/20.9.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard TK, Pallister KB, Zurek OW, Voyich JM. The impact of α-toxin on host cell plasma membrane permeability and cytokine expression during human blood infection by CA-MRSA USA300. J Leukoc Biol. 2013;94:971–979. doi: 10.1189/jlb.0213080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura CY, Nizet V. Subterfuge and sabotage: evasion of host innate defenses by invasive gram-positive bacterial pathogens. Annu Rev Microbiol. 2014;68:439–458. doi: 10.1146/annurev-micro-092412-155711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parimon T, Li Z, Bolz DD, McIndoo ER, Bayer CR, Stevens DL, Bryant AE. Staphylococcus aureus α-hemolysin promotes platelet-neutrophil aggregate formation. J Infect Dis. 2013;208:761–770. doi: 10.1093/infdis/jit235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanowska-Grabowska R, Wallace K, Field JJ, Chen L, Marshall MA, Figler R, Gear ARL, Linden J. P-selectin-mediated platelet-neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arterioscler Thromb Vasc Biol. 2010;30:2392–2399. doi: 10.1161/ATVBAHA.110.211615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers ME, Bubeck Wardenburg J. Igniting the fire: Staphylococcus aureus virulence factors in the pathogenesis of sepsis. PLoS Pathog. 2014;10:e1003871. doi: 10.1371/journal.ppat.1003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers ME, Kim HK, Wang Y, Bubeck Wardenburg J. ADAM10 mediates vascular injury induced by Staphylococcus aureus α-he-molysin. J Infect Dis. 2012;206:352–356. doi: 10.1093/infdis/jis192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaiah SK, Jaeschke H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol. 2007;35:757–766. doi: 10.1080/01926230701584163. [DOI] [PubMed] [Google Scholar]
- Reiss K, Saftig P. The “a disintegrin and metalloprotease” (ADAM) family of sheddases: physiological and cellular functions. Semin Cell Dev Biol. 2009;20:126–137. doi: 10.1016/j.semcdb.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Sampedro GR, DeDent AC, Becker RE, Berube BJ, Gebhardt MJ, Cao H, Bubeck Wardenburg J. Targeting Staphylococcus aureus α-toxin as a novel approach toreduce severity ofrecurrent skin and soft-tissue infections. J Infect Dis. 2014;210:1012–1018. doi: 10.1093/infdis/jiu223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer G, Urakami H, Specian RD, Stokes KY, Granger DN. Platelet recruitment in the murine hepatic microvasculature during experimental sepsis: role of neutrophils. Microcirculation. 2006;13:89–97. doi: 10.1080/10739680500466343. [DOI] [PubMed] [Google Scholar]
- Spaan AN, Surewaard BGJ, Nijland R, van Strijp JAG. Neutrophils versus Staphylococcus aureus: a biological tug of war. Annu Rev Microbiol. 2013;67:629–650. doi: 10.1146/annurev-micro-092412-155746. [DOI] [PubMed] [Google Scholar]
- Thomer L, Schneewind O, Missiakas D. Multiple ligands of von Willebrand factor-binding protein (vWbp) promote Staphylococcus aureus clot formation in human plasma. J Biol Chem. 2013;288:28283–28292. doi: 10.1074/jbc.M113.493122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronik-Le Roux D, Roullot V, Poujol C, Kortulewski T, Nurden P, Marguerie G. Thrombasthenic mice generated by replacement of the integrin alpha(IIb) gene: demonstration that transcriptional activation of this megakaryocytic locus precedes lineage commitment. Blood. 2000;96:1399–1408. [PubMed] [Google Scholar]
- Valeva A, Walev I, Pinkernell M, Walker B, Bayley H, Palmer M, Bhakdi S. Transmembrane beta-barrel of staphylococcal alpha-toxin forms in sensitive but not in resistant cells. Proc Natl Acad Sci USA. 1997;94:11607–11611. doi: 10.1073/pnas.94.21.11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyrich AS, Elstad MR, McEver RP, McIntyre TM, Moore KL, Morrissey JH, Prescott SM, Zimmerman GA. Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest. 1996;97:1525–1534. doi: 10.1172/JCI118575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke GA, Bubeck Wardenburg J. Role of a disintegrin and met-alloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc Natl Acad Sci USA. 2010;107:13473–13478. doi: 10.1073/pnas.1001815107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CH, Jenne CN, Petri B, Chrobok NL, Kubes P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol. 2013;14:785–792. doi: 10.1038/ni.2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaman MR. Platelets in defense against bacterial pathogens. Cell Mol Life Sci. 2010;67:525–544. doi: 10.1007/s00018-009-0210-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest. 2006;116:3211–3219. doi: 10.1172/JCI29499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.