

Abstract
While gene mutations in the amyloid precursor protein (APP) and the presenilins lead to an accumulation of the amyloid β-peptide (Aβ) in the brain causing neurodegeneration and familial Alzheimer's disease (AD), over 95% of all AD cases are sporadic. Despite the pathologies being indistinguishable, relatively little is known about the mechanisms affecting generation of Aβ in the sporadic cases. Vascular disorders such as ischaemia and stroke are well established risk factors for the development of neurodegenerative diseases and systemic hypoxic episodes have been shown to increase Aβ production and accumulation. We have previously shown that hypoxia causes a significant decrease in the expression of the major Aβ-degrading enzyme neprilysin (NEP) which might deregulate Aβ clearance. Aβ itself is derived from the transmembrane APP along with several other biologically active metabolites including the C-terminal fragment (CTF) termed the APP intracellular domain (AICD), which regulates the expression of NEP and some other genes in neuronal cells. Here we show that in hypoxia there is a significantly increased expression of caspase-3, 8, and 9 in human neuroblastoma NB7 cells, which can degrade AICD. Using chromatin immunoprecipitation we have revealed that there was also a reduction of AICD bound to the NEP promoter region which underlies the decreased expression and activity of the enzyme under hypoxic conditions. Incubation of the cells with a caspase-3 inhibitor Z-DEVD-FMK could rescue the effect of hypoxia on NEP activity protecting the levels of AICD capable of binding the NEP promoter. These data suggest that activation of caspases might play an important role in regulation of NEP levels in the brain under pathological conditions such as hypoxia and ischaemia leading to a deficit of Aβ clearance and increasing the risk of development of AD.
Keywords: neprilysin, hypoxia, caspases, AICD, APP
Introduction
Cerebral hypoxia is a condition in which the brain is deprived of oxygen and, as a result, cells elicit a wide range of adaptive responses and major metabolic alterations. Currently, vascular disorders such as ischaemia and stroke are considered as established risk factors for the development of neurodegenerative diseases, in particular of Alzheimer's disease (AD) (O'Brien and Markus, 2014). The pathology in familial AD is caused by overproduction and accumulation in the brain of abnormally high concentrations of the amyloid-β (Aβ) peptide and its oligomers causing synaptic loss and neuronal cell death (Hardy and Higgins, 1992; Walsh et al., 2002; Hardy, 2009). It is now becoming evident, however, that Aβ levels in the brain represent a dynamic equilibrium between its production from the amyloid precursor protein (APP) and removal by a cohort of amyloid clearance proteins which can be either enzymes (proteases) or binding/transport proteins. The group of enzymes capable of proteolytic degradation of Aβ currently embraces more than 20 members (for review, see Nalivaeva et al., 2012a, 2014). Several of the main amyloid-degrading enzymes (ADEs) are members of the neprilysin peptidase family: NEP, NEP2 and endothelin converting enzymes (ECE-1 and ECE-2) (Turner, 2003; Marr and Spencer, 2010; Nalivaeva et al., 2012a; Pacheco-Quinto and Eckman, 2013). Another metallopeptidase which plays an important role in Aβ metabolism is insulin-degrading enzyme (IDE) (Qiu and Folstein, 2006; Leissring and Turner, 2013). With aging and under pathological conditions expression levels and activity of these enzymes decline (Nalivaeva et al., 2004; Caccamo et al., 2005; Kochkina et al., 2015) leading to an amyloid clearance deficit, which is now considered to be one of the major factors of the sporadic form of AD (Selkoe, 2012; Pluta et al., 2013).
As we have shown earlier, hypoxia leads to reduced NEP levels and activity both in cellular models and in vivo (Nalivaeva et al., 2004, 2012b; Fisk et al., 2007). A prolonged exposure to a hypoxic environment has also been reported to increase Aβ levels significantly accelerating the hyperphosphorylation of tau and contributing to neuronal cell death (Jendroska et al., 1995; Li et al., 2009; Fang et al., 2010). Regulation of NEP expression is complex as the enzyme appears to have a constitutive regulatory pathway (D'Adamio et al., 1989; Li et al., 1995) as well as an epigenetically-regulated component (Pardossi-Piquard et al., 2005; Belyaev et al., 2009). The latter involves competitive binding of a transcription factor, namely the APP intracellular domain (AICD) produced in the β-secretase amyloidogenic pathway (Belyaev et al., 2010), to the NEP gene promoter leading to activation of mRNA synthesis while histone deacetylases inhibit this process. As the effects of hypoxia on NEP expression may represent an important pathological trigger in AD, the factors affecting NEP dysregulation under these conditions need to be better understood. AICD is an extremely labile peptide being a substrate of various intracellular peptidases including caspases (Bertrand et al., 2001) which might result in dysregulation of AICD-dependent NEP expression under various pathological conditions related to caspase activation. In particular, hypoxia was shown to be accompanied by increased levels of caspase expression and activity in the brain (Khurana et al., 2002). The aim of this study was to assess whether activation of caspases might be a factor leading to dysregulation of NEP gene expression and activity under hypoxic conditions. For this we have employed human neuroblastoma NB7 cells which possess high endogenous levels of NEP and, as has been demonstrated, are responsive to hypoxia (Fisk et al., 2007; Belyaev et al., 2009).
Methods
Cell culture and hypoxia treatment
The NB7 (SJ-N-CG) neuroblastoma cell line, which expresses high endogenous levels of NEP, was obtained from St Jude Children's Research Hospital (Memphis, USA, kind gift of Dr. Vincent J. Kidd). The NB7 cells were cultured in RPMI-1640 media supplemented with 10% (v/v) fetal bovine serum, 50 units/ml penicillin, 50 μg/ml streptomycin and 2 mM glutamine (all from Cambrex Bio Science Ltd., Wokingham, Berkshire, UK) at 37°C in 5% (v/v) CO2 and sub-cultured every 7 days. After reaching the confluent stage, cells were incubated in an O2/CO2 incubator (MC0-175M, Sanyo) for 24 h under 1% O2. The cells were collected 24 (or 48 h) later, washed twice with 10 ml PBS, scraped into 10 ml of PBS (pH 7.2), pelleted at 3000 g for 5 min and used for mRNA and protein content analysis as well as for the activity assays.
Cell viability determination by trypan blue exclusion
Cells were washed twice in PBS, incubated in trypsin/EDTA for 5 min at 37°C and knocked from the surface of the flask prior to adding 5 ml of media then pelleting at 400 g for 5 min. Pellets were resuspended in 1 ml of media and a 1:1 dilution of cell suspension in 4% trypan blue was prepared. Twenty microlitres of cell suspension was loaded under a cover slip on a haemocytometer and allowed to fill the chambers by capillary action. The number of total and viable cells was determined by viewing under a light microscope at 40X magnification. Non-viable cells take up the trypan blue and appear dark (Strober, 2001).
Gene expression analysis
Cell RNA was prepared using the RNeasy extraction kit (Qiagen, Crawley, UK) according to the manufacturer's protocol. RNA was treated with DNase I (Invitrogen, Paisley, UK) and cDNA was prepared using the iScript cDNA kit (BioRad, UK). cDNA was amplified using conventional PCR or real-time PCR as in Zuccato et al. (2007). DNA amplified by conventional PCR was analyzed in 2% agarose gels containing ethidium bromide (1 μg/ml) and visualized on a Molecular Imager Gel Doc XR System with Quantity One 4.6.1 programme (BioRad). Image densitometry was performed using Aida Array Analyzer 4.15 software. Real-time PCR was performed in an iCycler Thermal Cycler with Multicolour PCR detection system, (Biorad, Hercules, CA) using SYBR Green (BioRad) incorporation and expression reported relative to actin mRNA. Primer sequences used were as follows:
NEP F- CCTGGAGATTCATAATGGATCTTGT
R- AAAGGGCCTTGCGGAAAG
Caspase-1 F- TGCTTTCTGCTCTTCAACACC
R-CACAAGACCAGGCATATTCTTTC
Caspase-3 F-GAGGCCGACTTCTTGTATGC
R-AATTCTGTTGCCACCTTTCG
Caspase-8 F-AGAGCCTGAGGGAAAGATGTC
R-TCACATCATAGTTCACGCCAGT
Caspase-9 F-CGTGGTGGTCATCCTCTCTC
R-GAGCATCCATCTGTGCCATA
U6 F- CTCGCTTCGGCAGCACA
R- AACGCTTCACGAATTTGCGT
Actin F- CGCAGCAGTCAGGGACATTT
R- TTCACATACAGCTTGGGAAGC
Preparation of cell lysates
Cells were washed twice with phosphate-buffered saline (PBS: 1.5 mM KH2PO4, 2.7 mM Na2HPO4, 150 mM NaCl, pH 7.4), harvested and pelleted by centrifugation. For detection of proteins from cell lysates, cells were lysed in RIPA buffer [10 mM Tris/HCl pH 8.0, 150 mM NaCl, 1% (v/v) Nonidet P-40, 0.5% (w/v) sodium deoxycholate, 5 mM EDTA, 1X complete inhibitor mix (Roche diagnostics)] on ice for 20 min. Lysates were homogenized through 22 G needles 10 times then clarified by centrifugation at 2800 g for 10 min. Protein concentration of lysates was determined using the bicinchoninic acid (BCA) assay (Sigma Aldrich).
Electrophoresis and western blotting
Samples (30 μg or, for AICD detection, 50 μg, protein) were resolved on 10% polyacrylamide gels or, for AICD detection, 10–20% tricine gels (Invitrogen, Paisley, UK), and transferred onto Hybond-P poly(vinylidene) difluoride (PVDF) membranes (Amersham Life Sciences, Buckinghamshire, UK). The membranes were blocked overnight at 4°C in Tris-buffered saline (TBS: 50 mM Tris, 150 mM NaCl) containing 0.1% (v/v) Tween-20 (TBST) and 5% (w/v) skimmed milk powder. Membranes were incubated with the primary antibodies for 3 h. Primary antibodies used for blotting included: NEP (1:100, mouse monoclonal, NCL-CD10-270, Novocastra, Newcastle, UK or rabbit anti-rat polyclonal antibody, US Biological); GLUT-1 (1:500, rabbit polyclonal, from Prof. S. Baldwin, University of Leeds); anti- β-actin (1:500, rabbit polyclonal, Sigma-Aldrich Co., Poole, Dorset, UK); anti-APP C-Terminal fragment A8717 and anti-actin (both Sigma-Aldrich) used at 1:1000 and 1:10,000, respectively. Membranes were washed in TBST for 1 h before incubation with peroxidase-conjugated rabbit anti-mouse or donkey anti-rabbit secondary antibodies (GE Healthcare, Bucks, UK) at a dilution of 1:4000. Membranes were washed further before detection of proteins using the enhanced chemiluminescence method. Western blots were quantified using AIDA image analyzer v.4.22 (Straubenhardt, Germany).
Neprilysin activity assay
NEP activity was measured in a two-step coupled fluorescence assay. NEP cleaves between Ala-Phe of the fluorogenic substrate succinoyl-Ala-Ala-Phe-7-amido-4-methylcoumarin (Mumford et al., 1980) and, in the presence of leucine aminopeptidase, releases the fluorophore (AMC) from Phe and prevents quenching allowing fluorescence to be detected. Protein samples (1 μg) were loaded into opaque 96-well plates in 100 mM Tris-HCl (pH 7.4) with or without 20 μM of the NEP inhibitor thiorphan and incubated at 37°C for 20 min. The substrate Suc-AAF-AMC (50 μM) and Leu-aminopeptidase (40 munits/ml) were added to each well (100 μl total volume/well) and fluorescence was observed on a FLUOstar Omega multidetection microplate reader (excitation = 355 nm, emission = 460 nm) at suitably frequent time points. NEP activity was determined as the difference between fluorescence measurements in the absence and presence of 20 μM thiorphan.
Caspase 3/7 activity assay
Caspase-3 and caspase-7 activities were determined using a luminescent Caspase-Glo® 3/7 activity assay (Promega, Southampton, UK) as per manufacturers' instruction. Briefly, approximately 20,000 cells were added to each well of a 96 well, white-walled plate. At room temperature, the Caspase-Glo reagent was made up by mixing the Caspase-Glo substrate with the Caspase-Glo buffer and 100 μl was added to each well. Contents were mixed by shaking for 1 min then incubated at 37°C for 1 h. Luminescence was measured on a MicroLumat Plus LB 96V (Berthold Technologies).
Chromatin immunoprecipitation analysis
ChIP was performed as described previously (Zuccato et al., 2007; Belyaev et al., 2009). Cells were fixed, extracts sonicated and, following incubation with protein-G-Sepharose, primary antibodies were applied overnight. After de-crosslinking and DNA extraction, analysis was performed by real time PCR. Data are represented as the fold enrichment of DNA pulled down with the specific antibody over that immunoprecipitated with IgG. Antibodies used were anti-AICD (Covance, Harrogate) and anti-IgG (mouse) (negative control) (Abcam). Promoter primer sequences used were as follows:
NEP F- GGTGCGGGTCGGAGGGATGC
R- CTCCCAGCGCCCTGGGCGCTCG
Statistics
All results are given as mean ± SEM from at least three experiments. Results were compared using unpaired two-tailed Student's t-test with a threshold of p < 0.05.
Results
Cell viability and GLUT1 expression in NB7 cells after hypoxia
Hypoxia elicits a wide range of cellular adaptations and the severity and duration of a hypoxic episode can significantly affect hypoxia-induced apoptosis (Banasiak and Haddad, 1998). In order to show that the NB7 cells were responding to hypoxic conditions and were still viable, a cell viability assay was carried out as well as a Western blot analysis of the established hypoxia marker, GLUT1. Acting through the hypoxia- inducible factor-1, GLUT-1 expression is enhanced in hypoxia, stimulating glucose transport and maintaining cell homeostasis (Ouiddir et al., 1999; Zhang et al., 1999). After incubation at 1% O2 for 24 h, the viability of NB7 cells was not significantly different from those incubated under normal conditions (Figure 1A). In both conditions cell viability was above 90%. Although GLUT1 protein content was detected at high levels in both normoxia and hypoxia incubated cells, 24 h of hypoxia increased GLUT-1 protein levels significantly suggesting that the cells were responding to hypoxic insult by changing the level of expression of hypoxia-related genes (Figure 1B).
Figure 1.
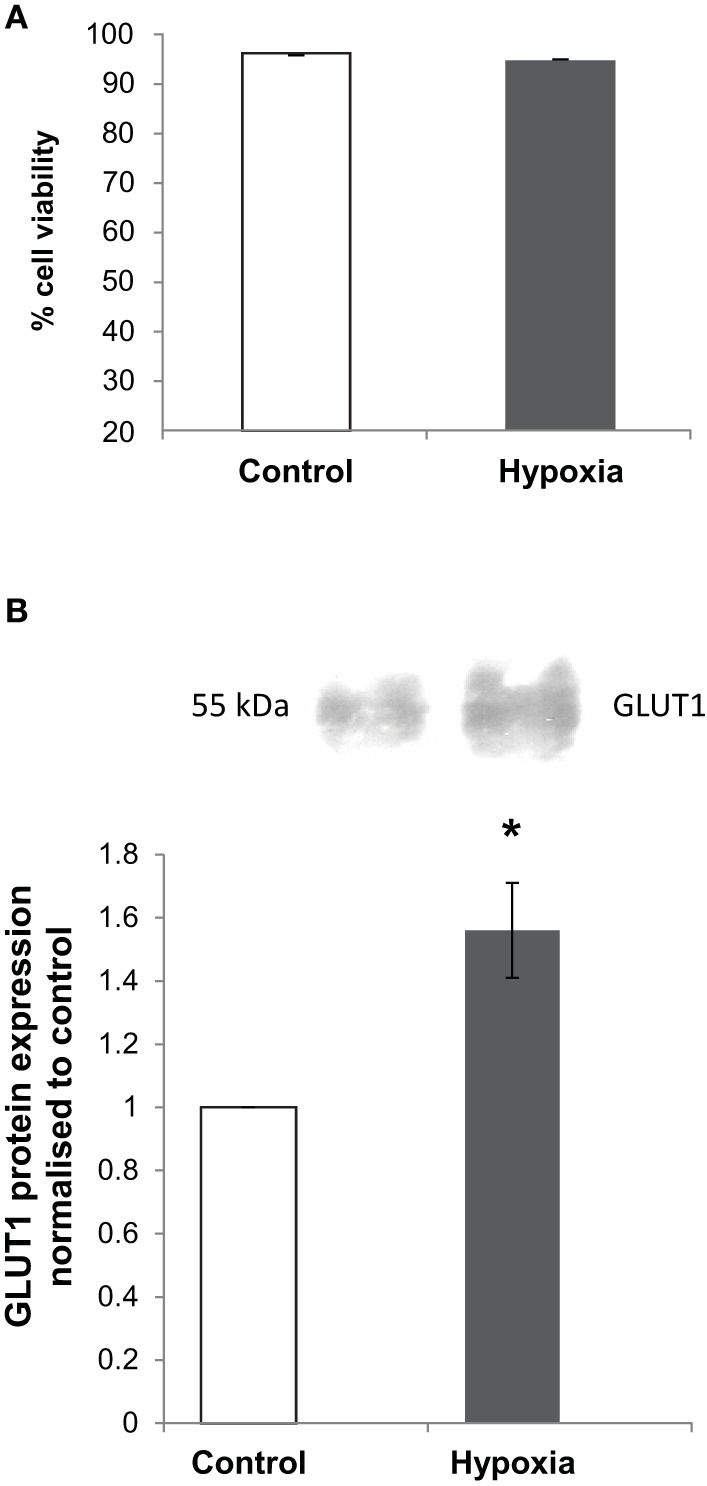
The effects of hypoxia on cell viability and GLUT1 protein levels in NB7 cells. (A) A cell viability assay carried out in NB7 cells subjected to 24 h normoxia or hypoxia (1% O2). (B) Representative Western blot and calculation of GLUT1 levels in whole cell lysates of NB7 cells subjected to 24 h normoxic or hypoxic (1% O2) conditions. Bars represent mean ± SEM, *p < 0.05 (n = 3).
The effects of hypoxia on NEP mRNA expression, protein level, and activity
Assessing the effect of hypoxia on NEP expression in NB7 cells we have confirmed that both NEP mRNA and protein levels in the cells subjected to hypoxia for 24 h were reduced by approximately 30% compared to the control cells (Figures 2A,B). Similarly, a fluorogenic NEP enzyme activity assay has demonstrated that NEP activity was reduced in hypoxia by approximately 40% (Figure 2C).
Figure 2.

The effects of hypoxia on NEP expression and activity. (A) NEP mRNA expression levels normalized to U6; (B) NEP protein levels showing a representative blot with CD10 antibody against human NEP and β-actin as loading control; (C) NEP enzyme activity (nmol/min/mg) in NB7 cells subjected to 24 h normoxia or 24 h hypoxia (1% O2). Bars represent mean ± SEM, *p < 0.05 (n = 5).
The effects of hypoxia on mRNA expression and activity of members of the caspase family
Caspases are a family of cysteine proteases that are crucial mediators of apoptosis. Hypoxic conditions are known to induce cellular apoptosis and the family of caspases are participants in the hypoxia-induced apoptotic cascade (Khurana et al., 2002; Li et al., 2005). In order to investigate the effect of hypoxia on the expression of caspases in our cellular model, mRNA levels for caspases-1, -8, and -9 were determined by real-time PCR and a caspase-3/-7 activity assay was also carried out. A three to five-fold increase in mRNA was detected for caspase-1 and -9 (Figures 3A,C). In accordance with other studies (Finlay et al., 2010), caspase-8 expression was extremely low in the NB7 cells. However, despite endogenously low levels, hypoxia also increased its mRNA expression (Figure 3B). Since caspase-3 and caspase-7 exhibit similar substrate specificity, the analysis of their activity was combined. Total caspase -3/-7 activity was increased by approximately four-fold in the NB7 cells incubated in hypoxic conditions compared with control (Figure 3D).
Figure 3.

The effect of hypoxia on caspase-1,-8, and -9 mRNA expression and caspase -3/-7 activity levels. Expression levels of (A) caspase-1, (B) caspase-8, (C) caspase-9 normalized to U6 in NB7 cells subjected to 24 h normoxia or 24 h hypoxia (1% O2). (D) Caspase-3/-7 activity levels in NB7 cells subjected to 24 h normoxia or 24 h hypoxia (1% O2). Bars represent mean ± SEM, *p < 0.05, **p < 0.01.
The effect of hypoxia on AICD enrichment of the NEP promoter
The APP C-terminal fragments (CTFs) have been identified as a target for caspase cleavage due to the presence of a caspase-specific cleavage site at aspartate residue 664 (for APP695 isoform) in the APP molecule (Weidemann et al., 1999). As caspases appear to be significantly elevated in hypoxia, ChIP analysis was used to determine whether the reduction in NEP may be due to reduced AICD binding to the NEP promoter region. After 24 h hypoxic incubation, AICD enrichment on the NEP promoter region was found to be significantly reduced by approximately five-fold (Figure 4B).
Figure 4.
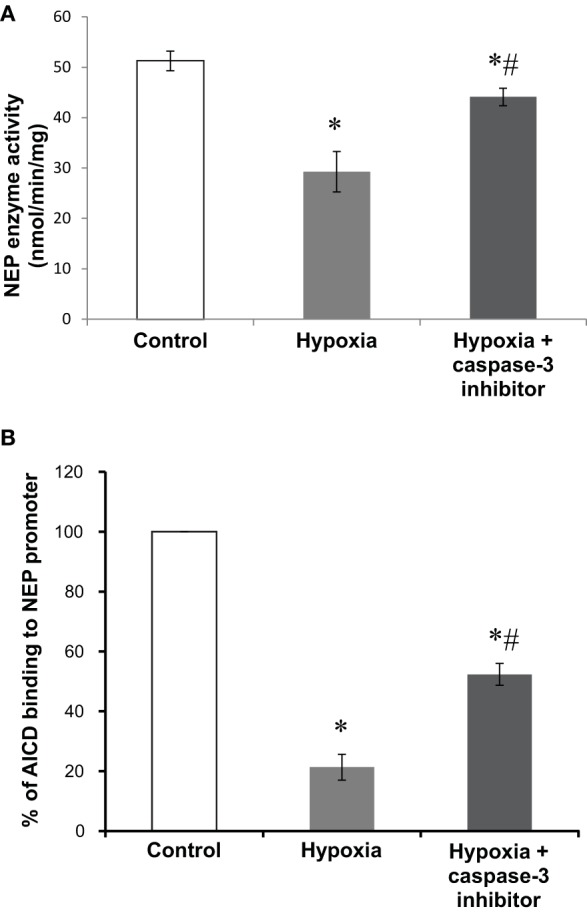
The effect of a caspase-3 inhibitor on NEP activity and AICD binding to the NEP promoter in hypoxia. (A) NEP enzyme activity (nmol/min/mg) and (B) enrichment of AICD on the NEP promoter analyzed by ChIP in NB7 cells subjected to 24 h normoxia or 24 h hypoxia (1% O2) with or without the caspase-3 inhibitor, Z-DEVD-FMK (100 μM, 24 h). Bars represent n of 3 ± SEM, *p < 0.05 compared to control, #p < 0.05 compared to hypoxia.
The effects of caspase inhibition in hypoxia on NEP activity and AICD enrichment on the NEP promoter
Caspase-3 has been identified as the predominant caspase involved in the cleavage of the APP-CTF, including AICD (Gervais et al., 1999), therefore in order to investigate whether caspase-3 is involved in the reduction of AICD-induced NEP expression, cells were subjected to hypoxia with or without the caspase-3 inhibitor, Z-DEVD-FMK. As previously shown, NEP activity was significantly reduced in hypoxia compared to control, but cell treatment with the caspase-3 inhibitor restored NEP activity to approximately 90% of the control level (Figure 4A). Similarly, ChIP analysis has revealed that caspase inhibitor treatment to a significant extent protected the level of AICD binding to the NEP gene promoter (Figure 4B). This testifies to the role of caspases, and especially of caspase-3, in reduction of NEP expression in neuronal cells subjected to hypoxic conditions.
Discussion
Hypoxia is an established factor for AD and brain deprivation of oxygen has been associated with an increase in Aβ levels and plaque deposition (Jendroska et al., 1995; Zhang and Le, 2010). The leading factor in Aβ accumulation might be deficit of its major degrading peptidase, NEP. Indeed, dysregulation of NEP after exposure to hypoxia has been reported in many areas of the body including the lungs (Carpenter and Stenmark, 2001; Wick et al., 2011), carotid body (Kumar et al., 1990), eye (Hara et al., 2006), and brain (Nalivaeva et al., 2004, 2012b; Oh-hashi et al., 2005). The data of the present study report significantly reduced NEP expression and activity in the human neuroblastoma NB7 cell line subjected to hypoxia, which are consistent with other reports.
Oxygen homeostasis is regulated by the hypoxia inducible factor (HIF)-1α (for review see Semenza, 2006). HIF-1α binds specifically to the hypoxia-responsive element (HRE) in gene promoter regions and in hypoxic conditions which can significantly alter cellular gene expression. The effects of hypoxia on APP processing have been widely studied and it is now well established that hypoxia increases the amyloidogenic pathway by facilitating the up-regulation of expression of β-secretase (BACE1), as well as components of the γ-secretase complex resulting in increased production both of Aβ40 and Aβ42 (Sun et al., 2006; Wang et al., 2006; Zhang et al., 2007; Li et al., 2009). Sequence analysis, mutagenesis and gel shift studies have revealed the binding of HIF-1α to both the BACE1 and APH-1A promoter regions and Notch processing has also been increased after hypoxic incubation (Wang et al., 2006; Zhang et al., 2007). Hypoxia has also been shown to decrease levels of metalloproteinases ADAM10 and ADAM17 expression and consequently reduce APP processing through the non-amyloidogenic, α-secretase pathway (Webster et al., 2004; Rybnikova et al., 2012). A reduction in mature ADAM10 and a concomitant increase in immature ADAM10 has also been reported to alter the regulation of α-secretase APP processing in hypoxia (Auerbach and Vinters, 2006).
The data in this paper provide evidence for an alternative mechanism by which hypoxia may increase Aβ levels. The hypoxia-induced activation of caspases may cause an increased cleavage of the APP-CTF and thus reduce AICD levels. This then results in the down-regulation of the AICD-mediated gene NEP and reduction of enzyme expression and activity which inevitably will lead to increase of Aβ content. It has been proven both in cell and animal models that NEP deficit is one of the factors leading to increased Aβ production and load in the brain (Iwata et al., 2000; Hanson et al., 2011) and up-regulation of NEP, on the contrary, leads to reduction of Aβ levels (Mohajeri et al., 2004; Kerridge et al., 2014). We have also shown that caspase-3 inhibition using the potent DEVD caspase inhibitor can protect NEP activity in the hypoxic conditions. Similar findings have been reported in dying motor-neurons deprived of trophic support. This type of cellular stress was shown to cause an increase in the expression of APP and the cell death protease caspase-3, as well as a concomitant increase in neurotoxic Aβ (Barnes et al., 1998). Although AICD and NEP levels were not investigated, treatment with a caspase-3 inhibitor significantly reduced the Aβ peptide content in these types of cells. Apoptotic stimuli activating the caspase-3 protease family have also been reported to cleave both PS1 and PS2 (Kim et al., 1997). As such, an alternative mechanism could involve an altered γ-secretase cleavage of APP thus generating APP-CTF incapable of translocating to the nucleus and/or regulating the transcription of NEP.
It was demonstrated that hypoxia affects NEP gene expression via histone modifications on its promoter region (Wang et al., 2011) which can affect accessibility of AICD to its binding site. As we have shown here, AICD binding to the NEP gene promoter is indeed reduced under hypoxic conditions but this decrease can be prevented by a caspase inhibitor. Because inhibition of caspase activity does not restore completely the level of AICD binding to the NEP promoter observed in the control cells, it is reasonable to suggest that either there are other peptidases active under hypoxic conditions that degrade AICD, or changes in the NEP promoter structure due to histone deacetylation are still interfering with its activation by AICD. As we have shown previously (Belyaev et al., 2009; Nalivaeva et al., 2012b), inhibition of histone deacetylases by valproic acid leads to significant but not complete restoration of NEP gene expression and NEP enzyme activity after hypoxia which confirms the role of histone modifications in NEP regulation under reduced oxygen supply.
AICD was also shown to regulate HIF-1α expression (Kaufmann et al., 2013) and, therefore, decreased levels of this transcription factor under hypoxic conditions might result in a modified response of cells to hypoxic insults. As we have established, the Aβ-clearing protein TTR is also AICD-regulated (Kerridge et al., 2014) and is shown to be responsive to hypoxia (Ahmad et al., 2014). Alterations of the levels of this protein under hypoxic conditions may also affect amyloid metabolism leading to dysbalance of its production and clearance.
There is an increasing body of evidence that AICD levels can be regulated by various pharmacological agents (Eisele et al., 2007; Bauer et al., 2011; Kerridge et al., 2014) which opens an avenue for designing a therapeutic strategy for regulating important AICD-dependent genes. Because AICD-regulated genes have different roles in amyloid metabolism and are cell- and tissue-specific (for review see Beckett et al., 2012; Pardossi-Piquard and Checler, 2012) understanding the effect of various physiological conditions on its ability to regulate expression of the major amyloid-degrading enzyme is of great importance. This work extends further our knowledge about the role of hypoxia in NEP regulation via AICD and shows that caspase inhibitors can be potentially beneficial for protecting the brain against harmful effects of low oxygen supply.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by U.K. Medical Research Council, Alzheimer's Research UK (ART/PhD2009/4), RFBR 13-04-00388, Programme of Russian Academy of Science “Fundamental Sciences to Medicine.”
References
- Ahmad Y., Sharma N. K., Ahmad M. F., Sharma M., Garg I., Bhargava K. (2014). Proteomic identification of novel differentiation plasma protein markers in hypobaric hypoxia-induced rat model. PLoS ONE 9:e98027. 10.1371/journal.pone.0098027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach I. D., Vinters H. V. (2006). Effects of anoxia and hypoxia on amyloid precursor protein processing in cerebral microvascular smooth muscle cells. J. Neuropathol. Exp. Neurol. 65, 610–620. 10.1097/00005072-200606000-00009 [DOI] [PubMed] [Google Scholar]
- Banasiak K. J., Haddad G. G. (1998). Hypoxia-induced apoptosis: effect of hypoxic severity and role of p53 in neuronal cell death. Brain Res. 797, 295–304. [DOI] [PubMed] [Google Scholar]
- Barnes N. Y., Li L., Yoshikawa K., Schwartz L. M., Oppenheim R. W., Milligan C. E. (1998). Increased production of amyloid precursor protein provides a substrate for caspase-3 in dying motoneurons. J. Neurosci. 18, 5869–5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer C., Pardossi-Piquard R., Dunys J., Roy M., Checler F. (2011). γ-Secretase-mediated regulation of neprilysin: influence of cell density and aging and modulation by imatinib. J. Alzheimers Dis. 27, 511–520. 10.3233/JAD-2011-110746 [DOI] [PubMed] [Google Scholar]
- Beckett C., Nalivaeva N. N., Belyaev N. D., Turner A. J. (2012). Nuclear signalling by membrane protein intracellular domains: the AICD enigma. Cell. Signal. 24, 402–409. 10.1016/j.cellsig.2011.10.007 [DOI] [PubMed] [Google Scholar]
- Belyaev N. D., Kellett K. A., Beckett C., Makova N. Z., Revett T. J., Nalivaeva N. N., et al. (2010). The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a β-secretase-dependent pathway. J. Biol. Chem. 285, 41443–41454. 10.1074/jbc.M110.141390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyaev N. D., Nalivaeva N. N., Makova N. Z., Turner A. J. (2009). Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: implications for Alzheimer disease. EMBO Rep. 10, 94–100. 10.1038/embor.2008.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand E., Brouillet E., Caillé I., Bouillot C., Cole G. M., Prochiantz A., et al. (2001). A short cytoplasmic domain of the amyloid precursor protein induces apoptosis in vitro and in vivo. Mol. Cell. Neurosci. 18, 503–511. 10.1006/mcne.2001.1030 [DOI] [PubMed] [Google Scholar]
- Caccamo A., Oddo S., Sugarman M. C., Akbari Y., LaFerla F. M. (2005). Age- and region-dependent alterations in Aβ-degrading enzymes: implications for Aβ-induced disorders. Neurobiol. Aging 26, 645–654. 10.1016/j.neurobiolaging.2004.06.013 [DOI] [PubMed] [Google Scholar]
- Carpenter T. C., Stenmark K. R. (2001). Hypoxia decreases lung neprilysin expression and increases pulmonary vascular leak. Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L941–L948. Available online at: http://ajplung.physiology.org/content/281/4/L941.long [DOI] [PubMed] [Google Scholar]
- D'Adamio L., Shipp M. A., Masteller E. L., Reinherz E. L. (1989). Organization of the gene encoding common acute lymphoblastic leukemia antigen (neutral endopeptidase 24.11): multiple miniexons and separate 5′ untranslated regions. Proc. Natl. Acad. Sci. U.S.A. 86, 7103–7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele Y. S., Baumann M., Klebl B., Nordhammer C., Jucker M., Kilger E. (2007). Gleevec increases levels of the amyloid precursor protein intracellular domain and of the amyloid-beta degrading enzyme neprilysin. Mol. Biol. Cell. 18, 3591–3600. 10.1091/mbc.E07-01-0035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H., Zhang L. F., Meng F. T., Du X., Zhou J. N. (2010). Acute hypoxia promote the phosphorylation of tau via ERK pathway. Neurosci. Lett. 474, 173–177. 10.1016/j.neulet.2010.03.037 [DOI] [PubMed] [Google Scholar]
- Finlay D., Richardson R. D., Landberg L. K., Howes A. L., Vuori K. (2010). Novel HTS strategy identifies TRAIL-sensitizing compounds acting specifically through the caspase-8 apoptotic axis. PLoS ONE 5:e13375. 10.1371/journal.pone.0013375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk L., Nalivaeva N. N., Boyle J. P., Peers C. S., Turner A. J. (2007). Effects of hypoxia and oxidative stress on expression of neprilysin in human neuroblastoma cells and rat cortical neurones and astrocytes. Neurochem. Res. 32, 1741–1748. 10.1007/s11064-007-9349-2 [DOI] [PubMed] [Google Scholar]
- Gervais F. G., Xu D., Robertson G. S., Vaillancourt J. P., Zhu Y., Huang J., et al. (1999). Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-β precursor protein and amyloidogenic Aβ peptide formation. Cell 97, 395–406. [DOI] [PubMed] [Google Scholar]
- Hanson L. R., Hafez D., Svitak A. L., Burns R. B., Li X., Frey W. H., et al. (2011). Intranasal phosphoramidon increases beta-amyloid levels in wild-type and NEP/NEP2-deficient mice. J. Mol. Neurosci. 43, 424–427. 10.1007/s12031-010-9460-8 [DOI] [PubMed] [Google Scholar]
- Hara H., Oh-hashi K., Yoneda S., Shimazawa M., Inatani M., Tanihara H., et al. (2006). Elevated neprilysin activity in vitreous of patients with proliferative diabetic retinopathy. Mol. Vis. 12, 977–982. Available online at: http://www.molvis.org/molvis/v12/a109/ [PubMed] [Google Scholar]
- Hardy J. (2009). The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J. Neurochem. 110, 1129–1134. 10.1111/j.1471-4159.2009.06181.x [DOI] [PubMed] [Google Scholar]
- Hardy J. A., Higgins G. A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185. [DOI] [PubMed] [Google Scholar]
- Iwata N., Tsubuki S., Takaki Y., Watanabe K., Sekiguchi M., Hosoki E., et al. (2000). Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat. Med. 6, 143–150. 10.1038/77399 [DOI] [PubMed] [Google Scholar]
- Jendroska K., Poewe W., Daniel S. E., Pluess J., Iwerssen-Schmidt H., Paulsen J., et al. (1995). Ischemic stress induces deposition of amyloid β immunoreactivity in human brain. Acta Neuropathol. 90, 461–466. [DOI] [PubMed] [Google Scholar]
- Kaufmann M. R., Barth S., Konietzko U., Wu B., Egger S., Kunze R., et al. (2013). Dysregulation of hypoxia-inducible factor by presenilin/γ-secretase loss-of-function mutations. J. Neurosci. 33, 1915–1926. 10.1523/JNEUROSCI.3402-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerridge C., Belyaev N. D., Nalivaeva N. N., Turner A. J. (2014). The Aβ-clearance protein transthyretin, like neprilysin, is epigenetically regulated by the amyloid precursor protein intracellular domain. J. Neurochem. 130, 419–431. 10.1111/jnc.12680 [DOI] [PubMed] [Google Scholar]
- Khurana P., Ashraf Q. M., Mishra O. P., Delivoria-Papadopoulos M. (2002). Effect of hypoxia on caspase-3, -8, and -9 activity and expression in the cerebral cortex of newborn piglets. Neurochem. Res. 27, 931–938. 10.1023/A:1020347732741 [DOI] [PubMed] [Google Scholar]
- Kim T. W., Pettingell W. H., Jung Y. K., Kovacs D. M., Tanzi R. E. (1997). Alternative cleavage of Alzheimer-associated presenilins during apoptosis by a caspase-3 family protease. Science 277, 373–376. [DOI] [PubMed] [Google Scholar]
- Kochkina E. G., Plesneva S. A., Vasilev D. S., Zhuravin I. A., Turner A. J., Nalivaeva N. N. (2015). Effects of ageing and experimental diabetes on insulin-degrading enzyme expression in male rat tissues. Biogerontology 16, 473–484. 10.1007/s10522-015-9569-9 [DOI] [PubMed] [Google Scholar]
- Kumar G. K., Runold M., Ghai R. D., Cherniack N. S., Prabhakar N. R. (1990). Occurrence of neutral endopeptidase activity in the cat carotid body and its significance in chemoreception. Brain Res. 517, 341–343. [DOI] [PubMed] [Google Scholar]
- Leissring M. A., Turner A. J. (2013). Regulation of distinct pools of amyloid β-protein by multiple cellular proteases. Alzheimers Res. Ther. 5, 37. 10.1186/alzrt194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Booze R. M., Hersh L. B. (1995). Tissue-specific expression of rat neutral endopeptidase (neprilysin) mRNAs. J. Biol. Chem. 270, 5723–5728. [DOI] [PubMed] [Google Scholar]
- Li L., Zhang X., Yang D., Luo G., Chen S., Le W. (2009). Hypoxia increases Aβ generation by altering β- and γ-cleavage of APP. Neurobiol. Aging 30, 1091–1098. 10.1016/j.neurobiolaging.2007.10.011 [DOI] [PubMed] [Google Scholar]
- Li Y., Zhou C., Calvert J. W., Colohan A. R., Zhang J. H. (2005). Multiple effects of hyperbaric oxygen on the expression of HIF-1 alpha and apoptotic genes in a global ischemia-hypotension rat model. Exp. Neurol. 191, 198–210. 10.1016/j.expneurol.2004.08.036 [DOI] [PubMed] [Google Scholar]
- Marr R. A., Spencer B. J. (2010). NEP-like endopeptidases and Alzheimer's disease. Curr. Alzheimer Res. 7, 223–229. 10.2174/156720510791050849 [DOI] [PubMed] [Google Scholar]
- Mohajeri M. H., Kuehnle K., Li H., Poirier R., Tracy J., Nitsch R. M. (2004). Anti-amyloid activity of neprilysin in plaque-bearing mouse models of Alzheimer's disease. FEBS Lett. 562, 16–21. 10.1016/S0014-5793(04)00169-3 [DOI] [PubMed] [Google Scholar]
- Mumford R. A., Strauss A. W., Powers J. C., Pierzchala P. A., Nishino N., Zimmerman M. (1980). A zinc metalloendopeptidase associated with dog pancreatic membranes. J. Biol. Chem. 255, 2227–2230. [PubMed] [Google Scholar]
- Nalivaeva N. N., Beckett C., Belyaev N. D., Turner A. J. (2012a). Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer's disease? J. Neurochem. 120(Suppl. 1), 167–185. 10.1111/j.1471-4159.2011.07510.x [DOI] [PubMed] [Google Scholar]
- Nalivaeva N. N., Belyaev N. D., Kerridge C., Turner A. J. (2014). Amyloid-clearing proteins and their epigenetic regulation as a therapeutic target in Alzheimer's disease. Front. Aging Neurosci. 6:235. 10.3389/fnagi.2014.00235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalivaeva N. N., Belyaev N. D., Lewis D. I., Pickles A. R., Makova N. Z., Bagrova D. I., et al. (2012b). Effect of sodium valproate administration on brain neprilysin expression and memory in rats. J. Mol. Neurosci. 46, 569–577. 10.1007/s12031-011-9644-x [DOI] [PubMed] [Google Scholar]
- Nalivaeva N. N., Fisk L., Kochkina E. G., Plesneva S. A., Zhuravin I. A., Babusikova E., et al. (2004). Effect of hypoxia/ischemia and hypoxic preconditioning/reperfusion on expression of some amyloid-degrading enzymes. Ann. N.Y. Acad. Sci. 1035, 21–33. 10.1196/annals.1332.002 [DOI] [PubMed] [Google Scholar]
- O'Brien J. T., Markus H. S. (2014). Vascular risk factors and Alzheimer's disease. BMC Med. 12:218. 10.1186/s12916-014-0218-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh-hashi K., Nagai T., Tanaka T., Yu H., Hirata Y., Kiuchi K. (2005). Determination of hypoxic effect on neprilysin activity in human neuroblastoma SH-SY5Y cells using a novel HPLC method. Biochem. Biophys. Res. Commun. 334, 380–385. 10.1016/j.bbrc.2005.06.095 [DOI] [PubMed] [Google Scholar]
- Ouiddir A., Planès C., Fernandes I., VanHesse A., Clerici C. (1999). Hypoxia upregulates activity and expression of the glucose transporter GLUT1 in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 21, 710–718. [DOI] [PubMed] [Google Scholar]
- Pacheco-Quinto J., Eckman E. A. (2013). Endothelin-converting enzymes degrade intracellular β-amyloid produced within the endosomal/lysosomal pathway and autophagosomes. J. Biol. Chem. 288, 5606–5615. 10.1074/jbc.M112.422964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardossi-Piquard R., Checler F. (2012). The physiology of the β-amyloid precursor protein intracellular domain AICD. J. Neurochem. 120, 109–124. 10.1111/j.1471-4159.2011.07475.x [DOI] [PubMed] [Google Scholar]
- Pardossi-Piquard R., Petit A., Kawarai T., Sunyach C., Alves da Costa C., Vincent B., et al. (2005). Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron 46, 541–554. 10.1016/j.neuron.2005.04.008 [DOI] [PubMed] [Google Scholar]
- Pluta R., Jabłoński M., Ułamek-Kozioł M., Kocki J., Brzozowska J., Januszewski S., et al. (2013). Sporadic Alzheimer's disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer's disease genes. Mol. Neurobiol. 48, 500–515. 10.1007/s12035-013-8439-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W. Q., Folstein M. F. (2006). Insulin, insulin-degrading enzyme and amyloid-βpeptide in Alzheimer's disease: review and hypothesis. Neurobiol. Aging 27, 190–198. 10.1016/j.neurobiolaging.2005.01.004 [DOI] [PubMed] [Google Scholar]
- Rybnikova E., Gluschenko T., Galeeva A., Tulkova E., Nalivaeva N. N., Makova N. Z., et al. (2012). Differential expression of ADAM15 and ADAM17 metalloproteases in the rat brain after severe hypobaric hypoxia and hypoxic preconditioning. Neurosci. Res. 72, 364–373. 10.1016/j.neures.2011.12.010 [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (2012). Preventing Alzheimer's disease. Science 337, 1488–1492. 10.1126/science.1228541 [DOI] [PubMed] [Google Scholar]
- Semenza G. L. (2006). Regulation of physiological responses to continuous and intermittent hypoxia by hypoxia-inducible factor 1. Exp. Physiol. 91, 803–806. 10.1113/expphysiol.2006.033498 [DOI] [PubMed] [Google Scholar]
- Strober W. (2001). Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. Appendix 3:Appendix 3B. 10.1002/0471142735.ima03bs21 [DOI] [PubMed] [Google Scholar]
- Sun X., He G., Qing H., Zhou W., Dobie F., Cai F., et al. (2006). Hypoxia facilitates Alzheimer's disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. U.S.A. 103, 18727–18732. 10.1073/pnas.0606298103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner A. J. (2003). Exploring the structure and function of zinc metallopeptidases: old enzymes and new discoveries. Biochem. Soc. Trans. 31, 723–727. 10.1042/bst0310723 [DOI] [PubMed] [Google Scholar]
- Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe M. S., et al. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. 10.1038/416535a [DOI] [PubMed] [Google Scholar]
- Wang R., Zhang Y. W., Zhang X., Liu R., Zhang X., Hong S., et al. (2006). Transcriptional regulation of APH-1A and increased γ-secretase cleavage of APP and Notch by HIF-1 and hypoxia. FASEB J. 20, 1275–1277. 10.1096/fj.06-5839fje [DOI] [PubMed] [Google Scholar]
- Wang Z., Yang D., Zhang X., Li T., Li J., Tang Y., et al. (2011). Hypoxia-induced down-regulation of neprilysin by histone modification in mouse primary cortical and hippocampal neurons. PLoS ONE 6:e19229. 10.1371/journal.pone.0019229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster N. J., Green K. N., Settle V. J., Peers C., Vaughan P. F. (2004). Altered processing of the amyloid precursor protein and decreased expression of ADAM 10 by chronic hypoxia in SH-SY5Y: no role for the stress-activated JNK and p38 signalling pathways. Brain Res. Mol. Brain Res. 130, 161–169. 10.1016/j.molbrainres.2004.06.042 [DOI] [PubMed] [Google Scholar]
- Weidemann A., Paliga K., Dürrwang U., Reinhard F. B., Schuckert O., Evin G., et al. (1999). Proteolytic processing of the Alzheimer's disease amyloid precursor protein within its cytoplasmic domain by caspase-like proteases. J. Biol. Chem. 274, 5823–5829. [DOI] [PubMed] [Google Scholar]
- Wick M. J., Buesing E. J., Wehling C. A., Loomis Z. L., Cool C. D., Zamora M. R., et al. (2011). Decreased neprilysin and pulmonary vascular remodeling in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 183, 330–340. 10.1164/rccm.201002-0154OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Z., Behrooz A., Ismail-Beigi F. (1999). Regulation of glucose transport by hypoxia. Am. J. Kidney Dis. 34, 189–202. [DOI] [PubMed] [Google Scholar]
- Zhang X., Le W. (2010). Pathological role of hypoxia in Alzheimer's disease. Exp. Neurol. 223, 299–303. 10.1016/j.expneurol.2009.07.033 [DOI] [PubMed] [Google Scholar]
- Zhang X., Zhou K., Wang R., Cui J., Lipton S. A., Liao F. F., et al. (2007). Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and β-amyloid generation. J. Biol. Chem. 282, 10873–10880. 10.1074/jbc.M608856200 [DOI] [PubMed] [Google Scholar]
- Zuccato C., Belyaev N., Conforti P., Ooi L., Tartari M., Papadimou E., et al. (2007). Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington's disease. J. Neurosci. 27, 6972–6983. 10.1523/JNEUROSCI.4278-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]