

Abstract
Two enantioselective total syntheses of the nortriterpenoid natural product rubriflordilactone A are described, which use palladium- or cobalt-catalyzed cyclizations to form the CDE rings, and converge on a late-stage synthetic intermediate. These key processes are set up through the convergent coupling of a common diyne component with appropriate AB-ring aldehydes, a strategy that sets the stage for the synthetic exploration of other members of this family of natural products.
Keywords: cyclotrimerization, domino reactions, natural products, total synthesis, transition metal catalysis
Chinese herbal plants of the Schisandra and Kadsura genera have afforded a rich diversity of structurally related nortriterpenoid natural products, which are characterized by complex fused ring systems, a high degree of oxygenation, and densely arrayed stereochemistry.[1] Many of these have been found to exhibit bioactive properties, including promising levels of anti-HIV activity. Their attractive architectures also represent a formidable synthetic challenge, first met in 2011 by Yang and co-workers in their synthesis of schindilactone A.[2] This landmark achievement has recently been complemented by an elegant asymmetric synthesis of rubriflordilactone A (1, Scheme 1) by Li et al., where a 6π-electrocyclization was used to assemble the challenging pentasubstituted D-ring arene;[3] and syntheses of the related family members schilancitrilactones B and C, and propindilactone G.[4] Herein, we describe two convergent enantioselective total syntheses of rubriflordilactone A,[5] which are distinct from previous work in that the CDE ring system at the heart of the natural product framework is formed in a single tricyclization step.[6, 7] The two syntheses differ in the method used to construct this CDE framework, which is achieved under either palladium or cobalt catalysis; the products of these key cyclizations converge on a common late-stage intermediate.
Scheme 1.
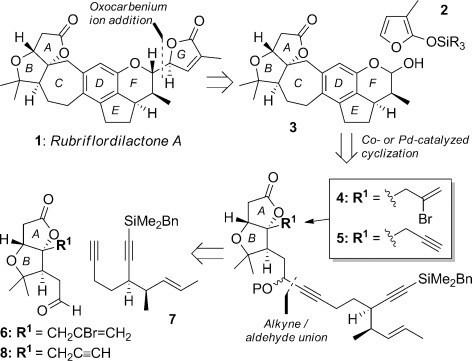
Retrosynthetic analysis of rubriflordilactone A.
Both strategies (Scheme 1) envisaged introduction of the butenolide G ring through addition of a siloxyfuran nucleophile 2 to an oxocarbenium ion in the final step. The latter synthon would arise from lactol 3, which in turn would be constructed from a bromoendiyne (4) or triyne (5) under palladium or cobalt catalysis, respectively. Bromoendiyne 4 could be formed from the union of aldehyde 6[6a] with diyne 7, the latter of which features two challenging contiguous stereocenters. The synthesis of triyne 5 would also require 7 and the alkyne-bearing aldehyde 8. These two strategies present different challenges: whilst palladium-catalyzed bromoendiyne cyclizations are established as efficient methods for tricycle synthesis,[8, 9] no applications of this method to natural product total synthesis have been reported; and whilst cobalt-catalyzed alkyne cyclotrimerization has a rich synthetic history,[10] its use in the formation of seven-membered rings (as in the C ring of rubriflordilactone A) is rare.
The synthesis of diyne 7 began with esterification of (S,E)-pent-3-en-2-ol[11] with carboxylic acid 9 (Scheme 2).[12] Ireland–Claisen rearrangement of the resultant ester 10 led to acid 11, a reaction that gave higher yield and diastereoselectivity with the free lithium enolate (96 %, >20:1 d.r.)[13] than with the silyl ketene acetal (92 %, 9:1 d.r.).[12] Sequential manipulations to convert the carboxylic acid in 11 into benzyldimethylsilyl alkyne 12 (where Stork–Zhao olefination[14]/elimination proved the most effective means for alkynylation), and then the para-methoxybenzyl ether into a terminal alkyne gave 7, which is primed for addition to AB-ring aldehydes 6 or 8.
Scheme 2.
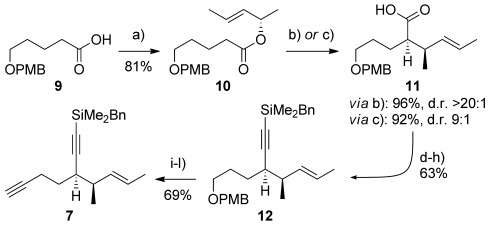
Reagents and conditions: a) (S,E)-pent-3-en-2-ol, EDC⋅HCl, Et3N, DMAP, THF, RT, 16 h, 81 %; b) LiHMDS, Et3N/toluene (3:1), −78 °C→RT, 5 h, 95 %, d.r.>20:1; c) LDA, TMSCl/Et3N (1:1), THF, −78 °C→0 °C, 3 h, 92 %, d.r. 9:1; d) TMSCHN2, toluene/MeOH (5:1), RT, 30 min, 88 %; e) DIBALH, CH2Cl2, −78 °C→−30 °C, 2 h, 97 %; f) DMP, NaHCO3, CH2Cl2, 0 °C→RT, 1 h, 90 %; g) [Ph3PCH2I]+I−, NaHMDS, THF, −78 °C→RT; then NaHMDS, −78 °C→RT, 84 %; h) LiHMDS, THF, −78 °C, 30 min; then BnMe2SiCl, −78 °C→RT, 3 h, 98 %; i) DDQ, CH2Cl2/H2O (4:1), RT, 1 h; j) DMP, NaHCO3, CH2Cl2, 0 °C→RT, 30 min, 83 % (2 steps); k) CBr4, PPh3, CH2Cl2 −30 °C→0 °C, 1 h, 85 %; l) nBuLi, THF, −78 °C→RT, 40 min, 98 %. Bn=benzyl, DIBALH=diisobutylaluminium hydride, DDQ=2,3-dichloro-5,6-dicyano-1,4-benzoquinone, DMAP=4-dimethylaminopyridine, DMP=Dess–Martin periodinane, EDC=1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide, HMDS=1,1,1,3,3,3-hexamethyldisilazane, LDA=lithium diisopropylamide, PMB=para-methoxybenzyl, TMS=trimethylsilyl.
The preparation of the alkyne-bearing aldehyde 8 offered an opportunity to avoid the Stille coupling we had used to install the bromoalkene sidechain in aldehyde 6.[6a] Its synthesis commenced with ester 13 (Scheme 3), which was converted into epoxide 14 through alkyne carbocupration[15] with 3-trimethylsilylpropynylmagnesium bromide,[16] ester reduction, and Sharpless asymmetric epoxidation (83 % over 3 steps, 92 % ee).[17] Ring-opening of 14 with allylmagnesium chloride,[18] followed by oxidation and β-lactone formation, afforded 15 in good yield (74 % over 4 steps).[19] Double nucleophilic addition of methylmagnesium bromide to the β-lactone smoothly installed the gem-dimethyl group of the B ring (16);[20] oxidative cleavage of the terminal alkene in 16, methyl acetal formation, and oxidation of the remaining primary alcohol afforded aldehyde regioisomers 17 and 18 (1.9:1 ratio, 72 % yield from 16). The formation of this separable mixture of aldehydes is inconsequential since both were found to be suitable for conversion to the AB-ring aldehyde 8. In the case of aldehyde 17, this was achieved through Ando olefination/A-ring lactonization[21] and acetal hydrolysis to give lactol 19. Pleasingly, exposure of this lactol to methanolic potassium carbonate gave the AB-ring aldehyde 8 in quantitative yield through oxy-Michael addition. From aldehyde 18, a similar sequence could be applied, which proceeded via enoate 20 (Z/E=2.5:1); acidic deprotection of the acetal in 20 led to spontaneous lactonization to 19.
Scheme 3.
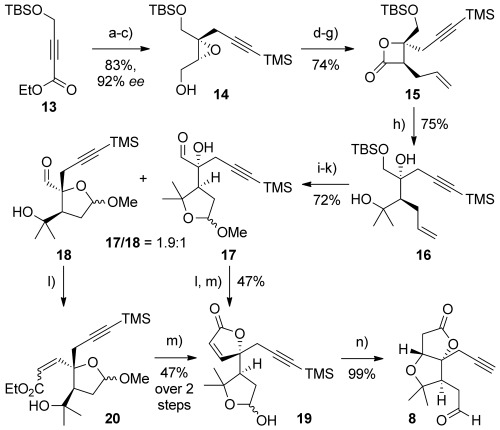
Reagents and conditions: a) TMSCαCCH2MgBr, CuBr⋅SMe2, THF, −78 °C→−40 °C, 40 min; 13, −78 °C; b) DIBALH, CH2Cl2, −78 °C→RT, 3 h, 90 % (2 steps); c) Ti(OiPr)4, d-(−)-diethyl tartrate, tBuOOH, 4 Å MS, CH2Cl2, −20 °C, 22 h, 92 %, 92 % ee; d) AllylMgBr, THF, 0 °C, 10 min, 97 %; e) SO3⋅py, DMSO, iPr2EtN, CH2Cl2, 0 °C→RT, 2 h; f) NaOCl, NaH2PO4, 2-methylbut-2-ene, tBuOH/H2O (3:1), RT, 18 h, 92 % (2 steps); g) BOPCl, py, MeCN, RT, 3 h, 83 %; h) MeMgBr, THF, −5 °C→RT, 1.5 h, 64 %+31 % ketone, recycled to give 75 % overall; i) OsO4, NaIO4, 2,6-lutidine, dioxane/H2O (4.6:1), RT, 2 h, 88 %; j) (±)-camphorsulfonic acid, MeOH, RT, 18 h, 98 %; k) SO3⋅py, DMSO, iPr2EtN, CH2Cl2, 0–10 °C, 1 h, 84 %; l) (PhO)2POCH2CO2Et, KHMDS, THF, 0 °C; m) TFA, CH2Cl2, 0 °C, 15 min, 47 % (from 17, and 18); n) K2CO3, MeOH, RT, 2 h, 99 %. BOPCl=bis(2-oxo-3-oxazolidinyl)phosphinic chloride, MS=molecular sieves, py=pyridine, TBS=tert-butyldimethylsilyl, TFA=trifluoroacetic acid.
With the key diyne and aldehyde components in hand, we now investigated their union and cyclizations to the ABCDE ring system of rubriflordilactone A (Scheme 4). We first chose to explore the palladium-catalyzed route, which began with addition of diyne 7 to bromoalkenyl aldehyde 6 to give alcohol 21 (67 %). Prior work in the group[6b] had shown that protection of the propargylic alcohol would be required to achieve high yields in the ensuing cyclization, and accordingly a TBS ether was installed at this position. The resultant bromoendiyne was then cyclized by treatment with [Pd(PPh3)4] (10 mol %) and triethylamine in acetonitrile, which to our delight afforded the ABCDE-ring pentacycle 22 in excellent yield (91 %). Oxidation of the aryl benzyldimethylsilane to the corresponding phenol proceeded smoothly,[22] and after benzylic deoxygenation, the fully functionalized ABCDE framework 23 was revealed.
Scheme 4.
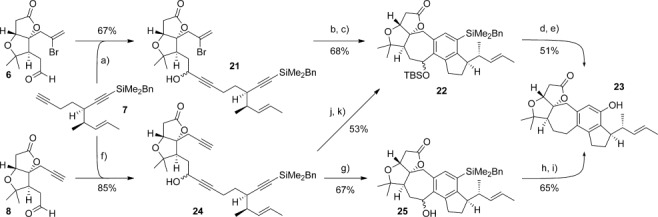
Reagents and conditions: a) nBuLi, 7, −78 °C; then add 6, −78 °C→−10 °C, 2 h, 67 %; b) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C→RT, 4 h 75 %; c) [Pd(PPh3)4] (10 mol %), Et3N, MeCN, 80 °C, 18 h, 91 %; d) TBAF, THF, RT, 30 min; then H2O2, KHCO3, MeOH, RT, 12 h; e) Et3SiH, ZnCl2, CH2Cl2, RT, 3 h; then TBAF, THF, RT, 20 min, 51 % (2 steps); f) nBuLi, 7, −78 °C; then add 8, −78 °C→−10 °C, 4 h, 85 %; g) [CpCo(CO)2] (20 mol %), PPh3 (40 mol %), PhCl, MW (300 W), 150 °C, 25 min, 67 %; h) TBAF, THF, RT, 30 min; then H2O2, KHCO3, MeOH, RT, 12 h, 84 %; i) Et3SiH, ZnCl2, CH2Cl2, RT, 3 h, 77 %; j) TBSCl, imid., DMAP, CH2Cl2, RT, 6 h, 98 %; k) [CpCo(CO)2] (20 mol %), PPh3 (40 mol %), PhCl, MW (300 W), 150 °C, 25 min, 54 %. Cp=cyclopentadienyl, MW=microwave irradiation, OTf=trifluoromethanesulfonate, TBAF=tetra-n-butylammonium fluoride.
At this juncture, we elected to compare the palladium-catalyzed cyclization route with the alternative cobalt-catalyzed cyclotrimerization approach. Diyne 7 was therefore instead combined with alkynyl aldehyde 8 to give triyne 24 (85 %). We were excited to find that cyclotrimerization of 24 under microwave heating[6b, 23] successfully afforded pentacycle 25 (67 %); this product could also be advanced to the same ABCDE-ring structure 23 prepared via the palladium-catalyzed route, through Tamao oxidation followed by benzylic deoxygenation.[22] Notably, silylation of the propargylic alcohol in 24 provides an alternative point of convergence between the two routes, since the product of cyclotrimerization of the resultant silyl ether is pentacycle 22, albeit obtained with slightly reduced efficiency compared to cyclization of the free alcohol 24.[24]
Our two strategies had now converged on the late-stage intermediate 23, and all that remained was elaboration of the FG ring system. This was achieved in four steps (Scheme 5), beginning with a two-step oxidative cleavage of the pendent alkene in 23 to give the lactol 26 (in equilibrium with the open-chain aldehyde). This lactol intercepts with the synthetic route reported by the Li group,[3] who progressed 26 to the natural product through the formation of a fluoropyran, stereoselective coupling of which with a furanyl stannane installed the butenolide G ring. With a view to avoiding the use of a tin-based nucleophile, we explored alternative methods to activate the lactol. This proved challenging, but after some experimentation we found that chloropyran 27 could be prepared by treating lactol 26 with a mixture of thionyl chloride and zinc(II) chloride.[25] This seemingly straightforward transformation in fact proceeds via initial and rapid formation of dimer 28, which over a period of 3 h is converted into the targeted chloropyran 27 (see graph in Scheme 5). This unstable intermediate was then reacted directly with siloxyfuran 28 in the presence of zinc(II) chloride and, to our delight, this reaction afforded rubriflordilactone A along with its C23-epimer 30 in 71 % yield (d.r.≈1:1). It is notable that the facial selectivity of the addition of the furan to the incipient oxocarbenium ion is excellent, since these two separable diastereomers were the predominant products formed in this addition. The spectroscopic data for the synthetic rubriflordilactone A were found to be identical to those for the natural product, with the exception of the specific rotation, which showed an equal and opposite value, thus indicating that 1 is the unnatural enantiomer (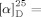 +58.3 (c=0.114, MeOH); lit.
+58.3 (c=0.114, MeOH); lit. 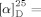 −58.1 (c=0.114, MeOH)).[5, 26]
−58.1 (c=0.114, MeOH)).[5, 26]
Scheme 5.
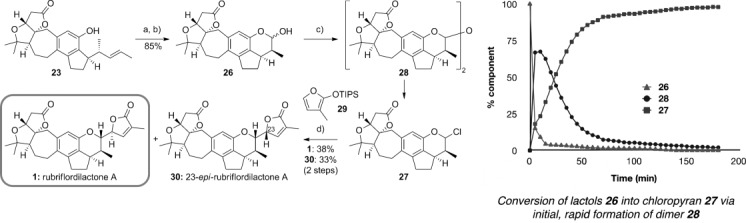
Reagents and conditions: a) OsO4 (2 mol %), NMO, acetone/H2O (3:1), RT, 3 h; b) NaIO4/SiO2, CH2Cl2, RT, 15 min, 85 % (2 steps); c) ZnCl2, SOCl2, CDCl3, RT, 3 h; d) 29, ZnCl2, CH2Cl2, −30 °C→RT, 12 h, 38 % of 1 and 33 % of 30 (2 steps). NMO=N-methylmorpholine-N-oxide, TIPS=triisopropylsilyl.
In conclusion, we have developed two synthetic strategies that achieve enantioselective syntheses of rubriflordilactone A. These employ palladium or cobalt catalysis to assemble the ABCDE ring system as the key framework-construction step. The routes are strategically highly convergent because their common late-stage intermediate is just four steps from the end of the synthesis. The modular nature of the coupling between a functionalized diyne and AB-ring aldehydes to assemble the cyclization substrates enables a unified approach to other members of this fascinating family of natural products, and offers a high degree of flexibility for the synthesis of rubriflordilactone analogues.
Acknowledgments
We thank the EPSRC for an Advanced Research Fellowship (EAA) (EP/E055273/1) and additional funding (EP/K005391/1), A*STAR for a National Science Scholarship (SSG), and postdoctoral support through a fellowship from the German Academic Exchange Service (BG).
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1a.Shi Y-M, Xiao W-L, Pu J-X, Sun H-D. Nat. Prod. Rep. 2015;32:367. doi: 10.1039/c4np00117f. Reviews on Schisandraceae natural products. [DOI] [PubMed] [Google Scholar]
- 1b.Xia Y-G, Yang B-Y, Kuang H-X. Phytochem. Rev. 2015;14:155. For reviews, see. [Google Scholar]
- 1c.Xiao WL, Li RT, Huang SX, Pu JX, Sun HD. Nat. Prod. Rep. 2008;25:871. doi: 10.1039/b719905h. [DOI] [PubMed] [Google Scholar]
- 2a.Xiao Q, Ren WW, Chen ZX, Sun TW, Li Y, Ye QD, Gong JX, Meng FK, You L, Liu YF, Zhao MZ, Xu LM, Shan ZH, Shi Y, Tang YF, Chen JH, Yang Z. Angew. Chem. Int. Ed. 2011;50:7373. doi: 10.1002/anie.201103088. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2011;123:7511. [Google Scholar]
- 2b.Sun T-W, Ren W-W, Xiao Q, Tang Y-F, Zhang Y-D, Li Y, Meng F-K, Liu Y-F, Zhao M-Z, Xu L-M, Chen J-H, Yang Z. Chem. Asian J. 2012;7:2321. doi: 10.1002/asia.201200363. [DOI] [PubMed] [Google Scholar]
- 2c.Li Y, Chen Z-X, Xiao Q, Ye Q-D, Sun T-W, Meng F-K, Ren W-W, You L, Xu L-M, Wang Y-F, Chen J-H, Yang Z. Chem. Asian J. 2012;7:2334. doi: 10.1002/asia.201200364. [DOI] [PubMed] [Google Scholar]
- 2d.Ren W-W, Chen Z-X, Xiao Q, Li Y, Sun T-W, Zhang Z-Y, Ye Q-D, Meng F-K, You L, Zhao M-Z, Xu L-M, Tang Y-F, Chen J-H, Yang Z. Chem. Asian J. 2012;7:2341. doi: 10.1002/asia.201200365. [DOI] [PubMed] [Google Scholar]
- 3.Li J, Yang P, Yao M, Deng J, Li A. J. Am. Chem. Soc. 2014;136:16477. doi: 10.1021/ja5092563. [DOI] [PubMed] [Google Scholar]
- 4a.Wang L, Wang H, Li Y, Tang P. Angew. Chem. Int. Ed. 2015;54:5732. doi: 10.1002/anie.201501169. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2015;127:5824. [Google Scholar]
- 4b.You L, Liang X-T, Xu L-M, Wang Y-F, Zhang J-J, Su Q, Li Y-H, Zhang B, Yang S-L, Chen J-H, Yang Z. J. Am. Chem. Soc. 2015;137:10120. doi: 10.1021/jacs.5b06480. [DOI] [PubMed] [Google Scholar]
- 5.Xiao WL, Yang LM, Gong NB, Wu L, Wang RR, Pu JX, Li XL, Huang SX, Zheng YT, Li RT, Lu Y, Zheng QT, Sun HD. Org. Lett. 2006;8:991. doi: 10.1021/ol060062f. [DOI] [PubMed] [Google Scholar]
- 6a.Gockel B, Goh SS, Puttock EJ, Baars H, Chaubet G, Anderson EA. Org. Lett. 2014;16:4480. doi: 10.1021/ol502027m. Previous work from our group towards the Schisandraceae natural products. [DOI] [PubMed] [Google Scholar]
- 6b.Goh SS, Baars H, Gockel B, Anderson EA. Org. Lett. 2012;14:6278. doi: 10.1021/ol303041j. [DOI] [PubMed] [Google Scholar]
- 6c.Cordonnier M-CA, Kan SBJ, Anderson EA. Chem. Commun. 2008:5818. doi: 10.1039/b814360a. [DOI] [PubMed] [Google Scholar]
- 7a.Peng Y, Duan S-M, Wang Y-W. Tetrahedron Lett. 2015;56:4509. Recent synthetic work from other groups. [Google Scholar]
- 7b.Shi H, De S, Wang Q, Gao S, Wang X, Chen C. Tetrahedron Lett. 2015;56:3225. doi: 10.1016/j.tetlet.2014.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c.Mehta G, Yaragorla S. Tetrahedron Lett. 2013;54:549. For synthetic studies, see. [Google Scholar]
- 7d.Bartoli A, Chouraqui GL, Parrain J-L. Org. Lett. 2012;14:122. doi: 10.1021/ol2029146. [DOI] [PubMed] [Google Scholar]
- 7e.Mehta G, Yaragorla S. Tetrahedron Lett. 2011;52:4485. [Google Scholar]
- 7f.Firoj Hossain M, Matcha K, Ghosh S. Tetrahedron Lett. 2011;52:6473. [Google Scholar]
- 7g.Maity S, Matcha K, Ghosh S. J. Org. Chem. 2010;75:4192. doi: 10.1021/jo1006448. [DOI] [PubMed] [Google Scholar]
- 7h.Zhang YD, Ren WW, Lan Y, Xiao Q, Wang K, Xu J, Chen JH, Yang Z. Org. Lett. 2008;10:665. doi: 10.1021/ol703126q. [DOI] [PubMed] [Google Scholar]
- 7i.Paquette LA, Lai KW. Org. Lett. 2008;10:2111. doi: 10.1021/ol800418m. [DOI] [PubMed] [Google Scholar]
- 8a.de Meijere A, von Zezschwitz P, Brase S. Acc. Chem. Res. 2005;38:413. doi: 10.1021/ar980025r. [DOI] [PubMed] [Google Scholar]
- 8b.Tokan WM, Meyer FE, Schweizer S, Parsons PJ, de Meijere A. Eur. J. Org. Chem. 2008:6152. [Google Scholar]
- 8c.Campbell CD, Greenaway RL, Holton OT, Chapman HA, Anderson EA. Chem. Commun. 2014;50:5187. doi: 10.1039/c3cc45634j. For recent work on ynamides from our group, see. [DOI] [PubMed] [Google Scholar]
- 8d.Blond G, Bour C, Salem B, Suffert J. Org. Lett. 2008;10:1075. doi: 10.1021/ol702855h. Suffert has also noted the utility of silylalkynes in similar cascades. [DOI] [PubMed] [Google Scholar]
- 8e.Negishi E, Harring LS, Owczarczyk Z, Mohamud MM, Ay M. Tetrahedron Lett. 1992;33:3253. Seminal work on this reactivity. [Google Scholar]
- 8f.Meyer FE, de Meijere A. Synlett. 1991:777. [Google Scholar]
- 8g.Leibeling M, Koester DC, Pawliczek M, Schild SC, Werz DB. Nat. Chem. Biol. 2010;6:199. doi: 10.1038/nchembio.302. [DOI] [PubMed] [Google Scholar]
- 9a.Leibeling M, Werz DB. Chem. Eur. J. 2012;18:6138. doi: 10.1002/chem.201200175. Recent examples of this method in the synthesis of complex fused polyaromatic ring systems. [DOI] [PubMed] [Google Scholar]
- 9b.Wallbaum J, Neufeld R, Stalke D, Werz DB. Angew. Chem. Int. Ed. 2013;52:13243. doi: 10.1002/anie.201307793. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2013;125:13485. [Google Scholar]
- 9c.Tietze LF, Eichhorst C, Hungerland T, Steinert M. Chem. Eur. J. 2014;20:12553. doi: 10.1002/chem.201402961. [DOI] [PubMed] [Google Scholar]
- 10a.Gandon V, Aubert C, Malacria M. Chem. Commun. 2006:2209. doi: 10.1039/b517696b. [DOI] [PubMed] [Google Scholar]
- 10b.Chouraqui G, Petit M, Phansavath P, Aubert C, Malacria M. Eur. J. Org. Chem. 2006:1413. doi: 10.1021/ol025623r. [DOI] [PubMed] [Google Scholar]
- 10c.Vollhardt KPC. Angew. Chem. Int. Ed. Engl. 1984;23:539. [Google Scholar]
- Angew. Chem. 1984;96:525. [Google Scholar]
- 11.Kocienski PJ, Christopher JA, Bell R, Otto B. Synthesis. 2005:75. [Google Scholar]
- 12.Wilson MS, Woo JCS, Dake GR. J. Org. Chem. 2006;71:4237. doi: 10.1021/jo0604585. [DOI] [PubMed] [Google Scholar]
- 13.Godenschwager PF, Collum DB. J. Am. Chem. Soc. 2008;130:8726. doi: 10.1021/ja800250q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stork G, Zhao K. Tetrahedron Lett. 1989;30:2173. [Google Scholar]
- 15.Anderson RJ, Corbin VL, Cotterrell G, Cox GR, Henrick CA, Schaub F, Siddall JB. J. Am. Chem. Soc. 1975;97:1197. doi: 10.1021/ja00838a039. [DOI] [PubMed] [Google Scholar]
- 16.Hernandez E, Soderquist JA. Org. Lett. 2005;7:5397. doi: 10.1021/ol051886k. [DOI] [PubMed] [Google Scholar]
- 17a.Gao Y, Klunder JM, Hanson RM, Masamune H, Ko SY, Sharpless KB. J. Am. Chem. Soc. 1987;109:5765. [Google Scholar]
- 17b.Larrosa I, Da Silva MI, Gómez PM, Hannen P, Ko E, Lenger SR, Linke SR, White AJP, Wilton D, Barrett AGM. J. Am. Chem. Soc. 2006;128:14042. doi: 10.1021/ja0662671. For a closely related substrate, see. [DOI] [PubMed] [Google Scholar]
- 18a.Morokuma K, Taira Y, Uehara Y, Shibahara S, Takahashi K, Ishihara J, Hatakeyama S. Tetrahedron Lett. 2008;49:6043. [Google Scholar]
- 18b.Taber DF, Green JH, Geremia JM. J. Org. Chem. 1997;62:9342. [Google Scholar]
- 19. The formation of a β-lactone is essential for high yields in the subsequent double methylation step.
- 20. 31 % of the methyl ketone that arises from a single addition of Grignard reagent to 15 was isolated, along with alcohol 16. Isolation and addition of MeMgBr to this ketone delivered a further 10 % of 16, giving an overall yield of 75 %.
- 21.Ando K. J. Org. Chem. 1997;62:1934. doi: 10.1021/jo970057c. [DOI] [PubMed] [Google Scholar]
- 22a.Tamao K. Proc. Jpn. Acad. Ser. B. 2008;84:123. doi: 10.2183/pjab.84.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b.Bracegirdle S, Anderson EA. Chem. Commun. 2010;46:3454. doi: 10.1039/b924135c. [DOI] [PubMed] [Google Scholar]
- 22c.Rayment EJ, Summerhill N, Anderson EA. J. Org. Chem. 2012;77:7052. doi: 10.1021/jo301363h. [DOI] [PubMed] [Google Scholar]
- 22d.Suginome M, Ihara H. J. Am. Chem. Soc. 2009;131:7502. doi: 10.1021/ja902314v. [DOI] [PubMed] [Google Scholar]
- 23.Nicolaus N, Strauss S, Neudorfl JM, Prokop A, Schmalz HG. Org. Lett. 2009;11:341. doi: 10.1021/ol802542c. [DOI] [PubMed] [Google Scholar]
- 24.Ajamian A, Gleason JL. Org. Lett. 2003;5:2409. doi: 10.1021/ol034541f. The reduced yield of this reaction is due to the isolation of a significant quantity (22 %) of a product in which the crotyl side-chain alkene has been isomerized to a terminal alkene; a similar cobalt-mediated alkene isomerizations can be found in. [DOI] [PubMed] [Google Scholar]
- 25a.Grob VD, Squires TG, Vercellotti JR. Carbohydr. Res. 1969;10:595. [Google Scholar]
- 25b.Egan LP, Squires TG, Vercellotti JR. Carbohydr. Res. 1970;14:263. [Google Scholar]
- 26. The specific rotation is also of opposite sign to that reported by the Li group (see Ref. [3]), although both our group and the Li group describe the synthesis of the same enantiomer of the natural product. Personal communications with Prof. Li indicate that, following re-measurement of the specific rotation of the Li group’s synthetic sample, the specific rotation reported in this manuscript is correct.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information