

Abstract
Pathological modification of α-synuclein is believed to be an important event in pathogenesis of Parkinson’s disease and several other neurodegenerative diseases. In normal cells this protein has been linked to many intracellular processes and pathways. However, neither normal function of α-synuclein in neuronal and certain other types of cells nor its exact role in the disease pathogenesis is well understood, which is largely due to limitations of animal models used for studying this protein. We produced and validated several novel mouse lines for manipulating expression of the endogenous Snca gene coding for α-synuclein. These include a line for conditional Cre-recombinase-driven inactivation of the gene; a line for conditional Flp-driven restoration of a neo-cassete-blocked α-synuclein expression; a new line with a “clean” constituent knockout of the gene as well as a line carrying this knockout locus and Rosa26-stop-lacZ reporter locus linked at the same mouse chromosome 6. Altogether these lines represent a set of new useful tools for studies of α-synuclein normal function and the role of this protein in disease pathogenesis.
Pathological aggregation of α-synuclein with formation of characteristic intracellular inclusions is a common feature of Parkinson’s disease (PD) and several neurodegenerative diseases, collectively known as synucleinopathies1,2,3. Moreover, certain structural modifications or increased expression of this protein have been associated with the development of early onset forms of PD4,5,6,7,8,9, whereas polymorphisms in the encoding locus, SNCA, have been found to affect the risk of the development of PD and certain other synucleinopathies10,11,12,13,14. Therefore, it is feasible that substantial changes in α-synuclein metabolism lead to the development of aggressive forms of the disease, while more subtle modifications can be efficiently compensated for a long time. Only later in life, probably in combination with certain environmental factors and/or with a general functional decline of the ageing nervous system, these subtle modifications trigger pathological changes and clinical manifestation of the disease.
A well-documented mechanism of α-synuclein-induced neuronal dysfunction and death is the toxicity of intermediate products of α-synuclein aggregation, namely oligomers and protofibrils (reviewed in15,16,17,18). However, this gain-of-function mechanism might be accompanied by a loss-of-function developing as the result of α-synuclein sequestering in aggregates and consequent depletion of the functional pool of this protein.
α-synuclein is abundant in presynaptic terminals of many types of neurons where it might play an important role in neurotransmission via regulation of synthesis, release and re-uptake of various neurotransmitters19,20,21,22,23,24,25,26,27,28,29,30. Although α-synuclein deficiency in mouse models can be compensated for long time either due to the redundancy within the synuclein family or by switching on some other mechanism in the developing nervous system, ageing animals lacking α-synuclein develop neuronal and particularly, synaptic deficiency31,32,33,34. Depletion of α-synuclein in adult rat substantia nigra by injection of siRNA-encoding AAV viruses caused a rapid development of substantially more severe neuronal dysfunction than observed in any of the previously produced animal lines with constituent inactivation of the Snca gene35. Another limitation of conventional α-synuclein knockout mouse models is that inactivation of the gene takes place in all body cells. Despite a particular abundance of α-synuclein in neurons, it is not possible to exclude a systemic effect of its depletion in other cells, e.g. erythrocytes, on animal physiology, which might hamper interpretation of the observed phenotypes. Therefore, the ability to targetedly and conditionally deplete specific cell population of α-synuclein is important for better understanding of this protein normal function and its role in human pathologies.
Having this in mind, we produce mice for conditional Cre-recombinase-driven inactivation of the Snca gene. In the process of producing and characterisation of these mice we created additional mouse lines, namely mice carrying inactivated Snca gene which expression can be conditionally restored by FLP-recombinase-driven deletion of the neo-cassette; “clean KO” mice, a novel line with constituent inactivation of Snca gene that carry neither neo-cassette nor other ectopic sequences that might interfere with the genome function; mice with the same “clean KO” linked with Rosa26-stop-lacZ reporter locus, which is located at the same mouse Chr 6.
Results and Discussion
Using traditional gene targeting approaches schematically illustrated in Fig. 1, we produced a novel line of mice than can be used for conditional inactivation of α-synuclein coding gene by Cre-driven recombination within transcriptionally active SncafloxΔneo locus. The normal level of α-synuclein in the neural tissues of homozygous SncafloxΔneo/floxΔneo mice was confirmed by Western blot analysis (Fig. 2). To prove that deletion of the exon II by Cre recombination leads to complete inactivation of the gene we bred SncafloxΔneo/floxΔneo mice with an “early deletor” Cre mice (see Material and Methods) to achieve germline deletion of the floxed exon II and surrounding sequences. Homozygous SncaΔflox/Δflox mice were produced and Western blot analysis did not reveal any α-synuclein protein in neural tissues of these animals (Fig. 3). Therefore, successful Cre recombination within the engineered SncafloxΔneo/floxΔneo locus efficiently inactivates α-synuclein expression. The line of mice carrying SncafloxΔneo allele has been deposited to The Jackson Laboratory (C57BL/6-Snca<tm1.1 Vlb>/J; JAX Stock#025636). Breeding of these animals with various Cre transgenic mice or stereotaxic injection of Cre-expressing viral vectors shall make it possible to deplete specific cell population or brain areas of α-synuclein. By using tamoxifen-inducible Cre-ERT2 transgenes this inactivation process can be initiated at any period of the animal development.
Figure 1. Gene targeting strategy for production of mice for conditional inactivation of α-synuclein gene and confirmation of introduced genomic modifications by Southern hybridisation.

See Supplementary Fig. S1 for sequences of the wildtype (WT) and modified Snca loci.
Figure 2. Restoration of α-synuclein expression in the cerebral cortex of floxed mice following germline deletion of neo-cassette.
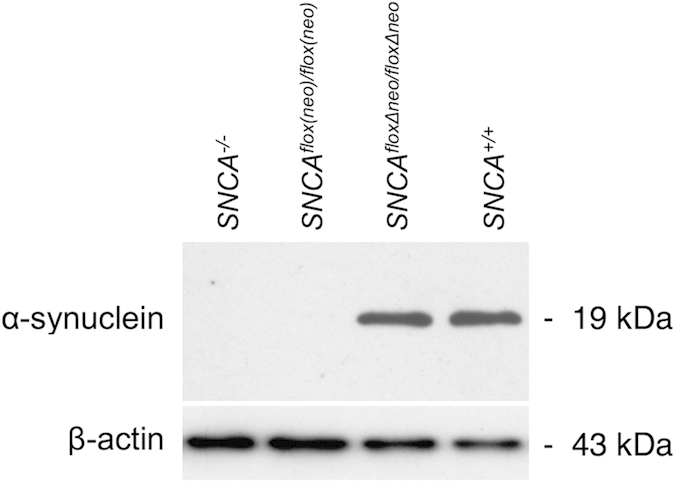
Western blot analysis of total protein samples extracted from the cerebral cortex of wild type mice (SNCA+/+), α-synuclein knockout mice (SNCA−/−, described previously19), floxed mice before deletion of neo-cassette (SNCAflox(neo)/flox(neo)) and floxed mice after deletion of neo-cassette (SNCAfloxΔneo/floxΔneo).
Figure 3. Depletion of α-synuclein in the brain tissues of homozygous mice following germline Cre-recombination.
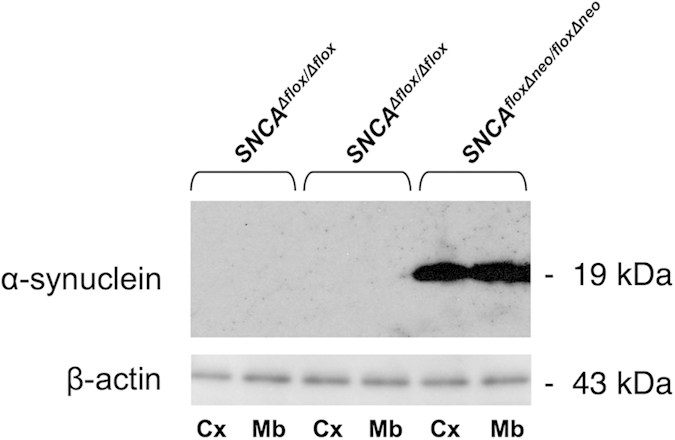
Western blot analysis of total protein samples extracted from the cerebral cortex (Cx) and midbrain (Mb) of a homozygous (SNCAfloxΔneo/floxΔneo) α-synuclein floxed mouse after deletion of the neo-cassette and two homozygous α-synuclein floxed mice after deletion of the exon II by Cre/loxP recombination (SNCAΔflox/Δflox).
However, conditional inactivation of a gene in a selected cell population commonly requires a reporter for monitoring efficiency of Cre recombination. This is particularly important for α-synuclein that in most neurons is localised not in the perikarion of targeted neurons but in presynaptic terminals, which makes it difficult to confirm its selective and efficient depletion. Rosa26-stop-lacZ reporter36 is widely used for this purpose. However, in the mouse genome both Snca and Rosa26 loci are located at chromosome 6 (Chr6) approximately 50 Mb apart. To make possible the use of Rosa26-stop-lacZ reporter in experiments with conditional inactivation of Snca gene we produced a mouse line carrying a completely inactivated SncaΔflox gene and Rosa26-stop-lacZ reporter linked at the same mouse Chr6 (see Materials and Methods). When control for the efficiency of Cre recombination is required, these mice and SncafloxΔneo mice could be used for production of an experimental cohort carrying an active floxed copy of Snca gene at one Chr6, and a constitutively inactive copy and Rosa26-stop-lacZ reporter on another Chr6. In addition to the possibility of monitoring the process of Cre recombination, the efficiency of the gene inactivation process is increased when such heterozygous animals are used, because to achieve a complete knockout in a given cell, only one allele needs to be inactivated. It should be noted that there is no evidence for any noticeable effects of Snca gene haploinsufficiency in mice.
The line of mice with constituent inactivation of Snca gene (SncaΔflox/Δflox) has a value of its own because for the best of our knowledge, it represents the most “clean” full α-synuclein knockout produced so far. Other published mouse lines still carry various selection cassettes in the vicinity of the inactivated Snca gene19,20,37,38 and the natural mutant line from Harlan UK39 possesses a deletion of approximately 350 kb fragment that includes Snca along with other genes. In contrast, the only ectopic sequence remained in the new SncaΔflox/Δflox line is a single loxP site and the deletion is restricted to the 1164 bp fragment containing exon II and adjacent intronic sequences of the Snca gene. The importance of using animals with less invasive genetic manipulations for accurate revealing the effects of α-synuclein depletion can be illustrated by the fact that in a widely used mouse knockout line19 the level of neuronal expression of the Mmrn1 gene (encoding multimerin 1) located close to the modified Snca gene is increased when compared to wild type or our SncaΔflox/Δflox mice (our unpublished observations), while in the Harlan UK mutant line this gene is completely absent. In both cases potential effects of these expression changes on neuronal physiology can be erroneously attributed to the absence of α-synuclein.
Analysis of α-synuclein expression in the nervous system of homozygous Sncaflox(neo)/flox(neo) mice generated at the first step of the targeting programme (see Fig. 1) revealed that the presence of the neo-cassette in the gene intron completely abolished α-synuclein production (Fig. 2). However, consequent Flp-driven deletion of this cassette restored normal expression of the protein in SncafloxΔneo/floxΔneo mice (Fig. 2). Therefore, Sncaflox(neo)/flox(neo) line can be used for conditional switching on endogenous α-synuclein expression in specific cell populations and/or developmental periods. This might be achieved by breeding mice of this line with transgenic mice expressing Flp recombinase under control of specitic promoters or by using stereotaxic injections of Flp expressing viral vectors.
In summary, we have produced a set of unique mouse lines for conditional switching off or switching on expression of endogenous α-synuclein as well as a novel improved line with constituent inactivation of the α-synuclein encoding gene. These lines represent new useful tools for studies of the normal function of α-synuclein in selected populations of neuronal and non-neuronal cells as well as the role of this protein in the development of neurodegenerative processes and transmission of α-synuclein pathology through the nervous system.
Materials and Methods
Targeting construct
To create arms for homologous recombination (for sequences see Supplementary Materials and Methods), fragments from the region of the exon II of mouse Snca gene (encoding amino acids 1–41 of α-synuclein) were PCR amplified using DNA extracted from C57Bl6-derived ES cells (line JM8A3.N1, gift of Dr. William Skarnes, Sanger Institute, Cambridge, UK) as a template. A high fidelity AccuPrime Pfx SuperMix (Invitrogen) and primers carrying desired additional sequences (loxP, FRT and/or restriction endonuclease cleavage sites) were used. PCR fragments were then cloned into the pCR-Blunt II-TOPO vector (Invitrogen) and verified by sequencing. Following digestion with appropriate restriction endonucleases, resulting engineered DNA fragments were cloned into pPNT1 vector plasmid to produce final targeting construct schematically shown in Fig. 1.
Gene targeting in mouse ES cells and generation of mouse founders
Mouse ES JM8A3.N1 cells40 were electroporated with the NotI-linearised targeting plasmid followed by selection of G418 and gancyclovir resistant clones. DNA extracted from individual clones was analysed for homologous recombination by Southern hybridisation with two outside probes (for sequences see Supplementary Fig. S1) as shown in Fig. 1. DNA extraction, Southern blotting, 32P-labeling of probes, hybridisation and washes were carried out according to protocols described previously41. Positive clones were expanded and used for injection into blastocysts of C57Bl6 cells followed by the embryotransfer into the uterus of pseudopregnant CD1 mice. Resulting chimeras were bred with C57Bl6 mice and successful germline transfer of the modified locus was confirmed by PCR analysis of DNA from ear biopsies.
Production of experimental mouse lines
Mice carrying a targeted α-synuclein locus (Sncaflox(neo)/+) were backcrossed with C57BL/6 mice for five generations and then intercrossed to produce animals homozygous for this locus (Sncaflox(neo)/ flox(neo)) and wild type littermates (Snca+/+).
To remove the neo-cassette, mice carrying a targeted α-synuclein locus were bred to C57BL/6 mice for two generations and then crossed to transgenic mice expressing FLP recombinase under control of beta-actin promoter (B6.Cg-Tg(ACTFLPe)9205Dym/H, on the C57BL/6 genetic background, The Jackson Laboratory). Successful deletion of FRT-flanked neo-cassette was confirmed by Southern hybridisation and PCR analysis of DNA from ear biopsies.
Mice with deleted neo-cassette (SncafloxΔneo/+) were further bred to C57BL/6 J mice and selection was applied to remove the FLP recombinase transgene. Homozygous (SncafloxΔneo/floxΔneo) and wild type littermates were produced by the intercrossing of mice that went through seven generations of the backcrossing.
To produce mice with constituent inactivation of α-synuclein gene SncafloxΔneo/floxΔneo mice were crossed with transgenic mice (on C57Bl6 background) expressing Cre recombinase under control of CMV promoter42. Mice with deleted neo-cassette (SncaΔflox/+) were further backcrossed to C57BL/6J mice and those lacking the Cre recombinase transgene were selected. Intercrosses of these mice produced homozygous (SncaΔflox/Δflox) animals.
Mice carrying SncaΔflox and Rosa26-stop-lacZ reporter36 loci linked at the same chromosome as the result of natural crossover were selected from a pool of animals generated during crossbreeding of two mouse lines (both on C57Bl6 background), each bearing one of these modified loci.
Animal genotyping
Animal genotypes were verified by PCR analysis of DNA from ear biopsies using combinations of primers and amplification conditions described in Supplementary Materials and Methods.
Western blotting
Total protein samples were prepared by homogenisation of mouse tissues in the SDS-PAGE loading buffer following incubation for 10 min at 100 oC. Gel electrophoresis, transfer to PVDF membrane, incubation with primary and secondary HRT-conjugated antibodies, and chemiluminescent-based detection were performed as described previously32. For detection of α-synuclein mouse monoclonal antibody (BD 610786 from BD Biosciences) were used in 1:1000 dilution. To confirm the equal loading membranes were re-probed with mouse monoclonal anti-beta-actin antibody (AC-15 from Sigma-Aldrich) in 1:10000 dilution.
All animal work was carried out in accordance with the United Kingdom (Scientific Procedures) Act (1986) and European Directive EC 86/609, and has been approved by the Cardiff University Ethical Review Committee and the Home Office (Project Licence 30/2844).
Additional Information
How to cite this article: Ninkina, N. et al. A novel resource for studying function and dysfunction of α-synuclein: mouse lines for modulation of endogenous Snca gene expression. Sci. Rep. 5, 16615; doi: 10.1038/srep16615 (2015).
Supplementary Material
Acknowledgments
This work was supported by grants from Parkinson’s UK (Project Grant G-1006), The Michael J. Fox Foundation for Parkinson’s Research (Rapid Response Innovation Award 2013 and Research Grant 8116.01) and The Welcome Trust (Programme Grant 075615/Z/04/z) to VLB, RFBR KOMFI 13-04-40379-H and the RF Ministry of Education and Science RFMEFI60414 × 0144 to NN and equipment of Chernogolovka’s Resources Share Centre, Contract N14.621.21.0008 (RFMEFI62114 × 0008).
Footnotes
Author Contributions N.N. and V.L.B. conceived the study, designed the experiments and wrote the main manuscript text. N.C.-R., A.A.U., T.V.T., T.A.S., N.N. and V.L.B. conducted the experiments.
References
- Galvin J. E., Lee V. M. & Trojanowski J. Q. Synucleinopathies: clinical and pathological implications. Arch. Neurol. 58, 186–190 (2001). [DOI] [PubMed] [Google Scholar]
- Goedert M. Filamentous nerve cell inclusions in neurodegenerative diseases: tauopathies and alpha-synucleinopathies. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 1101–1118 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini M. G. & Goedert M. The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann. N. .Y Acad. Sci. 920, 16–27 (2000). [DOI] [PubMed] [Google Scholar]
- Polymeropoulos M. H. et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047 (1997). [DOI] [PubMed] [Google Scholar]
- Kruger R. et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108 (1998). [DOI] [PubMed] [Google Scholar]
- Zarranz J. J. et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173 (2004). [DOI] [PubMed] [Google Scholar]
- Singleton A. B. et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841 (2003). [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin M. C. et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169 (2004). [DOI] [PubMed] [Google Scholar]
- Ibanez P. et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364, 1169–1171 (2004). [DOI] [PubMed] [Google Scholar]
- Kay D. M. et al. Genetic association between alpha-synuclein and idiopathic Parkinson’s disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, (2008). [DOI] [PubMed] [Google Scholar]
- Mizuta I. et al. Calbindin 1, fibroblast growth factor 20, and alpha-synuclein in sporadic Parkinson’s disease. Hum. Genet. 124, 89–94 (2008). [DOI] [PubMed] [Google Scholar]
- Scholz S. W. et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann. Neurol. 65, 610–614 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland G. T. et al. Do polymorphisms in the familial Parkinsonism genes contribute to risk for sporadic Parkinson’s disease? Mov. Disord. 24, 833–838 (2009). [DOI] [PubMed] [Google Scholar]
- Pankratz N. et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum. Genet. 124, 593–605 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B. & Lansbury P. T. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26, 267–298 (2003). [DOI] [PubMed] [Google Scholar]
- Dev K. K., Hofele K., Barbieri S., Buchman V. L. & Van der Putten H. Part II: alpha-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology 45, 14–44 (2003). [DOI] [PubMed] [Google Scholar]
- Fink A. L. The aggregation and fibrillation of alpha-synuclein. Acc. Chem. Res. 39, 628–634 (2006). [DOI] [PubMed] [Google Scholar]
- Uversky V. N. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J. Neurochem. 103, 17–37 (2007). [DOI] [PubMed] [Google Scholar]
- Abeliovich A. et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252 (2000). [DOI] [PubMed] [Google Scholar]
- Cabin D. E. et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 22, 8797–8807 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee F. J., Liu F., Pristupa Z. B. & Niznik H. B. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. Faseb J. 15, 916–926 (2001). [DOI] [PubMed] [Google Scholar]
- Perez R. G. et al. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 22, 3090–3099 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu A., Wersinger C. & Vernier P. alpha-Synuclein regulation of the dopaminergic transporter: a possible role in the pathogenesis of Parkinson’s disease. FEBS Lett. 565, 1–5 (2004). [DOI] [PubMed] [Google Scholar]
- Wersinger C. & Sidhu A. Attenuation of dopamine transporter activity by alpha-synuclein. Neurosci. Lett. 340, 189–192 (2003). [DOI] [PubMed] [Google Scholar]
- Chandra S. et al. Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc. Natl. Acad. Sci. USA 101, 14966–14971 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S., Gallardo G., Fernandez-Chacon R., Schluter O. M. & Sudhof T. C. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396 (2005). [DOI] [PubMed] [Google Scholar]
- Fountaine T. M. et al. The effect of alpha-synuclein knockdown on MPP+ toxicity in models of human neurons. Eur. J. Neurosci. 28, 2459–247 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fountaine T. M. & Wade-Martins R. RNA interference-mediated knockdown of alpha-synuclein protects human dopaminergic neuroblastoma cells from MPP(+) toxicity and reduces dopamine transport. J. Neurosci. Res. 85, 351–363 (2007). [DOI] [PubMed] [Google Scholar]
- Gallardo G., Schluter O. M. & Sudhof T. C. A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nat. Neurosci. 11, 301–308 (2008). [DOI] [PubMed] [Google Scholar]
- Senior S. L. et al. Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-synuclein and gamma-synuclein. Eur. J. Neurosci. 27, 947–957 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Wandi A. et al. Absence of alpha-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol. Aging 31, 796–804 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar S. et al. Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J. Neurosci. 31, 7264–7274 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J. et al. alpha-Synuclein promotes SNARE-complex cssembly in vivo and in vitro. Science 329, 1663–1667 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten-Harrison B. et al. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA 107, 19573–19578 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk O. S. et al. In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol. Ther. 18, 1450–1457 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70–71 (1999). [DOI] [PubMed] [Google Scholar]
- Dauer W. et al. Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. USA 99, 14524–14529 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter O. M. et al. Role of alpha-synuclein in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in mice. Neuroscience 118, 985–1002 (2003). [DOI] [PubMed] [Google Scholar]
- Specht C. G. & Schoepfer R. Deletion of the alpha-synuclein locus in a subpopulation of C57BL/6J inbred mice. BMC Neurosci. 2, 11 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt S. J. et al. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nat. Methods 6, 493–495 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman V. L. et al. Simultaneous and independent detection of C9ORF72 alleles with low and high number of GGGGCC repeats using an optimised protocol of Southern blot hybridisation. Mol. Neurodegener. 8, 12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk F., Baron U. & Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucl. Acids Res. 23, 5080–5081 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.