

Abstract
Innate immunity is part of the early response of the body to deal with tissue damage and infections. Due to the early nature of the innate immune inflammatory response, this inflammatory reaction represents an attractive option as a therapeutic target. The inflammasome is a component of the innate immune response involved in the activation of caspase-1 and the processing of pro-interleukin-1β. In this article we discuss the therapeutic potential of the inflammasome after central nervous system (CNS) injury and stoke, as well as the basic knowledge we have gained so far regarding inflammasome activation in the CNS. In addition, we discuss some of the therapies available or under investigation for the treatment of brain injury, spinal cord injury and stroke.
Keywords: Inflammasome, therapy, spinal cord injury, brain injury, stroke
1. Introduction
Inflammation is a critical response of the immune system to infection and disruption of tissue homeostasis. Generally, the inflammatory response diminishes after removal of the infectious pathogens or after tissue damage has been repaired. However, persistent inflammation may result if the inflammatory inducers are not eliminated, leading to tissue damage and chronic inflammatory disease. Emerging studies have revealed that inflammasome complexes comprised of large molecular platforms for caspase-1 activation and downstream inflammatory cytokine production play a critical role in innate immune inflammatory responses and contribute to various pathologies and metabolic dysfunctions. Therefore, an understanding of the mechanisms of inflammasome activation and regulation will aid in the development of novel therapeutics for treating diverse inflammatory diseases associated with persistent inflammasome hyper-activation.
Recent knowledge regarding the inflammasome, its structure and the different mechanisms of inflammasome activation offer a variety of therapeutic targets to modulate this innate immune complex. In the present review we cover a brief introduction on the inflammasome and its mechanisms of activation in the central nervous system (CNS). We also discuss current therapies available for the treatment of brain injury, spinal cord injury and stroke, and describe different therapies that may be used to inhibit inflammasome activation in the CNS after injury.
2. What is the Inflammasome
The inflammasome is a multi-protein complex involved in the activation of the inflammatory cysteine aspartase caspase-1. Once activated, caspase-1 processes the pro-inflammatory cytokines interleukin (IL)-1β and IL-18 (1). IL-1β and IL-18 belong to the family of IL-1 cytokines and have been shown to play a detrimental role after CNS injury (2-9).
The inflammasome was first described by Tschopp and colleagues in THP1 cells as a multi-protein complex comprised of caspase-1, the adaptor protein apoptosis-associated speck-like protein containing a caspase activating recruitment domain (CARD) (ASC) and a NOD-like receptor (NLR) such as NLRP1 or NLRP3 (1). Other inflammasome components initially described include caspase-5 in humans and caspase-11 in rodents (1). However, recent evidence indicates that caspase-11 forms a caspase-1-independent inflammasome, which is referred to as the non-canonical inflammasome (10). This non-canonical inflammasome is yet to be thoroughly studied in the CNS.
Overall, NLRP3 is the most studied inflammasome. In the CNS the NLRP1, NLR2 and AIM2 inflammasomes have been well characterized after injury to the brain and the spinal cord (11-14). The NLRP1 inflammasome in the CNS is comprised of NLRP1, caspase-1, caspase-11, ASC and the inhibitor of apoptosis protein XIAP (13). In addition to XIAP, other inhibitor of apoptosis proteins have been described to inhibit inflammasome activation such as cIAP-1 and cIAP-2 (15), but their involvement in CNS inflammasomes is yet to be determined. The NLRP1 inflammasome is present in motor neurons of the spinal cord as well as in brain cortical neurons (11-13, 16). The NLRP2 inflammasome is present in astrocytes (14) and is comprised of NLRP2, caspase-1 and ASC (14). Neurons also express the AIM2 inflammasome, containing AIM2, ASC and caspase-1. The AIM2 inflammasome is involved in the activation of caspase-1 as well as the program cell death process of pyroptosis (11).
Recently, inflammasome activation has been associated with downregulation of TH2 responses and IL-33 production in a model of house dust mite-induced experimental allergic airway inflammation (17). In the CNS, IL-33 is present in oligodendrocytes and when secreted it acts on microglia and astrocytes to induce recruitment of monocytes (18). After injury, IL-33 is responsible for the recruitment of M2 macrophages (18). Taken together, it could be hypothesized that after injury there is activation of the inflammasome, which inhibits IL-33 production, resulting in downregulation of a TH2 response and decreased recruitment of M2 macrophages into the tissue, resulting in impaired recovery after CNS injury.
3. What is the Mechanism of Inflammasome Activation in the Central Nervous System?
There are several mechanisms that have been described regarding inflammasome activation. In general, the innate immune response is activated by danger/damage-associated molecular patterns (DAMPs) or by pathogen-associated molecular patterns (PAMPs) that stimulate pattern recognition receptors (PRRs) (19). PAMPs are proteins derived from pathogens such as bacteria (lipopolysaccharide, flagellin or muramyl dipeptide) or viruses (double stranded RNA) (20). In contrast, DAMPs are endogenous proteins that when sensed by PRRs trigger an innate immune response (19). PRR include toll-like receptors (TLRs), Rig-like receptors (RLRs), C-type lectin receptors (CLRs) (21) or NOD-like receptors (NLRs). NLRs such as NLRP1 or NLRP2 inflammasome components have recently been reviewed elsewhere (22).
In general, PRRs sense a DAMP or PAMP, resulting in the activation of a PRR, which results in production of inflammatory cytokines. In the case of the inflammasome, the PRR is an NLR that forms protein-protein interactions with the pro-inflammatory caspase caspase-1 and with the adaptor protein ASC. Once these proteins associate as a holoenzyme, caspase-1 is cleaved. Once cleaved, caspase-1 has the catalytic activity necessary to process the pro-inflammatory cytokines pro-IL-1β and pro-IL-18 into their respective active forms (1).
There are several unanswered questions regarding the activation of the inflammasome. For instance, how does the inflammasome sense DAMPs or PAMPs? Do DAMPs/PAMPs directly bind to NLRs? If there is no direct contact between a NLR and a DAMP/PAMP, what is the upstream signaling event that triggers the oligomerization of the different inflammasome components? How does the upstream activating signal communicate with the inflammasome?
Evidence indicates that inflammasomes composition may vary in different tissues. For instance, the NLRP1 inflammasome in the CNS is a pre-assembled multi-protein complex (12, 13). However, that is not the case in cells outside the CNS where inflammasome components assemble into a complex upon activation (23). This diversity in inflammasome-activating signaling is consistent with different activating mechanisms that have been described. These mechanisms include potassium efflux (23) and reactive oxygen species (24), but whether these play a role in the CNS has not been clearly determined. The current accepted mechanism of inflammasome activation in the CNS involves signaling through purinergic receptors and the pannexin-1 channel (25).
The series of events leading to inflammasome activation in the CNS is as follows (Figure 1): 1) The purinergic receptor P2X4 is activated by ATP that is released from dying cells (26); 2) Once P2X4 is activate, it causes an efflux in potassium that results in increased extracellular potassium concentration in the vicinity of pannexin-1 (25); 3) The increase in extracellular potassium results in pannexin-1 activation (25); 4) Pannexin-1 forms protein-protein interactions with P2X7 and the inflammasome (25), and upon activation of pannexin-1, there is an efflux of ATP that then activates P2X7 (14, 25). P2X7 interacts with the inflammasome (14, 25); however, the mechanism by which P2X7 is involved in activation of the inflammasome remains unresolved. 5) The inflammasome in the CNS is preassembled and under the inhibition of the inhibitor of apoptosis protein XIAP. Upon inflammasome activation, XIAP is cleaved, thus decreasing its inhibition on caspase-1 (12, 13); 6) Caspse-1 and ASC form prion-like filaments in the cytosol, leading to activation of caspase-1 and processing of pro-IL1b and pro-IL-18. 7) These pro-inflammatory cytokines are secreted, thus initiating an inflammatory cytokine response in neighboring cells, and 8) Caspase-1 and ASC prion-like filaments are secreted into the extracellular space where they remain active and recruit pro-caspase-1 and pro-IL-1β, leading to inflammation and pyroptotic cell death.
Figure 1. Schematic of Inflammasome Activation in the CNS.
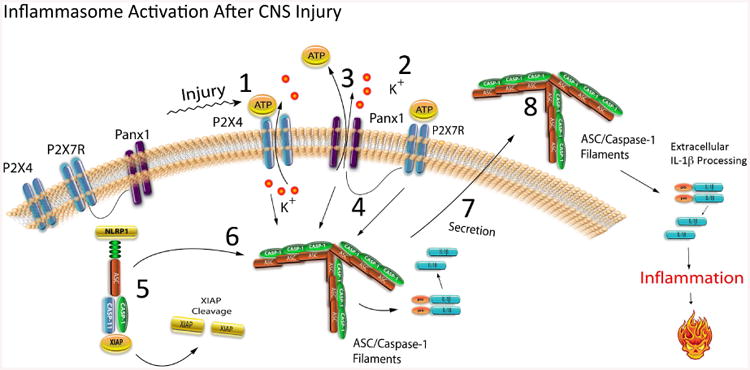
Upon activation the inflammasome triggers inflammation and pyroptotic cell death that is dependent on the formation of ASC filaments/specks.
4. What are the current options for the treatment of spinal cord injury?
The incidence of spinal cord injury (SCI) in the United States is more than 10,000 per year, resulting in 720 per million persons and enduring permanent disability each year (27). The economic impact of SCI is estimated to be more than four billion dollars annually (28). To date, no proven therapeutic modality exists that has demonstrated a positive effect in neurological outcome, emphasizing the need for continuous research in the pathophysiology and treatment of this serious clinical problem (29). Although acute methylprednisolone (MP) therapy has been approved and shown to be protective in some situations, its efficacy is limited and has been shown to have serious side effects that have limited its clinical use more recently (30).
Current ongoing studies have emphasized the complex pathophysiology of SCI and the need for considering various therapeutic strategies in preclinical models as well as moving effective agents to the clinic (31). A recent publication has provided a comprehensive summary of the current therapeutic strategies for SCI including agents that target secondary injury mechanisms including neuroinflammation, free radical damage, excitotoxicity, and ionic changes (27). Encouraging, many treatment studies have reported significant improvements in sensory and motor function that indicate the translational potential for treating clinical SCI. To this end, several pharmacological therapies including Nimodipine, Gacyclidine, Thyrotropin Releasing Hormone, GM1 Ganglioside and the Rho-antagonist Cethrin have been tested as acute neuroprotective strategies. In addition, reparative strategies including Anti-Nogo antibodies, Fibroblast Growth Factor and cell transplantation therapies have also been tested in animal models to enhance reparative strategies (32).
In terms of spinal cord clinical studies, several trials have been completed with limited degrees of beneficial improvements reported (27, 29). As previously mentioned, MP has been evaluated in four clinical trials, the first being the National Acute Spinal Cord Injury study 1 (NASCIS1). As stated, the observed positive results generated in preclinical studies were not matched by the effectiveness of MP in a clinical setting. In other studies Nimodipine with or without MP also failed to show any beneficial effects (33). Gacyclidine, an N-methyl-D-Aspartate antagonist, was tested in acute SCI patients early after injury (34). That study, although initially promising, failed to show beneficial effects at the one-year follow-up. Other studies including TRH have shown some degree of benefit in limited patient populations but these need to be followed-up in large randomized multicenter trials (35). GM1-ganglioside, which is found in cell membranes, has also shown promise in preclinical studies. However, in one multicenter double blind study, GM1 failed to establish efficacy, although a positive trend was reported in some outcome measures (36). Most recently Cethrin which blocks Rho A was tested after SCI with an application applied to the surface of the exposed dura (37). This safety trial showed promise, and further clinical trials to test efficacy are planned. In addition to these studies that target acute injury pathomechanisms, other treatment strategies that may enhance axon sprouting or regeneration, leading to increased circuit plasticity are also currently being evaluated in the lab and in clinical trials. For instance, according to clinicaltrials.gov, the Rick Hansen Institute Canada Inc. is sponsoring a minocycline study in acute spinal cord injured patients: Minocyline in Acute Spinal Cord Injury (MASC). Thus, it is encouraging that many clinical trials are being conducted or planned for the near future to target the detrimental consequences of SCI. Never-the-less there remains a great need to continue to investigate novel injury targets and therapeutic treatments including anti-inflammatory agents that may target clinically relevant secondary mechanisms such as the inflammasome.
In reference to inflammatory processes after experimental SCI, our group first reported evidence for a molecular platform in neurons regulating abnormal inflammasome signaling (13). Importantly, neutralization of ASC with a novel antibody treatment regimen after SCI reduced indicators of inflammasome activation and improved behavioral and histopathological outcomes. More recently, abnormal inflammasome activation in the sperm of men with SCI was also reported with neutralization improving sperm motility (38, 39). Together, these studies emphasize multiple roles of inflammasome activation in neuroinflammation after SCI and a target for treatment strategies to improve function and quality of life issues in people living with paralysis.
5. What are the current options available for the treatment of brain injury?
Traumatic brain injury (TBI) is a major cause of death and disability to humans and is a global problem. The incidence in TBI in the United States is at least 1.7 million annually with an estimated 5 million patients experiencing long-term complications (Centers for Disease Control (CDC) and Prevention) (40). In addition to injury to civilians, the use of explosive devices in recent wars has resulted in increasing numbers of blast-related head injury in military personnel (41). According to the CDC, the annual estimated direct and indirect medical cost of TBI is close to 76.5 billion in the United States. The pathophysiology of TBI like other neurological disorders is multifactorial (42-44), a situation that has increased the complexity of developing therapeutic interventions to target secondary injury mechanisms.
A recent publication has provided an excellent summary of neuroprotective strategies for TBI (43). A large number of preclinical studies have shown encouraging results with a variety of treatment strategies (45). However, more than 30 phase III clinical trials based on preclinical data have failed to generate positive results in severe TBI patients (43). In the past, various treatments have been tested for its efficacy in improving outcomes after TBI, including corticosteroids, progesterone, thyrotropin-releasing hormone (TRH), erythropoietin, statins and cyclosporin A. Regarding progesterone, two randomized double-blinded placebo-controlled Phase II clinical trials have been concluded that show trends toward improvement of outcome (46). However, recent phase III multicenter clinical trials including the ProTECT and SyNAPSE trials have been recently terminated because of lack of data supporting efficacy (47). Erythropoietin has been reported to exhibit multiple effects on excitotoxic, free radicals, edema and inflammatory processes after experimental TBI (48). In one randomized clinical study, erythropoietin failed to alter clinically relevant biomarkers associated with brain injury (49). Future clinical studies testing other interesting agents are underway in many countries.
In addition to pharmacological treatment strategies, other protocols such as targeted temperature management and induced therapeutic hypothermia are being conducted in single institutions as well as multicenter TBI trials (50). Temperature modifications after neurotrauma have the advantage of affecting multiple injury pathways and in many preclinical studies, shown beneficial effects in improving long-term functional outcome and reducing histopathological damage (51). Never-the-less multicenter, trials have failed to show significant differences in neurological recovery (52). Most recently, preclinical data has indicated the possibility of a selective group of severe TBI patients including individuals undergoing decompression surgery to have benefited from early hypothermic treatment. Multicenter trials to test this specific hypothesis are currently underway.
Statins or other inhibitors of cholesterol biosynthesis have been shown in preclinical models to have neuroprotective effects (53). A recent clinical trial with rosuvastatin in TBI patients reported improvements in some outcome measures and a phase II clinical trial is currently being planned. Cyclosporin A is an immunosuppressant that in multiple clinical studies has shown effectiveness (54). A randomized, double blind clinical trial of Cyclosporin A in TBI patients reported reduced CSF glutamate concentrations and improvements in cerebral perfusion pressure. With any new therapeutic strategy, while preclinical and early clinical findings are encouraging, well-designed multi-center trials are required to demonstrate beneficial effects across a very heterogeneous patient population.
Thus in TBI there is a great need of continued investigation, novel pathophysiological mechanisms and new treatments for this devastating disorder. In this regard, evidence for abnormal inflammasome activation has been reported in several models of brain injury (12, 55). Also, abnormal levels of inflammasome proteins have been seen in CSF samples taken from TBI patients that appear to correlate with injury severity (56). In terms of new treatment options for TBI, the therapeutic neutralization of the NLRP1 inflammasome reduced histopathological damage in one study (12). Finally, therapeutic hypothermia decreases inflammasome signaling, a result that emphasizes the importance of posttraumatic temperature on the innate immune response to brain injury (57).
6. What are the current options available for the treatment of stroke?
Stroke is the second leading cause of death in the United States. Despite decades of research, treatment options still remain limited (58, 59). Currently the only drug approved for the treatment of ischemic stroke is recombinant tissue plasminogen activator (rtPA), which has a limited time window for administration (60). Other options in addition to opening clotted blood vessels are being considered (61), including targeting inflammation, excitoxicity, oxidative stress, apoptosis and edema formation (62). Since 2000, 25 controlled clinical trials testing neuroprotective strategies have been completed based on encouraging preclinical data (59).
A recent publication has provided and an excellent summary of current clinical trials for Stroke (58). Magnesium sulfate, has been shown to improve recovery in some stroke patients and is currently being tested in pre-hospital settings (63). In a large clinical trial for subarachnoid hemorrhage, magnesium treatment was reported not to result in significant improvements (64). Other ongoing studies include the International Citicoline trial on Acute Stroke, which represents a nutritional supplemental strategy to improve memory retention. The efficacy of oral minocycline has been examined in a recent single, blinded, opened-label study (65). Patients receiving minocycline showed improvement at 30 days, thus leading to potential larger phase II and III trials. Cerebrolysin, which can mimic the actions of neurotrophic factors, has been shown in animal models to improve outcome. A double-blinded clinical trial was conducted and although no significant differences in the treatment groups at 90 days were seen, a positive trend was reported in a subpopulation of stroke patients (66). Finally, the calcium channel antagonist Ginsenoside-Rd showed significant improvements in outcomes at 15 and 90 days after stroke in one trial (67).
As discussed, several strategies to minimize the detrimental consequences of inflammation have been studied in various stroke models with variable results (58, 68). Recently the importance of inflammasome activation in the pathophysiology of global and focal cerebral ischemia has been discussed and demonstrated in the literature (16, 68-70). In an animal model of thromboembolic stroke, Abulafia and colleagues (16) reported evidence for the NLRP1 inflammasome complex forms after focal brain ischemia. Importantly, treatment with a neutralizing antibody against NLRP1 improved histopathological damage in this clinically relevant stroke model.
7. Is the Inflammasome A Promising Therapeutic Target For Central Nervous System Injury?
Inflammasome formation is regulated at various levels. First, general cellular conditions such as ion concentration and specific regulatory proteins such as heat shock protein (HSP)-90 and suppressor of G2 allele of skp1 (SGT1) influence inflammasome activation (71). Second, inflammasomes may be controlled at the level of sensor molecules or at the level of ASC and caspase-1 interactions through pyrinpyrin interactions or through death domain interactions. Third, assembly of inflammasomes may be negatively regulated by alternative splicing of ASC (72). Therefore, many proteins and protein interactions may contribute to different levels to activation of the inflammasome, offering a variety of control-points to interfere with signaling of these important signaling platforms.
Our studies show that antibody neutralization of ASC interferes with inflammasome signaling after TBI and SCI (12, 13). However, the precise mechanism of antibody blocking of ASC remains undetermined, but the most likely interpretation is that anti-ASC antibodies bind to ASC specks that are released into the extracellular space after inflammasome activation preventing the extracellular activation of caspase-1 and IL-1b (Figure 1) (73). In addition, an antibody against NLRP1 showed promise in reducing histopathological damage after stroke (16).
Inflammasome activation may also result in a unique form of cell death termed pyroptosis. Pyroptosis is characterized by the activation of caspase-1 and the formation of the pyroptosome, a complex of oligomerized ASC molecules. Since pyroptosis is caspase-1 dependent, it can be inhibited by the caspase-1 blocker YVAD (Tyr-Val-Ala-Asp) (74, 75). In cortical neurons, DNA is responsible for activating the AIM2 inflammasome, resulting in the formation of the pyroptosome and the opening of 2 to 3 nm membrane-pores that are permeable to the fluorescent dye YO-PRO and the cell-impermeant viability indicator ethidium homodimer-2 (Eth-D2). Inhibition with the pannexin-1 channel blockers Probenecid or brilliant blue FCF decreases pore formation, indicating that pannexin-1 is the pore that opens during the process of pyroptosis (76). These studies suggest pannexin-1 as a cell death effector channel in pyroptosis and demonstrate that inhibition of pannexin1 by Probenecid attenuates pyroptosis in neurons. Thus, pyroptosis may serve as a neuronal cell death mechanism induced by activation of the AIM2 inflammasome, providing additional evidence that neurons are not passive bystanders during injury or infection, but possess functional PRRs that recognize and respond to PAMPs or DAMPs.
The inflammasome proteins ASC, NLRP1 and caspase-1 are secreted into the cerebrospinal fluid (CSF) following TBI (56). Interestingly, the levels of these proteins were elevated in brain-injured patients that had poorer Glasgow outcome scale scores (GOS 1-3) when compared to less severely injured patients (GOS scores of 4-5) (56). GOS scores were obtained 5 months after injury but inflammasome protein levels were measured in CSF samples collected within then first week after injury. Thus, acute CSF concentrations of inflammasome proteins may be used as biomarkers to predict long-term outcomes or to guide therapeutic interventions with the goal of lowering inflammasome protein concentrations in the CSF. In support of these findings, two recent reports demonstrate that upon activation of caspase-1, oligomeric ASC-filaments/specks were released from macrophages (73, 77), resulting in pyroptotic cell death. After pyroptosis, ASC-filaments accumulate in the extracellular space, where they promote maturation of IL-1β. Oligomeric ASC-filaments were also present in the serum of patients with autoimmune pathologies and inherited autoinflammatory diseases (73, 77). These studies reveal extracellular functions of secreted inflammasome proteins that appear to amplify the innate immune inflammatory response (Figure 1).
Recently we showed that these inflammasome proteins in CSF of TBI and SCI subjects are packaged in exosomes (78). Exosomes are bioactive vesicles derived from the cell's endosomal membrane system and are secreted into surrounding body fluids. In terms of mechanisms for inducing an immune response, our studies suggest that exosomes carry bioactive cytokines such as IL-1β and inflammasome components (78). Thus, exosomes trigger an innate immune response and amplify such a response via the cargo of proteins, RNA or miRNA that transfers immune responsiveness to neighboring cells. Moreover, recent studies show that exosomes loaded with siRNA specifically deliver their cargo and silence ASC, a key component of inflammasome signaling, after SCI in CNS cells (78).
In addition, the therapeutic potential of exosome-mediated siRNA-delivery was first demonstrated by the knockdown of β-secretase 1 (BACE1), a therapeutic target in Alzheimer's disease (79). It has been proposed that exosomes containing pathological proteins mediate the spread of cell damage or alter the microenvironment in various metabolic and nervous system disorders (80, 81). However, the role of exosome-associated pathogenic proteins in disease etiology remains unclear and there is a paucity of information about the role of exosomes released after CNS trauma. A role of exosomes may have applicability to diverse pathologies, including CNS trauma, mental retardation, seizure, multiple sclerosis, Alzheimer's disease, HIV encephalitis, dementia, and ischemic injury (82-84). This understanding may give rise to new insights into biologically relevant targets to control inflammatory diseases or infections that will complement or replace existing therapies that are hindered by limited clinical efficacy, excessive adverse complications and failure to cross the blood-brain barrier.
8. What Therapies Can Be Used to Treat inflammasome-Related Immune Responses?
Prior to the discovery of the inflammasome, therapies intended to target the inflammatory response included blocking IL-1β and IL-18. In some conditions or diseases, these therapies, although efficient at times, have also failed to show long term beneficial effects. In addition, caspase-1 has also been identified as a target to decrease inflammation. However, all these therapies have failed to show a benefit in patients after CNS injury. With the discovery of the inflammasome, a new therapeutic target has been identified to treat damaging inflammation following CNS injury. In this regard, the adaptor protein-ASC offers great promise as a target since ASC is present in several different inflammasomes such as the NLRP1, NLRP2, NLRP3 and AIM2 inflammasomes. A neutralizing antibody against ASC has been shown to be anti-inflammatory after SCI (13) and TBI (12). Anti-ASC delivered after CNS injury results in decreased caspase-1 cleavage (decreased inflammasome activation) as well as improved histopathology and functional outcomes (12, 13). Similarly, a neutralizing antibody against NLRP1 has been shown to decrease inflammasome activation after stroke (16). These data indicate that biologics against inflammasome proteins offer a promising anti-inflammatory approach that may result in improved outcomes after CNS injury.
Another drug targeting the inflammasome component ASC is the cytokine release inhibitory drug CRID3. CRID3, also known as CP-456,773 is a Pfizer compound that prevents ASC oligomerization thus decreasing inflammasome activation (85). However, CRID3's effects in vivo have not been described in the literature. In addition, Harter at al. described a cysteinyl leukotriene receptor antagonist developed by Bayer Pharmaceuticals, that targets ASC oligomerization and is described in U.S Patent application number 7,498,460 (85). Cysteinyl leukotriene receptor antagonists have been shown to block myocardial ischemia/reperfusion injury in mice (85) and are neuroprotective after brain ischemia (86) and seizures (87).
Other potential therapies that target the inflammasome are those agents that block IL-1 (IL-1RA). For example, Anakinra, is a human recombinant protein against the receptor of IL-1β. It is commercialized under the name of Kineret by Amgen and is approved for the treatment of rheumatoid arthritis (88). Anakinra reverses the peripheral innate immune suppression during the acute phase of stroke (89). However, whether Anakinra improves stroke severity in patients is yet to be studied. Importantly, animal studies using IL-1RA show good penetration of this drug into the brain parenchyma within therapeutic levels (90). Animal studies demonstrate a therapeutic potential for Anakinra in stroke (91), but whether Anakinra is beneficial after TBI or SCI in humans has not been determined.
Another drug that targets IL-1β is Canakinumab, a human IgGκ monoclonal antibody developed by Novartis under the name of Ilaris and has been approved for the treatment of cryopyrin-associated periodic syndromes (CAPS) (92). Whether Canakinumab is effective in CNS injury is yet to be determined. However, there is promising data suggesting that Canakinumab may be beneficial for the treatment of cardiovascular events and traumatic brain injury (93-96).
In regards to IL-18, although IL-18 has been implicated in the pathogenesis of TBI and stroke (2, 3, 6, 8, 9), no animal data exist showing that direct inhibition of IL-18 improves outcomes after injury. Targeting IL-18, GlaxoSmithKline has recently completed a phase II clinical trial for the treatment of diabetes mellitus using an IL-18 neutralizing antibody, GSK1070806. However, to the best of our knowledge, this drug has not been tested in CNS indications (97).
Several caspase-1 inhibitors have been tested in inflammatory conditions such as rheumatoid arthritis and epilepsy. The small molecule Pralnacasan (VX-740), manufactured by Vertex Pharmaceuticals, has been shown to improve inflammatory outcomes in mouse models of arthritis (98), and has currently completed phase II clinical trials for the treatment of rheumatoid arthritis. This trial showed a concentration-dependent improvement in clinical signs of arthritis in the Pralnacasan-treated population (98, 99). In regards to the CNS, VRT-018858, the active metabolite of Pralnacasan has been shown to improve brain damage following transient brain ischemia in rats (100). Similarly, Belnacasan, (VX-765), another drug by Vertex Pharmaceuticals, has been shown to be effective as an anti-seizure medication in rodents (101). VX-765 has been tested in clinical trials for Treatment-Resistant Partial Epilepsy, but the study was terminated by the sponsor, according to clinicaltrials.gov.
Another caspase-1 inhibitor is Parthenolide. Parthenolide is the major sesquiterpene lactone present in Mexican medicinal plants and in Tanacetum parthenium or feverfew. It has a potent anti-inflammatory action and has been reported to be neuroprotective after stroke (102). However, it is not water-soluble and the bioavailability of Parthenolide limits its use as a drug, but efforts are currently underway to develop Parthenolide analogs for therapeutic use (103).
NLRs are also potential candidates that could be targeted with anti-inflammatory drugs. NLR biology has identified ATPase activity as necessary for NLRP3 inflammasome activation (104). The sulfonylurea Glyburide inhibits ATP-sensitive K+ (KATP) channels (105) and has been shown to inhibit NLRP3 inflammasome activation, without affecting the NLRP1 or NLRC4 inflammasomes (106). Glyburide is approved for the treatment of type II diabetes (107). Importantly, the hypoglycemic effects of Glyburide limit its therapeutic usage to other indications including CNS injury; however, 16673-34-0, an intermediate substrate free of the cyclohexylurea moiety of glyburide has been shown to be beneficial in a mouse model of myocardial infarction. Importantly, 16673-34-0 does not cause a hypoglycemic effect (108). Another inhibitor of NLR signaling is the NLRP3 inhibitor MCC950. MCC950 also prevents ASC oligomerization, and it has been shown to improve the clinical score in a rodent model of experimental autoimmune encephalomyelitis (109).
Finally, other protein targets that may inhibit inflammasomes in the CNS include P2X7, P2X4 and pannexin-1 (14, 25, 26). Probenecid, which is approved for the treatment of gout, inhibits inflammasome activation by blocking the pannexin-1 channel (25). Moreover, the P2X7 inhibitor AZD9056 has been tested for the treatment of rheumatoid arthritis with no clinical success (110). Moreover, this drug has not been tested for CNS indications. Other drugs targeting P2X7 have also shown similar failures in clinical trials (111). Finally, in regards to P2X4 no potent selective antagonists have been identified yet (112).
9. Conclusion
Over the last two decades, there has been a significant increase in our understanding of inflammasomes and inflammasome-related diseases. Inflammasome-mediated diseases include: common metabolic disorders, monogenic inflammatory disorders and trauma-induced disorders. However, many questions remain before therapeutics can be designed to regulate the innate immune response in inflammasome disease-states and conditions. We now know that inflammasomes are responsive to noninfectious triggers such as ion fluxes and cold temperatures as well as to microbial and viral metabolites. Recent studies also demonstrate that inflammasomes act outside the cell in the formation of ASC specks that accumulate in the extracellular space after pyroptotic cell death. ASC specks are found in tissue fluids from TBI and SCI patients and in patients with chronic inflammatory lung diseases, autoimmune diseases and CAPS.
Despite the immense impact of inflammasome biology since its discovery over a decade ago, no therapies that directly target the inflammasome have been approved. Ongoing discoveries will facilitate the novel therapeutics that directly target inflammasome components like ASC. Understanding the mechanisms regulating inflammasome function should help in the development of therapeutics for clinical management of inflammation-based disease processes and conditions.
Acknowledgments
This work was supported by grants to JPdRV (American Heart Association 12SDG11970010) and to RWK (NIH Grant NINDS 1R42NS086274) as well as The Miami Project to Cure Paralysis. All authors have read the journal's authorship agreement and the manuscript has been reviewed by and approved by all named authors.
Footnotes
Conflict of Interest: All authors have read the journal's policy on disclosure of potential conflicts of interest. The authors declare that they are co-founders of InflamaCORE, LLC.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular cell. 2002 Aug;10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 2.Piazza O, Scarpati G, Cotena S, Lonardo M, Tufano R. Thrombin antithrombin complex and IL-18 serum levels in stroke patients. Neurology international. 2010;2(1):e1. doi: 10.4081/ni.2010.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jefferis BJ, Whincup PH, Welsh P, Wannamethee SG, Rumley A, Ebrahim S, et al. Prospective study of IL-18 and risk of MI and stroke in men and women aged 60-79 years: a nested case-control study. Cytokine. 2013 Feb;61(2):513–20. doi: 10.1016/j.cyto.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cvetkovic JT, Wiklund PG, Ahmed E, Weinehall L, Hallmans G, Lefvert AK. Polymorphisms of IL-1beta, IL-1Ra, and TNF-alpha genes: a nested case-control study of their association with risk for stroke. Journal of stroke and cerebrovascular diseases : the official journal of National Stroke Association. 2005 Jan-Feb;14(1):29–35. doi: 10.1016/j.jstrokecerebrovasdis.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Hutchinson PJ, O'Connell MT, Rothwell NJ, Hopkins SJ, Nortje J, Carpenter KL, et al. Inflammation in human brain injury: intracerebral concentrations of IL-1alpha, IL-1beta, and their endogenous inhibitor IL-1ra. Journal of neurotrauma. 2007 Oct;24(10):1545–57. doi: 10.1089/neu.2007.0295. [DOI] [PubMed] [Google Scholar]
- 6.Ciaramella A, Della Vedova C, Salani F, Viganotti M, D'Ippolito M, Caltagirone C, et al. Increased levels of serum IL-18 are associated with the long-term outcome of severe traumatic brain injury. Neuroimmunomodulation. 2014;21(1):8–12. doi: 10.1159/000354764. [DOI] [PubMed] [Google Scholar]
- 7.Kostulas N, Pelidou SH, Kivisakk P, Kostulas V, Link H. Increased IL-1beta, IL-8, and IL-17 mRNA expression in blood mononuclear cells observed in a prospective ischemic stroke study. Stroke; a journal of cerebral circulation. 1999 Oct;30(10):2174–9. doi: 10.1161/01.str.30.10.2174. [DOI] [PubMed] [Google Scholar]
- 8.Yatsiv I, Morganti-Kossmann MC, Perez D, Dinarello CA, Novick D, Rubinstein M, et al. Elevated intracranial IL-18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL-18-binding protein after experimental closed head injury. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2002 Aug;22(8):971–8. doi: 10.1097/00004647-200208000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Bossu P, Salani F, Cacciari C, Picchetto L, Cao M, Bizzoni F, et al. Disease outcome, alexithymia and depression are differently associated with serum IL-18 levels in acute stroke. Current neurovascular research. 2009 Aug;6(3):163–70. doi: 10.2174/156720209788970036. [DOI] [PubMed] [Google Scholar]
- 10.Vigano E, Mortellaro A. Caspase-11: The driving factor for noncanonical inflammasomes. European journal of immunology. 2013 Sep;43(9):2240–5. doi: 10.1002/eji.201343800. [DOI] [PubMed] [Google Scholar]
- 11.Adamczak SE, de Rivero Vaccari JP, Dale G, Brand FJ, 3rd, Nonner D, Bullock MR, et al. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2014 Apr;34(4):621–9. doi: 10.1038/jcbfm.2013.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2009 Jul;29(7):1251–61. doi: 10.1038/jcbfm.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW. A molecular platform in neurons regulates inflammation after spinal cord injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008 Mar 26;28(13):3404–14. doi: 10.1523/JNEUROSCI.0157-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia. 2013 Jul;61(7):1113–21. doi: 10.1002/glia.22499. [DOI] [PubMed] [Google Scholar]
- 15.Beug ST, Cheung HH, LaCasse EC, Korneluk RG. Modulation of immune signalling by inhibitors of apoptosis. Trends in immunology. 2012 Nov;33(11):535–45. doi: 10.1016/j.it.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 16.Abulafia DP, de Rivero Vaccari JP, Lozano JD, Lotocki G, Keane RW, Dietrich WD. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2009 Mar;29(3):534–44. doi: 10.1038/jcbfm.2008.143. [DOI] [PubMed] [Google Scholar]
- 17.Madouri F, Guillou N, Fauconnier L, Marchiol T, Rouxel N, Chenuet P, et al. Caspase-1 activation by NLRP3 inflammasome dampens IL-33-dependent house dust mite-induced allergic lung inflammation. Journal of molecular cell biology. 2015 Feb 24; doi: 10.1093/jmcb/mjv012. [DOI] [PubMed] [Google Scholar]
- 18.Gadani SP, Walsh JT, Smirnov I, Zheng J, Kipnis J. The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron. 2015 Feb 18;85(4):703–9. doi: 10.1016/j.neuron.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. Journal of leukocyte biology. 2007 Jan;81(1):1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 20.Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunological reviews. 2012 Sep;249(1):158–75. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Rivero Vaccari JC, Brand FJ, 3rd, Berti AF, Alonso OF, Bullock MR, de Rivero Vaccari JP. Mincle signaling in the innate immune response after traumatic brain injury. Journal of neurotrauma. 2015 Feb 15;32(4):228–36. doi: 10.1089/neu.2014.3436. [DOI] [PubMed] [Google Scholar]
- 22.Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW. Pattern recognition receptors and central nervous system repair. Experimental neurology. 2014 Aug;258:5–16. doi: 10.1016/j.expneurol.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nature reviews Immunology. 2013 Jun;13(6):397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lane T, Flam B, Lockey R, Kolliputi N. TXNIP shuttling: missing link between oxidative stress and inflammasome activation. Frontiers in physiology. 2013;4:50. doi: 10.3389/fphys.2013.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silverman WR, de Rivero Vaccari JP, Locovei S, Qiu F, Carlsson SK, Scemes E, et al. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. The Journal of biological chemistry. 2009 Jul 3;284(27):18143–51. doi: 10.1074/jbc.M109.004804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Rivero Vaccari JP, Bastien D, Yurcisin G, Pineau I, Dietrich WD, De Koninck Y, et al. P2X4 receptors influence inflammasome activation after spinal cord injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012 Feb 29;32(9):3058–66. doi: 10.1523/JNEUROSCI.4930-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varma AK, Das A, Wallace Gt, Barry J, Vertegel AA, Ray SK, et al. Spinal cord injury: a review of current therapy, future treatments, and basic science frontiers. Neurochemical research. 2013 May;38(5):895–905. doi: 10.1007/s11064-013-0991-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ackery A, Tator C, Krassioukov A. A global perspective on spinal cord injury epidemiology. Journal of neurotrauma. 2004 Oct;21(10):1355–70. doi: 10.1089/neu.2004.21.1355. [DOI] [PubMed] [Google Scholar]
- 29.Tator CH. Review of treatment trials in human spinal cord injury: issues, difficulties, and recommendations. Neurosurgery. 2006 Nov;59(5):957–82. doi: 10.1227/01.NEU.0000245591.16087.89. [DOI] [PubMed] [Google Scholar]
- 30.Coleman WP, Benzel D, Cahill DW, Ducker T, Geisler F, Green B, et al. A critical appraisal of the reporting of the National Acute Spinal Cord Injury Studies (II and III) of methylprednisolone in acute spinal cord injury. Journal of spinal disorders. 2000 Jun;13(3):185–99. doi: 10.1097/00002517-200006000-00001. [DOI] [PubMed] [Google Scholar]
- 31.Coleman WP, Benzel D, Cahill DW, Ducker T, Geisler F, Green B, et al. A critical appraisal of the reporting of the National Acute Spinal Cord Injury Studies (II and III) of methylprednisolone in acute spinal cord injury. Journal of spinal disorders. 2000 Jun;13(3):185–99. doi: 10.1097/00002517-200006000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Kan EM, Ling EA, Lu J. Stem cell therapy for spinal cord injury. Current medicinal chemistry. 2010;17(36):4492–510. doi: 10.2174/092986710794182971. [DOI] [PubMed] [Google Scholar]
- 33.Pointillart V, Petitjean ME, Wiart L, Vital JM, Lassie P, Thicoipe M, et al. Pharmacological therapy of spinal cord injury during the acute phase. Spinal cord. 2000 Feb;38(2):71–6. doi: 10.1038/sj.sc.3100962. [DOI] [PubMed] [Google Scholar]
- 34.Fehlings MG, Baptiste DC. Current status of clinical trials for acute spinal cord injury. Injury. 2005 Jul;36(Suppl 2):B113–22. doi: 10.1016/j.injury.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 35.Faden AI. Opiate-receptor antagonists, thyrotropin-releasing hormone (TRH), and TRH analogs in the treatment of spinal cord injury. Central nervous system trauma : journal of the American Paralysis Association. 1987;4(4):217–26. doi: 10.1089/cns.1987.4.217. [DOI] [PubMed] [Google Scholar]
- 36.Chinnock P, Roberts I. Gangliosides for acute spinal cord injury. The Cochrane database of systematic reviews. 2005;(2):CD004444. doi: 10.1002/14651858.CD004444.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fehlings MG, Theodore N, Harrop J, Maurais G, Kuntz C, Shaffrey CI, et al. A phase I/IIa clinical trial of a recombinant Rho protein antagonist in acute spinal cord injury. Journal of neurotrauma. 2011 May;28(5):787–96. doi: 10.1089/neu.2011.1765. [DOI] [PubMed] [Google Scholar]
- 38.Ibrahim E, Castle SM, Aballa TC, Keane RW, de Rivero Vaccari JP, Lynne CM, et al. Neutralization of ASC improves sperm motility in men with spinal cord injury. Human reproduction. 2014 Nov;29(11):2368–73. doi: 10.1093/humrep/deu230. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X, Ibrahim E, de Rivero Vaccari JP, Lotocki G, Aballa TC, Dietrich WD, et al. Involvement of the inflammasome in abnormal semen quality of men with spinal cord injury. Fertility and sterility. 2013 Jan;99(1):118–24. doi: 10.1016/j.fertnstert.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 40.Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. The Lancet Neurology. 2008 Aug;7(8):728–41. doi: 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- 41.Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. The New England journal of medicine. 2008 Jan 31;358(5):453–63. doi: 10.1056/NEJMoa072972. [DOI] [PubMed] [Google Scholar]
- 42.Bramlett HM, Dietrich WD. Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Progress in brain research. 2007;161:125–41. doi: 10.1016/S0079-6123(06)61009-1. [DOI] [PubMed] [Google Scholar]
- 43.Kabadi SV, Faden AI. Neuroprotective strategies for traumatic brain injury: improving clinical translation. International journal of molecular sciences. 2014;15(1):1216–36. doi: 10.3390/ijms15011216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McIntosh TK, Smith DH, Garde E. Therapeutic approaches for the prevention of secondary brain injury. European journal of anaesthesiology. 1996 May;13(3):291–309. doi: 10.1046/j.1365-2346.1996.00981.x. [DOI] [PubMed] [Google Scholar]
- 45.Faden AI. Neuroprotection and traumatic brain injury: theoretical option or realistic proposition. Current opinion in neurology. 2002 Dec;15(6):707–12. doi: 10.1097/01.wco.0000044767.39452.bf. [DOI] [PubMed] [Google Scholar]
- 46.Stein DG. Progesterone in the treatment of acute traumatic brain injury: a clinical perspective and update. Neuroscience. 2011 Sep 15;191:101–6. doi: 10.1016/j.neuroscience.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 47.Wright DW, Kellermann AL, Hertzberg VS, Clark PL, Frankel M, Goldstein FC, et al. ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Annals of emergency medicine. 2007 Apr;49(4):391–402. e1–2. doi: 10.1016/j.annemergmed.2006.07.932. [DOI] [PubMed] [Google Scholar]
- 48.Ponce LL, Navarro JC, Ahmed O, Robertson CS. Erythropoietin neuroprotection with traumatic brain injury. Pathophysiology : the official journal of the International Society for Pathophysiology/ISP. 2013 Feb;20(1):31–8. doi: 10.1016/j.pathophys.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nirula R, Diaz-Arrastia R, Brasel K, Weigelt JA, Waxman K. Safety and efficacy of erythropoietin in traumatic brain injury patients: a pilot randomized trial. Critical care research and practice. 2010;2010 doi: 10.1155/2010/209848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marion DW. Decompressive craniectomy in diffuse traumatic brain injury. The Lancet Neurology. 2011 Jun;10(6):497–8. doi: 10.1016/S1474-4422(11)70098-9. [DOI] [PubMed] [Google Scholar]
- 51.Dietrich WD, Bramlett HM. The evidence for hypothermia as a neuroprotectant in traumatic brain injury. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2010 Jan;7(1):43–50. doi: 10.1016/j.nurt.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yokobori S, Hosein K, Burks S, Sharma I, Gajavelli S, Bullock R. Biomarkers for the clinical differential diagnosis in traumatic brain injury--a systematic review. CNS neuroscience & therapeutics. 2013 Aug;19(8):556–65. doi: 10.1111/cns.12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wible EF, Laskowitz DT. Statins in traumatic brain injury. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2010 Jan;7(1):62–73. doi: 10.1016/j.nurt.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mazzeo AT, Alves OL, Gilman CB, Hayes RL, Tolias C, Niki Kunene K, et al. Brain metabolic and hemodynamic effects of cyclosporin A after human severe traumatic brain injury: a microdialysis study. Acta neurochirurgica. 2008 Oct;150(10):1019–31. doi: 10.1007/s00701-008-0021-7. discussion 31. [DOI] [PubMed] [Google Scholar]
- 55.Liu HD, Li W, Chen ZR, Hu YC, Zhang DD, Shen W, et al. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochemical research. 2013 Oct;38(10):2072–83. doi: 10.1007/s11064-013-1115-z. [DOI] [PubMed] [Google Scholar]
- 56.Adamczak S, Dale G, de Rivero Vaccari JP, Bullock MR, Dietrich WD, Keane RW. Inflammasome proteins in cerebrospinal fluid of brain-injured patients as biomarkers of functional outcome: clinical article. Journal of neurosurgery. 2012 Dec;117(6):1119–25. doi: 10.3171/2012.9.JNS12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tomura S, de Rivero Vaccari JP, Keane RW, Bramlett HM, Dietrich WD. Effects of therapeutic hypothermia on inflammasome signaling after traumatic brain injury. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2012 Oct;32(10):1939–47. doi: 10.1038/jcbfm.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Majid A. Neuroprotection in stroke: past, present, and future. ISRN neurology. 2014;2014:515716. doi: 10.1155/2014/515716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tymianski M. Novel approaches to neuroprotection trials in acute ischemic stroke. Stroke; a journal of cerebral circulation. 2013 Oct;44(10):2942–50. doi: 10.1161/STROKEAHA.113.000731. [DOI] [PubMed] [Google Scholar]
- 60.Reed SD, Cramer SC, Blough DK, Meyer K, Jarvik JG. Treatment with tissue plasminogen activator and inpatient mortality rates for patients with ischemic stroke treated in community hospitals. Stroke; a journal of cerebral circulation. 2001 Aug;32(8):1832–40. doi: 10.1161/01.str.32.8.1832. [DOI] [PubMed] [Google Scholar]
- 61.Noorian AR, Gupta R, Nogueira RG. Stenting of complete vertebral artery ostial occlusion in a patient with medically refractory vertebrobasilar ischemia. Journal of neurointerventional surgery. 2012 Nov;4(6):e31. doi: 10.1136/neurintsurg-2011-010139. [DOI] [PubMed] [Google Scholar]
- 62.O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Annals of neurology. 2006 Mar;59(3):467–77. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- 63.Saver JL, Starkman S, Eckstein M, Stratton SJ, Pratt FD, Hamilton S, et al. Prehospital use of magnesium sulfate as neuroprotection in acute stroke. The New England journal of medicine. 2015 Feb 5;372(6):528–36. doi: 10.1056/NEJMoa1408827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dorhout Mees SM group M-Is. Magnesium in aneurysmal subarachnoid hemorrhage (MASH II) phase III clinical trial MASH-II study group. International journal of stroke : official journal of the International Stroke Society. 2008 Feb;3(1):63–5. doi: 10.1111/j.1747-4949.2008.00168.x. [DOI] [PubMed] [Google Scholar]
- 65.Davalos A, Alvarez-Sabin J, Castillo J, Diez-Tejedor E, Ferro J, Martinez-Vila E, et al. Citicoline in the treatment of acute ischaemic stroke: an international, randomised, multicentre, placebo-controlled study (ICTUS trial) Lancet. 2012 Jul 28;380(9839):349–57. doi: 10.1016/S0140-6736(12)60813-7. [DOI] [PubMed] [Google Scholar]
- 66.Heiss WD, Brainin M, Bornstein NM, Tuomilehto J, Hong Z. Cerebrolysin Acute Stroke Treatment in Asia I. Cerebrolysin in patients with acute ischemic stroke in Asia: results of a double-blind, placebo-controlled randomized trial. Stroke; a journal of cerebral circulation. 2012 Mar;43(3):630–6. doi: 10.1161/STROKEAHA.111.628537. [DOI] [PubMed] [Google Scholar]
- 67.Liu X, Wang L, Wen A, Yang J, Yan Y, Song Y, et al. Ginsenoside-Rd improves outcome of acute ischaemic stroke - a randomized, double-blind, placebo-controlled, multicenter trial. European journal of neurology : the official journal of the European Federation of Neurological Societies. 2012 Jun;19(6):855–63. doi: 10.1111/j.1468-1331.2011.03634.x. [DOI] [PubMed] [Google Scholar]
- 68.Mohamed IN, Ishrat T, Fagan SC, El-Remessy AB. Role of Inflammasome Activation in the Pathophysiology of Vascular Diseases of the Neurovascular Unit. Antioxidants & redox signaling. 2014 Nov 11; doi: 10.1089/ars.2014.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fann DY, Lee SY, Manzanero S, Tang SC, Gelderblom M, Chunduri P, et al. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell death & disease. 2013;4:e790. doi: 10.1038/cddis.2013.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma Q, Chen S, Hu Q, Feng H, Zhang JH, Tang J. NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Annals of neurology. 2014 Feb;75(2):209–19. doi: 10.1002/ana.24070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mayor A, Martinon F, De Smedt T, Petrilli V, Tschopp J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nature immunology. 2007 May;8(5):497–503. doi: 10.1038/ni1459. [DOI] [PubMed] [Google Scholar]
- 72.Bryan NB, Dorfleutner A, Kramer SJ, Yun C, Rojanasakul Y, Stehlik C. Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. Journal of inflammation. 2010;7:23. doi: 10.1186/1476-9255-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nature immunology. 2014 Aug;15(8):727–37. doi: 10.1038/ni.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell death and differentiation. 2007 Sep;14(9):1590–604. doi: 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 2008 Mar 18;105(11):4312–7. doi: 10.1073/pnas.0707370105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adamczak SE. Open Access Dissertations. 2012. Molecular Recognition of DNA by the AIM2 Inflammasome Induces Neuronal Pyroptosis: Implications in Infection and Host Tissue Damage. Paper 854. [Google Scholar]
- 77.Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nature immunology. 2014 Aug;15(8):738–48. doi: 10.1038/ni.2919. [DOI] [PubMed] [Google Scholar]
- 78.de Rivero Vaccari JP, Brand F, 3rd, Adamczak S, Lee SW, Barcena JP, Wang MY, et al. Exosome-mediated inflammasome signaling after central nervous system injury. Journal of neurochemistry. 2015 Jan 27; doi: 10.1111/jnc.13036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nature biotechnology. 2011 Apr;29(4):341–5. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 80.Fruhbeis C, Frohlich D, Kuo WP, Amphornrat J, Thilemann S, Saab AS, et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS biology. 2013 Jul;11(7):e1001604. doi: 10.1371/journal.pbio.1001604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pant S, Hilton H, Burczynski ME. The multifaceted exosome: biogenesis, role in normal and aberrant cellular function, and frontiers for pharmacological and biomarker opportunities. Biochemical pharmacology. 2012 Jun 1;83(11):1484–94. doi: 10.1016/j.bcp.2011.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Progress in neurobiology. 2005 Jun;76(2):77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 83.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends in neurosciences. 1999 Sep;22(9):391–7. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 84.Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. British journal of pharmacology. 2006 Jan;147(Suppl 1):S232–40. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Coll RC, Robertson A, Butler M, Cooper M, O'Neill LA. The cytokine release inhibitory drug CRID3 targets ASC oligomerisation in the NLRP3 and AIM2 inflammasomes. PloS one. 2011;6(12):e29539. doi: 10.1371/journal.pone.0029539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shi QJ, Xiao L, Zhao B, Zhang XY, Wang XR, Xu DM, et al. Intracerebroventricular injection of HAMI 3379, a selective cysteinyl leukotriene receptor 2 antagonist, protects against acute brain injury after focal cerebral ischemia in rats. Brain research. 2012 Nov 12;1484:57–67. doi: 10.1016/j.brainres.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 87.Lenz QF, Arroyo DS, Temp FR, Poersch AB, Masson CJ, Jesse AC, et al. Cysteinyl leukotriene receptor (CysLT) antagonists decrease pentylenetetrazol-induced seizures and blood-brain barrier dysfunction. Neuroscience. 2014 Sep 26;277:859–71. doi: 10.1016/j.neuroscience.2014.07.058. [DOI] [PubMed] [Google Scholar]
- 88.Cohen SB, Moreland LW, Cush JJ, Greenwald MW, Block S, Shergy WJ, et al. A multicentre, double blind, randomised, placebo controlled trial of anakinra (Kineret), a recombinant interleukin 1 receptor antagonist, in patients with rheumatoid arthritis treated with background methotrexate. Annals of the rheumatic diseases. 2004 Sep;63(9):1062–8. doi: 10.1136/ard.2003.016014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Smith CJ, Emsley HC, Udeh CT, Vail A, Hoadley ME, Rothwell NJ, et al. Interleukin-1 receptor antagonist reverses stroke-associated peripheral immune suppression. Cytokine. 2012 Jun;58(3):384–9. doi: 10.1016/j.cyto.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 90.Greenhalgh AD, Galea J, Denes A, Tyrrell PJ, Rothwell NJ. Rapid brain penetration of interleukin-1 receptor antagonist in rat cerebral ischaemia: pharmacokinetics, distribution, protection. British journal of pharmacology. 2010 May;160(1):153–9. doi: 10.1111/j.1476-5381.2010.00684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Banwell V, Sena ES, Macleod MR. Systematic review and stratified meta-analysis of the efficacy of interleukin-1 receptor antagonist in animal models of stroke. Journal of stroke and cerebrovascular diseases : the official journal of National Stroke Association. 2009 Jul-Aug;18(4):269–76. doi: 10.1016/j.jstrokecerebrovasdis.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 92.Kuemmerle-Deschner JB, Ramos E, Blank N, Roesler J, Felix SD, Jung T, et al. Canakinumab (ACZ885, a fully human IgG1 anti-IL-1beta mAb) induces sustained remission in pediatric patients with cryopyrin-associated periodic syndrome (CAPS) Arthritis research & therapy. 2011;13(1):R34. doi: 10.1186/ar3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Abbate A, Van Tassell BW, Biondi-Zoccai GG. Blocking interleukin-1 as a novel therapeutic strategy for secondary prevention of cardiovascular events. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy. 2012 Aug 1;26(4):217–33. doi: 10.1007/BF03261881. [DOI] [PubMed] [Google Scholar]
- 94.Clausen F, Hanell A, Bjork M, Hillered L, Mir AK, Gram H, et al. Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. The European journal of neuroscience. 2009 Aug;30(3):385–96. doi: 10.1111/j.1460-9568.2009.06820.x. [DOI] [PubMed] [Google Scholar]
- 95.Clausen F, Hanell A, Israelsson C, Hedin J, Ebendal T, Mir AK, et al. Neutralization of interleukin-1beta reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. The European journal of neuroscience. 2011 Jul;34(1):110–23. doi: 10.1111/j.1460-9568.2011.07723.x. [DOI] [PubMed] [Google Scholar]
- 96.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) American heart journal. 2011 Oct;162(4):597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 97.Mistry P, Reid J, Pouliquen I, McHugh S, Abberley L, DeWall S, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of single-dose antiinterleukin- 18 mAb GSK1070806 in healthy and obese subjects. International journal of clinical pharmacology and therapeutics. 2014 Oct;52(10):867–79. doi: 10.5414/CP202087. [DOI] [PubMed] [Google Scholar]
- 98.Rudolphi K, Gerwin N, Verzijl N, van der Kraan P, van den Berg W. Pralnacasan, an inhibitor of interleukin-1beta converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthritis and cartilage/OARS, Osteoarthritis Research Society. 2003 Oct;11(10):738–46. doi: 10.1016/s1063-4584(03)00153-5. [DOI] [PubMed] [Google Scholar]
- 99.Siegmund B, Zeitz M. Pralnacasan (vertex pharmaceuticals) IDrugs : the investigational drugs journal. 2003 Feb;6(2):154–8. [PubMed] [Google Scholar]
- 100.Ross J, Brough D, Gibson RM, Loddick SA, Rothwell NJ. A selective, non-peptide caspase-1 inhibitor, VRT-018858, markedly reduces brain damage induced by transient ischemia in the rat. Neuropharmacology. 2007 Oct;53(5):638–42. doi: 10.1016/j.neuropharm.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 101.Noe FM, Polascheck N, Frigerio F, Bankstahl M, Ravizza T, Marchini S, et al. Pharmacological blockade of IL-1beta/IL-1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiology of disease. 2013 Nov;59:183–93. doi: 10.1016/j.nbd.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 102.Dong L, Qiao H, Zhang X, Zhang X, Wang C, Wang L, et al. Parthenolide is neuroprotective in rat experimental stroke model: downregulating NF-kappaB, phospho-p38MAPK, and caspase-1 and ameliorating BBB permeability. Mediators of inflammation. 2013;2013:370804. doi: 10.1155/2013/370804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Guzman ML, Rossi RM, Neelakantan S, Li X, Corbett CA, Hassane DC, et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood. 2007 Dec 15;110(13):4427–35. doi: 10.1182/blood-2007-05-090621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006 Mar 9;440(7081):228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 105.Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. The Journal of clinical investigation. 2005 Aug;115(8):2047–58. doi: 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. The Journal of cell biology. 2009 Oct 5;187(1):61–70. doi: 10.1083/jcb.200903124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Neufeld ND, Harris M, Corbo LM, Koduri A. Effect of glyburide in type II diabetes mellitus. Studies of monocyte membrane fluidity, lipid composition, and insulin binding. Diabetes. 1987 Dec;36(12):1351–5. doi: 10.2337/diab.36.12.1351. [DOI] [PubMed] [Google Scholar]
- 108.Marchetti C, Chojnacki J, Toldo S, Mezzaroma E, Tranchida N, Rose SW, et al. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. Journal of cardiovascular pharmacology. 2014 Apr;63(4):316–22. doi: 10.1097/FJC.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature medicine. 2015 Feb 16; doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Keystone EC, Wang MM, Layton M, Hollis S, McInnes IB, Team DCS. Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Annals of the rheumatic diseases. 2012 Oct;71(10):1630–5. doi: 10.1136/annrheumdis-2011-143578. [DOI] [PubMed] [Google Scholar]
- 111.Ozaki E, Campbell M, Doyle SL. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. Journal of inflammation research. 2015;8:15–27. doi: 10.2147/JIR.S51250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.North RA, Jarvis MF. P2X receptors as drug targets. Molecular pharmacology. 2013 Apr;83(4):759–69. doi: 10.1124/mol.112.083758. [DOI] [PMC free article] [PubMed] [Google Scholar]