

Abstract
Constitutive activation of the extracellular-signal-regulated kinases 1 and 2 (ERK1/2) are central to regulating the proliferation and survival of many cancer cells. The current inhibitors of ERK1/2 target ATP binding or the catalytic site and are therefore limited in their utility for elucidating the complex biological roles of ERK1/2 through its phosphorylation and regulation of over 100 substrate proteins. To overcome this limitation, a combination of computational and experimental methods was used to identify low-molecular-mass inhibitors that are intended to target ERK1/2 substrate-docking domains and selectively interfere with ERK1/2 regulation of substrate proteins. In the present study, we report the identification and characterization of compounds with a thienyl benzenesulfonate scaffold that were designed to inhibit ERK1/2 substrates containing an F-site or DEF (docking site for ERK, FXF) motif. Experimental evidence shows the compounds inhibit the expression of F-site containing immediate early genes (IEGs) of the Fos family, including c-Fos and Fra1, and transcriptional regulation of the activator protein-1 (AP-1) complex. Moreover, this class of compounds selectively induces apoptosis in melanoma cells containing mutated BRaf and constitutively active ERK1/2 signalling, including melanoma cells that are inherently resistant to clinically relevant kinase inhibitors. These findings represent the identification and initial characterization of a novel class of compounds that inhibit ERK1/2 signalling functions and their potential utility for elucidating ERK1/2 and other signalling events that control the growth and survival of cancer cells containing elevated ERK1/2 activity.
Keywords: activator protein-1, ATP-independent inhibitors, extracellular-signal-regulated kinases 1 and 2 (ERK1/2), in silico design, melanoma, mitogen-activated protein kinase (MAPK)
INTRODUCTION
The mitogen-activated protein kinase (MAPK) family members, which include the extracellular-signal-regulated kinases 1 and 2 (ERK1/2), c-Jun N-terminal kinases (JNKs), p38 MAPKs and ERK5, are major regulators of cell proliferation, apoptosis, differentiation, migration and inflammation in response to a variety of intra- and extra-cellular signals [1,2]. MAPK proteins regulate cellular functions through phosphorylation of a diverse number of substrates. In particular, the ERK1/2 proteins have been implicated in the phosphorylation of well over 100 substrates, and stringent control over interactions of ERK1/2 with substrate proteins that allow efficient phosphate transfer is essential for cellular functions [3–5]. Unregulated activation of the ERK1/2 pathway is often observed in a variety of cancers, which contributes to uncontrolled cell proliferation, survival and resistance to anti-cancer drugs [6,7]. Thus, a better understanding of the mechanisms regulating ERK1/2 interactions with substrates may provide information on new approaches to selectively inhibit substrates involved in cancer cell proliferation and survival. Although several selective ATP-competitive and catalytic site inhibitors of ERK1/2 have been developed [8–10], these compounds have not advanced to the clinic and do not allow examination of select ERK functions that are dependent on interactions with specific substrate proteins.
Many of the ERK1/2 substrate and interacting proteins, including the Elk-1 transcription factor, p90 ribosomal S6 kinase-1 (RSK-1), the caspase 9 protease and the hematopoietic protein tyrosine phosphatase (hePTP), contain a DEJL (docking site for ERK and JNK, LXL) motif or D-domain that is involved in kinase recognition [11–14]. The D-domain consists of basic residues followed by a hydrophobic LL motif and interacts with acidic and hydrophobic regions in the C-terminus of ERK1/2, referred to as the common docking (CD) domain or D-recruitment site (DRS) [13–15]. A second docking domain, known as the F-site or DEF (docking site for ERK, FF) motif, has been identified on several ERK1/2 substrates, including Elk-1 and c-Fos transcription factors, A Raf kinase, the kinase suppressor of Ras-1 (KSR-1) scaffold protein and nuclear pore proteins such as NUP153 and NUP214 [16–18]. The F-site is typically separated from the phosphorylation site by six to ten amino acids, whereas the D-domain may be located 20 amino acids further from the phosphorylation site to accommodate the spatially separated hydrophobic interactions.
The DRS on ERK2 includes residues Asp316 and Asp319, adjacent hydrophobic amino acids and ED domain residues (Glu160/Asp161 for p38α MAPK and Thr157/Thr158 for ERK2) that facilitate selective interactions between D-domain-containing substrates and MAPKs [19]. F-site-containing substrates interact with hydrophobic regions on the ERK2 F-recruitment site (FRS) and include residues Leu198, Tyr231, Leu232, Leu235 and Tyr261 [20,21]. Other MAPKs, including p38α, may also utilize a FRS-like binding motif during substrate recognition [22,23]. Beyond the DRS and FRS, experimental studies using ERK2 mutants have implicated other residues that may be important for ERK interactions with substrates or other regulatory proteins [24]. Indeed, ERK2 and other MAPKs have a unique insert in the kinase homology region that may regulate its interactions with upstream activating MEK (MAPK/ERK kinase) proteins [25]. Given the large number of substrates regulated by ERK1/2 proteins, it is likely that additional docking sites on ERK1/2 proteins will be identified and shown to regulate specific protein– protein interactions.
In previous studies, we used computer-aided drug design (CADD) to identify low-molecular-mass compounds that inhibit ERK1/2-mediated phosphorylation of substrate proteins in an ATP-independent manner and inhibit cancer cell proliferation in in vitro and in vivo models [26,27]. This approach targeted the DRS region of ERK2, such that the identified inhibitors containing a thiazolidinedione scaffold that may selectively regulate distinct ERK2 signalling functions. From those efforts, several compounds were identified that inhibit phosphorylation of ERK1/2 substrates, including D-domain-containing substrates RSK-1 and caspase 9 and selectively inhibit cancer cell lines containing constitutively active ERK1/2 signalling [28–30].
The present study reports the identification of a new class of small molecules, based on a thienyl benzenesulfonate scaffold, which are putative inhibitors of substrates that interact with ERK1/2 through the FRS. The compounds were initially identified using virtual database screening followed by experimental assays. Validation of the biological activity of the scaffold was then achieved by designing analogues using the site identification by ligand competitive saturation (SILCS) approach [31,32], chemical synthesis and experimental evidence demonstrating inhibition of the expression of F-site-containing immediate early genes (IEGs) that comprise the activator protein-1 (AP-1) transcription factor complex. Correspondingly, melanoma cells containing mutated BRaf and active ERK1/2 signalling, including cells that are inherently resistant to currently available kinase inhibitors used in the clinic, were selectively inhibited by the new compounds. These results provide support for the feasibility of developing low-molecular-mass compounds that can selectively inhibit ERK1/2-mediated signalling functions that drive the proliferation and survival of cancer cells containing constitutively active ERK1/2 signalling.
EXPERIMENTAL
Materials
Initial test compounds were obtained from Chembridge Corporation. Lead compound analogues were synthesized using reagent grade chemicals and solvents purchased from Sigma– Aldrich, Alfa Aesar, Oakwood Chemicals and TCI America. 1H- and 13C-NMR spectra were recorded on Varian INOVA 400 MHz and Varian INOVA 500 MHz NMR spectrometers at 25°C. Chemical shifts are reported in parts per million (ppm). The residual solvent peak was used as an internal reference. The mass spectra were obtained on an Electrospray TOF (ESI–TOF) mass spectrometer (Bruker amaZon X). Prior to biological testing, final compounds were confirmed to be >95% pure by HPLC chromatography. The MEK1/2 inhibitor U0126 was purchased from EMD Millipore and the ATP-dependent pyrazolylpyrrole ERK2 inhibitor [9] and p38α/β MAPK inhibitor SB239063 were purchased from Santa Cruz Biotechnology. Unless indicated, all other chemicals listed below were purchased from Sigma–Aldrich and/or Fisher Scientific. HeLa cervical carcinoma cells, Jurkat T-csell leukaemia cells, A375 melanoma and mutated BRaf inhibitor-resistant RPMI7951 melanoma cells were purchased from the A.T.C.C. Additional details are provided in the Supplementary Methods. Antibodies recognizing total c-Fos, FosB, Fra1, c-Myc and c-Jun were purchased from Cell Signaling Technology. The phospho-specific ERK1/2 (pThr183/pTyr185) and α-tubulin antibodies were purchased from Sigma. Phosphorylation-specific antibodies against c-Fos (pSer32, pThr232, pThr325 or pSer374), RSK-1 (pThr573 or pSer380), MEK1/2 (pSer217/pSer221), EGFR (epidermal growth factor (EGF) receptor; pTyr1068), cRaf-1 (pSer289, pSer296, pSer301) or Elk-1 (pSer383) were purchased from Cell Signaling Technology. Antibodies against total early growth response-1 (Egr1), RSK-1, ERK2 and MEK1 were purchased from Santa Cruz Biotechnology.
Protein expression and purification
His6-tagged ERK2 wild-type was expressed in Escherichia coli and purified as described previously [27]. See the Supplementary Methods for more details.
Immunoblotting
Analysis of protein expression levels, activity and cell proliferation were done as previously described [28,29]. Cells were washed with ice-cold PBS (pH 7.2; Invitrogen) and protein lysates were collected with 2×SDS/PAGE sample buffer (4% SDS, 5.7 M β-mercaptoethanol, 0.2 M Tris/HCl, pH 6.8, 20% glycerol and, 5 mM EDTA). Proteins were separated by SDS/PAGE, analysed by immunoblotting and detected using ECL (GE Healthcare).
In vitro kinase assays
Assays were done with 10 nM of recombinant active forms of ERK2 or ERK5 (Abcam) and substrates including 100 nM c-Fos (Abcam) or ~0.2 μg/reaction GST–RSK1 (residues 386–752) or using previously described buffer conditions [33]. The p38α MAPK (0.43 μg) was activated with 75 ng of recombinant active MKK6 (S207E, S211E mutations; Life Technologies) and included recombinant ATF2 (2 μg) as the substrate. Compounds (0–60 μM) were pre-incubated with ERK or p38α MAPK proteins in the absence of substrates for 30 min at room temperature. Kinase assays were initiated with the addition of substrate and active MKK6 for p38α reactions, plus 100 μM ATP. Reactions were stopped after 30 min with an equal volume of 2× SDS/PAGE sample buffer and the proteins were analysed by immunoblotting for specific substrate phosphorylation sites and quantified by densitometry. Catalytic site inhibitors SCH772984 (Selleck Chemicals) and SB239063 were used as positive controls for ERK2 and p38α inhibition respectively.
Cell proliferation and apoptosis assays
Cells were seeded at 25 000 cells/well in 96-well plates, cultured overnight and treated for 24 h with the indicated dose of compounds. Cell viability or caspase 3/7 activity was measured according to the manufacturer’s instructions using the fluorescent Cell Titer Blue Assay or luminescent Caspase 3/7 Glo Assay, respectively (Promega).
AP-1/SRE promoter luciferase assays
HeLa or A375 cells were seeded in 24-well plates (40 000 cells/well) and incubated for 18 h to achieve ~60–70% confluence. Cells were transfected with AP-1 [pAP1(PMA)-TA-Luc; Clontech] or the serum-response element (SRE; pGL4.33-SRE; Promega) luciferase reporter plasmids (250 ng/well) using Lipofectamine™ (Invitrogen). After 16 h, cells were pre-treated with 1–100 μM compounds as indicated for 20 min, followed by stimulation with 25 ng/ml EGF or 200 nM PMA for 4.5 h. In some experiments, cells were transfected with the pAP1(PMA)-TA-Luc promoter and a constitutively active MEK1 mutant cDNA were treated with various amounts of compounds during the last 4 h of incubation. The luciferase activity in the cell extracts was determined with a Dual Luciferase Assay System (Promega) according to the manufacturer’s instructions. Human embryonic kidney (HEK) 293 cells (20 000 cells/well) were plated in white walled 96-well plates (Nunclon Delta Surface; Thermo Scientific) and then transfected with the luciferase reporter plasmids indicated above. Cells were treated with EGF or PMA for 5 h in the presence or absence of compounds (1–100 μM) and luminescence was measured using Bright-Glo Substrate (Promega). Luciferase activities were monitored with a Lumat LB 9507 luminometer (Berthold Technology) and data were normalized to the amount of protein in each sample.
Computational methods
Molecular dynamics (MD) simulations were performed using the programs Chemistry at HARvard Macromolecular Mechanics (CHARMM) [34] and NAnoscale Molecular Dynamics (NAMD) [35] with the CHARMM22/CMAP additive force field [36,37] with the Transferable Intermolecular Potential with 3 interaction sites (TIP3P) water model [38]. Simulations initiated with the 3D structures of ERK2 in both the phosphorylated (active) [39] and unphosphorylated (inactive) [40] states. SILCS simulations for the FRS (site 5) of ERK2 were initiated the unphosphorylated ERK2 structure and included 1 M benzene and 1 M propane, as previously described [31]. Binding site identification [41] and database screening was performed on diverse conformations from the simulations. Screening was performed in two steps against a virtual database of more than 1.5 million low-molecular-mass commercially available compounds. Final selection of the 150 compounds for experimental assay involved maximizing chemical and structural diversity, via fingerprint-based clustering [42,43] with the program Molecular Operating Environment (MOE) (Chemical Computing Group Inc.) as well as considering their potential bioavailability [44]. The purity of compounds shown to have biological activity was verified by MS.
Prediction of ligand modifications were performed using ligand grid free energy (LGFE) scores based on the SILCS methodology [32]. SILCS simulations were initiated from the 2.3.2–ERK2 crystal structure determined in the present work. Visualization of the SILCS FragMaps identified the region beyond the benzene ring of 2.3.2 as suitable for non-polar moieties. Subsequently, the analogues of 2.3.2 were modelled on to ERK2 starting from the 2.3.2–ERK2 crystal structure. Relative LGFE scores with respect to 2.3.2 were then determined and are presented in the Results’ section. Additional details of all the computational methods are included in the Supplementary Methods.
RESULTS
In silico approaches to identify lead compounds
The initial objective of these studies was to use in silico modelling and experimental assays to identify low-molecular-mass compounds that selectively inhibit ERK2 interactions with substrate proteins by targeting unique docking sites and do not interfere with ATP binding. Computational approaches were first applied to identify putative small-molecule-binding sites located on regions in the C-terminus of ERK2 previously implicated via mutagenesis studies to be important for substrate binding, as described in the Supplementary Online Data. The five sites targeted, with the residues shown on the structure of ERK2, include the DRS [19], which was previously targeted in our laboratory [27], the FRS known to be involved in interactions with ERK1/2 substrates containing the F-site or DEF motifs [20] and the focus of the present study and three putative sites that span the region between the DRS and FRS (Supplementary Table S1; Supplementary Figure S1). Database screening was then performed against these sites in which 50 000 compounds were selected following the initial round of docking with a second round of screening yielding 1000 compounds. Details of the screening approach, including the use of score normalization based on molecular mass [45], are given in the Supplementary Online Data (Supplementary Tables S2–S4; Supplementary Figure S2). Final compound selection for experimental assays from the 1000 compounds emphasized chemical and structural diversity to maximize the identification of active compounds [46]. Diversity was attained using fingerprint-based similarity clustering to group compounds into structurally similar clusters from which individual molecules were selected. This selection process also included bioavailability considerations [44].
Biological and structural evaluation of lead compounds
From the in silico screen, we focused on a group of seven structurally diverse compounds (Figure 1A) and their effects on ERK1/2-mediated phosphorylation and signalling events. Using lysates from EGF-treated HeLa cells, 100 μM compound 2.3.2 inhibited ERK1/2-mediated phosphorylation of the transcription factor Elk-1, which contains both a D-domain and an F-site (Figure 1B). Compounds 2.3.3, 2.3.4 and 2.3.5 also inhibited Elk-1 phosphorylation, but to a lesser extent (Figure 1B). Importantly, none of the compounds caused a corresponding inhibition of ERK1/2 activation, as measured by phosphorylation of the activation site residues, suggesting the compound’s mechanism of action involved targeting signalling downstream of ERK1/2. Whereas 2.3.2 inhibited ERK1/2-mediated Elk-1 phosphorylation, it appeared to be less potent at inhibiting ERK1/2-mediated phosphorylation of the D-domain-containing substrate RSK-1 (Figure 1C). Further support that 2.3.2 was acting on ERK1/2-mediated events was demonstrated by the lack of effects on EGF-induced phosphorylation of MEK1/2, which interact with ERK1/2 through their D-domains [25] or autophosphorylation of the EGFR (Figure 1C).
Figure 1. Selective inhibition of ERK-mediated phosphorylation of substrates by compounds.
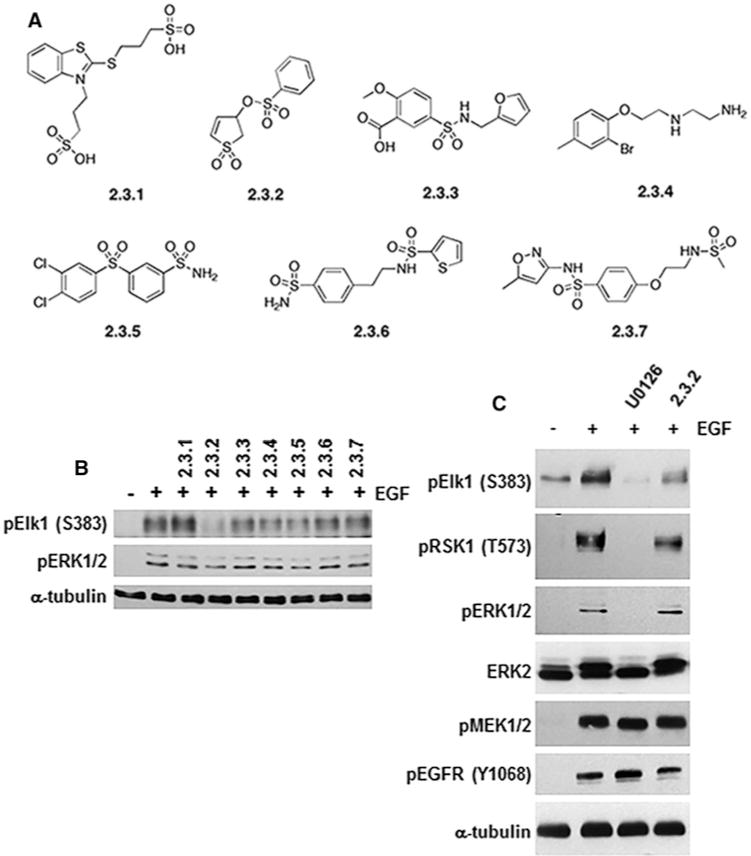
(A) Structures of the seven diverse compounds identified by CADD and initially tested. (B) HeLa cells were pre-incubated for 30 min with indicated compounds (100 μM) followed by treatment with EGF (25 ng/ml) for 10 min to activate ERK1/2 signalling. Lysates were immunoblotted for phosphorylated Elk-1 (pElk-1 Ser383) or active ERK1/2 (pERK1/2). (C) Following treatment as in (B), immunoblot analysis of lysates from cells treated with 2.3.2 suggested selective phosphorylation inhibition of Elk-1 as compared with RSK1 (pElk-1 Ser383 and pRSK1 Thr573). Phosphorylation of ERK1/2 and, MEK1/2 (pERK1/2 and pMEK1/2) or Tyr1068 autophosphorylation of EGFR (pEGFR Tyr1068) was not affected by 2.3.2. The MEK1/2 inhibitor U0126 (10 μM) was used to inhibit all ERK1/2 signalling, and α-tubulin expression was used as a protein loading control.
To provide additional evidence for the binding site of 2.3.2, crystallographic studies were undertaken and crystals of 2.3.2 bound to the inactive form of ERK2 were obtained (Supplementary Table S5). Upon inspection of the model that included protein and 46 water molecules, additional density was identified in a pocket vacated by Phe181/Leu182 located below the activation loop that includes residues Thr183 and Tyr185 (Supplementary Figure S3). Given that the densities of these two residues are well resolved, it was predicted that the elongated density filling this region was representative of 2.3.2 (Supplementary Figure S3). The irregular shape of the electron-density fragment was accounted for by assuming that 2.3.2 binds in at least two orientations. In one of the modelled conformations, 2.3.2 appears to make hydrogen bonds to the backbone amides of Phe181 and Leu182; however, this should be taken with caution given the disordered and partially occupied nature of the ligand in the binding site. Thus, the data suggest that 2.3.2 is the first molecule identified to interact with ERK2 in the FRS region involved with regulating interactions with substrates containing an F-site. Although the role of the FRS region in coordinating protein–protein interactions may be unique to ERK2, it is possible that other MAPKs utilize this region for determining substrate recognition. For example, a comparable FRS region has recently been identified on p38α/β MAPKs [23] as well as the kinase domain of ERK5 [47]. However, sequence alignment suggests that the ERK2 amino acids predicted to interact with 2.3.2 have differences with the corresponding residues on other major MAPKs, which could allow for FRS selectivity with small molecules (Supplementary Figure S4).
It has to be emphasized that the observed difference omit electron density (i.e. Fo − Fc map calculated in the absence of ligand) is by itself not conclusive of 2.3.2 binding to the FRS region of ERK2. However, the additional density observed with 2.3.2 was not observed for the ERK2 crystals obtained without ligand soaking. This indicates that the electron density is probably originating from 2.3.2 binding and not that of other components of the crystallization buffer (e.g. non-detergent sulfobetaines (NDSB)-256). Furthermore, it is clearly possible to place other molecules that were present in crystallization buffer (including NDSB-256) into the rather featureless electron density. Additional complication arises from the proximity of the crystal contact to the putative binding site, which may result in a binding mode that is somewhat distorted compared with what happens in solution. Thus, the crystallographic evidence presented can only suggest the possibility of the 2.3.2 binding in the detected location on the protein surface and does not unequivocally prove it. Therefore, additional support for 2.3.2 and chemically related analogues regulating ERK1/2 functions through the FRS comes from further computational and biochemical studies described in the following sections.
Optimization of lead compound by SILCS
Chemically similar analogues were obtained from SILCS 3D FragMaps obtained from SILCS simulations initiated from the putative 2.3.2–ERK2 crystal structure are shown overlaid on the crystal structure obtained (Figure 2A). The FragMaps represent the regions to which different type of functional groups bind and include contributions from protein flexibility, desolvation and functional group protein interactions [31]. Visual analysis revealed the overlap of the aliphatic and aromatic SILCS FragMaps with the phenyl moiety of 2.3.2, with those FragMaps extending beyond the crystallographic position of the phenyl ring. This indicated that the addition of non-polar moieties to this phenyl ring may improve affinity and subsequent potency. Synthetically accessible chemical modifications of the phenyl ring were identified, modelled on to 2.3.2 bound to ERK2, and the relative SILCS LGFE scores were calculated and presented as a change in LGFE scores (LGFE) relative to 2.3.2 and suggested that hydrophobic modifications may increase activity (Figure 2B). Accordingly, along with 2.3.2, which is from now on referred to as SF-3-026, analogues containing the p-CH3 (SF-3-027), biphenyl (SF-3-029) or naphthyl (SF-3-030) group were synthesized for evaluation in subsequent biochemical and cell-based assays.
Figure 2. SILCS identified analogues of 2.3.2 inhibit Elk-1 phosphorylation.
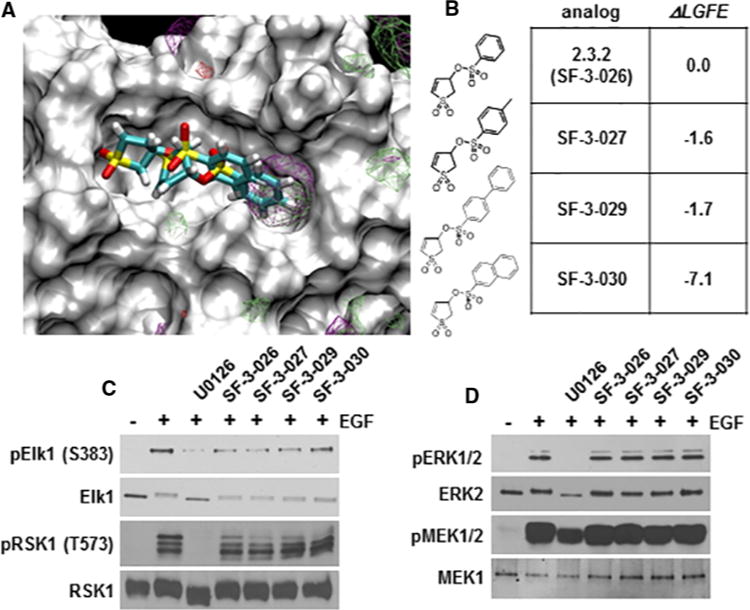
(A)SILCS FragMaps overlaid on to the 2.3.2–ERK2 crystallographic structure. 3D probability distributions are shown for aliphatic (green) and aromatic (purple) functional groups. (B) The structures and change in LGFE scores (LGFE) for 2.3.2 (now referred to as SF-3-026) and analogues (SF-2-027, SF-2-029 and SF-2-030). (C and D) Lysates from HeLa cells pre-treated with 50 μM of SF-3-026 or analogues followed by stimulation with EGF for 10 min were immunoblotted for phosphorylated Elk-1 (pElk-1 Ser383) and RSK1 (pRSK1 Thr573) in (C) or ERK1/2 (pERK1/2) and MEK1/2 (pMEK1/2) in (D). Total Elk-1 and RSK1 or ERK2 and MEK1 are shown as protein loading controls in (C) or (D) respectively. Results are representative of three independent experiments.
Lead compounds inhibit ERK1/2-mediated IEG promoter activity
ERK1/2 phosphorylation of Elk-1 regulates the SRE that drives the transcription of IEGs, such as c-fos, which is an F-site containing ERK1/2 substrate [48]. Initial testing of SF-3-026 and analogues at 50 μM in EGF-treated HeLa cells showed preferential inhibition of the phosphorylation of Elk-1 as compared with the D-domain-containing substrate RSK-1 (Figure 2C). In addition, the phosphorylation of ERK1/2 or MEK1/2 proteins was not affected by the compounds at the dose tested (Figure 2D). It was next determined whether the compounds could affect the transcription factor functions of Elk-1 or AP-1 proteins. HeLa cells expressing luciferase constructs driven by the SRE or AP-1 promoters, which are regulated by Elk-1 or Fos family proteins, respectively, were treated with SF-3-026 and analogues in the context of EGF stimulation. EGF alone caused a more than 20-fold increase in SRE- or AP-1-mediated luciferase activity, which could be inhibited by ~90% with 10 μM of the MEK1/2 inhibitor, U0126 (results not shown). Treatment with the test compounds caused a dose-dependent inhibition of the SRE and AP-1 promoters (Figures 3A and 3B). As predicted by SILCS, SF-3-029 and SF-3-030 were the most potent inhibitors especially with the AP-1 promoter where IC50 values were ~10 μM (Figure 3B). To further demonstrate the targeting of ERK1/2 mediated transcription, AP-1 promoter activity was measured in HeLa cells expressing an active MEK1 mutant, which only activates ERK1/2. As shown in Figure 3(C), SF-3-026 and its analogues caused dose-dependent inhibition of AP-1 promoter activity with SF-3-029 and SF-3-030 with IC50 values around 5–10 μM as compared with SF-3-026 and SF-3-027 with IC50 values of ~40 μM. As controls, MEK1-induced AP-1 promoter activity was inhibited by >80% with 10 μM U0126 or a previously reported ATP-competitive pyrazolylpyrrole ERK inhibitor [9] (results not shown). We further evaluated AP-1 or SRE promoter activity in HEK293 cells stimulated with EGF or PMA. As shown in Table 1, the compounds were more potent at inhibiting PMA-induced promoter activity compared with EGF and SF-3-030 was the most potent lending further support of the SILCS computational predictions.
Figure 3. Compound SF-3-026 and analogues inhibit SRE and AP-1 transcription factor function.

HeLa cells were transfected with constructs containing SRE (A) or AP-1 (B) promoters driving luciferase expression and then stimulated with EGF in the presence or absence of various doses of SF-3-026 and analogues for 4 h followed by measurements of luciferase activity. (C) HeLa cells were transfected with the AP-1 promoter construct and a constitutively active MEK1 mutant in the presence or absence of various doses of SF-3-026 and analogues for 4 h followed by measurements of luciferase activity. Data were normalized to EGF-only treatment or cells expressing constitutively active MEK1 and results for each condition represent the mean ± S.D. from three independent experiments.
Table 1. Effects of SF-3-026 and analogues on AP-1 or SRE promoter activity.
AP-1 or SRE promoter-mediated luciferase activity was determined in HEK293 cells treated with EGF or PMA in the presence of 0–100 μM of each test compound. IC50 values (μM) are shown.
| AP-1 (PMA) | AP-1 (EGF) | SRE (PMA) | SRE (EGF) | |
|---|---|---|---|---|
| SF-3-026 | 12.8 | 21.7 | 14.0 | 22.7 |
| SF-3-027 | 11.2 | 19.7 | 11.3 | 22 |
| SF-3-029 | 10.7 | 16.6 | 10.9 | 18.5 |
| SF-3-030 | 4.7 | 9.8 | 5.2 | 10.7 |
Lead compounds inhibit EGF or PMA-induced expression of IEG proteins
HeLa cells treated with EGF show a transient phosphorylation of Elk-1, c-Fos and Fra 1, followed by robust expression of c-Fos, FosB, FosB2 and Fra 1 after 30–60 min (Figure 4A). Although SF-3-026 and analogues had little effect on the initial phosphorylation of c-Fos and Fra 1 as was evident by gel shifts (Figure 4B), the robust induction of Fos family proteins by EGF at 1 h was inhibited in a dose-dependent manner with SF-3-030 (Figure 4C). Inhibition of c-Fos phosphorylation at Ser32 was observed prior to loss of protein, but phosphorylation of Ser374, which is the reported priming site for other phosphorylation events on c-Fos [49], was unaffected (Figure 4C). SF-3-030 also inhibited EGF-mediated expression of other IEGs including c-Myc, but was less effective at inhibiting c-Jun and Egr 1 protein induction (Figure 4C). These data suggest that SF-3-030 can selectively inhibit EGF-mediated induction of IEG proteins.
Figure 4. SF-3-026 and analogues inhibit EGF-induced expression of IEGs.
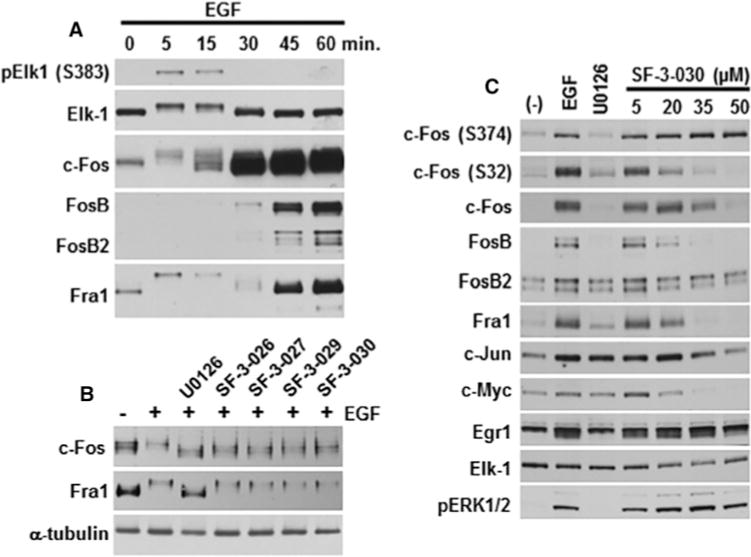
(A) HeLa cells treated with EGF (25 ng/ml) for up to 60 min show transient phosphorylation of Elk-1 (pElk-1 S383) and subsequent expression of c-Fos, FosB, FosB2 and Fra1. (B) Expression of c-Fos or Fra1 in untreated (−) HeLa cells or cells pre-treated with 50 μM of SF-3-026 or analogues and then treated with EGF for 15 min. (C) Expression of phosphorylated c-Fos (Ser374 or Ser32) or total c-Fos, FosB, FosB2, Fra1, c-Jun, c-Myc or Egr-1 in HeLa cells pre-treated with various doses of SF-3-030 and then treated with EGF for 60 min. Controls include cells treated only with EGF (EGF lane) or EGF treatment in the presence of U0126 (U0126 lane). Total Elk-1 and phosphorylated ERK1/2 expression were used as controls for protein loading and evaluating inhibitor effects. Results are representative of three independent experiments.
SF-3-030 also inhibited IEG expression in cells treated with the phorbol ester PMA. Like EGF, PMA caused a transient phosphorylation of Elk-1 followed by increased expression of c-Fos, FosB and Fra 1 after 60–90 min (Figure 5A). At a dose of 40 μM, SF-3-030 inhibited c-Fos phosphorylation at Thr232 and Ser32, which may be involved in transcription regulation, but, similar to EGF treatment, had no effect on phosphorylation of Ser374 (Figure 5B). The expression of total FosB, FosB2, Fra1 and F-site-containing c-Myc was also decreased at the 40 μM dose (Figure 5B). The inhibition of c-Fos phosphorylation at Thr232 and Ser32 by SF-3-030 corresponded to a loss of FosB and Fra 1 expression, suggesting that c-Fos is transcriptionally inactive (Figure 5B). At high doses (70 and 100 μM), SF-3-030 increased ERK1/2 phosphorylation (Figure 5B), which indicated potential off-target toxicity or inhibition of negative feedback mechanisms on MEK1/2 and Raf-1 [50,51]. The PMA-mediated IEG phosphorylation and expression was further evaluated at doses of less than 40 μM. As shown in Figure 5(C), a dose-dependent inhibition of c-Fos phosphorylation of Thr232 by SF-3-030 corresponded to decreased FosB, FosB2 and Fra1 expression and supported impaired c-Fos transcription activity. SF-3-030 at low doses inhibited the negative feedback phosphorylation sites of Raf-1 (Figure 5C), which appeared to increase with higher doses in a manner that correlated with increased ERK1/2 phosphorylation (Figure 5B). A Raf, but not Raf-1 or BRaf, has a consensus F-site [16] and the specific docking interactions between Raf-1 and ERK1/2 have not been defined.
Figure 5. SF-3-026 and analogues inhibit PMA-induced expression of Fos family IEGs.
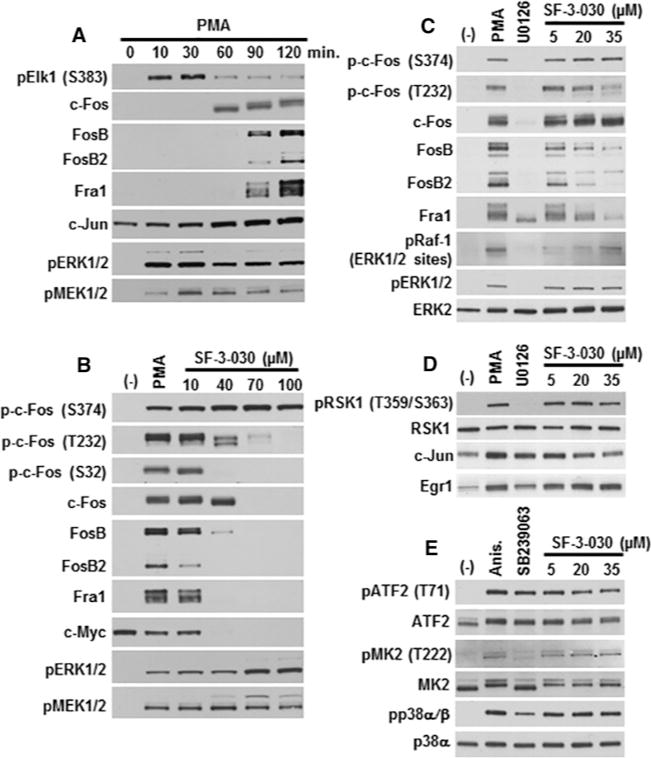
(A) HeLa cells treated with PMA (200 nM) for up to 120 min show transient phosphorylation of Elk-1 (pElk-1 S383) and subsequent expression of c-Fos, FosB, FosB2, Fra1 and c-Jun. (B) Expression of phosphorylated c-Fos (p-c-Fos Ser374, Thr232 or Ser32), total c-Fos, FosB, FosB2, Fra1 or c-Myc in HeLa cells pre-treated for 30 min with the indicated dose of SF-3-030 and then treated with PMA for 120 min. Phosphorylated ERK1/2 and MEK1/2 levels are shown in (A) and (B). (C) Cells were pre-treated as in (B) with the indicated dose of SF-3-030 or 10 μM U0126 and the levels of phosphorylated c-Fos (p-c-Fos Ser374 and Thr232), total c-Fos, FosB, FosB2, Fra1 or the negative feedback ERK1/2 phosphorylation sites (Ser289, Ser296 and Ser301) on Raf-1 were evaluated. Phosphorylated ERK1/2 and total ERK2 are shown for controls. Controls for (B–D) include cells treated only with PMA (PMA lane) or PMA treatment in the presence of U0126 (U0126 lane). (D) Cells treated as in (C) were immunoblotted for phosphorylated RSK1 (pRSK1 Thr359/Ser363), total RSK1, c-Jun or Egr1. (E) HeLa cells were pre-treated with or without SF-3-030 as in (D) or 10 μM SB239063 and then stimulated in the absence or presence of anisomycin (Anis.) for 1 h followed by analysis of phosphorylated ATF2 (pATF2 Thr71), MK2 (pMK2 Thr222) or p38α/β MAPK (pp38α/β). Total ATF2, MK2 or p38α MAPK expression levels are shown for loading controls.
The effects of SF-3-030 on the expression of Fos proteins appeared to be selective as ERK1/2-mediated phosphorylation of RSK-1 or the induction of other IEGs, such as Egr1 and c-Jun, were less affected (Figure 5D). SF-3-030 also had minimal effects on anisomycin-mediated p38 MAPK signalling (Figure 5E), including phosphorylation of p38 MAPK substrates ATF2 and MK2. The phosphorylation of ATF2 at Thr71 may also be achieved by ERK1/2 [52], which could explain the lack of effects of the p38 inhibitor SB239063 and some inhibition by SF-3-030 at doses >20 μM (Figure 5E). Together, these data provide additional support that SF-3-030 preferentially affects ERK1/2-mediated signalling events.
SF-3-030 selectively inhibits ERK2-mediated phosphorylation of c-Fos in vitro
The effects of SF-3-030 on ERK-mediate phosphorylation of c-Fos were evaluated using in vitro kinases assays. ERK5 was used for comparison as it has been reported to phosphorylate c-Fos at Thr232 and Ser32 and regulate protein stability and transcription factor function [53], although others suggest that ERK1/2 is the primary regulator of c-Fos phosphorylation and activity [54]. As shown in Figure 6(A), active ERK2 was the primary regulator of c-Fos as was evident by phosphorylation at the F-site-dependent Thr325 site, Ser374 and Thr232, as well as the c-Fos gel shift, which did not occur in the presence of active ERK5 (Figure 6A). Ser32 phosphorylation increased to a lesser degree with ERK2 as a result of high basal phosphorylation at this site, which was probably due to c-Fos expression and purification from eukaryotic cells (Figure 6A). Whereas SF-3-030 inhibited ERK2-mediated phosphorylation of Thr325 in a dose-dependent manner, it had little effect on the phosphorylation of Ser374, Thr232 or Ser32 (Figures 6A and 6B). However, SCH772984, a catalytic site inhibitor of ERK1/2, inhibited phosphorylation of all c-Fos sites (Figure 6A). SF-3-030 did not affect the modest induction of Ser374 phosphorylation by ERK5, but it did appear to cause a slight decrease in ERK5-mediated phosphorylation of Thr232 (Figure 6A), which is consistent with the cell data in Figure 5. Similarly, SF-3-030 had no apparent effects on ERK2 phosphorylation of the D-domain-containing substrate RSK1 or p38α-MAPK-mediated phosphorylation of the F-site-containing ATF2 protein, further supporting selective inhibition of ERK-mediated phosphorylation events by SF-3-030 (Figure 6C).
Figure 6. SF-3-030 selectively inhibits ERK2-mediated c-Fos phosphorylation in an in vitro kinase assay.
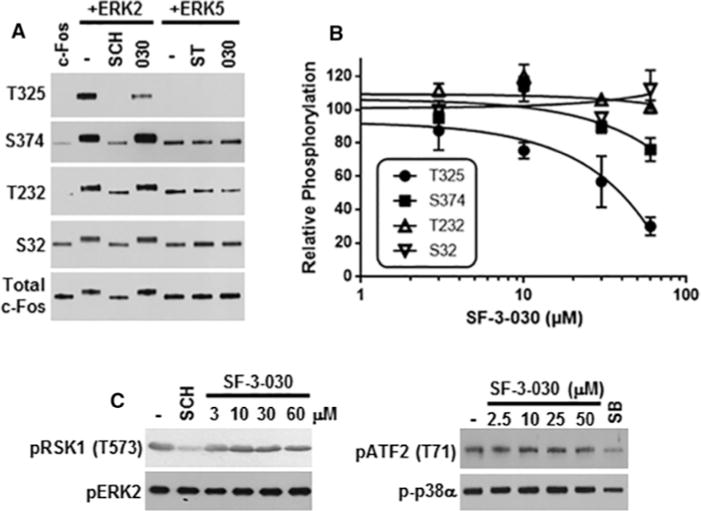
(A) Active ERK2 or ERK5 were pre-incubated in the absence or presence of 50 μM SF-3-030 (030), 10 μM SCH772984 (SCH) or 10 μM staurosporine (ST) followed by initiation of the kinase assay with the addition of c-Fos substrate and ATP. After 30 min of incubation, the reaction was stopped and the c-Fos phosphorylation sites indicated were evaluated by immunoblotting. (B) Densitometry analysis of ERK2-mediated c-Fos phosphorylation on Thr325, Ser374, Thr232 and Ser32 in the presence of 3–60 μM SF-3-030. Data are normalized to ERK2-mediated c-Fos phosphorylation in the presence of vehicle (DMSO) only. (C) Immunoblot analysis of in vitro kinase assays for ERK2-mediated RSK1 phosphorylation (pRSK1 Thr573) or p38α MAPK-mediated phosphorylation of ATF2 (pATF2 Thr71) in the absence or presence of 2.5–60 μM SF-3-030. Controls for ERK2 or p38α kinase activity include 1 μM SCH772984 (SCH) or 10 μM SB239063 (SB) respectively. The levels of phosphorylated ERK2 or p38α are shown for each condition.
Lead compounds inhibit AP-1 promoter activity in melanoma cells with constitutively active ERK1/2
The effects of SF-3-026 and analogues were next tested in A375 melanoma cells containing an activating valine to glutamate mutation on residue 600 (V600Q) of BRaf and constitutive ERK1/2 activation [55]. Similar to the EGF- or PMA-stimulated HeLa and HEK293 cells, SF-3-029 and SF-3-030 were more potent inhibitors of AP-1 activity in A375 cells (Figure 7A). Given that SF-3-030 appeared to be the most effective at inhibiting the basal expression of Fos family proteins in A375 cells (Figure 7B), the dose-dependent inhibition effects of SF-3-030 on the expression of AP-1 proteins and other IEGs were further examined. SF-3-030 showed a dose-dependent inhibition of c-Fos, FosB, FosB2, Fra1 and c-Myc, but Egr1 expression was unaffected (Figure 7C). SF-3-030 also caused an increase in phosphorylated ERK1/2 (Figure 7C), which others have observed in A375 cells treated with an ATP-competitive ERK1/2 inhibitor, VTX-11e, and attributed to effects on negative feedback pathways [10]. However, MEK1/2 phosphorylation is not increased with SF-3-030 (Figure 7C), suggesting that the effects on ERK1/2 negative feedback by SF-3-030 in cells expressing mutated BRaf are different from those in HeLa cells treated with EGF or PMA (Figures 4 and 5). The MEK1/2 inhibitor U0126 blocked Fra1 phosphorylation involved in transcription regulation (results not shown) but did not block the expression of Fra1 as was observed with other Fos proteins and c-Jun (Figures 7B and 7C). Additional selectivity of the compounds was shown where SF-3-026 and analogues did not affect basal expression of the D-domain-containing c-Jun protein or the phosphorylation of MEK1/2 (Figure 7D). As shown in Figure 7(C), all compounds at 50 μM increased phosphorylation of ERK1/2 and the D-domain substrate RSK1 (Figure 7D).
Figure 7. SF-3-026 and analogues inhibit Fos family proteins in melanoma cells.
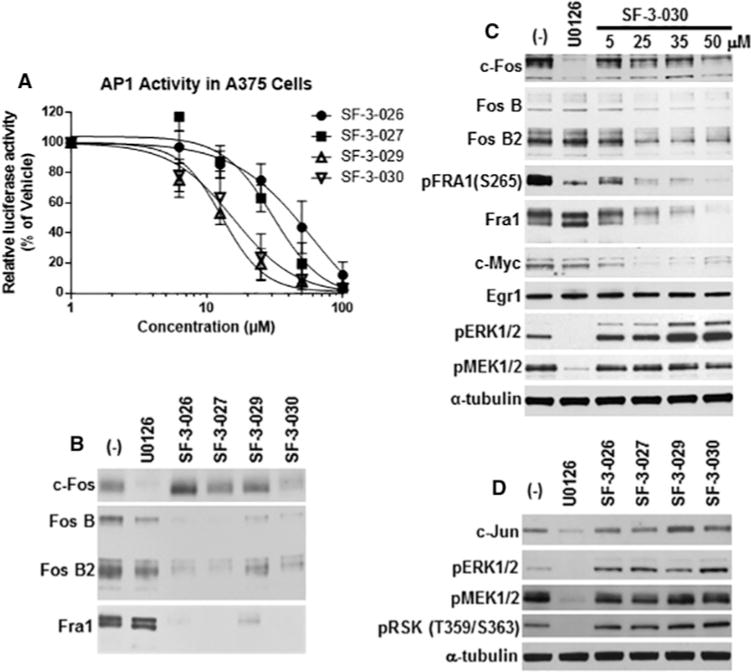
(A) A375 cells expressing the AP-1 promoter construct were treated with 0–100 μM SF-3-026 analogues for 4 h followed by measurements of luciferase activity. Data were normalized to untreated A375 cells and represent the means ± S.D. from three independent experiments. (B) A375 cells treated for 1 h in the absence or presence of 10 μM U0126 or 50 μM SF-3-026 and analogues were immunoblotted for total c-Fos, FosB, FosB2 or Fra1. (C) Expression of total c-Fos, FosB, FosB2, Fra1, c-Myc or Egr1 from A375 cells treated in the absence or presence of U0126 or indicated concentrations of SF-3-030. Controls for inhibition by U0126 include phosphorylated Fra1 (pFra1 Ser265), pERK1/2 and pMEK1/2. (D) Expression of c-Jun or phosphorylated RSK1, ERK1/2 and MEK1/2 from A375 cells treated as in (B). α-tubulin expression was used for a loading control.
Lead compounds selectively inhibit proliferation of melanoma cells with constitutively active ERK1/2
It was next determined whether SF-3-026 and analogues affected the proliferation of A375 and RPMI7951 melanoma cells driven by BRaf (V600Q) mutation. RPMI7951 cells are heterozygous for the BRaf (V600Q) mutation but are resistant to BRaf inhibitors due to overexpression of MAP3K8 that re-activates ERK1/2 signalling [56,57]. Both melanoma cells lines were treated with various doses of SF-3-026 or analogues and cell viability was compared with HeLa cervical carcinoma and Jurkat T-cell leukaemia cells, which are p53-defective but contain no known activating mutations in the ERK1/2 pathway [58]. Compound SF-3-026 and its analogues showed a dose-dependent inhibition of each cell line; however, the A375 and RPMI7951 melanoma cells were more sensitive to the test compounds with GI50 values in the low micromolar range and severalfold lower than the HeLa or Jurkat cells even when adjusted for individual cell growth rates (Table 2). As a control, the BRaf inhibitor PLX4032 was effective against the drug-sensitive A375 cells but not the RPMI7951, HeLa or Jurkat cells (Table 2).
Table 2. Effects of SF-3-026 and analogues on proliferation of cancer cell lines.
GI50 values (μM) for test compounds in A375, RPMI7951, HeLa or Jurkat cells. The BRaf inhibitor PLX4032 was used as a control.
| A375 | RPMI7951 | HeLa | Jurkat | |
|---|---|---|---|---|
| SF-3-026 | 7.8 | 5.1 | 32 | >50 |
| SF-3-027 | 5.5 | 3.1 | 22 | >50 |
| SF-3-029 | 7.9 | 4.9 | 25 | >50 |
| SF-3-030 | 7.1 | 4.6 | 29 | >50 |
| PLX4032 | 0.060 | >10 | >10 | >10 |
To test whether the inhibition of cell proliferation was due to induction of apoptotic markers, A375 and HeLa cells were treated with various doses of SF-3-030 and analogues, and cell viability along with caspase 3/7 activity was measured. Treatment with compounds caused a dose-dependent increase in caspase 3/7 activity in A375 cells but had little effect on caspase 3/7 activity in HeLa cells (Figure 8A).
Figure 8. Lead compounds induce caspase 3/7 activity in A375 melanoma cells.
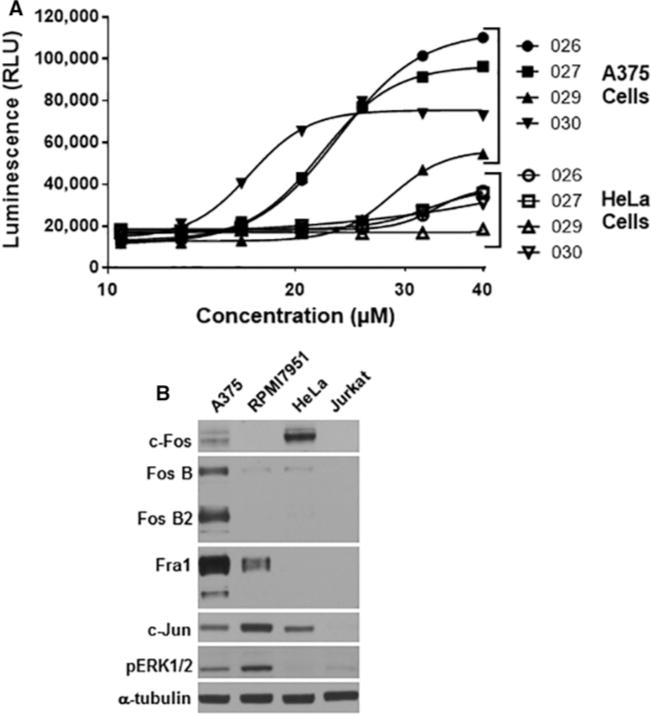
(A) A375 (closed symbols) or HeLa (open symbols) cells were treated for 24 h with the indicated concentration of SF-3-026, SF-2-027, SF-2-029 or SF-2-030, and caspase 3/7 activity was measured using the luminescent Caspase 3/7 Glo Assay. Data are representative of two experiments with each condition performed in duplicate. (B) Relative expression levels for AP-1 proteins in select cancer cell lines. Protein lysates from A375, RPMI7951, HeLa or Jurkat cells were immunoblotted for total c-Fos, Fra1, FosB, FosB2, c-Jun or phosphorylated ERK1/2 (pERK1/2). α-Tubulin expression is shown as a protein loading control.
Lastly, the expression of AP-1 proteins was evaluated in the four cell lines to evaluate whether differences in protein expression could explain why the test compounds selectively inhibit the proliferation of melanoma cells with active ERK1/2 signalling. As expected, phosphorylated and active ERK1/2 was elevated in the A375 and the drug-resistant RPMI7951 melanoma cells as compared with the HeLa or Jurkat cells (Figure 8B). When normalized to α-tubulin expression, all AP-1 proteins evaluated were expressed in A375 cells, which also contained the highest basal expression levels of Fra1 and FosB proteins as compared with the other cells (Figure 8B). Fra1 expression was also found in RPMI7951 cells but not in HeLa or Jurkat cells. Interestingly, HeLa cells showed the highest level of basal c-Fos expression, which could explain their increased sensitivity to growth inhibition when treated with the compounds compared with the Jurkat cells that have undetectable levels of the AP-1 proteins (Figure 8B). However, these data indicate that selective targeting of AP-1 heterodimer partners consisting of Fra1 by the lead compounds may play a role in sensitizing melanoma cells containing mutated BRaf and active ERK1/2 signalling to growth inhibition and apoptosis.
DISCUSSION
The findings of the present study describe a thienyl benzenesulfonate scaffold that can target ERK1/2 signalling pathways and selectively inhibit the expression of Fos family proteins involved in AP-1-regulated transcription. A variety of cancer types have been associated with dysregulated AP-1 transcription factor components [59,60]. Thus, inhibition of AP-1 functions may sensitize cancer cells with activated ERK1/2 signalling to growth inhibition and be used to augment the effects of existing anti-cancer drugs [61,62]. Targeted inhibition of the ERK1/2 signalling has received much attention given the presence of activating mutations in receptor tyrosine kinases, Ras G-proteins and BRaf [63–66]. However, therapies targeting mutated BRaf or MEK1/2 proteins invariably lead to drug resistance due to alternative mechanisms to activate ERK1/2 and recent efforts to directly inhibit ERK1/2 proteins are being tested [10,67,68]. Current small-molecule kinase inhibitors being used or evaluated in the clinic act by competing with ATP binding or targeting allosteric sites in the catalytic domain that causes complete kinase inhibition. A frequent cause of resistance to kinase inhibitors is the development of mutations in the ATP binding and catalytic site, which subsequently reduce inhibitor interactions and efficacy [69].
The present study provides evidence for a new class of compounds that selectively inhibit ERK1/2 signalling through F-site-containing substrates, including Fos family proteins and c-Myc, but have limited effects on D-domain-containing substrates, such as c-Jun or RSK1 (Figures 4, 5 and 7). In addition, other IEGs that contain undefined docking domains and are regulated by ERK1/2, such as Egr1, were also unaffected by the lead compounds (Figures 4, 5 and 7). Although the structural studies (Supplementary Online Data) and in vitro kinase assays (Figure 6) suggest the lead compounds target the FRS on ERK2, future studies will need to evaluate whether other proteins are targeted in cells. It was particularly encouraging to observe selective inhibition of the F-site-dependent Thr325 phosphorylation site with SF-3-030 in the in vitro kinase assays. Unfortunately, we were unable to detect Thr325 phosphorylation with endogenous c-Fos from cell lysates. Thus, validation that the test compounds inhibit F-site-mediated phosphorylation in cells remains to be obtained. Nonetheless, other phosphorylation sites on c-Fos (Ser32 and Thr232) and Fra1 (Ser265) that were inhibited by the MEK1/2 inhibitor U0126 are also inhibited by SF-3-030 prior to loss of total c-Fos and Fra1 protein (Figures 4, 5 and 7).
The data from the present study suggest that targeted inhibition of F-site-containing ERK1/2 substrates and AP-1-mediated transcription can selectively sensitize melanoma cells to growth inhibition and cell death. This response appears to correlate with the levels of activated ERK1/2 in the cells and is not related to the cell growth rates. It should be noted that the compound’s mechanism of action may be different with different types of melanoma. As shown in Table 2, the GI50s values for the compounds are similar for A375 and RPMI7951 cells, despite different expression levels of AP-1 proteins (Figure 8). However, the expression of Fra1 is common to both of these melanoma cell lines and future studies will be aimed at determining whether Fra1 expression is elevated in other ERK1/2-driven cancer cells and whether Fra1 inhibition is sufficient to sensitize these cells to growth inhibition and induction of apoptosis. Given that the compounds show selective inhibition of ERK1/2-mediated signalling events, we propose that specific targeting of ERK1/2 signalling functions could be an effective approach to inhibit the survival of ERK1/2-addicted cancer cells in a way that reduces off-target toxicity and the potential to develop resistance to these selective agents.
There is significant interest in developing new approaches to target protein–protein interactions to improve specificity and efficacy in treating cancers with constitutive ERK1/2 pathway activity due to Ras and Raf mutations [70]. Although the present study supports the identification of new molecules that disrupt ERK1/2-dependent signalling events, other targets of these compounds are likely to be identified. There are only a limited number of studies reporting the discovery of small-molecule inhibitors of MAPK substrate-docking sites. An earlier report using deuterium-exchange MS described the identification of substrate-selective inhibitors of the p38α MAPK as a potential approach to improve regulation of inflammatory responses [71]. These studies described a small-molecule inhibitor that targeted the active site of p38α MAPK and caused perturbations in residues in the DRS region of this kinase, which allowed the inhibitor to discriminate between the D-domain-containing substrates MK2 and ATF2. However, others suggested the differential effects of this compound on MK2 and ATF2 phosphorylation were a function of substrate:kinase stoichiometry and not due to the substrate selectivity of the compound [72]. A previous study indicated that the natural product zuonin A can target the DRS on JNK MAPK and inhibit phosphorylation of c-Jun [73]. Similarly, BI-78D3 was a small molecule identified to compete with a D-domain-containing peptide found on the JNK-interacting protein-1 (JIP1) and inhibits interactions with JNK1 [74]. Other screening approaches have identified novel compounds that bind to exposed allosteric sites on the JNK or p38 MAPK surface and are ATP-independent [75]. Although it is not known whether these compounds have differential effects on substrate proteins, this represents an alternative approach for identifying novel kinase inhibitors with improved selectivity over inhibitors that target highly conserved ATP-binding sites.
Although the present results indicate selective inhibition of the phosphorylation of substrate proteins by the studied inhibitors, selectivity is not absolute and may be stimulus dependent. This is evident in the results showing inhibition of phosphorylation or expression of the D-domain substrates RSK-1 and c-Jun at higher concentrations of test compound following treatment with EGF (Figures 1, 2 and 4). However, RSK-1 and c-Jun phosphorylation or expression was unaffected by the compounds in cells treated with PMA or in melanoma cells containing BRaf mutations (Figures 5 and 7). This highlights the complexity of the signalling pathways and the potential for differential effects of inhibitors that have a common target. As an example, it has been reported that catalytic site inhibitors of MEK have differences in efficacy and effects on ERK1/2 signalling depending on whether the cells contain BRaf or KRas mutations [76]. We have also observed partial specificity with putative DRS-targeted inhibitors, which inhibit ERK1/2-mediated caspase 9 and RSK-1 phosphorylation and sensitize transformed cells to undergo apoptosis [28]. Two factors may contribute to this lack of absolute specificity. First, the interaction between ERK2 and substrate may involve a relatively large portion of the docking domain such that interactions with more than one of the spatially adjacent sites may occur. Although the structure of a full substrate protein bound to ERK2 is not available, structures of D-domain peptides from haematopoietic protein tyrosine phosphatase and MAPK phosphatase-3 are available [77–79]. These peptides bind primarily to the DRS, although the C-terminal regions of the peptides extend towards other putative binding sites. Such additional interactions may be anticipated to lead to compounds binding to a given site on the docking domains to partially block interactions of substrate proteins not primarily interacting with those sites.
In the context of substrate interactions with the FRS on ERK1/2, conformational changes associated with the active phosphorylated form of ERK2 were reported to promote interactions with F-site substrates [21]. Recently, studies using D-domain- or F-site-specific peptides reported that there was little communication between the DRS and FRS on ERK2 when one site was occupied with a peptide [80]. Nonetheless, occupancy at either site with a substrate protein may lead to conformational perturbations on either the structural and/or dynamical levels that regulate the formation of signalling complexes.
In summary, a combination of computational and experimental methods has been used to identify a new class of low-molecular-mass compounds that regulate the expression and function of F-site-containing ERK1/2 substrates involved in AP-1-mediated transcription and cancer cell proliferation. The availability of these compounds will facilitate investigations into the biological function of ERK1/2-mediated signalling using chemical biology and potentially lead to the development of novel therapeutic agents. In addition, the successful identification of ATP-independent substrate-specific inhibitors of ERK1/2 is anticipated to open the door for similar ligand design strategies targeting protein–protein interactions in systems where a given protein has multiple interacting partners.
Supplementary Material
Acknowledgments
Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including grant number P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS, NCRR or NIH.
FUNDING: This work was supported by the National Institutes of Health [grant numbers R01CA120215 (to P.S. and A.D.M.) and F31 GM100693 (to R.S.)]; and the University of Maryland Computer-Aided Drug Design Center.
Abbreviations
- AP-1
activator protein-1
- CADD
computer-aided drug design
- DEF
docking site for ERK, FXF
- DRS
D-recruitment site
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- Egr 1
early growth response-1
- ERK
extracellular-signal-regulated kinase
- FRS
F-recruitment site
- HEK
human embryonic kidney
- IEG
immediate early gene
- JNK
c-Jun N-terminal kinase
- LGFE
ligand grid free energy
- MAPK
mitogen-activated protein kinase
- MEK
MAPK/ERK kinase
- RSK-1
p90 ribosomal S6 kinase-1
- SILCS
site identification by ligand competitive saturation
- SRE
serum-response element
Footnotes
AUTHOR CONTRIBUTION
Paul Shapiro and Alexander MacKerell, Jr, conceived and designed the studies and wrote the paper. Ramin Samadani and Jun Zhang performed most of the experiments. Amanda Brophy assisted with the experimental analyses. Taiji Oashi, Deva Priyakumar and Prabhu Raman performed the CADD analysis. Franz St John and Edwin Pozharski helped with setting of the X-ray crystallography studies and structural analysis. Kwan-Young Jung and Steven Fletcher synthesized SF-3-026 and analogue compounds.
The structure of MAPK ERK2 complexed with 1,1-dioxo-2,3-dihydrothiophen-3-yl benzenesulfonate has been deposited in the PDB under accession code 3QYI.
References
- 1.Pearson G, English JM, White MA, Cobb MH. ERK5 and ERK2 cooperate to regulate NF-kappaB and cell transformation. J Biol Chem. 2001;276:7927–7931. doi: 10.1074/jbc.M009764200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 3.Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- 4.von Kriegsheim A, Baiocchi D, Birtwistle M, Sumpton D, Bienvenut W, Morrice N, Yamada K, Lamond A, Kalna G, Orton R, et al. Cell fate decisions are specified by the dynamic ERK interactome. Nat Cell Biol. 2009;11:1458–1464. doi: 10.1038/ncb1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- 6.McCubrey JA, Milella M, Tafuri A, Martelli AM, Lunghi P, Bonati A, Cervello M, Lee JT, Steelman LS. Targeting the Raf/MEK/ERK pathway with small-molecule inhibitors. Curr Opin Investig Drugs. 2008;9:614–630. [PubMed] [Google Scholar]
- 7.Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res. 2008;14:342–346. doi: 10.1158/1078-0432.CCR-07-4790. [DOI] [PubMed] [Google Scholar]
- 8.Ohori M, Kinoshita T, Okubo M, Sato K, Yamazaki A, Arakawa H, Nishimura S, Inamura N, Nakajima H, Neya M, et al. Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex, Biochem. Biophys Res Commun. 2005;336:357–363. doi: 10.1016/j.bbrc.2005.08.082. [DOI] [PubMed] [Google Scholar]
- 9.Aronov AM, Baker C, Bemis GW, Cao J, Chen G, Ford PJ, Germann UA, Green J, Hale MR, Jacobs M, et al. Flipped out: structure-guided design of selective pyrazolylpyrrole ERK inhibitors. J Med Chem. 2007;50:1280–1287. doi: 10.1021/jm061381f. [DOI] [PubMed] [Google Scholar]
- 10.Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3:742–750. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- 11.Zuniga A, Torres J, Ubeda J, Pulido R. Interaction of mitogen-activated protein kinases with the kinase interaction motif of the tyrosine phosphatase PTP-SL provides substrate specificity and retains ERK2 in the cytoplasm. J Biol Chem. 1999;274:21900–21907. doi: 10.1074/jbc.274.31.21900. [DOI] [PubMed] [Google Scholar]
- 12.Akella R, Moon TM, Goldsmith EJ. Unique MAP Kinase binding sites. Biochim Biophys Acta. 2008;1784:48–55. doi: 10.1016/j.bbapap.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin MC, Allan LA, Mancini EJ, Clarke PR. The docking interaction of caspase-9 with ERK2 provides a mechanism for the selective inhibitory phosphorylation of caspase-9 at threonine 125. J Biol Chem. 2008;283:3854–3865. doi: 10.1074/jbc.M705647200. [DOI] [PubMed] [Google Scholar]
- 14.Dimitri CA, Dowdle W, MacKeigan JP, Blenis J, Murphy LO. Spatially separate docking sites on ERK2 regulate distinct signaling events in vivo. Curr Biol. 2005;15:1319–1324. doi: 10.1016/j.cub.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 15.Sharrocks AD, Yang SH, Galanis A. Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem Sci. 2000;25:448–453. doi: 10.1016/s0968-0004(00)01627-3. [DOI] [PubMed] [Google Scholar]
- 16.Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 1999;13:163–175. [PMC free article] [PubMed] [Google Scholar]
- 17.Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG. Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol Cell. 2004;14:43–55. doi: 10.1016/s1097-2765(04)00161-3. [DOI] [PubMed] [Google Scholar]
- 18.Polychronopoulos S, Verykokakis M, Yazicioglu MN, Sakarellos-Daitsiotis M, Cobb MH, Mavrothalassitis G. The transcriptional ETS2 repressor factor associates with active and inactive Erks through distinct FXF motifs. J Biol Chem. 2006;281:25601–25611. doi: 10.1074/jbc.M605185200. [DOI] [PubMed] [Google Scholar]
- 19.Tanoue T, Maeda R, Adachi M, Nishida E. Identification of a docking groove on ERK and p38 MAP kinases that regulates the specificity of docking interactions. EMBO J. 2001;20:466–479. doi: 10.1093/emboj/20.3.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fantz DA, Jacobs D, Glossip D, Kornfeld K. Docking sites on substrate proteins direct extracellular signal-regulated kinase to phosphorylate specific residues. J Biol Chem. 2001;276:27256–27265. doi: 10.1074/jbc.M102512200. [DOI] [PubMed] [Google Scholar]
- 21.Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG. Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol Cell. 2004;14:43–55. doi: 10.1016/s1097-2765(04)00161-3. [DOI] [PubMed] [Google Scholar]
- 22.Galanis A, Yang SH, Sharrocks AD. Selective targeting of MAPKs to the ETS domain transcription factor SAP-1. J Biol Chem. 2001;276:965–973. doi: 10.1074/jbc.M007697200. [DOI] [PubMed] [Google Scholar]
- 23.Tzarum N, Komornik N, Ben Chetrit D, Engelberg D, Livnah O. DEF pocket in p38alpha facilitates substrate selectivity and mediates autophosphorylation. J Biol Chem. 2013;288:19537–19547. doi: 10.1074/jbc.M113.464511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Zhou B, Zheng CF, Zhang ZY. A bipartite mechanism for ERK2 recognition by its cognate regulators and substrates. J Biol Chem. 2003;278:29901–29912. doi: 10.1074/jbc.M303909200. [DOI] [PubMed] [Google Scholar]
- 25.Robinson FL, Whitehurst AW, Raman M, Cobb MH. Identification of novel point mutations in ERK2 that selectively disrupt binding to MEK1. J Biol Chem. 2002;277:14844–14852. doi: 10.1074/jbc.M107776200. [DOI] [PubMed] [Google Scholar]
- 26.Chen F, Hancock CN, Macias AT, Joh J, Still K, Zhong S, MacKerell AD, Jr, Shapiro P. Characterization of ATP-independent ERK inhibitors identified through in silico analysis of the active ERK2 structure. Bioorg Med Chem Lett. 2006;16:6281–6287. doi: 10.1016/j.bmcl.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hancock CN, Macias A, Lee EK, Yu SY, Mackerell AD, Jr, Shapiro P. Identification of novel extracellular signal-regulated kinase docking domain inhibitors. J Med Chem. 2005;48:4586–4595. doi: 10.1021/jm0501174. [DOI] [PubMed] [Google Scholar]
- 28.Boston SR, Deshmukh R, Strome S, Priyakumar UD, MacKerell AD, Jr, Shapiro P. Characterization of ERK docking domain inhibitors that induce apoptosis by targeting Rsk-1 and caspase-9. BMC Cancer. 2011;11:7. doi: 10.1186/1471-2407-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jung KY, Samadani R, Chauhan J, Nevels K, Yap JL, Zhang J, Worlikar S, Lanning ME, Chen L, Ensey M, et al. Structural modifications of (Z)-3-(2-aminoethyl)-5-(4-ethoxybenzylidene)thiazolidine-2,4-dione that improve selectivity for inhibiting the proliferation of melanoma cells containing active ERK signaling. Org Biomol Chem. 2013;11:3706–3732. doi: 10.1039/c3ob40199e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yap JL, Worlikar S, MacKerell AD, Jr, Shapiro P, Fletcher S. Small-molecule inhibitors of the ERK signaling pathway: Towards novel anticancer therapeutics. ChemMedChem. 2011;6:38–48. doi: 10.1002/cmdc.201000354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guvench O, MacKerell AD., Jr Computational fragment-based binding site identification by ligand competitive saturation. PLoS Comput Biol. 2009;5:e1000435. doi: 10.1371/journal.pcbi.1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raman EP, Yu W, Guvench O, Mackerell AD. Reproducing crystal binding modes of ligand functional groups using Site-Identification by Ligand Competitive Saturation (SILCS) simulations. J Chem Inf Model. 2011;51:877–896. doi: 10.1021/ci100462t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burkhard KA, Chen F, Shapiro P. Quantitative analysis of ERK2 interactions with substrate proteins: roles for kinase docking domains and activity in determining binding affinity. J Biol Chem. 2011;286:2477–2485. doi: 10.1074/jbc.M110.177899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mackerell AD, Jr, Feig M, Brooks CL., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 37.MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck J, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-atom empirical potential for molecular modeling and dynamics studies of protein. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 38.Jorgensen WL, Jayaraman C, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 39.Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 1997;90:859–869. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 40.Zhang F, Strand A, Robbins D, Cobb MH, Goldsmith EJ. Atomic structure of the MAP kinase ERK2 at 2.3 A resolution. Nature. 1994;367:704–711. doi: 10.1038/367704a0. [DOI] [PubMed] [Google Scholar]
- 41.Zhong S, MacKerell AD., Jr Binding response: a descriptor for selecting ligand binding site on protein surfaces. J Chem Inf Model. 2007;47:2303–2315. doi: 10.1021/ci700149k. [DOI] [PubMed] [Google Scholar]
- 42.Furci LM, Lopes P, Eakanunkul S, Zhong S, MacKerell AD, Jr, Wilks A. Inhibition of the bacterial heme oxygenases from Pseudomonas aeruginosa and Neisseria meningitidis: novel antimicrobial targets. J Med Chem. 2007;50:3804–3813. doi: 10.1021/jm0700969. [DOI] [PubMed] [Google Scholar]
- 43.Yu WM, Guvench O, Mackerell AD, Qu CK. Identification of small molecular weight inhibitors of Src homology 2 domain-containing tyrosine phosphatase 2 (SHP-2) via in silico database screening combined with experimental assay. J Med Chem. 2008;51:7396–7404. doi: 10.1021/jm800229d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 45.Pan Y, Huang N, Cho S, MacKerell AD., Jr Consideration of molecular weight during compound selection in virtual target-based database screening. J Chem Inf Comput Sci. 2003;43:267–272. doi: 10.1021/ci020055f. [DOI] [PubMed] [Google Scholar]
- 46.Huang N, Nagarsekar A, Xia G, Hayashi J, MacKerell AD., Jr Identification of non-phosphate-containing small molecular weight inhibitors of the tyrosine kinase p56 Lck SH2 domain via in silico screening against the pY + 3 binding site. J Med Chem. 2004;47:3502–3511. doi: 10.1021/jm030470e. [DOI] [PubMed] [Google Scholar]
- 47.Elkins JM, Wang J, Deng X, Pattison MJ, Arthur JS, Erazo T, Gomez N, Lizcano JM, Gray NS, Knapp S. X-ray crystal structure of ERK5 (MAPK7) in complex with a specific inhibitor. J Med Chem. 2013;56:4413–4421. doi: 10.1021/jm4000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gille H, Kortenjann M, Thomae O, Moomaw C, Slaughter C, Cobb MH, Shaw PE. ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 1995;14:951–962. doi: 10.1002/j.1460-2075.1995.tb07076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4:556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- 50.Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, Morrison DK. Regulation of raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215–224. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 51.Shapiro PS, Ahn NG. Feedback regulation of Raf-1 and mitogen-activated protein kinase (MAP) kinase kinases 1 and 2 by MAP kinase phosphatase-1 (MKP-1) J Biol Chem. 1998;273:1788–1793. doi: 10.1074/jbc.273.3.1788. [DOI] [PubMed] [Google Scholar]
- 52.Morton S, Davis RJ, Cohen P. Signalling pathways involved in multisite phosphorylation of the transcription factor ATF-2. FEBS Lett. 2004;572:177–183. doi: 10.1016/j.febslet.2004.07.031. [DOI] [PubMed] [Google Scholar]
- 53.Sasaki T, Kojima H, Kishimoto R, Ikeda A, Kunimoto H, Nakajima K. Spatiotemporal regulation of c-Fos by ERK5 and the E3 ubiquitin ligase UBR1, and its biological role. Mol Cell. 2006;24:63–75. doi: 10.1016/j.molcel.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 54.Gilley R, March HN, Cook SJ. ERK1/2, but not ERK5, is necessary and sufficient for phosphorylation and activation of c-Fos. Cell Signal. 2009;21:969–977. doi: 10.1016/j.cellsig.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 55.Gopal YN, Deng W, Woodman SE, Komurov K, Ram P, Smith PD, Davies MA. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010;70:8736–8747. doi: 10.1158/0008-5472.CAN-10-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Poulikakos PI, Rosen N. Mutant BRAF melanomas–dependence and resistance. Cancer Cell. 2011;19:11–15. doi: 10.1016/j.ccr.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 58.Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O’Meara S, Santarius T, Avis T, Barthorpe S, Brackenbury L, et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther. 2006;5:2606–2612. doi: 10.1158/1535-7163.MCT-06-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milde-Langosch K. The Fos family of transcription factors and their role in tumourigenesis. Eur J Cancer. 2005;41:2449–2461. doi: 10.1016/j.ejca.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 60.Verde P, Casalino L, Talotta F, Yaniv M, Weitzman JB. Deciphering AP-1 function in tumorigenesis: fra-ternizing on target promoters. Cell Cycle. 2007;6:2633–2639. doi: 10.4161/cc.6.21.4850. [DOI] [PubMed] [Google Scholar]
- 61.Treisman R. Ternary complex factors: growth factor regulated transcriptional activators. Curr Opin Genet Dev. 1994;4:96–101. doi: 10.1016/0959-437x(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 62.Hsu T, Trojanowska M, Watson DK. Ets proteins in biological control and cancer. J Cell Biochem. 2004;91:896–903. doi: 10.1002/jcb.20012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arkenau HT, Kefford R, Long GV. Targeting BRAF for patients with melanoma. Br J Cancer. 2011;104:392–398. doi: 10.1038/sj.bjc.6606030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duffy A, Kummar S. Targeting mitogen-activated protein kinase kinase (MEK) in solid tumors. Target Oncol. 2009;4:267–273. doi: 10.1007/s11523-009-0125-x. [DOI] [PubMed] [Google Scholar]
- 65.Gee JM, Robertson JF, Gutteridge E, Ellis IO, Pinder SE, Rubini M, Nicholson RI. Epidermal growth factor receptor/HER2/insulin-like growth factor receptor signalling and oestrogen receptor activity in clinical breast cancer. Endocr Relat Cancer. 2005;12:S99–S111. doi: 10.1677/erc.1.01005. [DOI] [PubMed] [Google Scholar]
- 66.Kohno M, Pouyssegur J. Targeting the ERK signaling pathway in cancer therapy. Ann Med. 2006;38:200–211. doi: 10.1080/07853890600551037. [DOI] [PubMed] [Google Scholar]
- 67.Hatzivassiliou G, Liu B, O’Brien C, Spoerke JM, Hoeflich KP, Haverty PM, Soriano R, Forrest WF, Heldens S, Chen H, et al. ERK inhibition overcomes acquired resistance to MEK Inhibitors. Mol Cancer Ther. 2012;11:1143–1154. doi: 10.1158/1535-7163.MCT-11-1010. [DOI] [PubMed] [Google Scholar]
- 68.Corcoran RB, Settleman J, Engelman JA. Potential therapeutic strategies to overcome acquired resistance to BRAF or MEK inhibitors in BRAF mutant cancers. Oncotarget. 2011;2:336–346. doi: 10.18632/oncotarget.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krishnamurty R, Maly DJ. Biochemical mechanisms of resistance to small-molecule protein kinase inhibitors. ACS Chem Biol. 2010;5:121–138. doi: 10.1021/cb9002656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cseh B, Doma E, Baccarini M. “RAF” neighborhood: protein–protein interaction in the Raf/Mek/Erk pathway”. FEBS Lett. 2014;588:2398–2406. doi: 10.1016/j.febslet.2014.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Davidson W, Frego L, Peet GW, Kroe RR, Labadia ME, Lukas SM, Snow RJ, Jakes S, Grygon CA, Pargellis C, Werneburg BG. Discovery and characterization of a substrate selective p38alpha inhibitor. Biochemistry. 2004;43:11658–11671. doi: 10.1021/bi0495073. [DOI] [PubMed] [Google Scholar]
- 72.Hendriks BS, Seidl KM, Chabot JR. Two additive mechanisms impair the differentiation of ‘substrate-selective’ p38 inhibitors from classical p38 inhibitors in vitro. BMC Syst Biol. 2010;4:23. doi: 10.1186/1752-0509-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaoud TS, Park H, Mitra S, Yan C, Tseng CC, Shi Y, Jose J, Taliaferro JM, Lee K, Ren P, Hong J, Dalby KN. Manipulating JNK signaling with (–)-zuonin A. ACS Chem. Biol. 2012;7:1873–1883. doi: 10.1021/cb300261e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stebbins JL, De SK, Machleidt T, Becattini B, Vazquez J, Kuntzen C, Chen LH, Cellitti JF, Riel-Mehan M, Emdadi A, et al. Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc Natl Acad Sci USA. 2008;105:16809–16813. doi: 10.1073/pnas.0805677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Comess KM, Sun C, Abad-Zapatero C, Goedken ER, Gum RJ, Borhani DW, Argiriadi M, Groebe DR, Jia Y, Clampit JE, et al. Discovery and characterization of non-ATP site inhibitors of the mitogen activated protein (MAP) kinases. ACS Chem Biol. 2011;6:234–244. doi: 10.1021/cb1002619. [DOI] [PubMed] [Google Scholar]
- 76.Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, Hewitt JF, Zak M, Peck A, Orr C, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013;501:232–236. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- 77.Liu S, Sun JP, Zhou B, Zhang ZY. Structural basis of docking interactions between ERK2 and MAP kinase phosphatase 3. Proc Natl Acad Sci USA. 2006;103:5326–5331. doi: 10.1073/pnas.0510506103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Farooq A, Chaturvedi G, Mujtaba S, Plotnikova O, Zeng L, Dhalluin C, Ashton R, Zhou MM. Solution structure of ERK2 binding domain of MAPK phosphatase MKP-3: structural insights into MKP-3 activation by ERK2. Mol Cell. 2001;7:387–399. doi: 10.1016/s1097-2765(01)00186-1. [DOI] [PubMed] [Google Scholar]
- 79.Zhou T, Sun L, Humphreys J, Goldsmith EJ. Docking interactions induce exposure of activation loop in the MAP kinase ERK2. Structure. 2006;14:1011–1019. doi: 10.1016/j.str.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 80.Lee S, Warthaka M, Yan C, Kaoud TS, Ren P, Dalby KN. Examining docking interactions on ERK2 with modular peptide substrates. Biochemistry. 2011;50:9500–9510. doi: 10.1021/bi201103b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.