

Abstract
Background
Detection of BRAF mutations is an established standard of care to predict small-molecule inhibitor (vemurafenib) response in metastatic melanoma. Molecular assays should be designed to detect not only the most common p.V600E mutation, but also p.V600K and other non-p.V600E mutations.
Objective
The purpose of this study was to assess if tumor cellularity can function as a quality assurance (QA) measure in molecular diagnostics. Potential causes of discrepancy between the observed and predicted mutant allele percentage were also explored.
Methods
We correlated pathologist-generated estimates of tumor cellularity versus mutant allele percentage via pyrosequencing as a QA measure for BRAF mutation detection in formalin-fixed, paraffin-embedded melanoma specimens.
Results
BRAF mutations were seen in 27/62 (44 %) specimens, with 93 % p.V600E and 7 % non-p.V600E. Correlation between p.V600E mutant percentage and tumor cellularity was poor–moderate (r = −0.02; p = 0.8), primarily because six samples showed a low p.V600E signal despite high tumor cellularity. A QA investigation revealed that our initial pyrosequencing assay showed a false positive, weak p.V600E signal in specimens with a p.V600K mutation. A redesigned assay detected BRAF mutations in 50/131 (38 %) specimens, including 30 % non-p.V600E. This revised assay showed strong correlation between p.V600E BRAF mutant percentage and tumor cellularity (r = 0.76; p ≤ 0.01). Re-evaluation of the previously discordant samples by the revised assay confirmed a high level of p.V600K mutation in five specimens.
Conclusions
Pathologists play important roles in molecular diagnostics, beyond identification of correct cells for testing. Accurate evaluation of tumor cellularity not only ensures sufficient material for required analytic sensitivity, but also provides an independent QA measure of the molecular assays.
1 Introduction
Mutations of the BRAF gene often lead to constitutive activation of the mitogen-activated protein kinase (MAPK) pathway in a variety of human neoplasms [1, 2, 3]. Approximately 40–60 % of cutaneous melanomas carry a BRAF mutation [1, 4–6]. More than 90 % of all BRAF mutations in melanoma are comprised of a single amino acid substitution of valine by glutamic acid at codon 600 (p.V600E) as a result of c.1799T>A base variant [1, 7, 8]. However, non-p.V600E BRAF mutations may be more frequent than previously reported [4, 6, 9–11], particularly the p.V600K mutation (GTG>AAG). Vemurafenib (PLX4032) is a small-molecule inhibitor of mutated BRAF protein, shown to cause programmed cell death in melanoma cell lines [12]. Clinically, vemurafenib therapy has been shown to improve survival in previously treated or untreated metastatic melanoma patients, not only those with the BRAF p.V600E mutation but also those with non-p.V600E mutations such as p.V600K and p.V600R [13–16]. Dabrafenib (a BRAF inhibitor) and trametinib (an MEK inhibitor) have also been shown to improve the survival of melanoma patients [17–21].
Cobas 4800, a real-time polymerase chain reaction (PCR) mutational assay, was the first US FDA-approved companion diagnostic test to detect the BRAF p.V600E mutation. The Cobas 4800 assay was designed to detect the p.V600E mutation, however it also cross-reacts with non-p.V600E mutations, including 70 % of p.V600K mutations [22, 23]. The analytic sensitivity (or limit of detection) of Cobas 4800 for the p.V600E mutation may be different for non-p.V600E mutations; some patients with non-p.V600E mutations who might otherwise benefit from vemurafenib therapy could therefore be missed [11, 13, 16, 24]. A variety of other assays to detect BRAF mutations have been developed [22, 23, 25–28], including a clinical pyrosequencing assay used in our laboratory.
In an effort to ascertain the limitations of our clinical pyrosequencing assay, we reviewed our ability to detect both p.V600E and non-p.V600E BRAF mutations in melanoma. As a quality assurance (QA) measure, we correlated p.V600E mutation assay results with pathologist-generated estimates of tumor cellularity in specimens with indeterminate or low/borderline positive p.V600E signal. A discordant observation—for instance, a low p.V600E signal in specimens with relatively high tumor cellularity—triggered a QA follow-up. On follow-up, most specimens with low tumor cellularity showed a low positive signal consistent with a p.V600E mutation. However, a few specimens with high tumor cellularity showed an unexpectedly low p.V600E signal. The objectives of this study were to examine the performance of our BRAF pyrosequencing assay, to identify and provide a rationale for any unexpected/discrepant results, and to ascertain the utility of tumor cellularity as a QA measure.
2 Materials and Methods
2.1 Materials
We examined 194 formalin-fixed, paraffin-embedded (FFPE) specimens diagnosed as either primary or recurrent melanoma that were submitted for BRAF mutation testing between March 2010 and July 2013. This included 62 specimens tested with our initial pyrosequencing assay (March 2010–October 2011) and 132 specimens tested with our revised pyrosequencing assay (November 2011–July 2013), with both assays described further below. Pyrosequencing failed in only one (0.5 %) of the 194 specimens. This specimen was from a resection performed at an outside hospital, with unknown tissue fixation conditions. No melanin was observed on examination of the accompanying hematoxylin and eosin (H&E) slide. The remaining 193 specimens included 146 cutaneous melanomas (49 primary or locally recurrent tumors, as well as 97 metastatic tumors), 42 metastatic melanomas with an unknown primary tumor location, 4 mucosal melanomas, and 1 primary uveal melanoma. Tissue blocks with adequate tumor cellularity were selected by the pathologists who made the histologic diagnosis. One H&E slide followed by five unstained slides were prepared according to standard pre-PCR protocol. The H&E slide was examined and marked by the pathologist for subsequent macrodissection of the FFPE neoplastic tissues from two to five unstained slides of 5-micron thick sections. DNA was isolated from the area(s) designated by pathologists using the Pinpoint DNA Isolation System (Zymo Research, Irvine, CA, USA), followed by further purification via the QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA) [29].
The tumor cellularity was first evaluated by pathologists who requested the diagnostic BRAF testing and then blindly re-evaluated by a single pathologist (MTL). H&E-stained slides were available for 192 of 193 specimens with successful BRAF mutation testing by pyrosequencing. An additional three specimens were excluded because of significant crushing artifact. The estimated ratio of tumor versus non-tumor nuclei was separated into quantiles (1–20, 21–40, 41–60, 61–80 and 81–100 %). A tumor cellularity of 41–60 % was assigned when the number of neoplastic cells was approximately close to the non-neoplastic cells. For those specimens with a relatively higher number of neoplastic cells, tumor cellularity was assigned as 81–100 % for specimens with predominantly neoplastic cells and 61–80 % for the remaining specimens. For those specimens with a relatively higher number of non-neoplastic cells, tumor cellularity was assigned as 1–20 % for specimens with predominantly non-neoplastic cells and 21–40 % for the remaining specimens. The total number of neoplastic cells was counted to calculate the tumor cellularity in some specimens with scant overall cellularity or an estimated tumor cellularity near the analytic sensitivity of the pyrosequencing assay.
2.2 Pyrosequencing
Pyrosequencing was performed as previously described [30, 31]. The established limit of detection for the assay in our laboratory is 5 % mutant alleles or 10 % tumor cells [32]. The PCR primers used for the amplification step of both the initial and revised pyrosequencing assays were as follows: forward primer 5′-GAAGACCTCACAGTAAAAATAG-3′ and biotinylated reverse primer 5′-ATAGCCTCAATTCTTACCATCC-3′. The forward sequencing primer of our initial pyrosequencing assay (pre-November 2011; designed primarily for detection of BRAF p.V600E) was 5′-AGGTGATTTTGGTCTAGCTACAG-3′ (Fig. 1). The sequencing primer of our revised pyrosequencing assay (post-November 2011) was 5′-GACCTCACAGTAAAAATAGGTGATTTTG-3′ (Fig. 1). The differences between the two are shown in Fig. 1, but to briefly summarize, the 3′ end of the forward sequencing primer in the initial assay was at the first nucleotide of codon 600, whereas in the revised assay the 3′ end is at the first nucleotide of codon 596. PCR conditions were 95 °C for 15 min, 42 cycles of 95 °C for 20 s, 53 °C for 30 s and 72 °C for 20 s, and 72 °C for 5 min. The nucleotide dispensation order for pyrosequencing was: 5′-GCATCGATCTCGATGAGTG-3′ and 5′-GTAGCTAGCTATCAGCATCGACTCTCGATGAGTG-3′ for the initial and revised assays, respectively. A signal of 4–5 % mutant allele was designated as ‘indeterminate’ and a signal of less than 3 % mutant allele was reported as a negative result. The interpretation of complex pyrogram patterns, such as those seen in two or more nucleotide variants within the same allele, was resolved by a freely-available software designed in our laboratory (Pyromaker; http://pyromaker.pathology.jhmi.edu) and confirmed by Sanger sequencing, as previously described [30, 33].
Fig. 1.
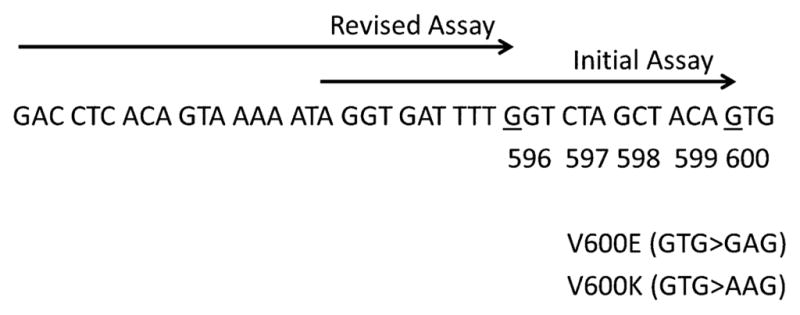
Assay design. The assays are based on a polymerase chain reaction-amplified sequence of 124 bp. The primer (→) for the initial pyrosequencing assay ends at the first nucleotide (underlined) of codon 600, while the primer of the revised assay ends at the first nucleotide (underlined) of codon 595
2.3 Next-Generation Sequencing
Specimens with complex pyrograms were also retrospectively tested by a next-generation sequencing (NGS) platform, conducted using AmpliSeq Cancer Hotspot Panel (v2) for targeted multi-gene amplification. The panel consists of 207 amplicons covering approximately 2,800 COSMIC mutations from 50 oncogenes and tumor suppressor genes. Briefly, we used Ion AmpliSeq Library Kit 2.0 for library preparation, Ion OneTouch 200 Template Kit v2 DL and Ion OneTouch Instrument for emulsion PCR and template preparation, and Ion PGM 200 Sequencing Kit with Ion 318 Chip and Personal Genome Machine (PGM) as the sequencing platform (Life Technologies, Carlsbad, CA, USA), all per manufacturers’ protocol. The DNA input for targeted multi-gene PCR was 10–30 ng. Eight specimens were barcoded using Ion Xpress Barcode Adapters (Life Technologies) for each Ion 318 chip. The background noise of p.V600E mutation, assessed by using 16 non-neoplastic FFPE tissues, was less than 0.3 % (mean ± 3 standard deviations[SD]) [data not shown].
2.4 Single-Nucleotide Polymorphism Array
Single-nucleotide polymorphism (SNP) array analysis was performed as previously described [34]. Briefly, DNA samples extracted from FFPE tissue (optimally 200 ng) were treated with the Infinium HD FFPE DNA restore kit before running on the Illumina Infinium II SNP array (HumanCytoSNP-12 v2.1 DNA Analysis BeadChip, Illumina Inc., San Diego, CA, USA) according to the manufacturer’s standard protocol. The B-allele frequency and log R ratio data were analyzed using Illumina KaryoStudio software version 2.0 and CNV (copy number variation) partition V2.4.4.0.
2.5 Statistics and Data Analysis
Statistical analyses were performed using the GraphPad Prism software, unless otherwise specified (GraphPad Software, ver5, La Jolla, CA, USA). Categorical study cohort data (% mutated BRAF, % p.V600E) were evaluated by Chi-squared test. Correlation between the observed % BRAF mutant versus the % predicted mutant based on tumor cellularity were examined by Spearman’s rank correlation coefficient and denoted as r. Error bars represent SD. For all analyses, differences were considered significant at *p < 0.05 or **p ≤ 0.01.
3 Results
3.1 Initial Pyrosequencing Assay: Tumor Cellularity as a Quality Assurance Measure
As a QA measure, we correlated the pathologist-generated estimates of tumor cellularity with the corresponding % p.V600E variant detected by our BRAF clinical pyrosequencing assay. In early October 2011, we noted a discrepant result: a specimen with 61–80 % tumor cellularity (Fig. 2a) but a p.V600E signal of only 5 % (Fig. 2b). The discrepancy was further evaluated by Sanger sequencing, which revealed mutations in the first two nucleotides (GTG>AAG at codon 600), leading to p.V600K if on the same allele (cis) or a double p.V600E and p.V600M if the mutations occurred on different alleles (trans) (Fig. 2c). This was later retrospectively confirmed by NGS analysis, which showed p.V600K with 28 % mutant allele, consistent with the predicted 31–40 % based on tumor cellularity (61–80 % tumor containing a heterozygous mutation, data not shown). This discrepant result prompted a detailed review of the adequacy of the first iteration of our pyrosequencing assay (henceforth designed ‘initial assay’) as well as subsequent development of the ‘revised’ pyrosequencing assay.
Fig. 2.

False p.V600E signal of the initial pyrosequencing assay. Hematoxylin and eosin for a case with high tumor cellularity (61–80 %) (a) but low false p.V600E signal (5 %) via initial pyrosequencing assay (b). p.V600K mutation was confirmed by Sanger sequencing (c) and revised pyrosequencing assays (33 %) (d). Subpanel inset shows the negative control [b, d]
3.2 Characterization of the Initial Pyrosequencing Assay
Sixty-two specimens were tested with the initial pyrosequencing assay from March 2010 to October 2011. BRAF mutations were detected in 27 of 62 specimens (44 %) [Table 1]. The specimens were analyzed in detail in two cohorts, according to % p.V600E mutant allele: 16 specimens showed a mutant allele of 5–50 %, while nine specimens showed a mutant allele of 53–74 %, suggesting loss of heterozygosity or amplification of mutant alleles. The results (tumor cellularity vs. % observed mutant allele) were designated as concordant if the observed mutant allele percentage was within 10 % of the predicted % mutant allele based on tumor cellularity (assuming a heterozygous mutation). Among specimens with BRAF mutant allele ≤50 %, only 8 of 16 (50 %) were concordant: a 10 % lower than predicted mutant allele percentage was observed in six specimens (5–14 % observed vs. 31–50 % predicted; cases 1–4, 6 and 7) and a 10-point higher than predicted mutant allele percentage was observed in two specimens (cases 14 and 16) [Table 2]. Overall, the Spearman correlation coefficient between % observed mutant allele and % predicted mutant allele based on tumor cellularity (r = −0.02; p = 0.8) was lower than might be otherwise expected, primarily because of the six specimens with low p.V600E signal despite high tumor cellularity (Fig. 3a).
Table 1.
Summary of BRAF mutations in melanoma using initial and revised assays
| Initial assay (n = 62) | Revised assay (n = 131) | |
|---|---|---|
| Positive, total | 27 | 50 |
| p.V600E | 25 (20)a | 35 |
| p.V600K | 0 (5)a | 13 |
| p.V600R | 0 | 1 |
| p.K601E | 1 (1)a | 0 |
| p.V600E K601IdelinsEI | 1 (1)a | 0 |
| p.V600_K601delinsE | 0 | 1 |
Number in parentheses indicates mutations re-evaluated by the revised assay
Table 2.
BRAF mutation rate by pyrosequencing vs. predicted tumor cellularity based on hematoxylin and eosin
| Initial assay (%) | Revised assay (%) | % Predicted mutanta | |
|---|---|---|---|
| 1 | V600E (5)b | V600K (33) | 31–40b |
| 2 | V600E (6)b | V600K (45) | 31–40b |
| 3 | V600E (6)b | V600K (44) | 31–40b |
| 4 | V600E (12)b | V600E (10)b | 41–50b |
| 5 | V600E (13) | V600E (13) | 21–30 |
| 6 | V600E (13)b | V600K (64) | 41–50b |
| 7 | V600E (14)b | V600K (67) | 41–50b |
| 8 | V600E (29) | V600E (24) | 31–40 |
| 9 | V600E (34) | V600E (34) | 31–40 |
| 10 | V600E (41) | V600E (39) | 31–40 |
| 11 | V600E (38) | V600E (31) | 21–30 |
| 12 | V600E (44) | V600E (40) | 41–50 |
| 13 | V600E (47) | V600E (44) | 41–50 |
| 14 | V600E (48) | V600E (45) | 11–20 |
| 15 | V600E (49) | V600E (48) | 41–50 |
| 16 | V600E (50) | V600E (48) | 21–30 |
| 17–25 | V600E (53–74) | V600E (39–74) | 31–40 or 41–50 |
All cases show a predicted tumor cellularity of less than 50 %, based on the assumption of heterozygosity of the mutant allele in tumor cells
Observed mutant allele frequency was 10 % less than the predicted mutant allele frequency
Fig. 3.

Correlations of predicted % mutant allele (based on tumor cellularity) and observed % mutant allele (detected by pyrosequencing). Initial pyrosequencing assay (a). Samples with low p.V600E BRAF mutations, despite high tumor cellularity (outliers) are circled. Squares represent v.600K-positive samples characterized as false, low positive p.V600E by the initial assay. Revised pyrosequencing assay (b): correlation for 35 p.V600E BRAF mutations. Repeat analysis of the 25 samples from the initial assay (c): re-run of the revised pyrosequencing assay. Significance *p < 0.05 or **p < 0.01
An additional concern raised by the retrospective review of our initial assay was the high rate of p.V600E versus non-p.V600E mutations. Among the 27 BRAF mutated samples, 25 (93 %) showed p.V600E (GTG>GAG), one case showed p.K601E (AAA>GAA), and one case showed p.V600_K601delinsEI (p.V600E, GTG>GAA at codon 600 and p.K601I AAA>ATA at codon 601) [Table 1]. The two non-p.V600E mutations were confirmed by Sanger sequencing. In the more complex case, we confirmed the presence of both p.V600E and p.K601I, but could not determine whether the mutations were located within the same allele (cis) or different allele (trans). Using a software tool called Pyromaker as well as NGS analysis, we confirmed that both 3-nucleotide mutations (p.V600E and p.K601I) occurred in cis within a single clone. The apparent difficulty in detection of non-p.V600E, together with above-mentioned low false-positive p.V600E identification in a case with p.V600K, cemented the need to redesign the pyrosequencing assay.
3.3 Design and Characterization of the Revised Pyrosequencing Assay
To appropriately detect a range of common non-p.V600E mutations, a new sequencing primer was designed that spanned to end at the first nucleotide of codon 596 instead of the first nucleotide of codon 600 in the initial assay (Fig. 1). As a case study, we used the revised assay in conjunction with Pyromaker to test the p.V600K-positive case mis-identified by our initial pyrosequencing assay as a p.V600E mutation. Indeed, with the revised assay we observed p.V600K (Fig. 2d)—the same result as Sanger sequencing and the NGS assays. Following validation in October 2011, this revised BRAF pyrosequencing assay replaced our initial assay. As part of the ongoing QA process, we continued to both track the pyrosequencing results for melanoma cases and to prospectively correlate these results with tumor cellularity. The results for the period November 2011–July 2013 are summarized in Tables 1 and 2. Briefly, BRAF mutations were detected in 50 of 131 specimens (38 %), comparable to the rate seen by the initial assay (27 of 62 specimens [44 %]; p = 0.48). However, unlike the initial assay, the revised assay detected a broader range of BRAF mutations, with p.V600E comprising 70 % rather than 93 % of the total (*p = 0.02). Specific BRAF mutations included 35 cases with p.V600E, 13 cases with p.V600K, one case with p.V600R (GTG>AGG) and one case with p.V600_K601delinsE (c.1799_1801del). Pyromaker analysis supported all of the identifications of non-p.V600E mutations.
In contrast to the initial assay, we also observed a strong correlation between the 35 p.V600E BRAF mutant specimens identified by the revised assay and the mutant allele % predicted by tumor cellularity. We again analyzed the specimens in two cohorts, according to % mutant allele: 27 specimens showed a mutant allele of 7–50 %, while eight specimens showed a mutant allele of 53–77 %. Among the cases with ≤50 % p.V600E mutant, the observed mutant allele was within 10 % of the predicted in 24 of 27 specimens (89 %), markedly higher than 50 % concordance observed in the initial assay. A 10 % lower than the predicted mutant allele was observed in two specimens (18 % observed vs. 31–40 % predicted, and 28 % observed vs. 41–50 % predicted). SNP array analysis of these two specimens revealed a gain of chromosome 7, suggesting that the gain of the wild-type allele may account for the discrepancy. A 10 % higher than predicted mutant allele percentage was observed in one specimen (35 % observed vs. 11–20 % predicted mutant allele). SNP array analysis of this specimen revealed copy number change of chromosome 7q22-q35, a region that includes the BRAF gene, and this amplification may largely account for the above discrepancy. The revised assay demonstrated improved correlation between observed mutant allele percentage and tumor cellularity (r = 0.76; p ≤ 0.01) [Fig. 3b].
3.4 Detailed Re-Evaluation of the 25 p.V600E Mutants Identified by the Initial Assay
All 25 melanoma specimens with p.V600E mutations identified by our initial pyrosequencing assay were reevaluated using the revised assay. p.V600E mutation was confirmed in 20 specimens (Table 1). The remaining five samples with p.V600E signal by the initial assay instead showed a markedly higher p.V600K via the revised assay. The % p.V600E allele detected by the initial and revised assays was highly concordant; however, in cases where the revised assay correctly identified p.V600K instead of p.V600E, the revised assay more closely approached the % predicted mutant (Table 2, rows 2 and 3). The remaining three cases with more than 10 % discrepancy between observed vs. predicted % p.V600E may again be largely accounted for by a gain or loss of BRAF gene on chromosome 7q. SNP array analysis of cases 14 and 16 (48 % observed vs. 11–20 % predicted, and 50 % observed vs. 21–30 % predicted) revealed a gain of chromosome 7q, suggesting amplification of the genomic region containing BRAF gene (data not shown). SNP array analysis of case 4 failed. Overall, the 25 p.V600E-mutated specimens reevaluated using the revised pyrosequencing assay demonstrated a stronger correlation between % observed mutant allele and tumor cellularity (r = 0.41; p ≤ 0.05) [Fig. 3c].
4 Discussion
Molecular testing for mutations in the BRAF gene is now a standard of care for patients with metastatic melanoma and is used to determine potential benefit from treatment with vemurafenib or other targeted therapy [13, 16, 26]. The assays must detect not only the most common and best-understood p.V600E mutation, but also non-p.V600E mutations, since clinical trials have shown that patients with the non-p.V600E BRAF mutant may also benefit from targeted therapy [11, 13–16]. In this study, pathologist-generated estimates of tumor cellularity served as a QA measure for a clinical BRAF pyrosequencing assay. Detection of a lower-than-expected p.V600E signal in specimens with a high tumor cellularity prompted an investigation, which revealed that our original pyrosequencing assay, similar to the Cobas 4800 BRAF V600 mutation test [22–24, 35, 36], may show a weak p.V600E signal in p.V600K positive specimens. Our revised pyrosequencing assay could distinguish p.V600E mutation from other non-p.V600E mutations and confirmed a higher incidence of p.V600K mutation than previously reported in melanomas [4, 6, 9–11].
Many real-time PCR-based BRAF assays, including Cobas 4800, utilize probes designed specifically to detect the p.V600E allele (GTG>GAT for codon 600). However, the probes have been shown to also cross-hybridize with the p.V600K allele and other non-p.V600E mutant alleles such as p.V600D [22–24, 35, 36]. In addition, Cobas 4800 and other PCR-based assays detect non-p.V600E mutations with a lower analytic sensitivity. This leads to a low false-positive p.V600E signal in non-p.V600E positive specimens with high tumor cellularity, or possibly a false-negative result when the tumor cellularity is low [22–24, 35]. The 3′ nucleotide of the sequencing primer of our initial pyrosequencing assay ended at the first nucleotide of codon 600. With retrospective and prospective review, we determined that a mutation involving this codon, such as p.V600K (GTG>AAT) or p.V600R (GTG>AGG), will lead to a weaker hybridization with the sequencing primer. This phenomenon appears to be similar to an allelic dropout caused by an SNP underlying a primer binding site [37–39]. Like the Cobas 4800 BRAF V600E test, the initial iteration of our pyrosequencing assay may also show either no signal or a low false-positive p.V600E signal in tumor specimens with non-p.V600E BRAF mutations.
In addition to the Sanger sequencing, pyrosequencing, and real-time-based assays (such as Cobas 4800), several molecular assays have been developed to detect BRAF mutations with various analytic sensitivity, such as the melt curve analysis, primer extension MassARRAY system, multiplex SNaPshot assay, allele-specific PCR, and NGS [9, 36, 40–45]. NGS not only provides a high analytic sensitivity, but also a high diagnostic sensitivity by simultaneous detection of all common and uncommon mutations of the BRAF gene. We have recently validated the PGM NGS platform for simultaneous detection of BRAF, NRAS, and KIT mutations for melanoma. Immunohistochemistry (IHC) is also a highly sensitive and cost-effective assay for detection of the BRAF p.V600E mutation [35, 36, 46–48]. The assay can be applied to specimens with low tumor cellularity, particularly those with neoplastic cells intermingled with a lymphocyte population. To date, VE1 and other monoclonal antibodies have been tested primarily in tumor specimens with p.V600E mutation [49]; a study focused specifically on non-p.V600E mutations has yet to be reported.
Pathologists play a critical role in the triage of cancer specimens for molecular testing [50, 51]. A pathologist must make the diagnosis, request the appropriate molecular tests, select the tissue blocks, and designate the region with adequate tumor cellularity. In this article, we stress the importance of estimating the tumor cellularity within the designated region, so there is sufficient material to ensure proper analytic sensitivity of the requested assay. Specimens with poor tumor cellularity may lead to false negative results, particularly if assays with lower analytic sensitivity are implemented. The role of surgical pathology in ensuring accurate and appropriate molecular testing cannot be overstated, particularly in the context of waning reimbursement for this important service.
Our work and several other studies point to multiple reasons for discrepancy between tumor cell percentage and observed % mutant allele by molecular testing. Estimation of tumor cell percentage by the pathologists may not always be precise or accurate. Substantial interobserver variation has been demonstrated [52–54]. The correlation between estimated tumor cellularity and mutant allele percentage has also been shown to be poor [22, 23]. Selection of appropriate tissue blocks and designation of adequate target areas by pathologists may facilitate the estimation of tumor cellularity and improve both precision and accuracy. In general, resection specimens may be better than the biopsy specimens due to the presence of more tumor cells, but they also present a potential difficulty in choosing an optimal block. Resection specimens following neoadjuvant therapy can also present a difficult scenario for testing, because there may be few, if any, viable tumor cells [55, 56]. Fine-needle aspiration specimens, particularly those from lymph nodes with metastasis, are often challenging when the tumor cells are intermingled with abundant small lymphocytes. Pathologists may also want to avoid areas with prominent desmoplastic reaction or inflammatory cell infiltration, which may significantly reduce the tumor cellularity and increase the difficulty for estimation, and areas with abundant mucin, necrosis, or melanin. For specimens with heterogeneous tumor cellularity, designating the whole area containing tumor cells for DNA extraction may also be challenging for the estimation of tumor cell percentage. Pathologists should mark multiple smaller areas of more than 2–3 mm × 2–3 mm and exclude areas with low tumor cellularity. Counting absolute number of neoplastic and non-neoplastic cells in each specimen may not be practical for daily clinical routine. However, it may be worthwhile to do so when the estimated tumor cellularity is close to the analytic sensitivity of the requested assay. Prospective or retrospective quality management processes correlating the estimated tumor cell percentages with observed mutant allele percentages will provide a training process for the pathologist to improve precision and accuracy for estimating tumor cellularity. Computerized image systems may provide an automated, objective determination of tumor cellularity for molecular testing in the near future [57].
To allow potential inter- and intraobserver variability, we arbitrarily designated a conservative ‘within 10 %’ as a set cut-off for congruent vs. discrepant results for comparison between the observed and predicted mutant allele percentage. PCR amplification bias, extremely low input or low quality of DNA, and chromosome anomalies with gain or loss of the mutant allele or the wild-type allele may affect the observed % mutant allele. The real-time PCR-based assays and our original pyrosequencing assay are two examples showing how a design bias of the assay can cause discrepancy between the observed and predicted mutant allele percentage in specimens with a p.V600K mutation. Extremely low input or low quality of DNA for PCR may lead to allele dropout and inconsistent observed mutant allele percentage between different runs. Loss of the wild-type allele (loss of heterozygosity) or amplification of the mutant allele will lead to an observed mutant allele percentage more than that expected. Although an observed mutant allele percentage lower than that expected may be caused by a gain of the wild-type allele, it is most likely due to overestimation of the tumor cellularity.
Another pre-analytical cause for the perceived discrepancy may center on how pathologists determine tumor cellularity as a % of the total specimen. In both training and clinical practice, pathologists use simple linear measurements for microscopic evaluation of tumors. The maximum tumor size, margins and extent of lymph node metastases are all such measures. Not surprisingly, pathologists therefore may estimate tumor cellularity by evaluating the surface area of tumor cells rather the number of tumor nuclei. Overall, this results in an overestimate; a sheet of large cancer cells, for instance, is visually more impressive than a tiny lymphocyte, yet the total DNA in both the tumor cell and lymphocyte is about the same (Fig. 4a). Another reason for discrepancies may be a physical characteristic of histological sections. When one generates routine 5 μm sections, it is likely that the majority of a compact nucleus of a small lymphocyte will be contained in that section, while it is much less likely that a large tumor-cell nucleus will be included (Fig. 4b).
Fig. 4.

Caveats of tumor cellularity estimation. Number of nuclei rather than surface area of nuclei or entire cells should dictate % tumor cellularity estimates (a). This may be difficult for human “eyeball” estimates—here, tumor cellularity is estimated at 50 % according to surface area, but the mutant allele (assuming heterozygosity) is 20 %. Nuclear truncation in tissue section also leads to overestimated tumor contents (b)
5 Conclusion
We used BRAF mutation detection by pyrosequencing to demonstrate that tumor cellularity estimated by pathologists can be a QA measure to improve accurate detection of non-p.V600E BRAF mutations. Using our revised assay, we confirm that the p.V600K mutation and other non-p.V600E mutations are not uncommon. The Cobas 4800 BRAF V600 mutation test and our original pyrosequencing assay may not be able to detect the non-p.V600E mutations, particularly when tumor cellularity in the specimens is low. Adequate designation to enrich tumor cellularity by pathologists, as well as regular prospective and retrospective quality management processes in the molecular diagnostics laboratory, may reduce the risk for a false negative result. Assays with higher analytic sensitivity will help identify mutations in specimens with lower tumor cellularity, and those with higher diagnostic sensitivity will help cover all the reported mutations in exon 11 and exon 15 of BRAF. NGS platforms may be ideally suited for this purpose. Such assays will likely prove worthwhile to identify all patients with p.V600E mutations and non-p.V600E mutations who may benefit from vemurafenib or other targeted therapies.
Key Points.
Pathologists play a critical role in molecular diagnostics, including tissue-based pathological diagnosis, requests for appropriate molecular tests, selection of optimal tissue block for testing, and designation of neoplastic tissues for macro- or micro-dissection, as well as estimates of tumor cellularity.
Pathologist-generated estimates of tumor cellularity can be used as a quality assurance measure in molecular diagnostics.
Molecular assays capable of identifying both p.V600E and non-p.V600E are needed for detection of the BRAF mutation in patients with malignant melanoma.
Acknowledgments
We would like to thank Molly Van Appledorn for statistical analysis consultation, and Norman J. Barker for assistance in generating figures.
Footnotes
Disclosure The authors have no conflicts of interest pertaining to this work.
This work will be presented, in part, at the 103rd Annual Meeting of the United States and Canadian Academy of Pathology (USCAP) in San Diego, CA, USA, in March 2014.
Contributor Information
Jonathan C. Dudley, Department of Pathology, Johns Hopkins University School of Medicine, Johns Hopkins Hospital, Park SB202, 600 North Wolfe St., Baltimore, MD 21287, USA
Grzegorz T. Gurda, Department of Pathology, Johns Hopkins University School of Medicine, Johns Hopkins Hospital, Park SB202, 600 North Wolfe St., Baltimore, MD 21287, USA
Li-Hui Tseng, Department of Pathology, Johns Hopkins University School of Medicine, Johns Hopkins Hospital, Park SB202, 600 North Wolfe St., Baltimore, MD 21287, USA. Department of Medical Genetics, National Taiwan University Hospital, Taipei, Taiwan.
Derek A. Anderson, Department of Pathology, Johns Hopkins University School of Medicine, Johns Hopkins Hospital, Park SB202, 600 North Wolfe St., Baltimore, MD 21287, USA
Guoli Chen, Department of Pathology, Johns Hopkins University School of Medicine, Johns Hopkins Hospital, Park SB202, 600 North Wolfe St., Baltimore, MD 21287, USA.
Janis M. Taube, Department of Pathology, Johns Hopkins University School of Medicine, Johns Hopkins Hospital, Park SB202, 600 North Wolfe St., Baltimore, MD 21287, USA
Christopher D. Gocke, Department of Pathology, Johns Hopkins University School of Medicine, Johns Hopkins Hospital, Park SB202, 600 North Wolfe St., Baltimore, MD 21287, USA. Department of Oncology, Johns Hopkins University School of Medicine, Johns Hopkins Hospital, Baltimore, MD, USA
James R. Eshleman, Department of Pathology, Johns Hopkins University School of Medicine, Johns Hopkins Hospital, Park SB202, 600 North Wolfe St., Baltimore, MD 21287, USA. Department of Oncology, Johns Hopkins University School of Medicine, Johns Hopkins Hospital, Baltimore, MD, USA
Ming-Tseh Lin, Email: mlin36@jhmi.edu, Department of Pathology, Johns Hopkins University School of Medicine, Johns Hopkins Hospital, Park SB202, 600 North Wolfe St., Baltimore, MD 21287, USA.
References
- 1.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 2.Kong Y, Kumar SM, Xu X. Molecular pathogenesis of sporadic melanoma and melanoma-initiating cells. Arch Pathol Lab Med. 2010;134(12):1740–9. doi: 10.1043/2009-0418-RAR.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roring M, Brummer T. Aberrant B-Raf signaling in human cancer: 10 years from bench to bedside. Crit Rev Oncog. 2012;17(1):97–121. doi: 10.1615/critrevoncog.v17.i1.70. [DOI] [PubMed] [Google Scholar]
- 4.Amanuel B, Grieu F, Kular J, Millward M, Iacopetta B. Incidence of BRAF p.Val600Glu and p.Val600Lys mutations in a consecutive series of 183 metastatic melanoma patients from a high incidence region. Pathology. 2012;44(4):357–9. doi: 10.1097/PAT.0b013e3283532565. [DOI] [PubMed] [Google Scholar]
- 5.Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353(20):2135–47. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 6.Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239–46. doi: 10.1200/JCO.2010.32.4327. [DOI] [PubMed] [Google Scholar]
- 7.Ellerhorst JA, Greene VR, Ekmekcioglu S, Warneke CL, Johnson MM, Cooke CP, et al. Clinical correlates of NRAS and BRAF mutations in primary human melanoma. Clin Cancer Res. 2011;17(2):229–35. doi: 10.1158/1078-0432.CCR-10-2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ugurel S, Thirumaran RK, Bloethner S, Gast A, Sucker A, Mueller-Berghaus J, et al. B-RAF and N-RAS mutations are preserved during short time in vitro propagation and differentially impact prognosis. PLoS One. 2007;2(2):e236. doi: 10.1371/journal.pone.0000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greaves WO, Verma S, Patel KP, Davies MA, Barkoh BA, Galbincea JM, et al. Frequency and spectrum of BRAF mutations in a retrospective, single-institution study of 1112 cases of melanoma. J Mol Diagn. 2013;15(2):220–6. doi: 10.1016/j.jmoldx.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menzies AM, Haydu LE, Visintin L, Carlino MS, Howle JR, Thompson JF, et al. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin Cancer Res. 2012;18(12):3242–9. doi: 10.1158/1078-0432.CCR-12-0052. [DOI] [PubMed] [Google Scholar]
- 11.Rubinstein JC, Sznol M, Pavlick AC, Ariyan S, Cheng E, Bacchiocchi A, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med. 2010;8:67. doi: 10.1186/1479-5876-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596–9. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein O, Clements A, Menzies AM, O’Toole S, Kefford RF, Long GV. BRAF inhibitor activity in V600R metastatic melanoma. Eur J Cancer. 2013;49(5):1073–9. doi: 10.1016/j.ejca.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 15.McArthur G, Hauschild A, Robert C, Larkin J, Haanen JB, Ribas A, et al. Efficacy of vemurafenib in BRAFV600K mutation-positive melanoma disease: results from phase 3 clinical study BRIM3. Pigment Cell Melanoma Res. 2012;25(6):871. [Google Scholar]
- 16.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–14. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ascierto PA, Minor D, Ribas A, Lebbe C, O’Hagan A, Arya N, et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol. 2013;31(26):3205–11. doi: 10.1200/JCO.2013.49.8691. [DOI] [PubMed] [Google Scholar]
- 18.Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):782–9. doi: 10.1016/S1470-2045(12)70269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379(9829):1893–901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–14. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 21.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 22.Anderson S, Bloom KJ, Vallera DU, Rueschoff J, Meldrum C, Schilling R, et al. Multisite analytic performance studies of a real-time polymerase chain reaction assay for the detection of BRAF V600E mutations in formalin-fixed, paraffin-embedded tissue specimens of malignant melanoma. Arch Pathol Lab Med. 2012;136(11):1385–91. doi: 10.5858/arpa.2011-0505-OA. [DOI] [PubMed] [Google Scholar]
- 23.Halait H, Demartin K, Shah S, Soviero S, Langland R, Cheng S, et al. Analytical performance of a real-time PCR-based assay for V600 mutations in the BRAF gene, used as the companion diagnostic test for the novel BRAF inhibitor vemurafenib in metastatic melanoma. Diagn Mol Pathol. 2012;21(1):1–8. doi: 10.1097/PDM.0b013e31823b216f. [DOI] [PubMed] [Google Scholar]
- 24.Qu K, Pan Q, Zhang X, Rodriguez L, Zhang K, Li H, et al. Detection of BRAF V600 mutations in metastatic melanoma: comparison of the Cobas 4800 and Sanger sequencing assays. J Mol Diagn. 2013;15(6):790–5. doi: 10.1016/j.jmoldx.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Fadhil W, Ibrahem S, Seth R, Ilyas M. Quick-multiplex-consensus (QMC)-PCR followed by high-resolution melting: a simple and robust method for mutation detection in formalin-fixed paraffin-embedded tissue. J Clin Pathol. 2010;63(2):134–40. doi: 10.1136/jcp.2009.070508. [DOI] [PubMed] [Google Scholar]
- 26.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kotoula V, Charalambous E, Biesmans B, Malousi A, Vrettou E, Fountzilas G, et al. Targeted KRAS mutation assessment on patient tumor histologic material in real time diagnostics. PLoS One. 2009;4(11):e7746. doi: 10.1371/journal.pone.0007746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan YH, Liu Y, Eu KW, Ang PW, Li WQ, Salto-Tellez M, et al. Detection of BRAF V600E mutation by pyrosequencing. Pathology. 2008;40(3):295–8. doi: 10.1080/00313020801911512. [DOI] [PubMed] [Google Scholar]
- 29.Lin MT, Tseng LH, Rich RG, Hafez MJ, Harada S, Murphy KM, et al. Delta-PCR, a simple method to detect translocations and insertion/deletion mutations. J Mol Diagn. 2011;13(1):85–92. doi: 10.1016/j.jmoldx.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen G, Olson MT, O’Neill A, Norris A, Beierl K, Harada S, et al. A virtual pyrogram generator to resolve complex pyrosequencing results. J Mol Diagn. 2012;14(2):149–59. doi: 10.1016/j.jmoldx.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsiatis AC, Norris-Kirby A, Rich RG, Hafez MJ, Gocke CD, Eshleman JR, et al. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. J Mol Diagn. 2010;12(4):425–32. doi: 10.2353/jmoldx.2010.090188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olson MT, Harrington C, Beierl K, Chen G, Thiess M, O’Neill A, Taube J, Zeiger MA, Lin MT, Eshleman JR. BRAF pyrosequencing analysis aided by a lookup table. Am J Clin Pathol. 2014 doi: 10.1309/AJCPVWH1K2ZIHHTV. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin MT, Tseng LH, Beierl K, Hsieh A, Thiess M, Chase N, et al. Tandem duplication PCR: an ultrasensitive assay for the detection of internal tandem duplications of the FLT3 gene. Diagn Mol Pathol. 2013;22(3):149–55. doi: 10.1097/PDM.0b013e31828308a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harada S, Henderson LB, Eshleman JR, Gocke CD, Burger P, Griffin CA, et al. Genomic changes in gliomas detected using single nucleotide polymorphism array in formalin-fixed, paraffin-embedded tissue: superior results compared with microsatellite analysis. J Mol Diagn. 2011;13(5):541–8. doi: 10.1016/j.jmoldx.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colomba E, Helias-Rodzewicz Z, Von Deimling A, Marin C, Terrones N, Pechaud D, et al. Detection of BRAF p. V600E mutations in melanomas: comparison of four methods argues for sequential use of immunohistochemistry and pyrosequencing. J Mol Diagn. 2013;15(1):94–100. doi: 10.1016/j.jmoldx.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 36.Ihle MA, Fassunke J, Konig K, Grunewald I, Schlaak M, Kreuzberg N, et al. Comparison of high resolution melting analysis, pyrosequencing, next generation sequencing and immunohistochemistry to conventional Sanger sequencing for the detection of p. V600E and non-p.V600E BRAF mutations. BMC Cancer. 2014;14(1):13. doi: 10.1186/1471-2407-14-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mullins FM, Dietz L, Lay M, Zehnder JL, Ford J, Chun N, et al. Identification of an intronic single nucleotide polymorphism leading to allele dropout during validation of a CDH1 sequencing assay: implications for designing polymerase chain reaction-based assays. Genet Med. 2007;9(11):752–60. doi: 10.1097/gim.0b013e318159a369. [DOI] [PubMed] [Google Scholar]
- 38.Schwartz KM, Pike-Buchanan LL, Muralidharan K, Redman JB, Wilson JA, Jarvis M, et al. Identification of cystic fibrosis variants by polymerase chain reaction/oligonucleotide ligation assay. J Mol Diagn. 2009;11(3):211–5. doi: 10.2353/jmoldx.2009.080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ward KJ, Ellard S, Yajnik CS, Frayling TM, Hattersley AT, Venigalla PN, et al. Allelic drop-out may occur with a primer binding site polymorphism for the commonly used RFLP assay for the -1131T>C polymorphism of the Apolipoprotein AV gene. Lipids Health Dis. 2006;5:11. doi: 10.1186/1476-511X-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lade-Keller J, Romer KM, Guldberg P, Riber-Hansen R, Hansen LL, Steiniche T, et al. Evaluation of BRAF mutation testing methodologies in formalin-fixed, paraffin-embedded cutaneous melanomas. J Mol Diagn. 2013;15(1):70–80. doi: 10.1016/j.jmoldx.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 41.Magnin S, Viel E, Baraquin A, Valmary-Degano S, Kantelip B, Pretet JL, et al. A multiplex SNaPshot assay as a rapid method for detecting KRAS and BRAF mutations in advanced colorectal cancers. J Mol Diagn. 2011;13(5):485–92. doi: 10.1016/j.jmoldx.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCourt CM, McArt DG, Mills K, Catherwood MA, Maxwell P, Waugh DJ, et al. Validation of next generation sequencing technologies in comparison to current diagnostic gold standards for BRAF, EGFR and KRAS mutational analysis. PLoS One. 2013;8(7):e69604. doi: 10.1371/journal.pone.0069604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rechsteiner M, von Teichman A, Ruschoff JH, Fankhauser N, Pestalozzi B, Schraml P, et al. KRAS, BRAF, and TP53 deep sequencing for colorectal carcinoma patient diagnostics. J Mol Diagn. 2013;15(3):299–311. doi: 10.1016/j.jmoldx.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 44.Szankasi P, Reading NS, Vaughn CP, Prchal JT, Bahler DW, Kelley TW. A quantitative allele-specific PCR test for the BRAF V600E mutation using a single heterozygous control plasmid for quantitation: a model for qPCR testing without standard curves. J Mol Diagn. 2013;15(2):248–54. doi: 10.1016/j.jmoldx.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tuononen K, Maki-Nevala S, Sarhadi VK, Wirtanen A, Ronty M, Salmenkivi K, et al. Comparison of targeted next-generation sequencing (NGS) and real-time PCR in the detection of EGFR, KRAS, and BRAF mutations on formalin-fixed, paraffin-embedded tumor material of non-small cell lung carcinoma-superiority of NGS. Genes Chromosomes Cancer. 2013;52(5):503–11. doi: 10.1002/gcc.22047. [DOI] [PubMed] [Google Scholar]
- 46.Busam KJ, Hedvat C, Pulitzer M, von Deimling A, Jungbluth AA. Immunohistochemical analysis of BRAF(V600E) expression of primary and metastatic melanoma and comparison with mutation status and melanocyte differentiation antigens of metastatic lesions. Am J Surg Pathol. 2013;37(3):413–20. doi: 10.1097/PAS.0b013e318271249e. [DOI] [PubMed] [Google Scholar]
- 47.Long GV, Wilmott JS, Capper D, Preusser M, Zhang YE, Thompson JF, et al. Immunohistochemistry is highly sensitive and specific for the detection of V600E BRAF mutation in melanoma. Am J Surg Pathol. 2013;37(1):61–5. doi: 10.1097/PAS.0b013e31826485c0. [DOI] [PubMed] [Google Scholar]
- 48.Routhier CA, Mochel MC, Lynch K, Dias-Santagata D, Louis DN, Hoang MP. Comparison of 2 monoclonal antibodies for immunohistochemical detection of BRAF V600E mutation in malignant melanoma, pulmonary carcinoma, gastrointestinal carcinoma, thyroid carcinoma, and gliomas. Hum Pathol. 2013;44(11):2563–70. doi: 10.1016/j.humpath.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 49.Marin C, Beauchet A, Capper D, Zimmermann U, Julie C, Ilie M, et al. Detection of BRAF p. V600E mutations in melanoma by immunohistochemistry has a good interobserver reproducibility. Arch Pathol Lab Med. 2014;138(1):71–5. doi: 10.5858/arpa.2013-0031-OA. [DOI] [PubMed] [Google Scholar]
- 50.Hoorens A, Jouret-Mourin A, Sempoux C, Demetter P, De Hertogh G, Teugels E. Accurate KRAS mutation testing for EGFR-targeted therapy in colorectal cancer: emphasis on the key role and responsibility of pathologists. Acta Gastroenterol Belg. 2010;73(4):497–503. [PubMed] [Google Scholar]
- 51.van Krieken JH, Jung A, Kirchner T, Carneiro F, Seruca R, Bosman FT, et al. KRAS mutation testing for predicting response to anti-EGFR therapy for colorectal carcinoma: proposal for an European quality assurance program. Virchows Arch. 2008;453(5):417–31. doi: 10.1007/s00428-008-0665-y. [DOI] [PubMed] [Google Scholar]
- 52.Bellon E, Ligtenberg MJ, Tejpar S, Cox K, de Hertogh G, de Stricker K, et al. External quality assessment for KRAS testing is needed: setup of a European program and report of the first joined regional quality assessment rounds. Oncologist. 2011;16(4):467–78. doi: 10.1634/theoncologist.2010-0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smits AJ, Kummer JA, de Bruin PC, Bol M, van den Tweel JG, Seldenrijk KA, et al. The estimation of tumor cell percentage for molecular testing by pathologists is not accurate. Mod Pathol. 2014;27(2):168–74. doi: 10.1038/modpathol.2013.134. [DOI] [PubMed] [Google Scholar]
- 54.Viray H, Li K, Long TA, Vasalos P, Bridge JA, Jennings LJ, et al. A prospective, multi-institutional diagnostic trial to determine pathologist accuracy in estimation of percentage of malignant cells. Arch Pathol Lab Med. 2013;137(11):1545–9. doi: 10.5858/arpa.2012-0561-CP. [DOI] [PubMed] [Google Scholar]
- 55.Boissiere-Michot F, Lopez-Crapez E, Frugier H, Berthe ML, Ho-Pun-Cheung A, Assenat E, et al. KRAS genotyping in rectal adenocarcinoma specimens with low tumor cellularity after neoadjuvant treatment. Mod Pathol. 2012;25(5):731–9. doi: 10.1038/modpathol.2011.210. [DOI] [PubMed] [Google Scholar]
- 56.Ondrejka SL, Schaeffer DF, Jakubowski MA, Owen DA, Bronner MP. Does neoadjuvant therapy alter KRAS and/or MSI results in rectal adenocarcinoma testing? Am J Surg Pathol. 2011;35(9):1327–30. doi: 10.1097/PAS.0b013e3182253800. [DOI] [PubMed] [Google Scholar]
- 57.Viray H, Coulter M, Li K, Lane K, Madan A, Mitchell K, et al. Automated objective determination of percentage of malignant nuclei for mutation testing. Appl Immunohistochem Mol Morphol. doi: 10.1097/PAI.0b013e318299a1f6. Epub 24 Oct 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]