

Aberrant glutathione homeostasis is implicated in the pathology of many neurodegenerative diseases. Hong et al. reveal the molecular mechanism by which oxidative stress induces S-glutathionylation of the TRPC5 channel, and show that abnormal TRPC5 activation leads to death of striatal neurons in Huntington’s disease.
Keywords: Ca2+, cysteine, GSSG, neurodegeneration, TRPC

Aberrant glutathione homeostasis is implicated in the pathology of many neurodegenerative diseases. Hong et al. reveal the molecular mechanism by which oxidative stress induces S-glutathionylation of the TRPC5 channel, and show that abnormal TRPC5 activation leads to death of striatal neurons in Huntington’s disease.
Abstract
Aberrant glutathione or Ca2+ homeostasis due to oxidative stress is associated with the pathogenesis of neurodegenerative disorders. The Ca2+-permeable transient receptor potential cation (TRPC) channel is predominantly expressed in the brain, which is sensitive to oxidative stress. However, the role of the TRPC channel in neurodegeneration is not known. Here, we report a mechanism of TRPC5 activation by oxidants and the effect of glutathionylated TRPC5 on striatal neurons in Huntington’s disease. Intracellular oxidized glutathione leads to TRPC5 activation via TRPC5 S-glutathionylation at Cys176/Cys178 residues. The oxidized glutathione-activated TRPC5-like current results in a sustained increase in cytosolic Ca2+, activated calmodulin-dependent protein kinase and the calpain-caspase pathway, ultimately inducing striatal neuronal cell death. We observed an abnormal glutathione pool indicative of an oxidized state in the striatum of Huntington’s disease transgenic (YAC128) mice. Increased levels of endogenous TRPC5 S-glutathionylation were observed in the striatum in both transgenic mice and patients with Huntington’s disease. Both knockdown and inhibition of TRPC5 significantly attenuated oxidation-induced striatal neuronal cell death. Moreover, a TRPC5 blocker improved rearing behaviour in Huntington’s disease transgenic mice and motor behavioural symptoms in littermate control mice by increasing striatal neuron survival. Notably, low levels of TRPC1 increased the formation of TRPC5 homotetramer, a highly Ca2+-permeable channel, and stimulated Ca2+-dependent apoptosis in Huntington’s disease cells (STHdhQ111/111). Taken together, these novel findings indicate that increased TRPC5 S-glutathionylation by oxidative stress and decreased TRPC1 expression contribute to neuronal damage in the striatum and may underlie neurodegeneration in Huntington’s disease.
Introduction
Age-related neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease and Huntington’s disease, are characterized by progressive loss of neurons due to neuronal cell death associated with oxidative stress (Bredesen et al., 2006). Although oxidation reactions are critical for life, they can also be highly toxic, and thus cells rapidly detoxify oxidants using multiple enzymatic and non-enzymatic antioxidative systems.
Among the various antioxidants, reduced glutathione (GSH) is a ubiquitous, non-protein, thiol-containing molecule found at millimolar concentrations in eukaryotic cells. GSH is a tripeptide composed of cysteine, glutamate, and glycine; the cysteine (-SH) residue provides the reducing power. During oxidative stress, GSH is converted to oxidized glutathione (GSSG), which is restored to GSH by GSSG reductase. The ratio of GSH/GSSG determines the cellular redox potential and, consequently, redox homeostasis (Meister and Anderson, 1983). Oxidative stress leads to a decreased GSH/GSSG ratio and a reduction of redox-sensitive cysteine residues within proteins (Hurd et al., 2005; Cooper et al., 2011). Reactive cysteines can be modified at the sulphydryl group, resulting in S-glutathionylation, which is involved in gene expression, cell death or survival (Dalle-Donne et al., 2011). Signalling proteins and ion channels mediate cellular sensing of and response to oxidative stress (Kashio et al., 2012). Whether this sensing and response mediate S-glutathionylation in TRPC (canonical or classical transient receptor potential) channels is not known but may be of particular significance because of the Ca2+ permeability of these channels.
The brain is particularly vulnerable to oxidative stress due to high energy demands, the paucity of antioxidants, and high lipid content (Floyd, 1999). The neurodegenerative disorder Huntington’s disease is best known for its effect on motor control. Mood disturbances (Leroi and Michalon, 1998) such as depression, anxiety, and irritability also have a high prevalence in patients with Huntington’s disease and often begin before the onset of motor symptoms. The disease is caused by an abnormal expansion of glutamine in the N-terminal region of the 350-kDa huntingtin protein (encoded by HTT). Mutant HTT in striatal neurons can cause mitochondrial defects including the reduction of Ca2+ buffering capacity, loss of membrane potential, or decreased expression of oxidative phosphorylation enzymes. In particular, mitochondrial dysfunction facilitates impaired Ca2+ homeostasis linked to the glutamate receptor-mediated excitotoxicity (Damiano et al., 2010). However, the source or mediator of elevated Ca2+ remains unclear.
Transient receptor potential channel, subfamily C, member 5 (TRPC5) is predominantly expressed in the brain where it can form heterotetrameric complexes with TRPC1 and TRPC4 channel subunits. TRPC5 channels exhibit constitutive activity and voltage dependence and are also stimulated by oxidation and a range of factors, including G protein-coupled receptors, lysophospholipids and acidification. TRPC5 is stimulated by nitric oxide (NO) through a mechanism that requires oxidation (S-nitrosylation) at extracellular cysteines (Yoshida et al., 2006). However, we have demonstrated that intracellular oxidation rather than extracellular reaction regulates TRPC5 activation depending on the cytosolic redox state (extracellular GSH versus GSSG). TRPC5 knockout mice exhibit diminished innate fear levels in response to innately aversive stimuli. Moreover, mutant mice exhibit significant reductions in responses mediated by synaptic activation of G protein coupled receptors in neurons of the amygdala (Riccio et al., 2009).
In this study, we report that TRPC5 channels are novel components of the pathological mechanism that controls neuronal damage in Huntington’s disease. The activity of recombinant and endogenous TRPC5 is regulated by the cellular redox state. Extensive analysis revealed that TRPC5 (Cys176 and Cys178) is specifically glutathionylated in response to GSSG, resulting in channel activation and sustained Ca2+ influx. TRPC5 S-glutathionylation is constitutive, occurs at higher levels in striatal cells of a Huntington’s disease mouse model, and leads toneurodegeneration. These findings reveal the mechanism of cellular redox sensing through TRPC5 and its potential roles in Huntington’s disease and neurodegeneration.
Materials and methods
Cell culture, transient transfection, plasmids and chemicals
Human embryonic kidney (HEK)-293 cells were purchased from American Type Culture Collection (ATCC). Clonal striatal cells from wild-type (STHdhQ7/7) or mutant Htt knock-in (STHdhQ111/111) mice were obtained from Dr H. Ryu (Jeon et al., 2012a). The cells were maintained according to the supplier’s recommendations. Plasmids containing human TRPC5 or mouse TRPC5 were kindly donated by Dr S. Kaneko and Dr Y. Mori (Yoshida et al., 2006), respectively. Multiple cysteine mutants were changed using QuikChange® site-directed mutagenesis (Agilent Technologies). For transient transfection, we used FuGENE® 6 (Roche Molecular Biochemicals) or Lipofectamine® 2000 (Invitrogen) according to the manufacturer’s protocol. All experiments were performed 20–30 h after transfection.
All chemicals were purchased from Sigma Aldrich except tris(2-carboxyethyl)phosphine (TCEP), which was purchased from Thermo Scientific.
Electrophysiology
For current recording of TRPC5, transfected cells in 12-well plates were trypsinized and transferred into a recording chamber equipped to treat the cells with various solutions. Whole-cell currents were recorded using an Axopatch 200B amplifier (Axon Instruments). Currents were filtered at 5 kHz (−3 dB, 4-pole Bessel), digitized using a Digidata 1440A Interface (Axon Instruments), and analysed using a personal computer equipped with pClamp 10.2 software (Axon Instruments) and Origin software (Microcal origin v.8.0). The solutions for current recording were exactly as detailed in Hong et al. (2014) and the Supplementary material. All current traces were drawn from the selected values at −60 or +80 mV of the ramp pulses. For all bar graphs, inward current amplitudes at −60 mV are summarized.
Ca2+ measurement by Cameleon YC6.1 FRET and Fura-2
Foerster (fluorescence) resonance energy transfer (FRET) images of CFP, YFP, and raw FRET were obtained with a pE-1 Main Unit equipped with three FRET cubes and recorded at a resolution of 1.5 s/FRET image. The FRET images were used to calculate the FRET efficiency according to (Erickson, 2001). Further details of FRET and Fura-2 recording to determine [Ca2+]i are provided in the Supplementary material.
Western blot analysis
Cells were plated in 6-well dishes. Lysates were prepared in lysis buffer (0.5% Triton™ X-100, 50 mM Tris-Cl, 150 mM NaCl, 1 mM EDTA, pH 7.5) by passage 7–10 times through a 26-gauge needle after sonication. Lysates were centrifuged at 13 300g for 10 min at 4°C, and the protein concentration in the supernatants was determined. The proteins extracted in sample buffer were loaded onto 6 or 8% Tris-glycine sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) gels unless otherwise stated under non-reducing conditions; the lanes were marked with dotted lines. The proteins were transferred onto polyvinyl difluoride membranes, and the membranes were probed with TRPC5 and TRPC1 (NeuroMab and Alomone labs), caspase-3 and cleaved caspase-3 (Cell Signaling), and calpain1 (Santa Cruz) antibodies. GFP antibody (Invitrogen) was used to detect GFP-tagged TRPC5 protein as indicated. Co-immunoprecipitation and surface biotinylation were performed according to standard methods and are described in the Supplemental material.
MTT cell death assay
Q7 or Q111 cells were grown in 12-well plates. The MTT assay was used to assess cell viability according to the manufacturer's instructions (Sigma-Aldrich). The assay was quantified by measuring the absorbance at 570 nm using a micro plate reader (Biochrom).
Flow cytometry analysis
After treatment, the cells were harvested and incubated in fluorescence-activated cell sorter (FACS) buffer (phosphate-buffered saline with 0.1% bovine calf serum, 0.05% sodium azide) at 4°C for 30 min. After washing with FACS buffer, the cells were analysed using a FACSCalibur flow cytometer equipped with Cell Quest software (BD Pharmingen).
RT-PCR, siRNA silencing and cellular glutathione assay
Reverse transcriptase-PCR, siTNA silencing, and cellular glutathione assays were performed according to standard methods and are described in the Supplementary material.
Biochemical or behavioural experiments using the Huntington’s disease transgenic YAC128 mouse model
YAC128 mutant Huntington’s disease transgenic mice were purchased from Jackson Laboratory [FVB-Tg(YAC128)53Hay/J]. The animal experiments were conducted using standard methods. GSH/GSSG assay, immunohistochemistry, and behaviour assessments (rotarod test, open-field test, etc.) are described in the Supplementary material.
Statistics
Results are presented as the mean ± standard error of the mean (SEM). The results were compared using Student’s t-test for two groups or by ANOVA followed by post hoc test for three groups or more. P < 0.05 was considered statistically significant. The number of cell electrical recordings is given by n in bar graphs.
Results
TRPC5 activation by intracellular GSSG is reversed by GSH and dithiothreitol
Two contradictory studies have reported oxidation (Yoshida et al., 2006) or reduction (Xu et al., 2008) of TRPC5 homomer (also as a heteromer with TRPC1) currents. We first sought to determine whether oxidation/reduction of TRPC5 regulates TRPC5 activity and through what mechanism. Supplementary Fig. 1A shows that the membrane-permeable oxidant pyridyl disulphide (PDS) 2,2`-dithiobis 5-nitropyridine (DTNP) activated the TRPC5 current (DTNP, 120 ± 26 pA/pF) in HEK293 cells expressing TRPC5. Other membrane-permeable reactive pyridyl disulphides also activated TRPC5 such as 2,2`-dithiodipyridine (2-PDS, 122 ± 58 pA/pF, Supplementary Fig. 1B). Significantly, the membrane-impermeable PDS 5,5`-dithiobis(2-nitrobenzoic acid) (DTNB) had no effect when applied externally (Supplementary Fig. 1D) but markedly activated TRPC5 when applied intracellularly (Supplementary Fig. 1C).
Among cellular antioxidants, GSH plays an essential role in protecting against reactive oxygen species. Thus, the presence of GSH is important for preventing oxidative damage. Next, we investigated whether the TRPC5 channel is activated and induces Ca2+ influx according to changes in glutathione redox states (Dalle-Donne et al., 2011). When infused with a 3 mM GSH pipette solution, TRPC5 activation by DTNP was markedly attenuated from 108 ± 19 pA/pF to 16 ± 9 pA/pF (Fig. 1A). Elevated GSSG can modulate cellular oxidation by forming disulphide bonds with protein thiols (PS-SG) (Morgan et al., 2012). GSSG equally activated TRPC5 in a dose-dependent manner but only when applied intracellularly (Fig. 1B). The current increased shortly after patch rupture; infusion of 5 or 8 mM GSSG activated TRPC5 current by 74 ± 13 or 132 ± 19 pA/pF, respectively.
Figure 1.
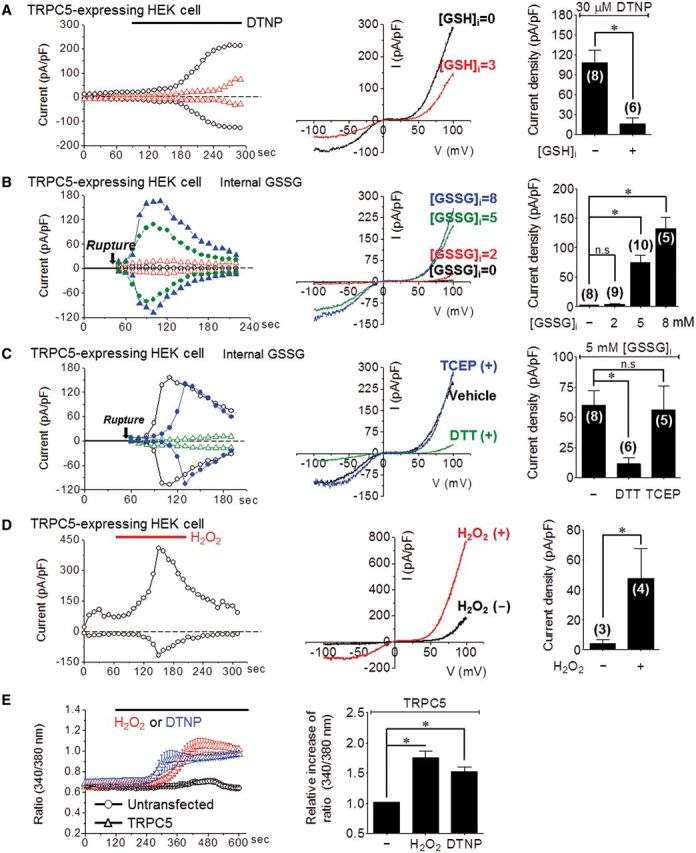
TRPC5 activation by intracellular GSSG is reversed by GSH and dithiothreitol. In all panels, compounds were applied at the time indicated by the bars. The typical current (I)–voltage (V) relationship is shown before and after maximal current activation. The bar graphs list the mean ± SEM of current density (pA/pF) at −60 mV of the indicated number of experiments. *P < 0.05 and n.s. = not significant. (A) Internal 3 mM GSH-attenuated TRPC5 current activated by the strong oxidizing agent DTNP (30 µM). (B) TRPC5 activation by intracellular GSSG in a dose (mM)-dependent manner. (C) Inhibition of 5 mM GSSG-activated TRPC5 current by the reducing agent dithiothreitol (DTT, 10 mM) but not by the cell-impermeable agent TCEP (1 mM). (D) TRPC5 activation by 1 mM H2O2. (E) Cytoplasmic Ca2+ increase by 1 mM H2O2 and 30 µM DTNP.
The activating effect of intracellular GSSG was unexpected considering previous reports that oxidation acts on extracellular cysteine to activate (Yoshida et al., 2006) or inhibit (Xu et al., 2008) the TRPC5 channel. Therefore, we further examined the sidedness of the effect of the reductants. The cell-permeable molecule dithiothreitol prevented TRPC5 activation by GSSG, while the cell-impermeable strong reductant TCEP had no effect (Fig. 1C). Similarly, dithiothreitol but not TCEP reversed the activation of TRPC5 by DTNP (Supplementary Fig. 1E and F). We further assessed the specificity of the oxidants and reductants by evaluating their effects on the redox-sensitive TRPM2 channel, which is activated by hydrogen peroxide (H2O2) (Kaneko, 2006). TRPM2 was activated by its ligand, cyclic adenosine diphosphate ribose (cADPR), but was completely resistant to GSSG and DTNP, which glutathionylates proteins (Supplementary Fig. 2A). Notably, TRPM2 oxidation occurs via oxidation of Met214 and not a cysteine residue.
These results suggest that intracellular GSSG is a novel activator of TRPC5. Such activation is likely to be highly toxic if it results in a sustained elevation of Ca2+. To test the effect of a cytoplasmic Ca2+ by oxidant, we investigated whether H2O2 and DTNP activate TRPC5 and cause an increase in Ca2+. H2O2 increased the TRPC5 current to 47 ± 19 pA/pF (Fig. 1D). As shown in Fig. 1E, 1 mM H2O2 or 30 µM DTNP induced a sustained increase in cytoplasmic Ca2+ in cells expressing TRPC5 that was highly toxic.
TRPC5 S-glutathionylation by GSSG at Cys176 and Cys178
GSSG can post-translationally modify proteins by S-glutathionylation in the presence of an appropriate GSH/GSSG ratio (Meister and Anderson, 1983). Among the TRPCs, TRPC1, C4, and C5 have high sequence homology and highly conserved cysteines (Supplementary Table 1). The locations of the conserved cysteines in TRPC5 are indicated in Fig. 2A. We used systematic deletions and mutations to determine whether GSSG covalently modifies the sulphydryl (-SH) groups of the cysteines in TRPC5. Mutation of all cytoplasmic and all N-terminal cysteines eliminated current activation by GSSG (Fig. 2B, 1.2 ± 0.3 pA/pF and 0.7 ± 0.2 pA/pF, respectively). However, mutation of all C-terminus cysteines had no effect on TRPC5 activation by GSSG (Fig. 2B, 109 ± 26 pA/pF). Similarly, deletion of the portion of the TRPC5 C-terminus that includes eight cysteines at positions 804, 809, 877, 895, 920, 924, 930, and 935 did not prevent activation by GSSG. By contrast, single mutations of Cys176S, Cys178S and Cys181S almost eliminated the activation of TRPC5 by GSSG. The Cys181S mutation inhibited the channel even when activated by GTPγS and thus was not considered further (Supplementary Fig. 3A). Additional analysis revealed that the Cys176S-Cys178S double mutant was also not activated by GSSG (3.8 ± 1.0 pA/pF) (Fig. 2C) or by DTNP (Supplementary Fig. 3B). The double mutation Cys176S-Cys178S also inhibited TRPC5-mediated Ca2+ influx activated by H2O2 (Supplementary Fig. 3E).
Figure 2.
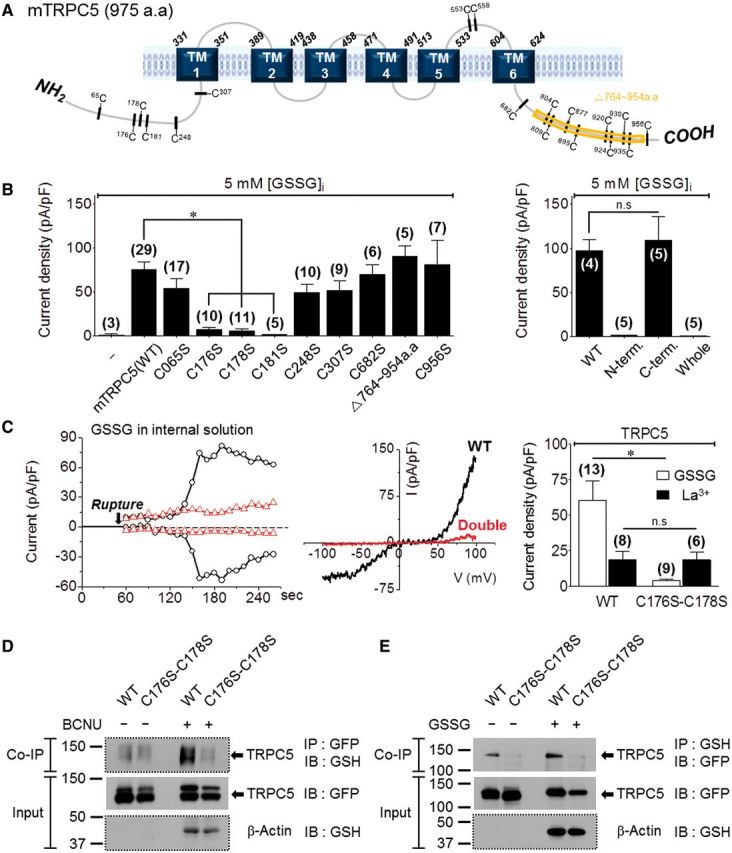
TRPC5 S-glutathionylation by GSSG at Cys176 and Cys178. The number of experiments is provided in parentheses. (A) Location of cysteine residues in mouse TRPC5. (B) Activation of TRPC5 [wild-type (WT) or mutants] by 5 mM GSSG. (C) Effect of the C176S and C178S double mutant on TRPC5 activation by 5 mM GSSG or 200 µM La3+. The figure shows a time course (left), I/V (middle) and the summary (right). (D) Direct S-glutathionylation of TRPC5 C176S-C178S after treatment with 100 µM BCNU (6 h). After immunoprecipitation (IP) with GSH, TRPC5 proteins were detected by anti-GFP without β-mercaptoethanol (reductant) to assay glutathionylated TRPC5. (E) Direct S- glutathionylationof TRPC5 after treatment of cell lysates 2 mM GSSG (1 h). Note that treatment with BCNU resulted in a marked increase in TRPC5 glutathionylation that was eliminated by the double cysteine mutation. (D and E) The blots with dotted lines were performed under non-reducing condition. IB = immunoblot.
To determine whether TRPC5 is directly glutathionylated by GSSG and the effect of the double mutant (Cys176S-Cys178S) on S-glutathionylation, we assayed the interaction of TRPC5 with glutathione by co-immunoprecipitation. Cells were treated with carmustine [1,3-bis(2-chloroethyl)-N-nitrosourea, BCNU], which shifts the intracellular GSH/GSSG redox balance towards oxidized GSSG. Elevation of cellular GSSG induced S-glutathionylation of TRPC5 (wild-type) but not TRPC5 (Cys176S-Cys178S) (Fig. 2D). Importantly, Fig. 2E shows that endogenous glutathione was bound to TRPC5 (wild-type) but not to TRPC5 (Cys176S-Cys178S), and this binding was increased by treating the transfected cell lysates with GSSG.
To determine whether the oxidized cysteines play a role in other modes of TRPC5 activation, we tested the effects of the mutants on TRPC5 activation by lanthanides (Jung et al., 2003) and GTPγS (Kanki et al., 2001). Figure 2C and Supplementary Fig. 3D show that TRPC5 (wild-type) and TRPC5 (Cys176S-Cys178S) were similarly activated by La3+. By contrast, GTPγS fully activated the Cys176S mutant and partially activated the Cys178S and Cys176S-Cys178S mutants, suggesting that S-glutathionylation of Cys178 may contribute to Gα-induced TRPC5 activation by GTPγS (Jeon et al., 2012b). Importantly, the Cys176 and Cys178 mutations had no effect on TRPC5 surface expression (Supplementary Fig. 3C).
These results imply that TRPC5 is directly S-glutathionylated by GSSG to activate the channel and that the gating is mediated by the S-glutathionylation of Cys176 and Cys178, which are located near the ankyrin repeat domain in the N-terminus of TRPC5.
Induction of Ca2+-dependent Huntington’s disease striatal cell toxicity by reduced redox potential and increased GSSG
Reduced antioxidant activity induces the apoptosis of neuronal cells in neurodegenerative diseases (Rao and Balachandran, 2002). TRPC5 and TRPC1 are densely expressed in the hippocampus, prefrontal cortex and lateral septum (Greka et al., 2003; Fowler et al., 2007). Thus, S-glutathionylation of TRPC5 may be involved in the pathogenesis of Huntington’s disease. In neurons, TRPC1 forms TRPC1/C5 heteromultimers in which TRPC1 decreases Ca2+ permeation of TRPC5 (Strubing et al., 2001; Storch et al., 2012). Thus, TRPC1 may prevent excessive Ca2+ influx via S-glutathionylated TRPC5.
To study Huntington’s disease, striatal cell lines obtained from wild-type (STHdhQ7/7, Q7) and mutant HTT knock-in (STHdhQ111/111, Q111) mice are commonly used (Jeon et al., 2012a; Ribeiro et al., 2012, 2013; Lee et al., 2013). In these cell lines, we analysed native TRPC5 or TRPC1 expression by western blot analysis using monoclonal TRPC5 and TRPC1 antibodies (Fig. 3A and B). To verify the specificity of the TRPC5 antibody, we performed a peptide competition assay in which the blot was incubated with TRPC5 peptides, which confirmed that the TRPC5 antibody detects the TRPC5-specific band (Supplementary Fig. 4A). Endogenous TRPC5 mRNA was also detected by reverse transcriptase-PCR (Fig. 3A). Interestingly, western blot and mRNA analysis revealed the expression of endogenous TRPC1 in Q7 cells and, at much lower levels, in Q111 cells (Fig. 3B and C).
Figure 3.
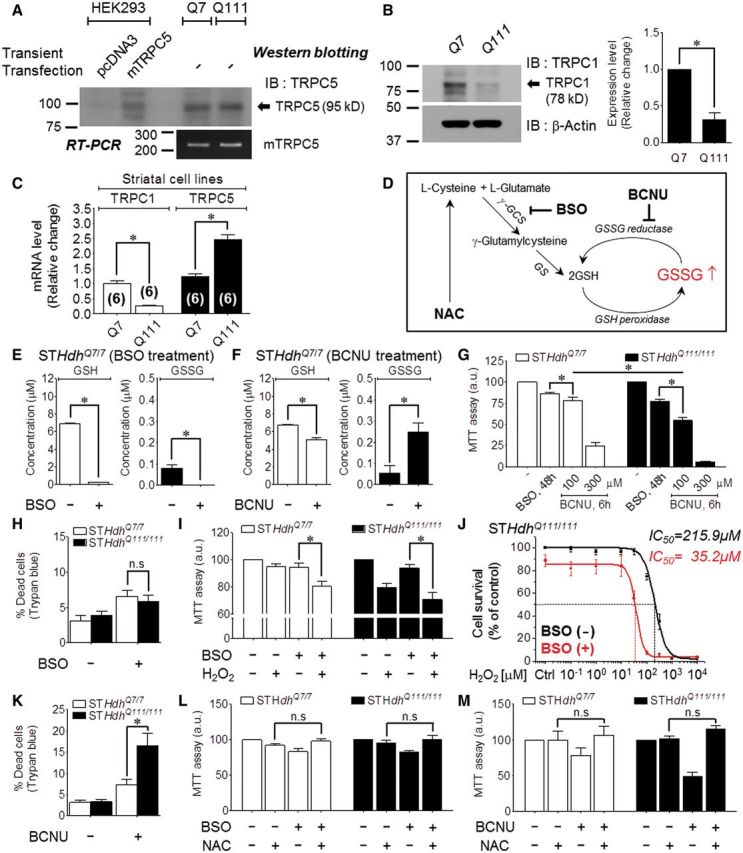
Increasing toxic effects of BCNU and BSO in Q111 Huntington’s disease striatal cells. All experiments were performed with Q7 and Q111 cells. *P < 0.05 and n.s. = not significant. (A) Analysis of the expression pattern of native TRPC5 (95 kD) in Q7 and Q111 cells using a monoclonal TRPC5 antibody. The reverse transcriptase-PCR bands indicated the presence of endogenous TRPC5. (B) Analysis of the expression level of native TRPC1 (78 kD) in Q7 and Q111 cells using a monoclonal TRPC1 antibody (n = 5). (C) mRNA levels of TRPC1 or TRPC5 in Q7 and Q111 cells (Q7; n = 6, Q111; n = 6). (D) Schematic diagram of the glutathione pathway and the sites of drug action (BCNU, BSO, NAC). (E) Depletion of GSH/GSSG levels after treatment with 100 µM BSO (24 h). (F) Elevation of the GSSG concentration after treatment with 100 µM BCNU (6 h). (G) MTT analysis of cell viability after treatment with 100 µM BSO (48 h) or with 100 µM or 300 µM BCNU (6 h) (BSO; n = 22, 100 µM BCNU; n = 9, 300 µM BCNU; n = 9). (H) Percentage dead cells as determined by Trypan blue staining after treatment with 100 µM BSO (24 h) (n = 12). (I) MTT analysis of cell viability after pretreatment with 20 µM H2O2 (24 h) after BSO application (n = 8). (J) Dose-dependent changes in Q111 cell viability induced by treatment with H2O2 (24 h) before and after BSO (24 h) application (n = 9). (K) Percentage dead cells as determined by Trypan blue staining after treatment with 100 µM BCNU (6 h) (n = 12). (L and M) Pretreatment with 5 mM NAC (24 h) prior to treatment with 100 µM BSO (24 h) (L, Q7, n = 6; Q111, n = 9) or 100 µM BCNU (6 h) (M, n = 10). NAC = N-acetyl-L-cysteine; IB = immunoblot; IP = immunoprecipitation.
To investigate glutathione-dependent cell viability in Huntington’s disease, we used several protocols to manipulate cellular GSSG, as illustrated in Fig. 3D. We used the membrane-permeable γ-GCS inhibitor L-buthionine (S,R) sulphoximine (BSO) to deplete glutathione and the GSSG reductase inhibitor BCNU to elevate cellular GSSG (Harkan et al., 1984; Park et al., 2009) in Q7 and Q111 cells. As expected, BSO completely depleted total glutathione (GSH and GSSG) (Fig. 3E). BCNU significantly decreased GSH and increased GSSG levels by inhibiting the reversion of GSSG to GSH (Fig. 3F). By applying the two drugs (BSO and BCNU) to wild-type (Q7) and mutant (Q111) Huntington’s disease cell lines, we compared the effects of reduced redox potential or increased GSSG on cell viability. A 6-h exposure to 100 µM BCNU or 48-h exposure to 100 µM BSO decreased the viability of the Q7 and Q111 cells, as determined by the MTT assay, although the decrease was greater for Q111 cells (Fig. 3G). Similar results were obtained for BCNU when cell viability was assayed with Trypan blue staining (Fig. 3K), whereas no difference was observed in the viability of Q7 and Q111 cells treated with BSO (Fig. 3H). We next examined whether reducing the redox potential via GSH depletion increases the vulnerability of cells to oxidative stress by H2O2 under conditions of GSH depletion by BSO. Both Q7 and Q111 cells treated with 20 µM H2O2 for 24 h after preincubation with 100 µM BSO for 24 h to deplete intracellular glutathione exhibited reduced viability, and this reduction was greater in Q111 cells (Fig. 3I). To calculate the inhibitory concentration 50% (IC50), we pretreated cells with a series of concentration of H2O2. In Q111 cells, the IC50 value for H2O2 decreased from 216 µM in the absence of BSO to 35 µM in the presence of BSO (Fig. 3J). Significantly, the effect of BCNU and BSO was prevented by the glutathione precursor N-acetyl-L-cysteine (NAC), which increases intracellular GSH levels (Fig. 3L and M). Moreover, treatment with BCNU increased Q111 apoptotic cell death as determined by flow cytometry of cells stained with propidium iodide and annexin V. Figure 4A and B show that BCNU had no effect on Q7 cells but increased the apoptosis of Q111 cells from 5.9 ± 1.2% to 12.7 ± 1.5%.
Figure 4.
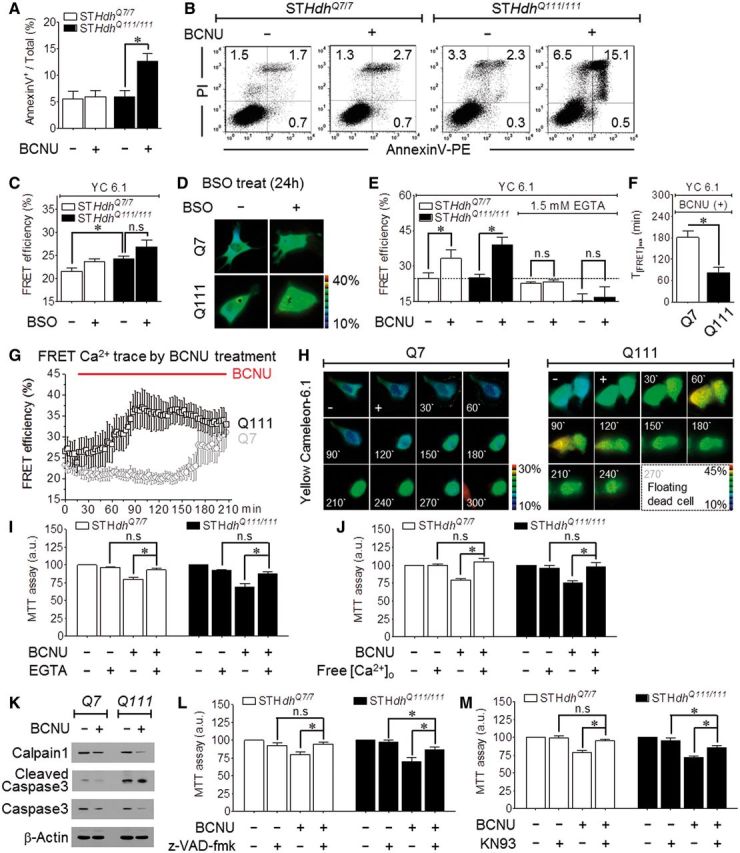
Enhancement of apoptotic Huntington’s disease cell death by GSSG is relevant to Ca2+-induced pathway. (A) Analysis of annexin V-stained Q7 and Q111 apoptotic cell after treatment with 100 µM BCNU (6 h) (n = 4). (B) Representative FACS analysis scatter grams of annexin V and propidium iodide -stained Q111 cells treated with BCNU. (C–H) Measurement of Ca2+in Q7 and Q111 cells by FRET-based YC6.1 after treatment with BSO or BCNU. (C) Maximum peak Ca2 + values after treatment with 100 µM BSO (24 h) (n = 19). (D) Fluorescence images of YC6.1-transfected cells as indicated above. (E) Maximum peak Ca2+ values after treatment with 100 µM BCNU (n = 6). (F) Time required for maximum Ca2+ increase (n = 6). (G) Time course change of FRET Ca2+ trace at 3-min intervals (n = 6). (H) Fluorescence images of YC6.1-transfected cells after treatment with BCNU for the indicated times. Floating Q111 dead cells detached from the bottom of the imaging dish after 270 min. (I and J) MTT analysis of the viability of cells pretreated with 1.5 mM EGTA (12 h) (I: Q7, n = 8; Q111, n = 10) or free Ca2 + medium (12 h) (J: Q7, n = 3; Q111, n = 3) prior to treatment with 100 µM BCNU (6 h). (K) Expression pattern of the indicated endogenous apoptotic proteins in Q7 and Q111 cells treated with 300 µM BCNU (6 h). Calpain 1 (90 kD), cleaved caspase-3 (17, 19 kD), caspase-3 (35 kD). (L and M) Pretreatment with the caspase inhibitor 10 µM z-VAD-fmk (2 h) (L, n = 8) or the potent CaMKII kinases inhibitor 10 µM KN93 (2 h) (M, n = 9) prior to treatment with 100 µM BCNU (6 h) protects against cell death analysed by MTT.
Huntington’s disease is associated with Ca2+-dependent cleavage of mutant huntingtin (Goffredo, 2002). The measurement of Ca2+ with the FRET-based cytoplasmic Ca2+ sensor Yellow Cameleon 6.1 (YC 6.1) indicated differences in basal Ca2+ changes between BCNU and BSO. Basal Ca2+ levels did not differ significantly in Q7 and Q111 cells preincubated with BSO (Fig. 4C and D). However, BCNU caused a sustained increase in basal Ca2+ levels that was faster and larger in Q111 (39.0 ± 3.1% in 83 min) than in Q7 cells (33.3 ± 3.7% in 181 min) (Fig. 4E–H). Significantly, maintaining the cells in Ca2+-free solution eliminated the difference in cytoplasmic Ca2+ levels between the two cell lines treated with BCNU (Fig. 4E), indicating that the sustained Ca2+ increase is mediated by Ca2+ influx. Accordingly, incubating the cells in Ca2+-containing medium with 1.5 mM EGTA, a Ca2+ chelator, prevented BCNU-induced cell death (Fig. 4I). The cells in Ca2+-free medium without the Ca2+ chelator were completely resistant to BCNU-induced cell death (Fig. 4J).
Ca2+-dependent neurotoxicity involves activation of proteolytic pathways and apoptosis. Indeed, treating the cells with 300 µM BCNU decreased the level of pro-calpain 1 to increase proteolytic potential and increased the level of the active cleaved caspase 3, particularly in Q111 cells (Fig. 4K). The role of activated caspase 3 in BCNU toxicity was confirmed by ability of 10 µM z-VAD-fmk, a broad-spectrum caspase inhibitor, to inhibit BCNU-induced cell death. BCNU-induced apoptosis was blocked in Q7 cells (79.9 ± 3.4% → 94.5 ± 3.4%) and attenuated in Q111 cells (69.6 ± 5.8% → 86.8 ± 3.6%, Fig. 4L). In addition, Q7 and Q111 cell toxicity induced by BCNU was partially rescued by 10 µM KN93, a Ca2+/calmodulin-dependent protein kinase (CaMK) inhibitor (Fig. 4M).
The results in Figs 3 and 4 suggest that Huntington’s disease Q111 cells are more susceptible than wild-type Q7 cells to oxidative damage due to reduced redox potential. This involves a sustained increase in cytoplasmic Ca2+ and activation of proteolytic activity to induce caspase activation and apoptosis.
Increased TRPC5 S-glutathionylation in huntingtin striatal cell
The activation of TRPC5 by S-glutathionylation raises the possibility that cytosolic GSSG directly activates TRPC5 to cause a sustained increase in Ca2+ and associated neuronal damage in Huntington’s disease. We attempted to record TRPC5 current in striatal cells of Huntington’s disease by activation with internal GSSG. The whole-cell currents recorded immediately following pipette break-in with a pipette containing 5 mM GSSG are presented in Fig. 5A–C for Q7 and 5D–F for Q111 cells. While the GSSG-induced current in Q7 cells was predominantly outward rectifying, similar to the TRPC1/C5 heteromeric current, GSSG activated a small current that tended to be more TRPC5-like, double-rectifying current in Q111.
Figure 5.
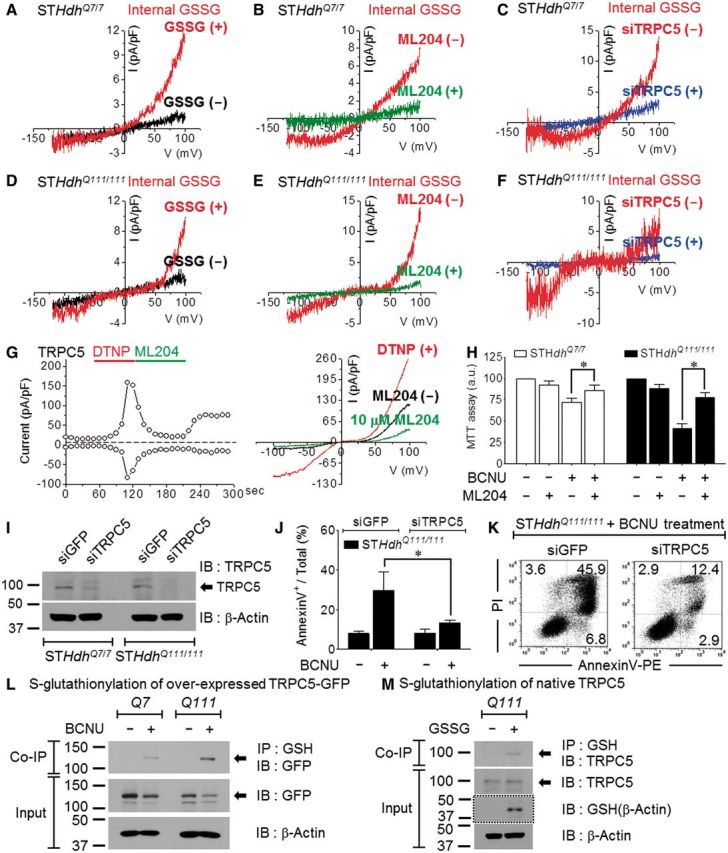
Increased TRPC5 S-glutathionylation in huntingtin striatal cells. (A–C) Outwardly rectifying current of Q7 cells activated by internal 5 mM GSSG (A) that was inhibited by 10 µM ML204, TRPC5 inhibitor (B), and by TRPC5 knockdown (C). (D–F) A doubly rectifying current of Q111 cells activated by internal 5 mM GSSG (D), inhibition by ML204 (E) and by TRPC5 knockdown (F). (G) ML204 (10 µM) inhibits 30 µM DTNP-activated TRPC5 current. (H) Inhibition of TRPC5 by 24-h pretreatment with ML204 blocks death of Q7 and Q111 cells caused by 100 µM BCNU (6 h), as evaluated by MTT assay (Q7, n = 5; Q111, n = 5). (I) Efficiency of knockdown of TRPC5 mRNA by siRNA as assayed by western blot. (J) TRPC5 knockdown eliminates Q111 apoptotic cell death by 100 µM BCNU (6 h) (n = 4). (K) Representative FACS analysis scatter grams of annexin V- and propidium iodide-stained Q111 cells treated with siGFP or siTRPC5 (48 h). (L) Transfected GFP-tagged mouse TRPC5 is S-glutathionylated in Q7 and Q111 cells treated with BCNU. (M) Native TRPC5 is S-glutathionylated in Q111 cell lysates reacted with 5 mM GSSG (30 min). The input blot with the dotted line using anti-GSH was performed under non-reducing conditions. IB = immunoblot; IP = immunoprecipitation.
We used several protocols to identify the current as mediated by TRPC5 under our recording conditions. TRPC5 is activated by a constitutively active form of Gαi3 protein (Gαi3 Q204L) (Jeon et al., 2012b). First, Gαi3 Q204L expression augmented the current in Q7 and Q111 cells (Supplementary Fig. 4B and C). Second, the selective and potent antagonist of TRPC5, ML204 [4-methyl-2-(piperidin-1-yl)quinolone; Miller et al., 2011], blocked the endogenous current activated by GSSG (Fig. 5B and E) as well as the current activation by DTNP and GSSG in cells transfected with TRPC5 (Fig. 5G and Supplementary Fig. 4D). Third, the current was eliminated by treating the cells with siTRPC5 (Fig. 5C and F). Western blotting (Fig. 5I) and reverse transcriptase-PCR (Supplementary Fig. 4E) assays demonstrated the efficiency of TRPC5 knockdown in the Q7 and Q111 cells.
Notably, inhibiting TRPC5 with 10 µM ML204 was sufficient to prevent BCNU-induced cell death in Q7 and Q111 cells (Fig. 5H). Moreover, knockdown of TRPC5 nearly eliminated the apoptosis induced by treating Q111 cells with 100 µM BCNU (Fig. 5J and K). The activation of endogenous TRPC5 by BCNU is likely the result of glutathionylation of TRPC5 due to the increase in GSSG induced by BCNU. We evaluated S-glutathionylation of TRPC5 in Q7 and Q111 cells upon BCNU induction or GSSG reaction. We transiently expressed GFP-tagged TRPC5 due to its low abundance and observed increased toxicity of BCNU treatment in Q7 and Q111 cells. Treating the cells with BCNU induced partial degradation of TRPC5 (Fig. 5L), particularly in Q111 cells, consistent with increased proteolytic activity (Fig. 4). Nevertheless, immunoprecipitation blotting demonstrated that treatment with BCNU resulted in a high level of glutathionylation of TRPC5 only in Q111 cells. To evaluate stable native TRPC5, we induced S-glutathionylation in Q111 cell lysates using 5 mM GSSG and detected glutathionylated TRPC5 using a monoclonal antibody (Fig. 5M). Immunoprecipitation blotting demonstrated that the GSSG application induced S-glutathionylation of native TRPC5 in Q111 cells. The immunoblotting with the GSH antibody confirmed that GSSG treatment properly induced protein S-glutathionylation via increased glutathionylation of endogenous β-actin under non-reducing conditions (Johansson and Lundberg, 2007).
The results in Fig. 5 imply that outwardly (in Q7) or doubly (in Q111) rectifying TRPC5 current is dependent on the TRPC1 expression level. Dominantly homomeric TRPC5 and oxidative conditions in Q111 cells facilitate Ca2+ influx. Thus, the activation of TRPC5 by glutathionylation mediates the sustained Ca2+ increase in oxidatively stressed striatal cells, leading to apoptotic cell death.
Protection of TRPC1 against excessive TRPC5-mediated Ca2+ influx
In Q111 striatal cells, increased cell toxicity by GSSG may be caused by attenuated TRPC1 activity due to decreased TRPC1 expression levels. Therefore, we further examined TRPC1 function in Q111 Huntington’s disease striatal cells. We first determined whether the TRPC1/C5 heteromeric channel is activated by DTNP or GSSG in HEK293 cells transfected with TRPC1 and TRPC5. DTNP (Fig. 6A) or GSSG (Fig. 6B) activated outwardly rectifying TRPC1/C5 current. Treatment with 200 µM La3+ as a control resulted in a typical outwardly rectifying current (Fig. 6A). To determine the protective role of TRPC1, the effect of TRPC1 knockdown on survival of Q7 and Q111 cells was determined. The identity of TRPC1 was verified by its marked reduction in cells treated with siTRPC1 (Fig. 6C). Knockdown of TRPC1 was sufficient to increase cell death in the two cell types (Fig. 6D and E). However, knockdown of TRPC1 increased cell death due to BCNU treatment in Q7 cells but less so in Q111 cells, which already express low levels of TRPC1. Knockdown of TRPC1 was sufficient to increase the number of apoptotic cells among Q111 cells. BCNU treatment further increased apoptosis in Q111 cells (Fig. 6F and G).
Figure 6.

Protection of TRPC1 against excessive TRPC5-mediated Ca2+ influx. (A and B) In heteromeric hTRPC1α/mTRPC5 channels, 30 µM DTNP (A) or 5 mM GSSG (B) activates a small constitutive current but strongly attenuates the current. La3+-activated current (green) was used as a control. (C) Analysis of knockdown of TRPC1 by siRNA treatment for 48 h in Q7 and Q111 cells by western blotting. Treatment with siGFP was used as a control. (D–G) TRPC1 knockdown exacerbates apoptotic cell death caused by treatment with 100 µM BCNU for 6 h, as assayed by MTT (D: Q7, n = 6; E: Q111; n = 9) or by FACS (F and G: n = 3) using annexin V. IB = immunoblot; IP = immunoprecipitation.
The results in Fig. 6 suggest that Q111 cells are less protected by TRPC1, consistent with the low TRPC1 level in these cells. Thus, although the residual TRPC1 can restrict apoptosis in Q111 cells, it is not sufficient to protect against oxidative cell damage and death.
The TRPC5 blocker ML204 improves striatal cell survival and rearing behaviour in Huntington’s disease mice
The YAC128 mouse model has been widely used to study the mechanism of Huntington’s disease. YAC128 mice that contain the entire mutant human Htt gene with 128 CAG repeats develop motor abnormalities and age-dependent brain atrophy, including cortical and striatal atrophy associated with striatal neuronal loss (Slow et al., 2003). These behaviour changes are associated with biphasic alterations in excitatory synaptic transmission to striatal medium-sized spiny neurons of the direct (expressing dopamine D1 receptors) and indirect (expressing dopamine D2 receptors) striatal output pathways (Andre et al., 2011). To determine the role of TRPC5 channel in the behavioural symptoms and biochemical changes of Huntington’s disease, we treated YAC128 mice with ML204 at 12–15 months of age. First, we identified endogenous TRPC1 and TRPC5 expression in the striatum of the YAC128 mice based on regional patterns of TRPC expression in the brain determined in previous studies (Greka et al., 2003; Fowler et al., 2007).
TRPC1 and TRPC5 transcript levels were quantified using quantitative reverse transcription-PCR in the striatum of YAC128 mice and wild-type mice (Fig. 7A). Using monoclonal TRPC1/C5 antibodies, we performed immunohistochemistry and detected TRPC1-positive and TRPC5-positive cells in the striatum of littermate control mice (wild-type) or YAC128 mice (Fig. 7B). To evaluate the presence of TRPC5 in YAC128 mice (Supplementary Fig. 5A) and human patients (Supplementary Fig. 6B), we performed a western blotting analysis that included a positive or a negative control. The immunoprecipitation blots in Fig 7C and Supplementary Fig. 6D show that the level of S-glutathionylation of endogenous TRPC5 was increased in the striatum of YAC128 mice and human patients with Huntington’s disease compared to each control. In addition, immunoblotting with the GSH antibody under non-reducing condition revealed increased glutathionylation of endogenous β-actin, indicating that oxidative stress may be responsible for overall glutathionylation under the neurodegenerative condition of Huntington’s disease (YAC128 mice and human patient).
Figure 7.
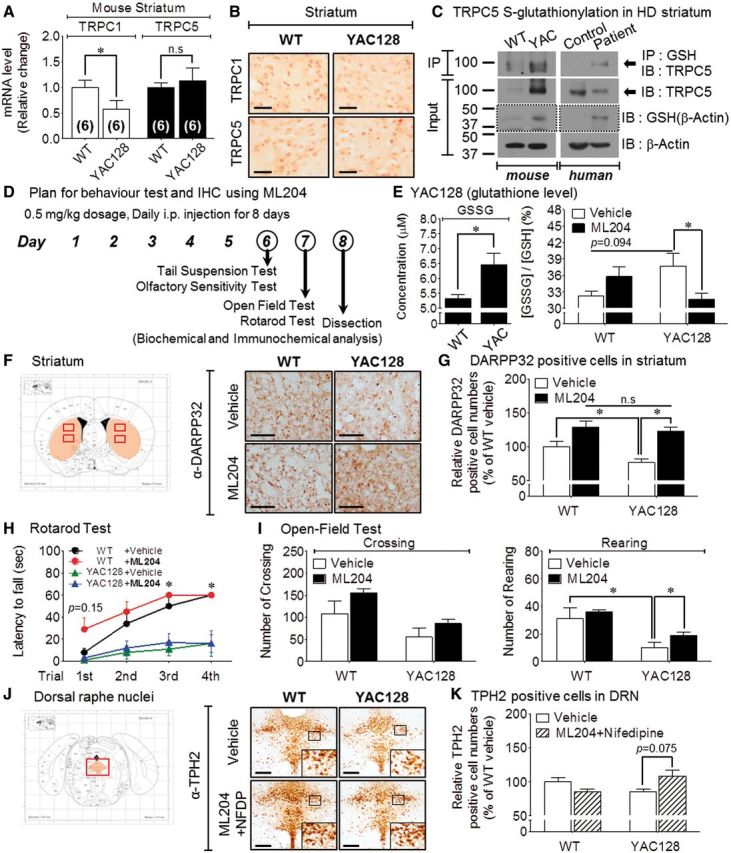
The TRPC5 blocker ML204 improves striatal cell survival and rearing behaviour in Huntington’s disease mice. All experiments were performed with littermate control (wild-type) and YAC128 Huntington’s disease mice (YAC128). *P < 0.05 and n.s. = not significant. (A) mRNA levels of Trpc1 or Trpc5 in the mouse Huntington’s disease model (YAC128) (wild-type, n = 6; YAC, n = 6). (B) Representative immunohistochemistry image of anti-TRPC1 (top) or anti-TRPC5 (bottom) in the striatum of wild-type and YAC128 Huntington’s disease mice. Scale bar = 50 μm. (C) Native TRPC5 is S-glutathionylated in the striatum tissue of YAC128 mice and a human patient with Huntington’s disease. The input blots with the dotted line using anti-GSH were performed under non-reducing conditions. (D) Outline of the behaviour study using transgenic Huntington’s disease mice responding to ML204 administration. (E) The effect of ML204 administration on the GSH/GSSG ratio in the striatum of YAC128 mice. (F and G) Representative immunohistochemistry image for anti-DARPP32 on the neuronal cell population in the striatum (F) of YAC128 mice after ML204 administration. Scale bar = 100 μm. Medium-sized spiny neurons in the striatum (G) decreased in YAC128 mice compared to wild-type. The number of these neuronal cells increased in YAC128 mice upon ML204 administration. (H–I) The effects of ML204 on the motor or non-motor behaviour of YAC128 mice: rotarod test (H), open-field test (I): (H) ML204 treatment improved the motor behaviour of wild-type mice on the rotarod test (wild-type; n = 10, YAC; n = 10). (I) ML204 significantly improved the rearing behaviour of wild-type and YAC128 mice in the open-field test (wild-type; n = 10, YAC; n = 10). (J and K) Representative immunohistochemistry image of anti-TPH2 on the neuronal cell population in the dorsal raphe nuclei (J) of YAC128 mice after treatment with a combination of ML204 and nifedipine (NFDP). Scale bar = 100 μm. Serotonergic neurons in the dorsal raphe nuclei (K) decreased in YAC128 mice compared to wild-type. The number of these neuronal cells increased in YAC128 mice upon the combination administration. WT = wild-type; IB = immunoblot; IP = immunoprecipitation.
ML204 inhibits the TRPC5-mediated intracellular Ca2+ rise and TRPC5 currents (Miller et al., 2011). ML204-treated paraventricular neuron exhibit suppression of the thyrotropin-releasing hormone-induced inward current (Zhang et al., 2013). To examine the role of the TRPC5 channel in the effect of ML204 in an animal model of Huntington’s disease, we administered 0.5 mg/kg of ML204 (i.p.) to Huntington’s disease transgenic (YAC128) and wild-type mice for 8 days and determined the behavioural symptoms and biochemical changes (Fig. 7D).
To determine if the intracellular redox imbalance was altered towards oxidative status in the striatum of YAC128 mice, we detected GSH and GSSG levels by fluorescence assay. Similar to the increased S-glutathionylation of TRPC5 or β-actin in Huntington’s disease mice and patients, the level of GSH was decreased in the striatum of YAC128 mice, while the level of GSSG and the ratio of GSSG/GSH were elevated compared to the vehicle group (Fig. 7E).
Next, we evaluated changes in neuronal cell population in several brain regions of YAC128 mice after ML204 injection. We detected specific neurons in several selected brain regions: DARPP32-positive GABAergic medium-sized spiny neurons in the striatum (ST), choline acetyltransferase (ChAT)-positive cholinergic neurons in the basolateral amygdala, TPH2-positive serotonergic neurons in the dorsal raphe nuclei, and TH-positive dopaminergic neurons in the substantia nigra pars compacta (SNpc). YAC128 mice exhibited decreased levels of ChAT-positive cells in basolateral amygdala and DARPP32-positive cells in striatum compared to wild-type mice. Administration of ML204 increased ChAT-positive cells in the basolateral amygdala (Supplementary Fig. 5B and C) and DARPP32-positive cells in the striatum (Fig. 7F and G) of YAC128 mice. The number of TPH2-positive cells in the dorsal raphe nuclei (Supplementary Fig. 5D and E) and TH-positive cells in the SNpc (Supplementary Fig. 5F and G) were not significantly altered by ML204 treatment in wild-type or YAC128 mice.
To assess the motor behaviour, we performed a rotarod test and an open-field test. The YAC128 mice exhibited significantly decreased latency to fall in the rotarod test compared to the wild-type mice. ML204 administration did not significantly alter the latency to fall in the YAC128 mice compared to the vehicle-treated group (Fig. 7H). In the open-field test, the YAC128 mice exhibited significantly decreased rearing counts compared to the wild-type mice. ML204 administration significantly increased rearing counts in YAC128 mice (Fig. 7I), but did not significantly alter crossing and grooming in any experimental group (Fig. 7I and Supplementary Fig. 5H). ML204-treated wild-type and YAC128 mice did not exhibit significant changes in olfactory function in the olfactory avoidance test or depression tendency in the tail suspension test (Supplementary Fig. 5I and J).
To rule out the effects of voltage-gated Ca2+ channels on neuronal survival in the YAC128 mice, we investigated the changes in the neuronal cell population in response to combination treatment of ML204 and nifedipine (NFDP, 1 mg/kg), an L-type calcium channel blocker (intraperitoneal). The combination treatment did not significantly alter the population of DARPP32-positive cells compared to treatment with ML204 alone (data not shown). Interestingly, the number of TPH2-positive serotonergic cells in the dorsal raphe nuclei exhibits an increased tendency by the combination treatment with nifedipine and ML204 (Fig. 7J and K).
The results in Fig. 7 suggest that the striatum of YAC128 mice is likely vulnerable to oxidative reactions, consistent with the increase in TRPC5 S-glutathionylation. As a result, downregulation of TRPC5 activity may improve the survival of striatal neurons and motor behavioural symptoms.
Discussion
GSH is the most abundant antioxidant in mammalian cells. GSSG glutathionylates cysteines in target proteins as a form of redox sensing and response. S-glutathionylation also plays a role in a variety of pathophysiological conditions that affect cell survival (Dalle-Donne et al., 2009). In the present study, we discovered a central role for TRPC5 glutathionylation in Huntington’s disease and determined its molecular mechanism. We demonstrated that glutathionylation potently activates TRPC5 in model systems and in vivo, resulting in a sustained [Ca2+]i increase and consequent cell toxicity and neurodegeneration.
Several studies have reported that TRPC5 is regulated by redox potential, although with contradictory results: cysteine S-nitrosylation (Yoshida et al., 2006) or breakage of a disulphide bridge (Xu et al., 2008) at Cys553/Cys558. We determined that Cys553 and Cys558 in TRPC5 are not S-glutathionylated, and mutation of these residues eliminates channel activity without affecting TRPC5 expression. Disulphide bond formation in extracellular Cys553 plays an important role in TRPC5 dimerization and membrane trafficking for functional activity (Hong et al., 2014). TRPC5 oxidation is reversibly modified by intracellular free cysteine (Fig. 1 and Supplementary Fig. 1). Moreover, Cys553 and Cys558 are located extracellularly and are not accessible to the intracellular redox environment. Collectively, our findings indicate that Cys176 and Cys178 of TRPC5, rather than Cys553 or Cys558, play prominent roles in the response of TRPC5 to cellular oxidative potential. Cytosolic oxidation by GSSG, NO, and H2O2 activate TRPC5 by glutathionylation, nitrosylation, and hydroxylation, respectively, most likely at Cys176 and Cys178 because mutation of these residues abrogated activation by all oxidants. This suggests that various oxidative insults may mediate their toxic effects through TRPC5 activation.
Due to its high energy demand and oxygen consumption, the brain is particularly vulnerable to oxidative stress as a normal by-product of cellular metabolism. Consequently, abnormal GSH homeostasis in neurons and GSSG accumulation are associated with several neurodegenerative conditions. Huntington’s disease is associated with proteolytic processing of mutant HTT and abnormal Ca2+ signalling, which is associated with disease progression and pathogenesis (Zeron et al., 2001). Several components of the Ca2+ signalling pathway have been implicated in abnormal Ca2+ signalling in Huntington’s disease, including decreased calbindin-D28K expression (Seto-Ohshima et al., 1988), sensitization of mGluR 1/5 (encoded by GRM1/5, respectively) to enhance Ca2+ influx (Sun et al., 2001), and enhanced activation of IP3R1 (encoded by ITPR1) by mGluR 1/5 (Tang et al., 2003). Mutant HTT function is unable to properly regulate calcium in the mitochondria, significantly decreasing the calcium threshold necessary to trigger mitochondrial permeability transition (MPT) pore opening (Pias and Aw, 2002; Choo et al., 2004; Lin and Beal, 2006). Furthermore, glutamate toxicity is a major contributor to pathological neuron cell death and is mediated by reactive oxygen species and GSH loss (Coyle and Puttfarcken, 1993; Herrera et al., 2007). The present study demonstrates that TRPC5 glutathionylation is involved in the molecular mechanism that mediates the toxic Ca2+ response to oxidative stress in Huntington’s disease. This oxidative damage aggravates mitochondrial dysfunction due to impaired Ca2+ homeostasis linked to TRPC5 S-glutathionylation.
Our search for the mechanism underlying the response to GSH-dependent oxidative stress was prompted by the reported depletion of cellular GSH in glutamate neurotoxicity. To further examine the in vivo effect of ML204, we used an animal model (YAC128) of Huntington’s disease. Importantly, ML204 administration ameliorated motor behaviours in wild-type control mice but not YAC128 mice (Fig. 7H). In particular, ML204 significantly improved rearing behaviour, a measure of general physical locomotive ability and, in part, anxiety. The protective effect of ML204 induces survival of neurons in the overall brain, indicating that TRPC5 downregulation increases cooperative neuronal networks.
Huntington’s disease usually begins at a later age. As an additional damaging mechanism, the expression of TRPC1 is suppressed in Huntington’s disease models (Q111 and YAC128). TRPC1 interacts with TRPC5 to reduce its activity and Ca2+ influx. The suppressed TRPC1 expression in Huntington’s disease further enhances TRPC5-dependent toxicity. Perturbation of GSH homeostasis and decreased TRPC1 expression may progress with ageing to cause Ca2+ signalling failure in age-related neurodegeneration. In the present study, we observed that transcript levels of TRPC1 and TRPC5 were not significantly changed compared to the control group in patients with Huntington’s disease (Supplementary Fig. 6A and B). Although the pattern of TRPC1 gene expression was differentially regulated between mice and humans with Huntington’s disease, the elevation of S-glutathionylation of TRPC5 (Fig. 7C and Supplementary Fig. 6C and D), a post-translational modification due to oxidation, was commonly correlated with neuronal damage in the mouse and human striatum in Huntington’s disease. Wu et al. (2011) determined that the store-operated calcium entry (SOC) activity is enhanced in neuronal cells expressing mutant HTT. Downregulation of the TRPC1-mediated SOC pathway protects Huntington’s disease neurons from toxicity. Further studies are needed to determine whether a severity of GSH redox state-dependent neuronal damage is correlated with the rate of clinical progression in Huntington’s disease.
Elevated Ca2+ levels can induce Ca2+-dependent proteolysis. Proteolytic cleavage of HTT is a key event in the pathogenesis of Huntington’s disease (Miller et al., 2010). The proteolytic pathways that have been implicated in HTT proteolysis in vivo include the caspases (Wellington et al., 2000; Ribeiro et al., 2012), calpains (Gafni et al., 2004), calcium/calmodulin-dependent protein kinase (McGinnis et al., 1998), and an unknown aspartic endopeptidase (Lunkes et al., 2002). Our findings demonstrate that activation of TRPC5 in Q111 cells is associated with activation of the caspases pathway. A recent study in TRPC5 null mice demonstrated that calpain cleaves and potentiates TRPC5 channel activity, resulting in changes in neuronal growth cone collapse (Kaczmarek et al., 2012). Thus, increased excitotoxicity of TRPC5 expressed in neurons leads to cell damage and cell death (e.g. ischaemia, developmental disorder).
Taken together, our data clarify the role of TRPC5 in Huntington’s disease by demonstrating that neuronal damage is caused by an abnormal sustained increase in cytosolic Ca2+ through the activation of TRPC5 by oxidative stress. Notably, this process involves in S-glutathionylation of TRPC5 at Cys176 and Cys178 and suppression of TRPC1 expression. Figure 8 depicts a model for the potential role of TRPC1 and TRPC5 in Huntington’s disease emerging from the present work. Because neuronal oxidative damage due to the deregulation of antioxidant scavenging systems is commonly observed in the pathogenesis of neurodegeneration, the pathological mechanism of oxidative stress-dependent TRPC5 activation may broadly contribute to neuronal damage in other neurodegenerative diseases.
Figure 8.

Schematic model of a pathological mechanism of TRPC5 and TRPC1/C5 S-glutathionylation and activation in Huntington’s disease. (A) Under normal conditions, TRPC5, a Ca2+-permeable cationic channel, can form homomeric channels or assemble with TRPC1 and TRPC4 to form heteromers that provide selective properties in the regulation of physiological levels of cytosolic Ca2+. (B) Under pathological conditions, such as in Huntington’s disease, suppression of TRPC1 expression and increased oxidative stress due to the deregulation of antioxidant scavenging systems lead to the elevation of TRPC5 S-glutathionylation at Cys176/Cys178 and the formation of TRPC5 homomeric channels. GSSG-activated TRPC5 current results in a sustained increase in cytosolic Ca2+. (C) The abnormal increase in a sustained cytosolic Ca2+ due to TRPC5 activation causes neuronal damage via the CaM kinase and calpain-caspase-dependent pathways in Huntington’s disease.
Funding
This study was supported by grants from the National Research Foundation of Korea, which is funded by the Ministry of Science, ICT (Information & Communication Technology) and Future Planning (MSIP) of the Korean government (2010-0019472 and 2012R1A2A1A01003073 to I.S., 2011-0030049, 2011-0030928, and 2012-003338 to H.S,, and 2013R1A1A1010783 to K. P. Lee), an NIH R01 grant (NS067283) and Flagship Grant of KIST (2E24380 to H.R.), and Korea Health Technology R&D project through KHDI, funded by the Ministry of Health & Welfare, ROK (HI14C1234 to E.C.Y.). C.H., M.K., and J.M. were supported by the BK21 plus program from the MSIP.
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- BCNU
1,3-bis(2-chloroethyl)-N-nitrosourea, carmustine
- BSO
L-buthionine (S,R)sulphoximine
- DTNP
2,2′-dithiobis(5-nitropyridine) / 5-nitro-2-PDS
- FRET
Foerster (fluorescence) resonance energy transfer
- GSH
reduced glutathione
- GSSG
oxidized glutathione, glutathione disulphide
- ML204
4-methyl-2-(piperidin-1-yl)quinolone
- TCEP
tris(2-carboxyethyl)phosphine
References
- Andre VM, Cepeda C, Fisher YE, Huynh M, Bardakjian N, Singh S, et al. Differential electrophysiological changes in striatal output neuronsin Huntington’s disease. J. Neurosci 2011; 31: 1180–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature 2006; 443: 796–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet 2004; 13: 1407–20. [DOI] [PubMed] [Google Scholar]
- Cooper AJ, Pinto JT, Callery PS. Reversible and irreversible protein glutathionylation by respiratory substrates. Expert Opin Drug Metab Toxicol 2011; 7: 891–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993; 262: 689–95. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci 2009; 34: 85–96. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Colombo G, Gagliano N, Colombo R, Giustarini D, Rossi R, et al. S-glutathionylation in life and death decisions of the cell. Free Radic Res 2011; 45: 3–15. [DOI] [PubMed] [Google Scholar]
- Damiano M, Galvan L, Deglon N, Brouillet E. Mitochondria in Huntington’s disease. Biochim Biophys Acta 2010; 1802: 52–61. [DOI] [PubMed] [Google Scholar]
- Erickson MG, Alseikhan BZ, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron 2001; 31: 973–85. [DOI] [PubMed] [Google Scholar]
- Floyd RA. Antioxidants, oxidative, and degenerative neurological disorders. Proc Soc Exp Biol Med 1999; 222: 236–45. [DOI] [PubMed] [Google Scholar]
- Fowler MA, Sidiropoulou K, Ozkan ED, Phillips CW, Cooper DC. Corticolimbic expression of TRPC4 and TRPC5 channels in the rodent brain. PLos One 2007; 27: e573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni J, Hermel E, Young JE, Wellington CL, Hayden MR, Ellerby LM. Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J Biol Chem 2004; 279: 20211–20. [DOI] [PubMed] [Google Scholar]
- Goffredo D, Rigamonti D, Tartari M, De Micheli A, Verderio C, Matteoli M, et al. Calcium-dependent cleavage of endogenous wild-type huntingtin in primary cortical neurons. J Biol Chem 2002; 277: 39594–8. [DOI] [PubMed] [Google Scholar]
- Greka A, Navarro B, Oancea E, Duggan A, Clapham DE. TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat Neurosci 2003; 6: 837–45. [DOI] [PubMed] [Google Scholar]
- Harkan JM, Levine JD, Callahan KS, Schwartz BR, Harker LA. Glutathione redox cycle protects cultured endothelial cells against lysis by extracellularly generated hydrogen peroxide. J Clin Invest 1984; 73: 706–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera F, Martin V, Garcia-Santos G, Rodriguez-Blanco J, Antolin I, Rodriguez C. Melatonin prevents glutamate-induced oxytosis in the HT22 mouse hippocampal cell line through an antioxidant effect specifically targeting mitochondria. J Neurochem 2007; 100: 736–46. [DOI] [PubMed] [Google Scholar]
- Hong C, Kwak M, Myeong J, Ha K, Wie J, Jeon JH, et al. Extracellular disulfide bridges stabilize TRPC5 dimerization, trafficking, and activity. Pflugers Arch 2014; 467: 703–12. [DOI] [PubMed] [Google Scholar]
- Hurd TR, Costa NJ, Dahm CC, Beer SM, Brown SE, Filipovska A, et al. Glutathionylation of mitochondrial proteins. Antioxid Redox Signal 2005; 7: 999–1010. [DOI] [PubMed] [Google Scholar]
- Jeon GS, Kim KY, Hwang YJ, Jung M, An S, Ouchi M, et al. Deregulation of BRCA1 leads to impaired spatiotemporal dynamics of γ-H2AX and DNA damage responses in Huntington’s disease. Mol Neurobiol 2012a; 45: 550–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon JP, Hong C, Park EJ, Jeon JH, Cho NH, Kim IG, et al. Selective Gai subunits as novel direct activators of transient receptor potential canonical (TRPC)4 and TRPC5 channels. J Biol Chem 2012b; 287: 17029–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M, Lundberg M. Glutathionylation of beta-actin via a cysteinyl sulfenic acid intermediary. BMC Biochem 2007; 8: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Muhle M, Schaefer M, Strotmann R, Schultz G, Plant TD. Lanthanides potentiate TRPC5 currents by an action at extracellular sites close to the pore mouth. J Biol Chem 2003; 278: 3562–71. [DOI] [PubMed] [Google Scholar]
- Kaczmarek JS, Riccio A, Clapham DE. Calpain cleaves and activates the TRPC5 channel to participate in semaphorin 3A-induced neuronal growth cone collapse. Proc Natl Acad Sci USA 2012; 109: 7888–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, Minami T, et al. A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J Pharmacol Sci 2006; 101: 66–76. [DOI] [PubMed] [Google Scholar]
- Kanki H, Kinoshita M, Akaike A, Satoh M, Mori Y, Kaneko S. Activation of inositol 1,4,5-trisphosphate receptor is essential for opening of mouse TRP5 channels. Mol Pharmacol 2001; 60: 989–98. [DOI] [PubMed] [Google Scholar]
- Kashio M, Sokabe T, Shintaku K, Uematsu T, Fukuta N, Kobayashi N, et al. Redox signal-mediated sensitization of transient receptor potential melastin 2 (TRPM2) to temperature affects macrophage functions. Proc Natl Acad Sci USA 2012; 109: 6745–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Hwang YJ, Shin JY, Lee WC, Wie J, Kim KY, et al. Epigenetic regulation of cholinergic receptor M1 (CHRM1) by histone H3K9me3 impairs Ca2+ signaling in Huntington’s disease. Acta Neuropathologica 2013; 125: 727–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroi I, Michalon M. Treatment of the psychiatric manifestations of Huntington’s disease: a review of the literature. Can J Psychiatry 1998; 43: 933–40. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006; 443: 787–95. [DOI] [PubMed] [Google Scholar]
- Lunkes A, Lindenberg KS, Ben-Haiem L, Weber C, Devys D, Landwehrmeyer GB, et al. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell 2002; 10: 259–69. [DOI] [PubMed] [Google Scholar]
- McGinnis KM, Whitton MM, Gnegy ME, Wang KK. Calcium/calmodulin-dependent protein kinase IV is cleaved by caspase-3 and calpain in SH-SY5Y human neuroblastoma cells undergoing apoptosis. J Biol Chem 1998; 273: 19993–20000. [DOI] [PubMed] [Google Scholar]
- Meister A, Anderson M. Glutathione. Annu Rev Biochem 1983; 52: 711–60. [DOI] [PubMed] [Google Scholar]
- Miller JP, Holcomb J, AI-Ramahi I, de Haro M, Gafni J, Zhang N, et al. Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in Huntington’s disease. Neuron 2010; 67: 199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M, Shi J, Zhu Y, Kustov M, Tian JB, Stevens A, et al. Identification of ML204, a novel potent antagonist that selectively modulates native TRPC4/C5 ion channels. J Biol Chem 2011; 286: 33436–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan B, Ezerina D, Amoako TNE, Riemer J, Dick TP. Multiple glutathione disulfide removal pathways mediate cytosolic redox homeostasis. Nat Chem Biol 2012; 9: 119–25. [DOI] [PubMed] [Google Scholar]
- Park HA, Khanna S, Rink C, Gnyawali S, Roy S, Sen CK. Glutathione disulfide induces neural cell death via a 12-lipoxygenase pathway. Cell Death Differ 2009; 16: 1167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pias EK, Aw TY. Apoptosis in mitotic competent undifferentiated cells is induced by cellular redox imbalance independent of reactive oxygen species production. FASEB J 2002; 16: 781–90. [DOI] [PubMed] [Google Scholar]
- Rao AV, Balachandran B. Role of oxidative stress and antioxidants in neurodegenerative diseases. Nutr Neurosci 2002; 5: 291–309. [DOI] [PubMed] [Google Scholar]
- Ribeiro M, Rosenstock TR, Cunha-Oliveira T, Ferreira IL, Oliveira CR, Rego AC. Glutathione redox cycle dysregulation in Huntington’s disease knock-in striatal cells. Free Radic Biol Med 2012; 53: 1857–67. [DOI] [PubMed] [Google Scholar]
- Ribeiro M, Silva AC, Rodrigues J, Naia L, Rego AC. Oxidizing effects of exogenous stressors in Huntington’s disease knock-in striatal cells-protective effect of cystamine and creatine. Toxicol Sci 2013; 136: 487–99. [DOI] [PubMed] [Google Scholar]
- Riccio A, Li Y, Moon J, Kim KS, Smith KS, Rudolph U, et al. Essential role for TRPC5 in amygdala function and fear-related behavior. Cell 2009; 137: 761–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto-Ohshima A, Emson PC, Lawson E, Mountjoy CQ, Carrasco LH. Loss of matrix calcium-binding protein-containing neurons in Huntington’s disease. Lancet 1988; 1: 1252–5. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet 2003; 12: 1555–67. [DOI] [PubMed] [Google Scholar]
- Storch U, Forst AL, Philipp M, Gudermann T, Mederos Schnitzler M. Transient receptor potential channel 1 (TRPC1) reduces calcium permeability in heteromeric channel complexes. J Biol Chem 2012; 287: 3530–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 2001; 29: 645–55. [DOI] [PubMed] [Google Scholar]
- Sun Y, Savanenin A, Reddy PH, Liu YF. Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-D-aspartate receptors via post-synaptic density 95. J Biol Chem 2001; 276: 24713–8. [DOI] [PubMed] [Google Scholar]
- Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, et al. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 2003; 39: 227–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellington CL, Singaraja R, Elleby L, Savill J, Roy S, Leavitt B, et al. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem 2000; 275: 19831–8. [DOI] [PubMed] [Google Scholar]
- Wu J, Shih HP, Vigont V, Hrdlicka L, Diggings L, Singh C, et al. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington’s disease treatment. Chem Biol 2011; 18: 777–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Sukumar P, Zeng F, Li J, Jairaman A, English A, et al. TRPC channel activation by extracellular thioredoxin. Nature 2008; 451: 69–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, et al. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol 2006; 2: 596–607. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Chen N, Moshaver A, Lee AT, Wellington CL, Raymond LA. Mutant huntingtin enhances excitotoxic cell death. Mol Cell Neurosci 2001; 17: 41–53. [DOI] [PubMed] [Google Scholar]
- Zhang L, Kolaj M, Renaud LP. GIRK-like and TRPC-like conductances mediate thyrotropin-releasing hormone-induced increases in excitability in thalamic paraventricular nucleus neurons. Neuropharmacology 2013; 72: 106–115. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.