

Synaptic dysfunction precedes neurodegeneration and cognitive impairment in Alzheimer’s disease. Portelius et al. show that CSF levels of the postsynaptic protein neurogranin are increased in early-stage Alzheimer’s disease, and that the increase predicts cognitive deterioration and disease-associated changes in metabolic and structural biomarkers over time.
Keywords: Alzheimer’s disease, neurogranin, cerebrospinal fluid, biomarker, mild cognitive impairment

Synaptic dysfunction precedes neurodegeneration and cognitive impairment in Alzheimer’s disease. Portelius et al. show that CSF levels of the postsynaptic protein neurogranin are increased in early-stage Alzheimer’s disease, and that the increase predicts cognitive deterioration and disease-associated changes in metabolic and structural biomarkers over time.
Abstract
Synaptic dysfunction is linked to cognitive symptoms in Alzheimer’s disease. Thus, measurement of synapse proteins in cerebrospinal fluid may be useful biomarkers to monitor synaptic degeneration. Cerebrospinal fluid levels of the postsynaptic protein neurogranin are increased in Alzheimer’s disease, including in the predementia stage of the disease. Here, we tested the performance of cerebrospinal fluid neurogranin to predict cognitive decline and brain injury in the Alzheimer’s Disease Neuroimaging Initiative study. An in-house immunoassay was used to analyse neurogranin in cerebrospinal fluid samples from a cohort of patients who at recruitment were diagnosed as having Alzheimer’s disease with dementia (n = 95) or mild cognitive impairment (n = 173), as well as in cognitively normal subjects (n = 110). Patients with mild cognitive impairment were grouped into those that remained cognitively stable for at least 2 years (stable mild cognitive impairment) and those who progressed to Alzheimer’s disease dementia during follow-up (progressive mild cognitive impairment). Correlations were tested between baseline cerebrospinal fluid neurogranin levels and baseline and longitudinal cognitive impairment, brain atrophy and glucose metabolism within each diagnostic group. Cerebrospinal fluid neurogranin was increased in patients with Alzheimer’s disease dementia (P < 0.001), progressive mild cognitive impairment (P < 0.001) and stable mild cognitive impairment (P < 0.05) compared with controls, and in Alzheimer’s disease dementia (P < 0.01) and progressive mild cognitive impairment (P < 0.05) compared with stable mild cognitive impairment. In the mild cognitive impairment group, high baseline cerebrospinal fluid neurogranin levels predicted cognitive decline as reflected by decreased Mini–Mental State Examination (P < 0.001) and increased Alzheimer’s Disease Assessment Scale–cognitive subscale (P < 0.001) scores at clinical follow-up. In addition, high baseline cerebrospinal fluid neurogranin levels in the mild cognitive impairment group correlated with longitudinal reductions in cortical glucose metabolism (P < 0.001) and hippocampal volume (P < 0.001) at clinical follow-up. Furthermore, within the progressive mild cognitive impairment group, elevated cerebrospinal fluid neurogranin levels were associated with accelerated deterioration in Alzheimer’s Disease Assessment Scale–cognitive subscale (β = 0.0017, P = 0.01). These data demonstrate that cerebrospinal fluid neurogranin is increased already at the early clinical stage of Alzheimer’s disease and predicts cognitive deterioration and disease-associated changes in metabolic and structural biomarkers over time.
Introduction
Alzheimer’s disease is the most common neurodegenerative disorder and a major health problem. Alzheimer’s disease is characterized by pathological hallmarks including neuronal and synaptic degeneration and loss together with deposits of aggregated amyloid-β and tau (Blennow et al., 2010). Biochemical (including CSF measurements of amyloid-β42 and tau) and neuroimaging (including PET and MRI) biomarkers may be used to monitor amyloid-β and tau pathology, cortical glucose metabolism and atrophy (Dubois et al., 2014). 18F-fluorodeoxyglucose (FDG) PET is a marker of cortical glucose metabolism that may be related to synaptic function (Mosconi et al., 2008), but until recently, no biomarker reflecting the synaptic pathology in Alzheimer’s disease has been available.
Synaptic dysfunction and loss are directly linked to memory disturbances and other cognitive symptoms already at the early stages of Alzheimer’s disease, and are believed to precede neuronal degeneration and loss (DeKosky et al., 1990; Bertoni-Freddari et al., 1996). Neuropathological studies have shown that synaptic loss is evident already at the mild cognitive impairment (MCI) stage of Alzheimer’s disease (Masliah et al., 2001; Scheff et al., 2007) and is more strongly correlated to the degree of cognitive dysfunction than plaque and tangle pathology (Davies et al., 1987; Blennow et al., 1996; Sze et al., 1997; Masliah et al., 2001). Thus, because synaptic loss occurs early and correlates with cognitive deficits in patients with Alzheimer’s disease, biomarkers reflecting this pathophysiological process could be useful for studies of disease mechanisms, to improve tools for early diagnosis and prognosis, and to monitor drug effects on synaptic degeneration in clinical trials of disease-modifying therapies for Alzheimer’s disease.
We have previously shown that several pre- and postsynaptic proteins, such as SNAP25, synaptotagmin and neurogranin, are detectable in CSF (Davidsson et al., 1996, 1999). Neurogranin is a post synaptic protein that is enriched in dendritic spines and plays an important role in long-term potentiation and memory consolidation (Gerendasy et al., 1997; Pak et al., 2000; Huang et al., 2004), which are abnormal in Alzheimer’s disease. It contains a calmodulin-binding IQ domain that interacts with and sequesters apo-calmodulin (the Ca2+-free form of calmodulin). On rise of the intracellular Ca2+ concentration, neurogranin releases calmodulin, freeing it to bind Ca2+ and activate downstream signalling molecules (Hayashi, 2009). In a pilot study, we showed by immunoprecipitation and semi-quantitative western blotting that CSF neurogranin is increased in Alzheimer’s disease dementia and correlates with abnormally elevated levels of CSF tau (Thorsell et al., 2010).
To investigate the potential of CSF neurogranin as a biomarker for Alzheimer’s disease, we recently produced monoclonal antibodies directed against neurogranin C-terminal regions and showed by mass spectrometry that neurogranin is metabolized into a series of endogenous peptides derived from the C-terminal part of the protein (Kvartsberg et al., 2014). We further developed and used a hybrid immunoaffinity-mass spectrometry method and an enzyme-linked immunosorbent assay (ELISA) to analyse and quantify CSF neurogranin (Kvartsberg et al., 2014). In three independent clinical cohorts, a marked increase in CSF neurogranin levels was found in patients with Alzheimer’s disease dementia as well as in the MCI stage of the disease (prodromal Alzheimer’s disease) compared to controls, with higher CSF neurogranin levels correlating with a more rapid change in cognition during clinical follow-up (Kvartsberg et al., 2014). These findings suggest that CSF neurogranin is altered in early stage Alzheimer’s disease and correlates with cognitive decline.
In this study, we present results on CSF neurogranin in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort of cognitively healthy controls, subjects with MCI and patients with Alzheimer’s disease dementia. We tested the specific hypotheses that CSF neurogranin is increased in Alzheimer’s disease compared to healthy control, that high CSF neurogranin predicts MCI conversion to Alzheimer’s disease dementia and that CSF neurogranin correlates with changes in cognition and Alzheimer’s disease molecular pathophysiology, as reflected by FDG-PET and MRI, over time.
Materials and methods
Data used in the preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). The ADNI was launched in 2004 by the National Institute on Aging, the Food and Drug Administration, private pharmaceutical companies and non-profit organizations as a highly innovative public–private partnership. The primary goal of ADNI has been to test whether serial MRI, PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early Alzheimer’s disease. The Principal Investigator of this initiative is Michael W. Weiner, MD, VA Medical Center and University of California – San Francisco. ADNI is the result of efforts of many co-investigators from a broad range of academic institutions and private corporations, and subjects have been recruited from over 50 sites across the USA and Canada. The initial goal of the ADNI was to recruit 800 subjects but it has been followed by ADNI-GO and ADNI-2. To date these three protocols have recruited >1500 adults, ages 55 to 90, to participate in the research, consisting of cognitively normal older individuals, people with early or late MCI, and people with early Alzheimer’s disease. The follow-up duration of each group is specified in the protocols for ADNI-1, ADNI-2 and ADNI-GO. Subjects originally recruited for ADNI-1 and ADNI-GO had the option to be followed in ADNI-2. For more information, see www.adni-info.org.
Subjects
Our study population consisted of all healthy control, MCI and Alzheimer’s disease dementia subjects with available baseline CSF samples from ADNI-1. Inclusion/exclusion criteria are described in detail at http://www.adni-info.org. Briefly, all subjects included in ADNI-1 were between the ages of 55 and 90 years, had completed at least 6 years of education, were fluent in Spanish or English, and were free of any significant neurological disease other than Alzheimer’s disease. Healthy control subjects had Mini-Mental State Examination (MMSE) score ≥ 24, and Clinical Dementia Rating scale score 0. MCI subjects had MMSE score ≥ 24, objective memory loss as shown on scores on delayed recall of the Wechsler Memory Scale Logical Memory II [>1 standard deviations (SD) below the normal mean], Clinical Dementia Rating scale 0.5, preserved activities of daily living, and absence of dementia. Patients with Alzheimer’s disease dementia fulfilled the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association criteria for probable Alzheimer’s disease, had MMSE scores between 20–26 and a Clinical Dementia Rating scale of 0.5 or 1.0. The evaluation was vigorous across all centres. Almost all of the centres participating in ADNI were experienced clinical trial sites and many were long-standing members of Alzheimer’s Disease Cooperative Studies. Study monitors frequently visited all sites to oversee uniform evaluations. The MCI group was stratified into stable MCI (no progression to Alzheimer’s disease dementia during at least 2 year follow-up) and progressive MCI (progression to Alzheimer’s disease dementia during follow-up). The main analyses therefore included four groups which at baseline included: healthy control (n = 110), stable MCI (n = 68), progressive MCI (n = 105) and Alzheimer’s disease dementia (n = 95). In a subanalysis, we also describe healthy control subjects who progressed to MCI (n = 24) or Alzheimer’s disease (n = 8) during follow-up. For details on the number of cases in each diagnostic group with data at different time-points, see Table 1.
Table 1.
Number of subjects included at baseline and at follow-ups for each parameter measured
| Biomarker | Months after baseline |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 6 | 12 | 18 | 24 | 36 | 48 | 60 | 72 | 84 | 96 | 108 | |
| Alzheimer’s disease | ||||||||||||
| Amyloid-β1–42 | 95 | |||||||||||
| Total tau | 95 | |||||||||||
| Phosphorylated tau | 95 | |||||||||||
| MMSE | 95 | 93 | 84 | 1 | 76 | 8 | 2 | |||||
| ADAS-Cog | 95 | 93 | 84 | 1 | 76 | 8 | 2 | |||||
| FDG-PET | 49 | 45 | 41 | 32 | ||||||||
| Hippocampal volume | 93 | 87 | 74 | 62 | ||||||||
| Healthy controls | ||||||||||||
| Amyloid-β1–42 | 110 | |||||||||||
| Total tau | 110 | |||||||||||
| Phosphorylated tau | 110 | |||||||||||
| MMSE | 110 | 108 | 106 | 99 | 92 | 64 | 54 | 58 | 47 | 36 | 10 | |
| ADAS-Cog | 110 | 108 | 106 | 99 | 92 | 64 | 54 | 58 | 47 | 36 | 11 | |
| FDG-PET | 50 | 51 | 44 | 44 | 38 | 32 | 26 | 20 | 18 | 1 | ||
| Hippocampal volume | 107 | 106 | 100 | 86 | 77 | 47 | 28 | 20 | ||||
| Progressive MCI | ||||||||||||
| Amyloid-β1–42 | 105 | |||||||||||
| Total tau | 105 | |||||||||||
| Phosphorylated tau | 105 | |||||||||||
| MMSE | 105 | 104 | 105 | 97 | 90 | 76 | 44 | 34 | 31 | 23 | 16 | 4 |
| ADAS-Cog | 105 | 104 | 105 | 97 | 90 | 76 | 44 | 34 | 31 | 23 | 16 | 5 |
| FDG-PET | 54 | 53 | 53 | 48 | 44 | 35 | 18 | 13 | 10 | 6 | 1 | |
| Hippocampal volume | 104 | 98 | 98 | 90 | 77 | 61 | 35 | 17 | 7 | |||
| Stable MCI | ||||||||||||
| Amyloid-β1–42 | 68 | |||||||||||
| Total tau | 68 | |||||||||||
| Phosphorylated tau | 68 | |||||||||||
| MMSE | 68 | 68 | 68 | 66 | 63 | 53 | 24 | 20 | 20 | 15 | 14 | 1 |
| ADAS-Cog | 68 | 68 | 68 | 66 | 63 | 53 | 24 | 20 | 20 | 15 | 14 | 1 |
| FDG-PET | 34 | 34 | 34 | 34 | 30 | 23 | 12 | 9 | 10 | 5 | ||
| Hippocampal volume | 66 | 66 | 64 | 55 | 53 | 36 | 18 | 15 | 2 | |||
CSF measurements
CSF collection, processing and storage procedures have been described previously (Shaw et al., 2009). CSF amyloid-β42, total tau and phosphorylated tau were measured using the multiplex xMAP® Luminex platform with the INNOBIA AlzBio3 kit (Innogenetics) as described previously (Shaw et al., 2009). Subjects were classified as amyloid-β positive or negative using a previously established cut-off (CSF amyloid-β42 < 192 pg/ml) that maximized the separation of autopsy-confirmed Alzheimer’s disease cases with amyloid-β pathology from controls without amyloid-β pathology (Shaw et al., 2009).
Electrochemiluminescence immunoassay for neurogranin
The in-house generated monoclonal antibody Ng7, which binds to an epitope including amino acids 52–65 (Kvartsberg et al., 2014), was used as capture antibody in the assay. Quickplex® 96-well plates (Mesoscale) were washed once with phosphate-buffered saline (PBS; 150 µl/well) before coating with Ng7 (25 µl/well) at a final concentration of 2.0 µg/ml in PBS overnight at room temperature. After washing with 300 µl PBS containing 0.05% Tween20 (PBS-Tween) four times for 30 s, the remaining protein binding sites were blocked with 5% MSD® Blocker A containing PBS for 1 h at room temperature (150 µl/well), while shaking at 700 rpm. Thereafter, plates were washed four times with 300 µl PBS-Tween for 30 s. Then the full-length neurogranin calibrators with concentrations ranging between 31.3–2000 pg/ml, blanks, and CSF samples (50 µl/well) were incubated in duplicate together with the detector antibody, polyclonal neurogranin anti-rabbit (ab 23570, Upstate) diluted 1:20 000 in 0.1% bovine serum albumin (BSA) in PBS-Tween, overnight at +4°C (50 µl/well). After washing four times, a MSD®Sulfotag goat anti-rabbit antibody diluted to 0.5 µg/ml in 0.1% BSA in PBS-Tween, was incubated for 1 h at room temperature while shaking at 700 rpm. After washing four times with 300 µl PBS-Tween for 30 s, 150 µl of 2× MSD Read Buffer T with surfactant was used for immediately reading on the Mesoscale instrument Quickplex SQ 120 reader. A fitted four-parameter logistic model with relative weighing (1/y2) was used as the calibration curve and the blank was included in the calibration curve as zero.
Magnetic resonance imaging
Structural MRI brain scans were acquired using 1.5 T MRI scanners (up to seven time points: screening, and 6, 12, 18, 24, 36 and 48 months) with a standardized protocol including T1-weighted MRI scans using a sagittal volumetric magnetization prepared rapid gradient echo sequence (Jack et al., 2008). In brief, automated volume measures were performed with FreeSurfer software package (http://surfer.nmr.mgh.harvard.edu/fswiki) (Fischl et al., 2002, 2004). We used averaged volume measurements for the right and left hippocampi for this study.
18F-Fluorodeoxyglucose-PET
ADNI PET image data were acquired (at up to seven times: screening, and at 6, 12, 18, 24, 36 and 48 months after baseline) and processed as described online (adni.loni.ucla.edu/about-data-samples/image-data/ and adni.loni.ucla.edu/research/pet-post-processing/, respectively). See Landau et al. (2012) for a detailed description. Briefly, we used mean PET counts of the lateral and medial frontal, anterior, and posterior cingulate, lateral parietal, and lateral temporal regions.
Cognition
Global cognition was assessed by MMSE (Folstein et al., 1975) and Alzheimer’s Disease Assessment Scale–cognitive subscale (ADAS-Cog; Rosen et al., 1984) (at up to seven times: screening, and at 6, 12, 18, 24, 36 and 48 months after baseline).
Statistical analysis
To evaluate potentially confounding factors, we tested associations between CSF neurogranin and demographic factors (age, sex, APOE ε4, education) using Mann-Whitney U and chi-square tests. Associations between the levels of CSF neurogranin and diagnostic groups were tested with linear regression, adjusted for age and sex. To test if amyloid-β influenced these associations, we included the interaction between diagnosis and amyloid-β positivity as a predictor in the model. For MMSE and hippocampal volume we derived intercepts (baseline values) and slopes (rates of change) using linear mixed effects models. The intercept and slopes were then used as outcomes in linear regression models with CSF neurogranin as predictor (adjusted for age and sex; and for intracranial volume for hippocampal volume; and for education for MMSE), within diagnostic groups. In the MCI group, associations between baseline CSF neurogranin levels and subsequent disease progression, as measured by MMSE, ADAS-Cog, hippocampal volume and average FDG-PET of angular, temporal, and posterior cingulate cortex were tested with linear mixed effects models, adjusted for age and sex (and education for cognitive measurements and intracranial volume for hippocampal volume). The impact of baseline CSF neurogranin levels was further illustrated by dividing the subjects with an MCI diagnosis at baseline into quartile groups according to CSF neurogranin levels at baseline and comparing linear regression models between the quartile groups. The linear regression was adjusted for age and sex (and education for cognitive measurements and intracranial volume for hippocampal volume). The associations of neurogranin with the other CSF biomarkers amyloid-β1–42, total tau and phosphorylated tau were investigated with Spearman’s correlation. All tests were two-sided, and significance was determined at P < 0.05. All statistics were done using R (v. 3.0.1, The R Foundation for Statistical Computing), SPSS version 20 (IBM).
Results
Baseline demographic and biomarker characteristics of the study participants are shown in Table 2 and the demographic information and biomarker characteristics at baseline of subjects followed to at least month 48 are shown in Supplementary Table 1.
Table 2.
Demographic and clinical characteristics of subjects included in the study at baseline
| Control | Alzheimer’s disease | Progressive MCI | Stable MCI | |
|---|---|---|---|---|
| Gender, n, female/male (% female) | 55/55 (50) | 42/53 (44) | 37/68 (35) | 22/46 (32) |
| Age at lumbar puncture, years | 76 (72–78) | 76 (70–80) | 75 (70–80) | 74 (70–80) |
| MMSE | 29 (29–30) | 24 (22–25)*** | 26 (25–28)*** | 28 (26–29)*** |
| ADAS-cog | 6.3 (4.0–8.0) | 17.7 (13.8–21.8)*** | 12.0 (9.67–15.3)*** | 9.67 (6.33–12.0)*** |
| Hippocampal volume | 3645 (3351–3854) | 2740 (2416–3208)*** | 2988 (2706–3271)*** | 3417 (3025–3764) |
| FDG-PET | 1.3 (1.2–1.4) | 1.1 (1.0–1.2)*** | 1.2 (1.1–1.3)*** | 1.2 (1.1–1.3) |
| CSF amyloid-β1–42, pg/ml | 220 (160–253) | 138 (122–159)*** | 139 (123–163)*** | 184 (133–245)*** |
| CSF total tau, pg/ml | 61 (50–86) | 111 (81–154)*** | 97 (75–129)*** | 69 (51–110)* |
| CSF phosphorylated tau, pg/ml | 20 (16–28) | 36 (29–49)*** | 38 (26–48)*** | 24 (18–39)* |
| CSF neurogranin, pg/ml | 304 (161–453) | 485 (349–744)*** | 492 (330–672)*** | 386 (190–582) |
The values presented are median (IQR).
*P < 0.05 versus controls; **P < 0.01 versus controls; ***P < 0.001 versus controls.
CSF neurogranin levels in different diagnostic groups
CSF neurogranin levels were significantly higher in patients with Alzheimer’s disease dementia and MCI as compared to healthy controls (P < 0.0001 for both groups). Higher CSF neurogranin levels were also found in both the stable MCI (P = 0.039) and progressive MCI (P < 0.001) subgroups as compared with healthy controls, and in Alzheimer’s disease dementia (P = 0.005) and progressive MCI (P = 0.015) compared with stable MCI (Fig. 1A). Among the healthy controls, 32 individuals progressed to MCI or Alzheimer’s disease dementia during follow-up (progressive healthy control group). These subjects had significantly higher CSF neurogranin (P = 0.025, adjusted for age and sex) [median (interquartile range, IQR), 382 pg/ml (256–523)] compared with cognitively stable healthy control [median (IQR), 287 pg/ml (138–377)] (Fig. 1B). For comparison, the CSF amyloid-β1–42 levels were similar between these two groups [median (IQR), 225 pg/ml (173–258)] and 210 pg/ml (139–234) for cognitively stable controls and healthy control who progressed to MCI or Alzheimer’s disease dementia, respectively.
Figure 1.

CSF neurogranin levels in different diagnostic groups. (A) Scatter plots showing CSF neurogranin levels in healthy control, stable MCI (sMCI), progressive MCI (pMCI) and Alzheimer’s disease (AD). Among the healthy control subjects, 32 individuals progressed to MCI or Alzheimer’s disease dementia during follow-up (progressive healthy controls) (B). The data are presented as medians and interquartile ranges. Differences between groups were assessed by linear regression, adjusted for age and sex. *P < 0.05; **P < 0.01; ***P < 0.0001.
CSF neurogranin levels in relation to amyloid-β pathology
Low CSF amyloid-β1–42 has been shown in several studies to correspond to cortical amyloid deposition as evaluated by amyloid-β amyloid PET, also within the ADNI study (Mattsson et al., 2014). To explore the association between CSF neurogranin and amyloid-β amyloid pathology, study subjects were dichotomized into CSF amyloid-β-positive and -negative using the previously established cut-off for CSF amyloid-β1–42 of 192 pg/ml (Shaw et al., 2009). CSF amyloid-β-positive stable MCI, progressive MCI and Alzheimer’s disease dementia patients all had increased CSF neurogranin as compared to CSF amyloid-β-negative healthy controls (P < 0.001 for all three groups, Fig. 2), whereas amyloid-β-negative stable MCI, progressive MCI and Alzheimer’s disease dementia patients had CSF neurogranin levels in the same range as amyloid-β-negative healthy control subjects. Furthermore, amyloid-β-positive progressive MCI, and Alzheimer’s disease dementia cases had increased CSF neurogranin compared to amyloid-β-negative stable MCI patients (P < 0.001 for both groups). There was no significant correlation between CSF amyloid-β42 and neurogranin in healthy control subjects. In stable MCI, progressive MCI, and Alzheimer’s disease dementia patients, CSF amyloid-β42 and neurogranin were negatively correlated (rs = −0.29, P = 0.019; rs = −0.28, P = 0.0037; and rs = −0.21, P = 0.038 for stable MCI, progressive MCI, and Alzheimer’s disease dementia, respectively).
Figure 2.
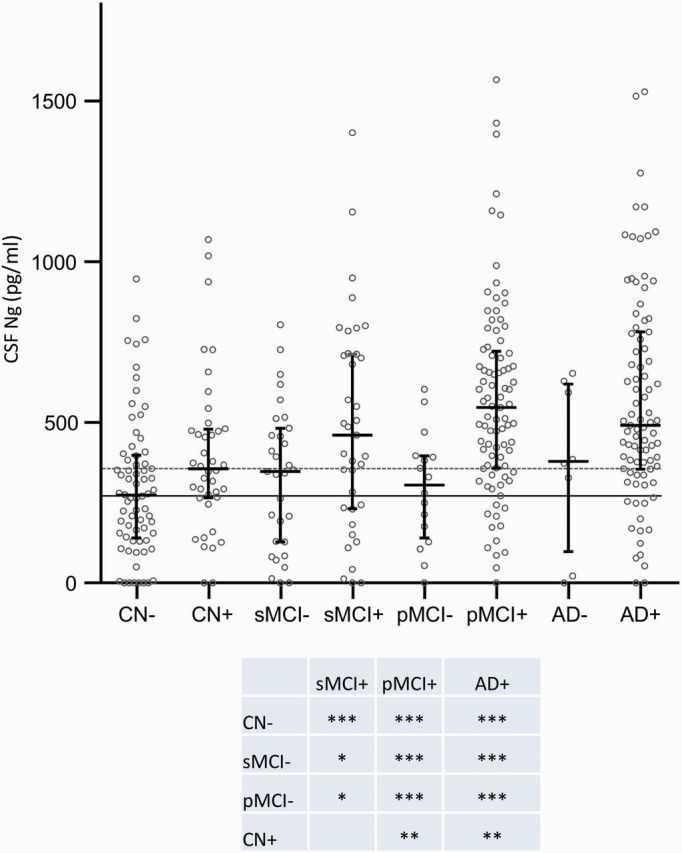
Scatter plot showing the subjects included in the study classified as amyloid positive (+) or negative (−) based on a previously established cut-off (CSF amyloid-β42 < 192 pg/ml). The data are presented as medians and interquartile ranges. Differences between groups were assessed by linear regression, adjusted for age and sex. *P < 0.05; **P < 0.01; ***P < 0.001. Ng = neurogranin.
CSF neurogranin levels in relation to tau biomarkers
Both total tau and phosphorylated tau were strongly correlated with neurogranin in healthy controls (rs = 0.75 for total tau and rs = 0.59 for phosphorylated tau, both P < 0.0001), stable MCI (rs = 0.82 for total tau and rs = 0.68 for phosphorylated tau, both P < 0.0001), progressive MCI (rs = 0.75 for total tau and rs = 0.58 for phosphorylated tau, both P < 0.0001) and Alzheimer’s disease dementia patients (rs = 0.76 for total tau and 0.75 for phosphorylated tau, both P < 0.0001) (Fig. 3A–D).
Figure 3.

CSF neurogranin levels in relation to tau biomarkers. Correlations between CSF neurogranin levels and total tau and phosphorylated tau in healthy control (A), stable MCI (B), progressive MCI (C) and Alzheimer’s disease (D). Open circles and the solid line represent the correlation between total tau and neurogranin while closed circles and the dashed line represents the correlation between phosphorylated tau and neurogranin. The association between neurogranin and total and phosphorylated tau was investigated with Spearman’s correlation. The regression lines in the figures are only for visualization.
CSF neurogranin in relation to cognition and future cognitive change
CSF neurogranin did not correlate with baseline MMSE or ADAS-Cog scores (Fig. 4A and B). In contrast, high CSF neurogranin levels correlated significantly with a more rapid increase in ADAS-Cog scores in progressive MCI during the clinical follow-up period (β = 0.0017, P = 0.01 for ADAS-Cog). There were also trends for associations between high CSF neurogranin and with more rapid change in MMSE (for progressive MCI) and ADAS-Cog (for progressive MCI and Alzheimer’s disease dementia) during follow-up, but these did not reach statistical significance (Fig. 4C and D).
Figure 4.
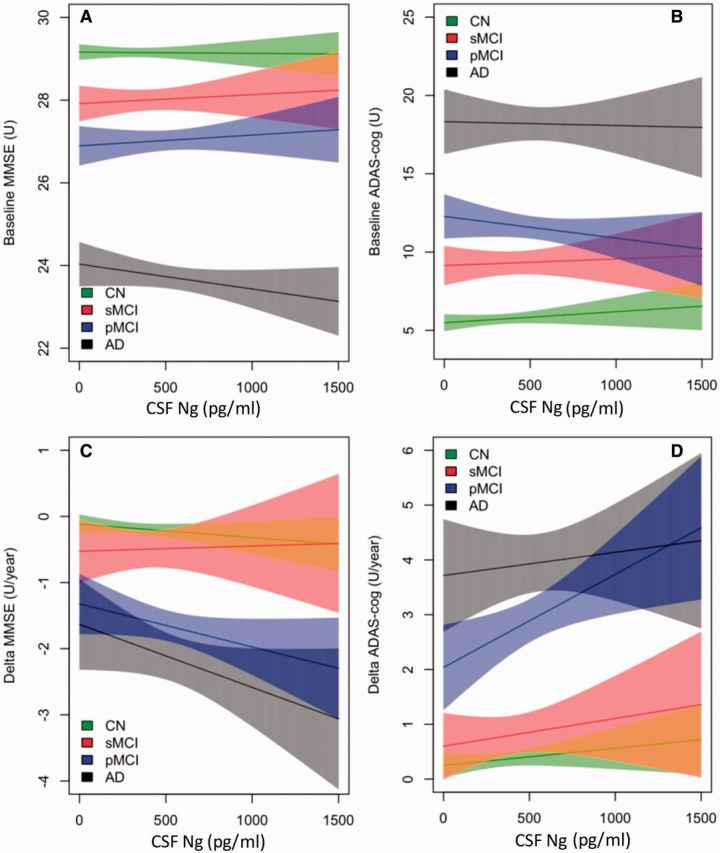
CSF neurogranin in relation to cognition and future cognitive change. MMSE and ADAS-Cog at baseline (A and B) and over time (C and D) as a function of baseline CSF neurogranin (Ng) in different diagnostic groups. Shaded areas indicate 95% confidence interval (CI) of the mean. CN = cognitively normal (green); sMCI = stable MCI (red); pMCI = progressive MCI (blue); AD = Alzheimer’s disease (grey).
CSF neurogranin in relation to brain structure and cognition
Next, we examined whether CSF neurogranin correlated with structural measures of hippocampal atrophy as measured with MRI or with functional measures of cortical glucose metabolism as measured using FDG-PET. CSF neurogranin was not associated with baseline hippocampal volume or FDG-PET in the Alzheimer’s disease dementia, progressive MCI, stable MCI or healthy control groups (Fig. 5A and B). However, CSF neurogranin was associated with rate of hippocampal atrophy during the follow-up period in the healthy control group (β = −0.016, P = 0.095) and the progressive MCI group (β = −0.017, P = 0.044) (Fig. 5C), whereas in the stable MCI and Alzheimer’s disease dementia groups, no such associations were found. In addition, CSF neurogranin was associated with rate of decrease in cortical glucose metabolism in the Alzheimer’s disease dementia group (β = −7.1 × 10−6, P = 0.014) (Fig. 5D).
Figure 5.
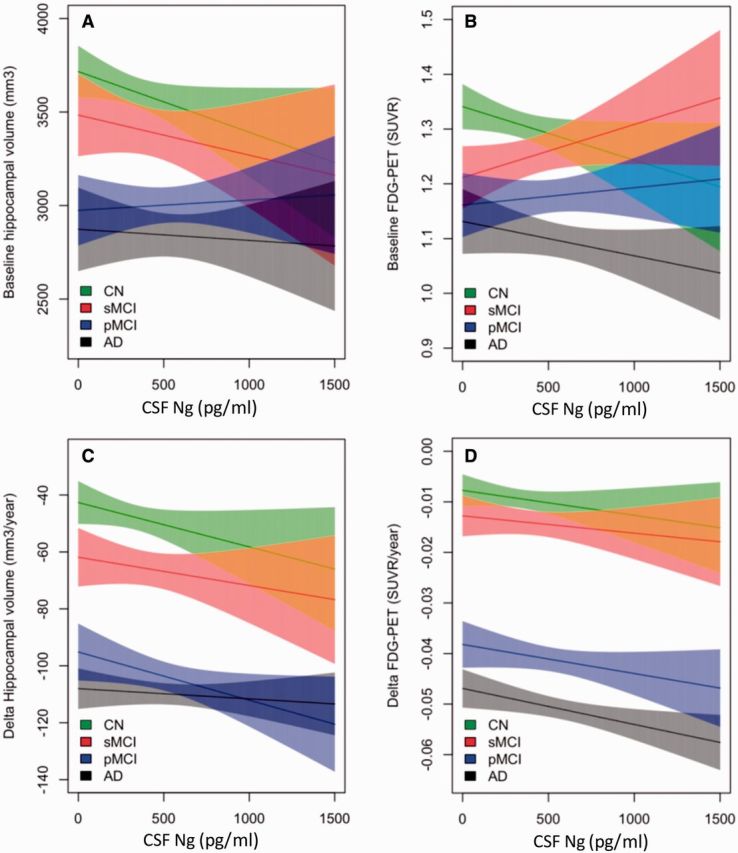
CSF neurogranin in relation to brain structure and cognition. Hippocampal volume and FDG-PET at baseline (A and B) and over time (C and D) as a function of baseline CSF neurogranin in different diagnostic groups. Shaded areas indicate 95% CI of the mean. CN = cognitively normal (green); sMCI = stable MCI (red); pMCI = progressive MCI (blue); AD = Alzheimer’s disease (grey).
CSF neurogranin as a predictor of disease progression
Based on the baseline CSF neurogranin levels, all subjects with an MCI diagnosis at baseline were classified into quartiles (low, low-medium, medium-high and high), which were tested for associations with disease progression as examined by longitudinal change in MMSE, ADAS-Cog, hippocampal volume, and cortical glucose metabolism. In a concentration-dependent manner, MMSE score decreased (P < 0.001 for all quartiles) over years from baseline for all quartiles (Q1 β = −0.366, Q2 β = −0.656, Q3 β = −0.834, Q4 β = −1.016), with a more pronounced decrease with higher baseline CSF neurogranin levels (Fig. 6A). A similar concentration-dependent change was evident for ADAS-Cog (P < 0.001 for all quartiles, Q1 β = 0.575, Q2 β = 0.811, Q3 β = 1.405, Q4 β = 2.275) (Fig. 6B). Expressed in an alternative way, the quartile of MCI subjects with highest CSF neurogranin decreased on average by 1.016 points on MMSE and increased 2.274 points on ADAS-Cog per year as compared with 0.366 and 0.575 points, respectively, for cases with the lowest neurogranin levels at baseline.
Figure 6.

CSF neurogranin as a predictor of disease progression. The patients were divided into quartiles according to CSF neurogranin levels at baseline. All trend lines were calculated using local regression. (A) Patients with higher levels of CSF neurogranin at baseline have faster disease progression over time as measured by change in MMSE scores. Quartile 1: n = 43, quartile 2: n = 43, quartile 3: n = 42 and quartile 4: n = 45. (B) Patients with higher levels of CSF neurogranin at baseline have faster disease progression over time as measured by change in ADAS-Cog scores. However, Q2 and Q1 overlap. Quartile 1: n = 43, quartile 2: n = 43, quartile 3: n = 42 and quartile 4: n = 45. (C) Generally, patients with higher levels of CSF neurogranin at baseline have faster disease progression over time as measured by hippocampal volume change. However, the patients with the highest levels of CSF neurogranin (Q4) progress slower than Q3. Quartile 1: n = 43, quartile 2: n = 42, quartile 3: n = 42 and quartile 4: n = 43. (D) Patients with higher levels of CSF neurogranin at baseline have faster disease progression over time as measured by change in FDG-PET signals. However, Q3 and Q4 overlap. Quartile 1: n = 19, quartile 2: n = 26, quartile 3: n = 21 and quartile 4: n = 22. SUVR=standardized uptake value ratio.
A more pronounced rate of loss of hippocampal volume was also found to depend on baseline CSF neurogranin levels, with high neurogranin predicting higher rate of atrophy (P < 0.001 for all quartiles, Q1 β = −66.0, Q2 β = −77.1, Q3 β = −107.4, Q4 β = −93.1) (Fig. 6C). Similarly, rate of decrease in cortical glucose metabolism as measured by FDG-PET depended on baseline CSF neurogranin levels (P < 0.001, Q1 β = −0.019, Q2 β = −0.022, Q3 β = −0.031, Q4 β = −0.036) (Fig. 6D). Thus, the quartile of MCI subjects with lowest CSF neurogranin at baseline lost 66.0 mm3 of hippocampal tissue volume per year, as compared with 93.1 mm3 for cases with the highest neurogranin levels at baseline.
The descriptive analysis above suggested that higher baseline CSF neurogranin was associated with deterioration in these four parameters (hippocampal volume, FDG-PET, MMSE and ADAS-Cog). To test this statistically we tested the interaction between CSF neurogranin and time in linear mixed models (adjusted for age and sex, and for education when analysing MMSE and ADAS-Cog, and intracranial volume when analysing hippocampal volume). The analyses showed that higher CSF neurogranin was significantly associated with longitudinal deterioration in all four parameters (hippocampal volume, βinteraction = −0.0364, P = 0.0051; FDG-PET, βinteraction = −0.0000335, P < 0.001; MMSE, βinteraction = −0.000862, P = 0.021; ADAS-Cog, βinteraction = 0.0024, P = 0.00020).
Discussion
Neuropathological studies have shown that synaptic loss is found in the very early stages of Alzheimer’s disease and may precede neuronal degeneration (DeKosky et al., 1990; Bertoni-Freddari et al., 1996; Masliah et al., 2001; Scheff et al., 2007). Even though there are several neuropathological characteristics of Alzheimer’s disease, including amyloid-β and tau pathology, synaptic pathology is not only a very early change, but is probably also the direct substrate of cognitive decline, showing a tight correlation with degree of cognitive symptoms and dementia (Davies et al., 1987; Blennow et al., 1996; Masliah et al., 2001; Sze et al., 1997). A biomarker reflecting this pathophysiologic process may be useful for studies of the underlying disease mechanisms contributing to Alzheimer’s disease, to predict progression of disease and may serve as a surrogate marker in treatment trials to monitor effects on reducing the rate of synaptic degeneration. Therefore, in the present study we evaluated CSF neurogranin levels in the ADNI cohort and investigated its performance to predict cognitive decline and indices of brain injury.
Neurogranin is highly expressed in the same grey matter brain regions that are affected in Alzheimer’s disease, i.e. the cerebral cortex, hippocampus, and amygdala, while it is almost absent in the thalamus, cerebellum, brainstem and the spinal cord (Represa et al., 1990). This synaptic protein is primarily expressed in excitatory neurons and is concentrated in distal dendrites and dendritic spines (Alvarez-Bolado et al., 1996), where it regulates calmodulin availability, thereby playing an important role in the regulation of synaptic signalling, long-term potentiation induction and memory consolidation (Baudier et al., 1991; Huang et al., 1993; Diez-Guerra, 2010). Consequently, this synaptic protein may thus serve as a biomarker to monitor an important aspect of Alzheimer’s disease molecular pathogenic mechanisms that previously has been difficult to assess in living patients.
CSF neurogranin was significantly increased in Alzheimer’s disease dementia and progressive MCI as compared with healthy control. These findings validate our previous study in which we showed an increase in CSF neurogranin levels in Alzheimer’s disease dementia and progressive MCI (Kvartsberg et al., 2014). In that study, we demonstrated that neurogranin is metabolized into a series of C-terminal peptides using hybrid immunoaffinity-mass spectrometry and that the endogenous peptide neurogranin48–76, which is the dominating endogenous peptide in human brain tissue, is significantly increased in Alzheimer’s disease compared to healthy control subjects (Kvartsberg et al., 2014). In two independent clinical cohorts including Alzheimer’s disease dementia and healthy control subjects, the samples were analysed by both hybrid immunoaffinity-mass spectrometry and an in-house developed immunoassay and the results were highly correlated in both clinical studies. In our previous study, we also showed that progressive MCI patients had increased CSF neurogranin levels compared to stable MCI (Kvartsberg et al., 2014), a finding that we confirmed in the present study. In conclusion, CSF neurogranin is significantly increased in Alzheimer’s disease dementia and progressive MCI as compared to healthy control and also in progressive MCI as compared to stable MCI.
Biomarkers for pathological changes that are present before overt clinical onset would be highly valuable in longitudinal clinical studies on Alzheimer’s disease. To investigate whether CSF neurogranin levels can be used to identify MCI patients with an underlying amyloid-β pathology, each group included in the study was dichotomized into either amyloid-positive (amyloid-β1–42 < 192 pg/ml) or amyloid-negative (amyloid-β1–42 > 192 pg/ml). As expected, the CSF neurogranin levels were significantly increased in the CSF amyloid-β-positive Alzheimer’s disease group. In addition, high CSF neurogranin levels were also found in the amyloid-positive progressive MCI group, suggesting that CSF neurogranin is an early pathophysiological marker of Alzheimer’s disease-related synaptic loss. This is further supported by the finding that the 32 healthy control subjects who converted to MCI or Alzheimer’s disease dementia during follow-up had increased CSF neurogranin levels compared to the other healthy control subjects and the finding that high CSF neurogranin correlated with a more rapid rate of cognitive decline in MCI patients.
When evaluating CSF biomarkers for Alzheimer’s disease, it is important to consider that there is a continuum of pathology, e.g. in amyloid plaque numbers, without any clear discrimination between patients with Alzheimer’s disease and cognitively intact elderly who died from other reasons (Curtis et al., 2015). Further, mixed pathology is very common in elderly patients with dementia. Indeed, elderly patients with Alzheimer’s disease have multiple pathologies; except for tangles and plaques a large proportion also have TDP-43 and α-synuclein as well as vascular pathologies (Kovacs et al., 2013). Cerebrovascular co-morbidities such as subtle hippocampal ischaemic injury and cerebral cortical microinfarcts cannot be consistently identified, even using high resolution MRI. This overlap in pathology may confound the diagnostic performance of a putative Alzheimer’s disease biomarker. In agreement with this reasoning, the best diagnostic separation for CSF neurogranin was found when dichotomizing the diagnostic groups into CSF amyloid-β-positive and negative subgroups.
The current data set also allowed us to examine the associations of CSF neurogranin levels with two other key biomarkers for Alzheimer’s disease: hippocampal atrophy rate measured by volumetric MRI and cortical glucose metabolism assessed by FDG-PET. We found associations between high CSF neurogranin levels and increased rate of hippocampal atrophy as well as reductions in cortical glucose metabolism, especially when testing the combined MCI group. Considering that synaptic dysfunction and loss probably is the main cause of the reduced cortical glucose metabolism in Alzheimer’s disease and that loss of neuropil (i.e. neurites, and pre-and postsynaptic elements) underlie hippocampal atrophy, we suggest that CSF neurogranin indeed is an independent novel biomarker for synaptic pathology in neurodegenerative diseases such as Alzheimer’s disease.
In conclusion, our findings support the use of CSF neurogranin as a biomarker for synaptic pathology in Alzheimer’s disease. CSF neurogranin was increased already in predementia stages of Alzheimer’s disease and higher concentrations correlated with a higher rate of cognitive deterioration, decreased glucose metabolism and higher hippocampal atrophy rates. We propose that CSF neurogranin levels are an independent measure of synaptic integrity which previously has been difficult to assess in living patients. Future studies may further explore this biomarker in relation to other aspects of Alzheimer’s disease, as well as in other neurodegenerative disorders, and for its potential roles in clinical research, trials and practice.
Funding
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the NIA, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. This research was also supported by NIH grants P30 AG010129 and K01 AG030514. The study was supported by the Swedish Research Council (project # 14002, K2010-63P-21562-01-4, K2011-61X-20401-05-6), the Knut and Alice Wallenberg Foundation, Stiftelsen Gamla Tjänarinnor, Magnus Bergvalls Stiftelse, Gun och Bertil Stohnes Stiftelse, Axel Linders stiftelse, the Swedish Brain foundation, the Alzheimer Foundation, Sweden, the Dementia Association, Sweden, the JPND Project BIOMARKAPD and the Swedish Brain Power consortium.
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- ADAS-Cog
Alzheimer’s Disease Assessment Scale–cognitive subscale
- ADNI
Alzheimer’s Disease Neuroimaging Initiative
- FDG
18F-fluorodeoxyglucose
- MCI
mild cognitive impairment
- MMSE
Mini-Mental State Examination
- PBS
phosphate-buffered saline
References
- Alvarez-Bolado G, Rodriguez-Sanchez P, Tejero-Diez P, Fairen A, Diez-Guerra FJ. Neurogranin in the development of the rat telencephalon. Neuroscience 1996; 73: 565–80. [DOI] [PubMed] [Google Scholar]
- Baudier J, Deloulme JC, Van Dorsselaer A, Black D, Matthes HW. Purification and characterization of a brain-specific protein kinase C substrate, neurogranin (p17). Identification of a consensus amino acid sequence between neurogranin and neuromodulin (GAP43) that corresponds to the protein kinase C phosphorylation site and the calmodulin-binding domain. J Biol Chem 1991; 266: 229–37. [PubMed] [Google Scholar]
- Bertoni-Freddari C, Fattoretti P, Casoli T, Caselli U, Meier-Ruge W. Deterioration threshold of synaptic morphology in aging and senile dementia of Alzheimer’s type. Anal Quant Cytol Histol 1996; 18: 209–13. [PubMed] [Google Scholar]
- Blennow K, Bogdanovic N, Alafuzoff I, Ekman R, Davidsson P. Synaptic pathology in Alzheimer’s disease: relation to severity of dementia, but not to senile plaques, neurofibrillary tangles, or the ApoE4 allele. J Neural Transm 1996; 103: 603–18. [DOI] [PubMed] [Google Scholar]
- Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 2010; 6: 131–44. [DOI] [PubMed] [Google Scholar]
- Curtis C, Gamez JE, Singh U, Sadowsky CH, Villena T, Sabbagh MN, et al. Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol 2015; 72: 287–94. [DOI] [PubMed] [Google Scholar]
- Davidsson P, Jahn R, Bergquist J, Ekman R, Blennow K. Synaptotagmin, a synaptic vesicle protein, is present in human cerebrospinal fluid: a new biochemical marker for synaptic pathology in Alzheimer disease?. Mol Chem Neuropathol 1996; 27: 195–210. [DOI] [PubMed] [Google Scholar]
- Davidsson P, Puchades M, Blennow K. Identification of synaptic vesicle, pre- and postsynaptic proteins in human cerebrospinal fluid using liquid-phase isoelectric focusing. Electrophoresis 1999; 20: 431–7. [DOI] [PubMed] [Google Scholar]
- Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J Neurol Sci 1987; 78: 151–64. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 1990; 27: 457–64. [DOI] [PubMed] [Google Scholar]
- Diez-Guerra FJ. Neurogranin, a link between calcium/calmodulin and protein kinase C signaling in synaptic plasticity. IUBMB Life 2010; 62: 597–606. [DOI] [PubMed] [Google Scholar]
- Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol 2014; 13: 614–29. [DOI] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002; 33: 341–55. [DOI] [PubMed] [Google Scholar]
- Fischl B, van der Kouwe A, Destrieux C, Halgren E, Segonne F, Salat DH, et al. Automatically parcellating the human cerebral cortex. Cereb Cortex 2004; 14: 11–22. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12: 189–98. [DOI] [PubMed] [Google Scholar]
- Gerendasy DD, Sutcliffe JG. RC3/neurogranin, a postsynaptic calpacitin for setting the response threshold to calcium influxes. Mol Neurobiol 1997; 15: 131–63. [DOI] [PubMed] [Google Scholar]
- Hayashi Y. Long-term potentiation: two pathways meet at neurogranin. EMBO J 2009; 28: 2859–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KP, Huang FL, Chen HC. Characterization of a 7.5-kDa protein kinase C substrate (RC3 protein, neurogranin) from rat brain. Arch Biochem Biophys 1993; 305: 570–80. [DOI] [PubMed] [Google Scholar]
- Huang KP, Huang FL, Jager T, Li J, Reymann KG, Balschun D. Neurogranin/RC3 enhances long-term potentiation and learning by promoting calcium-mediated signaling. J Neurosci 2004; 24: 10660–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging 2008; 27: 685–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Milenkovic I, Wohrer A, Hoftberger R, Gelpi E, Haberler C, et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol 2013; 126: 365–84. [DOI] [PubMed] [Google Scholar]
- Kvartsberg H, Duits FH, Ingelsson M, Andreasen N, Öhrfelt A, Andersson K, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlated with cognitive decline in prodromal Alzheimeŕs disease. Alzheimeŕs Dement 2014. http://dx.doi.org/10.1016/j.jalz.2014.10.009. [DOI] [PubMed] [Google Scholar]
- Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol 2012; 72: 578–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr, et al. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 2001; 56: 127–9. [DOI] [PubMed] [Google Scholar]
- Mattsson N, Insel PS, Landau S, Jagust W, Donohue M, Shaw LM, et al. Diagnostic accuracy of CSF Ab42 and florbetapir PET for Alzheimer’s disease. Ann Clin Transl Neurol 2014; 1: 534–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci 2008; 1147: 180–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak JH, Huang FL, Li J, Balschun D, Reymann KG, Chiang C, et al. Involvement of neurogranin in the modulation of calcium/calmodulin-dependent protein kinase II, synaptic plasticity, and spatial learning: a study with knockout mice. Proc Natl Acad Sci USA 2000; 97: 11232–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Represa A, Deloulme JC, Sensenbrenner M, Ben-Ari Y, Baudier J. Neurogranin: immunocytochemical localization of a brain-specific protein kinase C substrate. J Neurosci 1990; 10: 3782–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry 1984; 141: 1356–64. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007; 68: 1501–8. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol 2009; 65: 403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol 1997; 56: 933–44. [DOI] [PubMed] [Google Scholar]
- Thorsell A, Bjerke M, Gobom J, Brunhage E, Vanmechelen E, Andreasen N, et al. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer’s disease. Brain Res 2010; 1362: 13–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.