

Abstract
Chronic respiratory disorders are important contributors to the global burden of disease. Genome-wide association studies (GWASs) of lung function measures have identified several trait-associated loci, but explain only a modest portion of the phenotypic variability. We postulated that integrating pathway-based methods with GWASs of pulmonary function and airflow obstruction would identify a broader repertoire of genes and processes influencing these traits. We performed two independent GWASs of lung function and applied gene set enrichment analysis to one of the studies and validated the results using the second GWAS. We identified 131 significantly enriched gene sets associated with lung function and clustered them into larger biological modules involved in diverse processes including development, immunity, cell signaling, proliferation and arachidonic acid. We found that enrichment of gene sets was not driven by GWAS-significant variants or loci, but instead by those with less stringent association P-values. Next, we applied pathway enrichment analysis to a meta-analyzed GWAS of airflow obstruction. We identified several biologic modules that functionally overlapped with those associated with pulmonary function. However, differences were also noted, including enrichment of extracellular matrix (ECM) processes specifically in the airflow obstruction study. Network analysis of the ECM module implicated a candidate gene, matrix metalloproteinase 10 (MMP10), as a putative disease target. We used a knockout mouse model to functionally validate MMP10's role in influencing lung's susceptibility to cigarette smoke-induced emphysema. By integrating pathway analysis with population-based genomics, we unraveled biologic processes underlying pulmonary function traits and identified a candidate gene for obstructive lung disease.
Introduction
Spirometric measurement of pulmonary function is the most commonly used method to assess the lung's physiologic and pathophysiologic state. Forced expiratory volume in the first second (FEV1) and its ratio to forced vital capacity (FEV1/FVC) are heritable traits that reproducibly measure airflow obstruction and predict morbidity and mortality in the general population (1,2). Impairment of FEV1 and FEV1/FVC are key criteria for the diagnosis of chronic obstructive lung disease (COPD)—a highly morbid condition predicted to become the third leading cause of death worldwide by 2030 (3). We have previously reported on two independent, large genome-wide association studies (GWASs) of pulmonary function, each comprising over 20 000 individuals of European ancestry (4,5). These studies, in combination with our recent results based on larger joint meta-analyses, have identified multiple loci associated with pulmonary function (6,7) and airflow obstruction (8). However, genetic variants at these regions explain <5% of the observed variance in spirometric traits (6), strongly implying that a substantial portion of the available genetic contribution to variability in lung function is not identified using standard GWAS statistical thresholds.
One promising approach to uncover the genetic underpinnings of complex phenotypes is to reach beyond genome-wide significant hits to extract information from the entire association dataset and discover sets of pathways linked to a given clinical trait (9). Genes do not exert their effects in isolation, but rather cooperate within pathways and larger biologic modules to influence disease susceptibility and progression (10,11). Pathway-based analysis is built on the premise that complex, polygenic phenotypes arise from distinct molecular pathways, and that small genetic perturbations (i.e. single-nucleotide polymorphisms or SNPs) at multiple sites within these gene sets can lead to the observed traits and diseases. This paradigm allows implementation of statistical tests that substantially relax cutoffs for statistical significance, such that a pathway can be enriched in a phenotype even though the individual SNPs do not achieve genome-wide significance. Several publications have applied pathway-based approaches to previously generated GWAS data and demonstrated the power of this method in identifying previously undetected mechanisms (12–14).
This study was designed to test the hypothesis that the genetic basis of pulmonary function and airflow obstruction is orchestrated by identifiable sets of functionally coherent pathways. Initially, we implemented a robust methodology based on gene set enrichment analysis (GSEA) (15,16) to identify and validate pathways linked to lung function by step-wise leveraging of two independent pulmonary function GWAS datasets. We then applied this procedure to the largest available GWAS of airflow obstruction (8). Our integrative approach systematically mapped pathways associated with lung function and airflow obstruction to functionally distinct modules, and lead to the identification of a putative COPD candidate.
Results
Staged analysis of lung function GWAS identifies and confirms a large repertoire of enriched gene sets
We performed two separate GWASs of pulmonary function (FEV1 and FEV1/FVC) using two large consortia—CHARGE and SpiroMeta. Cohort details are provided in Supplementary Material, Table S1. Quantile–quantile (QQ) plots of observed versus expected association P-values showed significant deviations from the null hypothesis for each lung function measure, indicating that many SNPs were strongly linked to these traits (Supplementary Material, Fig. S1). As outlined in Figure 1, after completing standard GWAS for each consortium, we initially applied GSEA to the CHARGE pulmonary function GWAS and identified 444 overrepresented gene sets associated with FEV1 or FEV1/FVC (FDR < 0.05). We validated these findings by implementing the same procedure for the SpiroMeta GWAS. Of the 444 pathways identified in CHARGE, 131 were also enriched in SpiroMeta (FDR < 0.05). Further analysis was restricted to these 131 gene sets that were significantly enriched in both independent studies. A complete list of consortium-specific gene sets is available in Supplementary Material, Table S2. To assess robustness of our findings, we performed an identical two-step pathway enrichment method using a different program known as GSA-SNP (17) and found broad overlap between processes enriched in both approaches (see Supplementary Material, Table S4).
Figure 1.

Outline of the staged approach for pathway analysis of lung function. We performed standard GWAS of pulmonary function measures in two large, independent consortia: CHARGE and SpiroMeta. Gene set enrichment analysis was initially applied to the CHARGE consortium's pulmonary function GWAS (Step 1), and subsequently performed on SpiroMeta's GWAS (Step 2). Only pathways that were significantly enriched in Step 1 and replicated in Step 2 were used for further analysis.
Enrichment of pathways is driven by SNPs moderately associated with pulmonary function
To assess the relative contribution of SNPs to gene set enrichment, we performed sensitivity analyses to verify that our findings were not due to a disproportionate contribution by a few, highly significant SNPs. First, we repeated GSEA after excluding all GWAS-significant SNPs (i.e. those with association P < 5 × 10–8). No differences were found in the list of enriched pathways. Next, we expanded our analysis by additionally excluding all SNPs (regardless of P-value) within 100 Kb in either direction from all GWAS-significant loci. Again, we did not observe any appreciable change in the enrichment profile of gene sets. While genes in enriched pathways included several GWAS-significant loci, most genes were mapped to SNPs with moderate association P-values ranging from 10–2 to 10–6 (Supplementary Material, Fig. S2). These observations imply that a rich repository of genomic information rests within a stratum of association P-values that is ignored in standard GWAS procedures.
Pathway-associated SNPs are enriched in DNase I hypersensitive sites
We investigated whether sentinel SNPs associated with lung function in enriched pathways were overrepresented in regulatory regions as represented by DHSs. We found highly significant enrichment of DHSs among pathway-associated SNPs in relative to all lung function GWAS SNPs [odds ratio 1.86, 95% confidence interval (1.73–2.00), P-value 1.75 × 10–68]. A similar analysis of SNPs linked to enriched pathways associated with airflow obstruction revealed significant overrepresentation of DHSs relative to all airflow obstruction GWAS SNPs [odds ratio 1.65, 95% confidence interval (1.51–1.80), P-value 3.39 × 10–15].
Lung function-associated pathways aggregate within distinct biologic modules
We applied unsupervised cluster analysis to the enriched pathways and their associated genes to determine segregation patterns based on membership profiles and association P-values. We defined a module as a group of two or more gene sets (pathways) that clustered together based on sharing common gene members. We found that a majority of enriched gene sets mapped to biologically distinct functional modules (Fig. 2)—including processes involved in Development, Cell Adhesion, Cell Proliferation/Migration, Cell Signaling, Immunity, Ion Channel/Transport and Arachidonic Acid/Prostaglandin. However, most of the identified modules also shared member genes between them, with particularly large overlaps among Development, Immunity, Adhesion, Cell Signaling and Proliferation/Migration (Fig. 3).
Figure 2.

Unsupervised hierarchical clustering of enriched pathways (gene sets) associated with pulmonary function. Rows representing enriched gene sets (n = 131) were grouped based on overlapping gene membership (columns). Gene sets with many shared genes clustered together and define functional modules (e.g. cell signaling, immunity and development). Columns represent gene membership profiles of enriched pathways and are colored according to association P-values of member genes. Note that for any given gene set, the majority of pathway-associated genes are not members and are displayed as black bars. For each clustered group (module), only representative pathways have been labeled. Complete list is available in Supplementary Material, Table S2.
Figure 3.

Wiring diagram of key biologic modules associated with lung function. These modules were derived from membership cluster analysis of enriched gene sets (Fig. 2). Intermodular connectivity indicates overlapping genes between modules with the thickness of connections drawn proportional to the number of shared genes—for example, development and immunity have 150 gene members in common.
Published literature strongly links enriched gene sets derived from pulmonary function GWAS to lung biology
To assess whether the identified pulmonary function-associated pathways and genes were implicated in lung biology, we systematically searched each pathway and gene via a PubMed mining tool (PubMatrix) using the modifier term ‘pulmonary function’. As summarized in Supplementary Material, Figure S3, every overrepresented gene set had published reports for a role in pulmonary biology, with immune system having the largest supporting evidence (n = 57 847 citations) and several other pathways with significant publication records such as free radical pathway (n = 14 888), morphogenesis (n = 7324), epidermal growth factor signaling (n = 1388) and Wnt signaling (n = 487). Many of the pathway-associated genes were also highly cited, including tumor necrosis factor (n = 8961), interleukin 8 (IL8, n = 4376), epidermal growth factor receptor (n = 3549), transforming growth factor β1 (n = 1758) and cystic fibrosis transmembrane conductance regulator (n = 1630). Several genetic loci identified in our previous lung function GWASs were members of enriched pathways and cited in PubMed including advanced glycosylation end product-specific receptor (n = 196), hedgehog-interacting protein (n = 19) and patched 1 (n = 15).
Pathway analysis of airflow obstruction GWAS reveals many enriched gene sets including those involved in tissue remodeling
The promising results from gene set analysis of pulmonary function motivated us to expand this approach to airflow obstruction—a clinically relevant spirometric phenotype characteristic of obstructive lung disorders such as COPD. Applying the same computational procedures to the airflow obstruction GWAS, we identified 156 enriched gene sets at FDR < 0.001 (Supplementary Material, Table S3). Unlike the two distinct lung function GWASs, the airflow obstruction GWAS was a combined analysis of CHARGE and SpiroMeta cohorts and therefore a two-step validation strategy was impractical. Instead, we minimized false-positive findings by applying a highly stringent FDR cutoff to designate significance.
We grouped enriched gene sets into biologic modules based on membership and functional overlap. The results are summarized in Figure 4 and provide an overview of key pathways and processes associated with airflow limitation. Since most of the airflow obstruction cohorts had also participated in the lung function GWASs and airflow obstruction is defined based on spirometry, it was not surprising that many of these enriched modules were also identified in the pulmonary function analysis, including Development, Cell Signaling, Ion Channel, Cell Adhesion and Proliferation. However, we also identified pathways that were not enriched in the lung function analysis (Supplementary Material, Table S3). Prominent among these airflow obstruction-associated gene sets were processes involved in remodeling of the extracellular matrix (ECM), such as collagen, proteinaceous ECM and integrin pathway. Since airway remodeling is a key pathophysiologic characteristic of COPD, we further explored the ECM module by mapping relationships among its members using a comprehensive gene product interaction knowledgebase (Ingenuity) (18). This analysis identified several distinct networks within the ECM module, with the highest scoring network being comprised of 21 interconnected focus genes as depicted in Figure 4 (see Supplementary Material, Table S6). Since nodal connectivity in disease networks is a topologic property that can indicate biologic importance (19,20), we selected the most connected node in the ECM network, MMP10, as a candidate for further functional validation.
Figure 4.
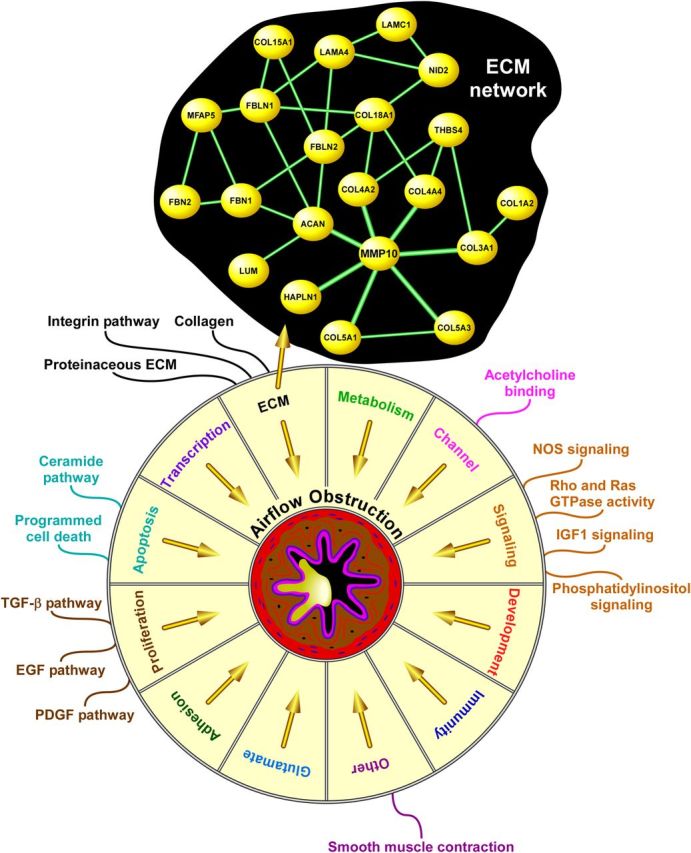
Graphical overview of enriched biological modules associated with airflow obstruction. Since the airflow obstruction GWAS was primarily based on the pulmonary function GWASs, several of these modules overlap with those identified in the lung function analysis (Fig. 3). However, some modules such as apoptosis and ECM were enriched only in airflow obstruction. Furthermore, even within common biologic modules, substantial differences in enriched gene sets were observed between the two phenotypes with several representative pathways associated with airflow obstruction being highlighted. Deeper exploration of the ECM module using gene product interaction network analysis identified MMP10 as the most interconnected node and a putative driver of ECM processes influencing airflow obstruction. NOS, nitric oxide synthase; IGF1, insulin-like growth factor 1; TGF-β, transforming growth factor beta; EGF, epidermal growth factor; PDGF, platelet-derived growth factor. Complete list is available in Supplementary Material, Table S3.
MMP10 is a COPD candidate gene
We assessed the role of MMP10 in obstructive lung disease by genetically targeting this gene in an established animal model of emphysema. After chronic exposure to cigarette smoke, wild-type mice developed extensive airway and airspace destruction characteristic of emphysema, whereas Mmp10–/– animals displayed only modest injury (Fig. 5A). Morphometric analysis of acinar airspaces using the mean linear intercept method confirmed the protective phenotype observed in Mmp10-null mice (Fig. 5B). To complement the morphometric assessment of lung injury in our model, we measured the expression of interleukin-1 beta (Il1b) and Mmp10. Il1b is a pro-inflammatory cytokine upregulated in the lungs patient with COPD and a robust biomarker for frequent exacerbations (21). Overexpression of Il1b causes abnormal airway remodeling and emphysema in mice (22). We found that Il1b was significantly increased in the lung of wild-type animals after chronic cigarette smoke injury but not in the emphysema-resistant Mmp10-null mice (Fig. 5C). Mmp10 expression was similarly upregulated in wild-type mice with emphysema, but remained undetectable in the knockouts as expected (Fig. 5C). Collectively, these observations implicate a pathophysiologic role for MMP10 in the development of cigarette smoke-induced lung disease.
Figure 5.

Mmp10–/– mice are resistant to cigarette smoke-induced emphysema. (A) Representative H&E lung sections of Mmp10–/– and wild-type animals exposed to 6 months of chronic cigarette smoke demonstrated extensive injury in wild types with significant loss of acinar and small airway structures (top row). In contrast, Mmp10-null mice were substantially less susceptible to smoke-induced emphysema (bottom row). Slides are ordered according to severity of lung injury in each genotype. (B) Morphometric analysis of airspaces using the mean linear intercept method confirmed the protective phenotype observed in Mmp10–/– mice (n = 10 for smoke exposed Mmp10–/– mice, n = 9 for all other groups). (C) Wild-type mice exposed to chronic cigarette smoke have significantly increased expression of the pro-inflammatory cytokine Il1b as well as Mmp10 in their lungs, whereas emphysema-resistant Mmp10–/– animals do not (n = 8 per group). All P-values based on two-tailed Student's t-test; NS, not significant.
Discussion
Understanding the genetic basis of normal and impaired lung function can provide new insights into the pathophysiology of pulmonary disorders. Chronic respiratory conditions are a leading cause of death worldwide (3) and there is a recognized need for new therapeutic targets (23). In this work, we initially leveraged two large, independent GWAS datasets totaling almost 50 000 subjects to systematically identify pathways associated with lung function. We found a diverse set of interconnected biologic modules linked to this trait. We extended this approach to airflow obstruction—a clinically relevant phenotype defined by spirometry—and identified additional pathways associated with obstructive lung disease. We functionally validated our approach by demonstrating that MMP10, a member of an enriched airflow obstruction module (i.e. ECM), influences the severity of cigarette smoke-induced emphysema using a genetic mouse model.
To date, over 2000 GWASs encompassing hundreds of complex traits and diseases have been published (24), with several pertaining to pulmonary phenotypes including asthma (25–27), idiopathic pulmonary fibrosis (28), COPD (29,30), sarcoidosis (31) and lung function (4,5,32). All of these studies have applied Bonferroni-type statistical cutoffs to minimize false-positive SNP-to-phenotype associations. For example, our previous pulmonary function GWASs identified numerous loci associated with FEV1, FEV1/FVC, FVC and airflow obstruction (4,6,8,32). We believe that those findings, while novel, did not comprehensively capture the complex biological processes underlying these traits because the standard GWAS approach is overly conservative and discards biologically informative yet statistically modest associations. Enrolling larger cohorts can increase the number of loci (6), but retains the individual SNP-focused structure of standard GWAS. To overcome this limitation, pathway-focused approaches can complement standard GWAS and allow deeper mining of genomic data (9). A key advantage of pathway enrichment methodologies is their ability to place trait-associated candidates within the context of biologically meaningful pathways and modules. However pathway approaches have important limitations, including the risk of false-positive findings and challenges in confirming results, especially at a phenotypic level (33).
In this work, we aimed to circumvent some of these shortcomings by implementing a step-wise gene set enrichment approach, whereby findings from one pulmonary function GWAS were confirmed using an independent and similarly powered GWAS (Fig. 1). We interrogated ∼2000 gene sets, but our two-step validation substantially narrowed down these pathways to a limited subset of 131 enriched pathways associated with pulmonary function. Since pathway selection was filtered by FDR cutoffs (<0.05) at each stage, false-positive findings were stringently controlled. Our approach of leveraging two independent analyses addresses instabilities reported during attempts to replicate gene set enrichment findings (34). The airflow obstruction GWAS was a combined study using cohorts from both CHARGE and SpiroMeta, precluding implementation of the step-wise GSEA strategy. Therefore, we applied a much stricter FDR threshold (<0.001) to identify significantly enriched gene sets and minimize false-positive associations. In both the lung function and airflow obstruction GSEA, the number of pathways identified as being enriched was <8% of the total gene sets surveyed, implying that the vast majority of the curated pathways were not significantly associated with these traits.
Our comprehensive, pathway-focused analysis of pulmonary function GWAS yielded several important results. First, we observed that SNPs driving gene sets linked to lung function were primarily those with moderate associations and not the GWAS-significant variants. These modestly significant SNPs did not reach genome-wide threshold, implying that standard GWAS analysis fails to capitalize on a substantial segment of the available genomic information (9) (Supplementary Material, Fig. S2). Secondly, enriched pathways clustered to a limited set of distinct modules with specific functional roles, including development, cell signaling and immunity (Fig. 2). The concept of a modular network built from specialized yet interconnected pathways is a fundamental property of complex biological systems (10,19). Our findings were consistent with this paradigm and show that each lung function-associated module—while comprised of functionally similar gene sets—also shared many gene members with other modules (Fig. 3). We corroborated the biologic relevance of the enriched gene sets by systematically literature-mining each term and observed that all pathways and many of their gene members had published links to ‘pulmonary function’, in some cases numbering in the thousands (Supplementary Material, Fig. S3). Furthermore, many SNPs linked to gene members of enriched pathways were within regulatory DHSs, implying that our approach captured functionally relevant genetic variants.
Pathway-based analysis of airflow obstruction GWAS revealed multiple processes potentially involved in the pathogenesis of COPD. While many of the airflow obstruction modules were similar to those associated with lung function, there were substantial differences between their respective gene sets (Supplementary Material, Tables S2 and S3). Several of these pathways such as transforming growth factor-β and phosphatidylinositol signaling have been linked to the development of obstructive lung disease (35–37). Another striking difference was selective enrichment of gene sets involved in ECM. This finding is biologically meaningful because COPD is characterized by structural destruction of respiratory acini and ECM remodeling of small airways (38). The network analysis of this module suggested that MMP10, a densely connected node, is a potential driver of ECM remodeling in airflow obstruction, and hence we chose to study its role further (Fig. 4).
MMPs are a family of ECM-associated proteins involved in diverse processes, including response to injury and inflammation, tumor metastasis and remodeling (39–41). Our finding that MMP10 may be associated with development of airflow obstruction is consistent with other reports implicating MMPs in COPD (30). For example, a functional variant in MMP12 has been associated with reduced risk of airflow obstruction in smokers (42), and mice lacking this gene are protected from cigarette smoke-induced emphysema (43). Furthermore, MMP10 was reported to be differentially expressed in airway and surrounding lung parenchyma of COPD patients (44).
It is important to note that in our discovery lung function GWAS the sentinel SNP in MMP10 (rs11225413) was far from being genome-wide significant (P = 0.03) and would have been excluded if standard, Bonferroni-type adjustments had been applied. A joint SNP and SNP-by-smoking meta-analysis of this SNP and other MMP10-associated variants did not reveal significant gene–environment interactions (7,45). Interestingly, integrating network analysis with pathway-based data mining of GWAS placed MMP10 within the context of other putative candidates. For example, another member of the ECM network (Fig. 4), Fibrillin 1, has been linked to development of early emphysema in humans (46), and mice with targeted knockout of this gene develop spontaneous emphysema (47). Finally, our functional validation of MMP10's role using an animal model of emphysema serves as a proof-of-concept that pathway-based approaches have the potential to reveal disease-associated processes and identify novel targets in complex disorders.
Our study has several limitations. We assumed that a given SNP affects the function of its proximally located gene. This is likely an over-simplification of the biologic effects of genetic variants since distal regulation can also occur (48). However, there is no accepted method to comprehensively assess the global influence of SNPs on gene function in humans. Analysis of expression quantitative trait loci (eQTL) can be useful for functional validation (49,50), but is based solely on the transcriptional effects of genetic variants, ignoring any post-transcriptional consequences. Furthermore, eQTLs exhibit significant tissue and even cell-type specificity (51,52) and require large sample sizes to achieve adequate power. Not surprisingly, many significant trait-associated SNPs do not have strong eQTLs. Thus, for our GSEA, we opted to not filter candidate SNPs based on limited eQTL information. However, we observed highly significant enrichment of DHSs among pathway-associated SNPs, implying overrepresentation of functional variants in our approach. Another inherent shortcoming of pathway-based methods is their reliance on known biologic processes and gene functions, impairing their ability to discover novel mechanisms or relationships. Nevertheless, previously unrecognized processes can be implicated in a given trait since enriched biologic modules are placed within a phenotype-specific context without bias. Our literature-mining effort was, by definition, based on available knowledge. In future studies, it will be of particular interest to investigate members of enriched gene sets that did not have published evidence of involvement in lung biology. Finally, since irreversible airflow obstruction is a required criterion for diagnosis of COPD and we did not assess bronchodilator response in our spirometric measurements, we have refrained from labeling individuals with airflow obstruction as having COPD.
In conclusion, by integrating pathway analysis with multiple GWASs, we have comprehensively mapped processes influencing lung function and airflow obstruction. While many of the identified gene sets have been previously linked to pulmonary biology in animal models or limited human tissue samples, our approach derived its genomic information from large human cohorts, making its findings broadly relevant to the general population. The proposed framework, therefore, may have particular applicability in dissecting disease mechanisms in complex lung diseases with available GWAS.
Materials and Methods
GWAS of lung function traits
CHARGE and SpiroMeta cohorts were independently meta-analyzed for GWAS of pulmonary function. All subjects were of European ancestry and underwent spirometry to assess lung function based on forced expiratory volume in 1 s (FEV1) and its ratio to forced vital capacity (FEV1/FVC). In the first phase, we analyzed data from seven cohorts totaling 25 366 subjects within the CHARGE consortium. For the validation step, we meta-analyzed data from 17 cohorts with 24 583 individuals within the SpiroMeta consortium.
The analysis of airflow obstruction was based on a combined GWAS meta-analysis of 14 CHARGE and SpiroMeta cohorts totaling 31 567 participants of European ancestry (3056 affected, 28 511 unaffected) (8). These participants constituted a subset of the total subjects analyzed in the CHARGE and SpiroMeta lung function GWASs described above. We used standardized definitions of airflow obstruction based on the lower limit of normal for FEV1 and FEV1/FVC from NHANES III prediction equations (53) across all cohorts. The presence of airflow obstruction was defined as an FEV1 and FEV1/FVC both less than the lower limit of normal (54), which is calculated from gender-specific equations for age, age2 and height2. Unaffected participants were defined by FEV1, FVC and FEV1/FVC all above the lower limit of normal. Individuals below the lower limit of normal for either FEV1 or FEV1/FVC but not both were excluded from these analyses. Therefore, unlike lung function, the presence of airflow obstruction was defined dichotomously.
GWAS procedures
Genotyping, imputation, genotype-phenotype association and meta-analysis procedures for lung function and airflow obstruction in CHARGE and SpiroMeta consortia have been previously described (4,5,8). Non-genotyped SNPs were imputed using MACH, IMPUTE or BIMBAM. Linear regression for age, age2, sex, height and ancestry principal components as covariates was performed on FEV1 (mm) and FEV1/FVC (%). Residuals were ranked and then transformed to z-scores and used for association testing under an additive genetic model stratified by ever-smoking and never-smoking status. Effect estimates were meta-analyzed using inverse-variance weighting across the cohorts in each consortium using R (version 2.9.2) or METAL (55) and genomic control correction (56) was applied. In the airflow obstruction GWAS, for each cohort logistic regression models were adjusted for current and former smoking dummy variables, pack-years of smoking, age, sex, standing height, center/cohort as needed, and principal components for genetic ancestry as needed. While heterogeneity was observed across cohorts, implementation of a fixed effects model was effective in extracting homogeneous findings. Genome-wide meta-analyses were performed using METAL with inverse-variance weighting to combine effect size estimates after applying a genomic control correction. QQ plots of expected and observed association P-values on a –log10 scale for CHARGE and SpiroMeta FEV1 and FEV1/FVC GWASs was created using local scripts in R (http://www.R-project.org/).
Gene set enrichment analysis of GWAS
We applied a methodology called improved GSEA for GWAS (i-GSEA4GWAS) to place variants associated with pulmonary function or airflow obstruction within curated pathways (16). Genotyped and imputed SNPs from lung function GWAS (n ≈ 2.5 × 106) were mapped to genes within a 100 kb distance (upstream or downstream). No filtering of SNPs based on LD structure or association P-value was performed prior to the enrichment analysis. For a given SNP, if multiple genes were located within this range, the closest gene was selected and assigned the association P-value. The SNP with the strongest association P-value was used to represent a gene. Since multiple SNPs can map to the same gene, an SNP label permutation was used to reduce potential biases caused by larger loci having disproportionately higher number of SNPs. Log-transformed association P-values (–log10P) were used to rank order the resulting gene list (∼18 000 genes) and calculate gene set enrichment scores. The i-GSEA4GWAS procedure calculates a significance proportional enrichment score that is based on the proportion of significant genes mapped to a given gene set relative to the proportion of significant genes across total genes in the GWAS. Approximately 2000 gene sets were obtained from the Molecular Signatures Database (http://www.broadinstitute.org/gsea/msigdb) (15,57). To maximize biologic relevance, gene sets were defined and limited to well-curated pathways derived from multiple resources such as KEGG, BioCarta, REACTOME and functional annotations extracted from the Gene Ontology database. Therefore, the terms ‘gene set’ and ‘pathway’ are used interchangeably.
Step-wise validation of functional enrichment analysis for pulmonary function
A two-step approach was taken to independently validate enriched pathways associated with lung function. The i-GSEA4GWAS algorithm was initially applied to the CHARGE pulmonary function GWAS and enriched gene sets were identified if they met an FDR < 0.05 for either FEV1 or FEV1/FVC. Next, the same procedure was implemented in the SpiroMeta consortium GWAS for FEV1 and FEV1/FVC. We restricted further analysis to those enriched pathways in CHARGE (FDR < 0.05) that were also significantly enriched in SpiroMeta (FDR < 0.05). Since these two large cohorts are independent, this requirement ensured strict control of false-positive findings. For the airflow obstruction pathway analysis, we used the available combined CHARGE and SpiroMeta meta-analyzed GWAS (8) and chose a much more stringent FDR < 0.001 to designate significant gene set enrichment.
Sensitivity analyses of gene set enrichment
Since several distinct analytical methods exist for pathway enrichment analysis and applying different algorithms to the same GWAS data may yield different results, we compared the performance of i-GSEA4GWAS against another pathway-based approach known as GSA-SNP (17). There are fundamental differences between the two methods, including the statistical framework used to assess enrichment as well as the size and content of the gene sets. We used the default z-statistic for enrichment and second-best SNP to randomly associated signals in GSA-SNP. We queried Gene Ontology and KEGG databases for gene sets. We performed an identical, two-stage analysis on CHARGE followed by SpiroMeta lung function GWASs using GSA-SNP. We identified over 300 enriched processes (at FDR < 0.05 in CHARGE that were also significant at FDR < 0.05 in SpiroMeta). While this was a larger number than identified using iGSEA4GWAS (n = 131), it likely reflected the fact that GSA-SNP sampled a much larger number of gene sets, whereas the proportion of identified significant gene sets is similar between the two approaches. Approximately one-quarter of the pathways identified by iGSEA4GWAS (32 of 131) were identical to those identified as significant by GSA-SNP even though iGSEA4GWAS included several data resources not sampled in GSA-SNP. Overall, there was substantial overlap between the two methods when the enriched gene sets were grouped based on broader functional modules as defined in Figures 2 and 3. A detailed comparison between the enriched gene sets identified by each approach is provided in Supplementary Material, Table S4.
We did not filter SNPs based on LD structure prior to initiating enrichment analysis in order to retain genome-wide coverage and prevent loss of information. The statistical structure of iGSEA4GWAS is based on associating a single sentinel SNP with its proximal gene locus (100 Kb window) without being influenced by its association with other potential SNPs in LD. Nevertheless, it was possible that the selected sentinel SNPs themselves may be in LD. Therefore, we systematically assessed pairwise LD between all 3307 pathway-associated SNPs using SNAP (https://www.broadinstitute.org/mpg/snap/ldsearchpw.php) at r2 thresholds of 0.2, 0.5 and 0.8. We found limited evidence for LD between the sentinel SNPs (Supplementary Material, Table S5).
Next, we repeated the entire iGSEA4GWAS analysis for lung function using a wider window (1 Mb versus 100 Kb) to associate proximal SNPs with loci. We observed over 70% identical matching between enriched pathways using the wider window, suggesting that the SNP to gene locus selection approach is robust to a range of selected windows. This observation is also consistent with our above finding that most pathway-associated SNPs are not in LD.
Cluster analysis
Two-way unsupervised hierarchical clustering was performed on enriched pathways in lung function based on the membership profile of gene sets and their associated genes' log-transformed P-values (–log10P) using Pearson's correlation metric (58). In this approach, gene sets were initially labeled by the presence or absence of any lung function-associated genes and then clustered together based on shared gene members to form larger, functionally coherent pathway groupings defined as ‘modules’.
Literature mining
We used PubMatrix (59), an online multiplex comparison tool for querying ‘search’ and ‘modifier’ terms within PubMed, to index published literature on the role of enriched gene sets and their associated gene members in influencing lung function. The search terms were either pathway-associated gene symbols (n = 3878) or pathway names (n = 131), and the modifier term was ‘pulmonary function’.
DNAse I hypersensitive site enrichment analysis
The frequency that SNPs selected for pathway analysis fell within DHS sites was compared with the frequency that all SNPs analyzed in the GWAS were located in DHSs. All SNPs were converted to their UCSC hg19 assembly positions using liftOver (http://genome.ucsc.edu). Sentinel SNPs associated with enriched pathways (one SNP per gene) and entire GWAS sets were intersected with the complete set of DHS hotspot regions (FDR < 0.05) identified in any of the 349 tissue or cell line samples available from Maurano et al. (48). The intersection was calculated using the BEDOPS software (60). The enrichment P-values were calculated using Fisher's exact test based on the total probability of two-tailed test. The frequency of lung function pathway-associated SNPs was compared with the union set of SNPs analyzed in the CHARGE and SpiroMeta lung function GWASs. The airflow obstruction pathway-associated SNPs were compared relative to all SNPs in the airflow obstruction GWAS.
Network analysis
We imported all 89 ECM-associated genes into Ingenuity software and used its network-generating algorithm to develop interaction networks built around these ‘focus genes’ using Ingenuity's knowledge base. We used only direct gene product interactions to link nodes, and excluded any non-focus genes added by the algorithm to grow the network. Networks were ranked based on Fisher's exact test of enrichment relative to networks generated from randomly selected genes from Ingenuity's knowledge base (Supplementary Material, Table S6). Subsequent analysis was based on the highest ranked ECM network that included the largest number of focused genes (n = 23 ECM-associated genes of which 21 had direct interactions with each other).
Animal experiments
The Institutional Animal Care and Use Committees at the University of Washington and Washington University in St. Louis approved all animal experiments. We generated Mmp10-null mice on a C57BL/6 background (61). These mice are healthy with no overt defects in fertility, litter size, gross appearance, organ structure or tissue histology. Adult male Mmp10–/– (n = 10) and wild-type (n = 9) mice were exposed to the smoke of four filtered cigarettes (2R4F, Kentucky Tobacco Research and Development Center, University of Kentucky) per day, 6 days per week for 6 months. An unexposed group of age and sex-matched Mmp10–/– (n = 9) and wild types (n = 9) were used as controls. The exposure experiments were performed in the Cigarette Smoke Exposure Core at Washington University, St. Louis. Upon completion of exposure experiments, all animals were killed by an intraperitoneal injection of a Tribromoethanol (Avertin) overdose and exsanguinated by cutting the caudal vena cava. Lungs were cannulated and inflated with neutral buffered formalin at a constant fluid pressure of 25 cm for 5 min. Lungs were removed, immersed in formalin overnight, washed with graded increasing concentrations of ethyl alcohol, and subsequently embedded in paraffin and sectioned. Structural changes caused by chronic cigarette smoke exposure between the genotypes were assessed in the Histology and Imaging Core at the University of Washington using morphometry based on a modification of the direct estimation of mean chord length from a set of random intercepts applied to H&E stained tissue sections (62). Whole slide images were acquired using a Nanozoomer (Hamamatsu model C9600) to scan entire right lung sections and were uploaded into Visiopharm image analysis software. The software provided 50 random images covering all lobes of the right lung, created two randomly oriented lines traversing the image, and provided the sampling tools necessary to manually measure distance along the test lines between airspace walls. Mean chord length was calculated for each mouse from all images counted.
Quantitative real-time PCR (qPCR) was performed on total RNA isolated using Trizol (Invitrogen, Carlsbad, CA, USA) from whole lung of wild-type and Mmp10–/– mice exposed to the chronic cigarette smoke protocol described above (n = 8 per group). RNA was quantified using a Nanodrop spectrophotometer (Thermo Scientific, Waltham, MA, USA) and 3–5 µg of total RNA was reverse transcribed using a High-Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA, USA). qPCR was performed in duplicates on an ABI HT7900 Fast Real-Time PCR System using TaqMan Gene Expression assays for Mmp10 and Il1b (Applied Biosystems) in duplicates. The threshold cycle (Ct) was determined by instrument software and data expressed as relative quantification calculated using 2–ΔΔCt for each gene and using hypoxanthine phosphoribosyltransferase (HPRT) as the reference gene. HPRT Ct levels ranged between 23 and 25 in all assays.
Supplementary Material
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
Funding
Supplementary Material
References
- 1.Hole D.J., Watt G.C., Davey-Smith G., Hart C.L., Gillis C.R., Hawthorne V.M. (1996) Impaired lung function and mortality risk in men and women: findings from the Renfrew and Paisley prospective population study. BMJ, 313, 711–715; discussion 715–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schunemann H.J., Dorn J., Grant B.J., Winkelstein W., Jr, Trevisan M. (2000) Pulmonary function is a long-term predictor of mortality in the general population: 29-year follow-up of the Buffalo Health Study. Chest, 118, 656–664. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization. (2011) World Health Statistics; WHO Press, Geneva, Switzerland. [Google Scholar]
- 4.Hancock D.B., Eijgelsheim M., Wilk J.B., Gharib S.A., Loehr L.R., Marciante K.D., Franceschini N., van Durme Y.M., Chen T.H., Barr R.G., et al. (2010) Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat. Genet., 42, 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Repapi E., Sayers I., Wain L.V., Burton P.R., Johnson T., Obeidat M., Zhao J.H., Ramasamy A., Zhai G., Vitart V., et al. (2010) Genome-wide association study identifies five loci associated with lung function. Nat. Genet., 42, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soler Artigas M., Loth D.W., Wain L.V., Gharib S.A., Obeidat M., Tang W., Zhai G., Zhao J.H., Smith A.V., Huffman J.E., et al. (2011) Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat. Genet., 43, 1082–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hancock D.B., Artigas M.S., Gharib S.A., Henry A., Manichaikul A., Ramasamy A., Loth D.W., Imboden M., Koch B., McArdle W.L., et al. (2012) Genome-wide joint meta-analysis of SNP and SNP-by-smoking interaction identifies novel loci for pulmonary function. PLoS Genet., 8, e1003098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilk J.B., Shrine N.R., Loehr L.R., Zhao J.H., Manichaikul A., Lopez L.M., Smith A.V., Heckbert S.R., Smolonska J., Tang W., et al. (2012) Genome-wide association studies identify CHRNA5/3 and HTR4 in the development of airflow obstruction. Am. J. Respir. Crit. Care Med., 186, 622–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang K., Li M., Hakonarson H. (2010) Analysing biological pathways in genome-wide association studies. Nat. Rev. Genet., 11, 843–854. [DOI] [PubMed] [Google Scholar]
- 10.Hartwell L.H., Hopfield J.J., Leibler S., Murray A.W. (1999) From molecular to modular cell biology. Nature, 402, C47–C52. [DOI] [PubMed] [Google Scholar]
- 11.Schadt E.E. (2009) Molecular networks as sensors and drivers of common human diseases. Nature, 461, 218–223. [DOI] [PubMed] [Google Scholar]
- 12.Wang K., Zhang H., Kugathasan S., Annese V., Bradfield J.P., Russell R.K., Sleiman P.M., Imielinski M., Glessner J., Hou C., et al. (2009) Diverse genome-wide association studies associate the IL12/IL23 pathway with Crohn Disease. Am. J. Hum. Genet., 84, 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang K., Li M., Bucan M. (2007) Pathway-based approaches for analysis of genomewide association studies. Am. J. Hum. Genet., 81, 1278–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lesnick T.G., Papapetropoulos S., Mash D.C., Ffrench-Mullen J., Shehadeh L., de Andrade M., Henley J.R., Rocca W.A., Ahlskog J.E., Maraganore D.M. (2007) A genomic pathway approach to a complex disease: axon guidance and Parkinson disease. PLoS Genet., 3, e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang K., Cui S., Chang S., Zhang L., Wang J. (2010) i-GSEA4GWAS: a web server for identification of pathways/gene sets associated with traits by applying an improved gene set enrichment analysis to genome-wide association study. Nucleic Acids Res., 38, W90–W95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nam D., Kim J., Kim S.Y., Kim S. (2010) GSA-SNP: a general approach for gene set analysis of polymorphisms. Nucleic Acids Res., 38, W749–W754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calvano S.E., Xiao W., Richards D.R., Felciano R.M., Baker H.V., Cho R.J., Chen R.O., Brownstein B.H., Cobb J.P., Tschoeke S.K., et al. (2005) A network-based analysis of systemic inflammation in humans. Nature, 437, 1032–1037. [DOI] [PubMed] [Google Scholar]
- 19.Vidal M., Cusick M.E., Barabasi A.L. (2011) Interactome networks and human disease. Cell, 144, 986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luscombe N.M., Babu M.M., Yu H., Snyder M., Teichmann S.A., Gerstein M. (2004) Genomic analysis of regulatory network dynamics reveals large topological changes. Nature, 431, 308–312. [DOI] [PubMed] [Google Scholar]
- 21.Bafadhel M., McKenna S., Terry S., Mistry V., Reid C., Haldar P., McCormick M., Haldar K., Kebadze T., Duvoix A., et al. (2011) Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am. J. Respir. Crit. Care Med., 184, 662–671. [DOI] [PubMed] [Google Scholar]
- 22.Lappalainen U., Whitsett J.A., Wert S.E., Tichelaar J.W., Bry K. (2005) Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am. J. Respir. Cell Mol. Biol., 32, 311–318. [DOI] [PubMed] [Google Scholar]
- 23.Martinez F.J., Donohue J.F., Rennard S.I. (2011) The future of chronic obstructive pulmonary disease treatment – difficulties of and barriers to drug development. Lancet, 378, 1027–1037. [DOI] [PubMed] [Google Scholar]
- 24.Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L., et al. (2014) The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res., 42, D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hancock D.B., Romieu I., Shi M., Sienra-Monge J.J., Wu H., Chiu G.Y., Li H., del Rio-Navarro B.E., Willis-Owen S.A., Weiss S.T., et al. (2009) Genome-wide association study implicates chromosome 9q21.31 as a susceptibility locus for asthma in mexican children. PLoS Genet., 5, e1000623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Himes B.E., Hunninghake G.M., Baurley J.W., Rafaels N.M., Sleiman P., Strachan D.P., Wilk J.B., Willis-Owen S.A., Klanderman B., Lasky-Su J., et al. (2009) Genome-wide association analysis identifies PDE4D as an asthma-susceptibility gene. Am. J. Hum. Genet., 84, 581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moffatt M.F., Kabesch M., Liang L., Dixon A.L., Strachan D., Heath S., Depner M., von Berg A., Bufe A., Rietschel E., et al. (2007) Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature, 448, 470–473. [DOI] [PubMed] [Google Scholar]
- 28.Mushiroda T., Wattanapokayakit S., Takahashi A., Nukiwa T., Kudoh S., Ogura T., Taniguchi H., Kubo M., Kamatani N., Nakamura Y. (2008) A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J. Med. Genet., 45, 654–656. [DOI] [PubMed] [Google Scholar]
- 29.Pillai S.G., Ge D., Zhu G., Kong X., Shianna K.V., Need A.C., Feng S., Hersh C.P., Bakke P., Gulsvik A., et al. (2009) A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet., 5, e1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho M.H., McDonald M.L., Zhou X., Mattheisen M., Castaldi P.J., Hersh C.P., Demeo D.L., Sylvia J.S., Ziniti J., Laird N.M., et al. (2014) Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med, 2, 214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hofmann S., Franke A., Fischer A., Jacobs G., Nothnagel M., Gaede K.I., Schurmann M., Muller-Quernheim J., Krawczak M., Rosenstiel P., et al. (2008) Genome-wide association study identifies ANXA11 as a new susceptibility locus for sarcoidosis. Nat. Genet., 40, 1103–1106. [DOI] [PubMed] [Google Scholar]
- 32.Loth D.W., Artigas M.S., Gharib S.A., Wain L.V., Franceschini N., Koch B., Pottinger T.D., Smith A.V., Duan Q., Oldmeadow C., et al. (2014) Genome-wide association analysis identifies six new loci associated with forced vital capacity. Nat. Genet., 46, 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khatri P., Sirota M., Butte A.J. (2012) Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput. Biol., 8, e1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaffe A.E., Storey J.D., Ji H., Leek J.T. (2013) Gene set bagging for estimating the probability a statistically significant result will replicate. BMC Bioinformatics, 14, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeda M., Ito W., Tanabe M., Ueki S., Kato H., Kihara J., Tanigai T., Chiba T., Yamaguchi K., Kayaba H., et al. (2009) Allergic airway hyperresponsiveness, inflammation, and remodeling do not develop in phosphoinositide 3-kinase gamma-deficient mice. J. Allergy Clin. Immunol., 123, 805–812. [DOI] [PubMed] [Google Scholar]
- 36.To Y., Ito K., Kizawa Y., Failla M., Ito M., Kusama T., Elliott W.M., Hogg J.C., Adcock I.M., Barnes P.J. (2010) Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med., 182, 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morty R.E., Konigshoff M., Eickelberg O. (2009) Transforming growth factor-beta signaling across ages: from distorted lung development to chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc., 6, 607–613. [DOI] [PubMed] [Google Scholar]
- 38.Annoni R., Lancas T., Yukimatsu Tanigawa R., de Medeiros Matsushita M., de Morais Fernezlian S., Bruno A., Fernando Ferraz da Silva L., Roughley P.J., Battaglia S., Dolhnikoff M., et al. (2012) Extracellular matrix composition in COPD. Eur. Respir. J., 40, 1362–1373. [DOI] [PubMed] [Google Scholar]
- 39.Parks W.C., Wilson C.L., Lopez-Boado Y.S. (2004) Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol., 4, 617–629. [DOI] [PubMed] [Google Scholar]
- 40.Overall C.M., Kleifeld O. (2006) Tumour microenvironment – opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer, 6, 227–239. [DOI] [PubMed] [Google Scholar]
- 41.Churg A., Zhou S., Wright J.L. (2012) Series “matrix metalloproteinases in lung health and disease”: matrix metalloproteinases in COPD. Eur. Respir. J., 39, 197–209. [DOI] [PubMed] [Google Scholar]
- 42.Hunninghake G.M., Cho M.H., Tesfaigzi Y., Soto-Quiros M.E., Avila L., Lasky-Su J., Stidley C., Melen E., Soderhall C., Hallberg J., et al. (2009) MMP12, lung function, and COPD in high-risk populations. N. Engl. J. Med., 361, 2599–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hautamaki R.D., Kobayashi D.K., Senior R.M., Shapiro S.D. (1997) Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science, 277, 2002–2004. [DOI] [PubMed] [Google Scholar]
- 44.Gosselink J.V., Hayashi S., Elliott W.M., Xing L., Chan B., Yang L., Wright C., Sin D., Pare P.D., Pierce J.A., et al. (2010) Differential expression of tissue repair genes in the pathogenesis of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med., 181, 1329–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manning A.K., LaValley M., Liu C.T., Rice K., An P., Liu Y., Miljkovic I., Rasmussen-Torvik L., Harris T.B., Province M.A., et al. (2011) Meta-analysis of gene-environment interaction: joint estimation of SNP and SNP x environment regression coefficients. Genet. Epidemiol., 35, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robbesom A.A., Koenders M.M., Smits N.C., Hafmans T., Versteeg E.M., Bulten J., Veerkamp J.H., Dekhuijzen P.N., van Kuppevelt T.H. (2008) Aberrant fibrillin-1 expression in early emphysematous human lung: a proposed predisposition for emphysema. Mod. Pathol., 21, 297–307. [DOI] [PubMed] [Google Scholar]
- 47.Pereira L., Andrikopoulos K., Tian J., Lee S.Y., Keene D.R., Ono R., Reinhardt D.P., Sakai L.Y., Biery N.J., Bunton T., et al. (1997) Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat. Genet., 17, 218–222. [DOI] [PubMed] [Google Scholar]
- 48.Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J., et al. (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science, 337, 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Westra H.J., Peters M.J., Esko T., Yaghootkar H., Schurmann C., Kettunen J., Christiansen M.W., Fairfax B.P., Schramm K., Powell J.E., et al. (2013) Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet., 45, 1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hao K., Bosse Y., Nickle D.C., Pare P.D., Postma D.S., Laviolette M., Sandford A., Hackett T.L., Daley D., Hogg J.C., et al. (2012) Lung eQTLs to help reveal the molecular underpinnings of asthma. PLoS Genet., 8, e1003029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heinzen E.L., Ge D., Cronin K.D., Maia J.M., Shianna K.V., Gabriel W.N., Welsh-Bohmer K.A., Hulette C.M., Denny T.N., Goldstein D.B. (2008) Tissue-specific genetic control of splicing: implications for the study of complex traits. PLoS Biol., 6, e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dimas A.S., Deutsch S., Stranger B.E., Montgomery S.B., Borel C., Attar-Cohen H., Ingle C., Beazley C., Gutierrez Arcelus M., Sekowska M., et al. (2009) Common regulatory variation impacts gene expression in a cell type-dependent manner. Science, 325, 1246–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hankinson J.L., Odencrantz J.R., Fedan K.B. (1999) Spirometric reference values from a sample of the general U.S. population. Am. J. Respir. Crit. Care Med., 159, 179–187. [DOI] [PubMed] [Google Scholar]
- 54.Swanney M.P., Ruppel G., Enright P.L., Pedersen O.F., Crapo R.O., Miller M.R., Jensen R.L., Falaschetti E., Schouten J.P., Hankinson J.L., et al. (2008) Using the lower limit of normal for the FEV1/FVC ratio reduces the misclassification of airway obstruction. Thorax, 63, 1046–1051. [DOI] [PubMed] [Google Scholar]
- 55.Willer C.J., Li Y., Abecasis G.R. (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Devlin B., Roeder K. (1999) Genomic control for association studies. Biometrics, 55, 997–1004. [DOI] [PubMed] [Google Scholar]
- 57.Subramanian A., Kuehn H., Gould J., Tamayo P., Mesirov J.P. (2007) GSEA-P: a desktop application for gene set enrichment analysis. Bioinformatics, 23, 3251–3253. [DOI] [PubMed] [Google Scholar]
- 58.Saeed A.I., Sharov V., White J., Li J., Liang W., Bhagabati N., Braisted J., Klapa M., Currier T., Thiagarajan M., et al. (2003) TM4: a free, open-source system for microarray data management and analysis. Biotechniques, 34, 374–378. [DOI] [PubMed] [Google Scholar]
- 59.Becker K.G., Hosack D.A., Dennis G., Jr., Lempicki R.A., Bright T.J., Cheadle C., Engel J. (2003) PubMatrix: a tool for multiplex literature mining. BMC Bioinformatics, 4, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Neph S., Kuehn M.S., Reynolds A.P., Haugen E., Thurman R.E., Johnson A.K., Rynes E., Maurano M.T., Vierstra J., Thomas S., et al. (2012) BEDOPS: high-performance genomic feature operations. Bioinformatics, 28, 1919–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kassim S.Y., Gharib S.A., Mecham B.H., Birkland T.P., Parks W.C., McGuire J.K. (2007) Individual matrix metalloproteinases control distinct transcriptional responses in airway epithelial cells infected with Pseudomonas aeruginosa. Infect. Immun., 75, 5640–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Knudsen L., Weibel E.R., Gundersen H.J., Weinstein F.V., Ochs M. (2010) Assessment of air space size characteristics by intercept (chord) measurement: an accurate and efficient stereological approach. J. Appl. Physiol., 108, 412–421. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.