

Abstract
Thymidylate synthase (TSase) catalyzes the de novo biosynthesis of thymidylate, a precursor for DNA, and is thus an important target for chemotherapeutics and antibiotics. Two sequential C-H bond cleavages catalyzed by TSase are of particular interest: a reversible proton abstraction from the 2′-deoxy-uridylate substrate, followed by an irreversible hydride transfer forming the thymidylate product. QM/MM calculations of the former predicted a mechanism where the abstraction of the proton leads to formation of a novel nucleotide-folate intermediate that is not covalently bound to the enzyme (Wang, Z.; Ferrer, S.; Moliner, V.; Kohen, A. Biochemistry 2013, 52, 2348–2358). Existence of such intermediate would hold promise as a target for a new class of drugs. Calculations of the subsequent hydride transfer predicted a concerted H-transfer and elimination of the enzymatic cysteine (Kanaan, N.; Ferrer, S.; Marti, S.; Garcia-Viloca, M.; Kohen, A.; Moliner, V. J. Am. Chem. Soc. 2011, 133, 6692–6702). A key to both C-H activations is a highly conserved arginine (R166) that stabilizes the transition state of both H-transfers. Here we test these predictions by studying the R166 to lysine mutant of E. coli TSase (R166K) using intrinsic kinetic isotope effects (KIEs) and their temperature dependence to assess effects of the mutation on both chemical steps. The findings confirmed the predictions made by the QM/MM calculations, implicate R166 as an integral component of both reaction coordinates, and thus provide critical support to the nucleotide-folate intermediate as a new target for rational drug design.
Keywords: Thymidylate Synthase, QM/MM calculations, Kinetic Isotope Effect, C-H bond activation, Phenomenological models, Donor and acceptor distances, Tunneling ready state
Enzymes enhance rate of reactions to an astounding extent, and a significant portion of the rate enhancements involve catalyzing formations and breakdowns of many otherwise stable chemical bonds including the covalent bond between a carbon and a hydrogen (C-H bond). The molecular and physical details underlying enzyme catalyzed C-H bond activations remain elusive, especially in enzymes that do not contain catalytic metals. Two such C-H bond activations are at the heart of the reaction catalyzed by thymidylate synthase (TSase), which is the de novo source of thymidylate (2′-deoxythymidine-5′-monophosphate, dTMP), one of the four DNA building blocks, in most organisms. TSase is a highly conserved enzyme, and 75% of 109 TSase sequences from pathogenic organisms were found to exhibit an overall identity of 40 to 80% with human TSase.1 Cancerous cells overexpress TSase, and inhibition of TSase causes thymineless cell death, which has attracted the development of many chemotherapeutic drugs targeting this protein.1–3 Derivatives of both pyrimidine (e.g., 5-fluorouracil) and folate (e.g., raltitrexed) have long been used as chemotherapeutic drugs.1,4 These drugs, however, exhibit toxicity, and their competency is limited due to the development of resistance.2,5,6 The need for a new class of drugs that would target TSase in malignant cells stimulates a detailed investigation of structures and mechanism and the correlation between them.1,3–5,7–10
TSase catalyzes a net transfer of a methyl group from its cofactor 5,10-methylene-5,6,7,8-tetrahydrofolate (CH2H4folate) to the substrate 2′-deoxyuridine-5′-monophosphate (dUMP) to form dTMP and 5,6-dihydrofolate (H2folate).11 In its traditionally proposed mechanism (Scheme 1),12,13 an active-site nucleophile cysteine (C146 in the E. coli TSase) initiates the reaction through Michael addition to C6 of dUMP (C6U), forming an enzyme-bound substrate enolate intermediate (compound B in Scheme 1), which then attacks the pre-activated CH2H4folate and forms a covalent ternary complex TSase-dUMP-CH2H4folate (compound C in Scheme 1). From this point, two chemical transformations lead to the formation of the final product dTMP: (i) a proton abstraction from the C5 of the pyrimidine base (C5U), and the elimination of H4folate from the bridging methylene, forming an exocyclic methylene intermediate (Compound D in Scheme 1), and (ii) a hydride transfer from the C6 of H4folate (C6F) to the C7 of the methylene intermediate (C7E) and the dissociation of the active site cysteine from the nucleotide, leading to the product dTMP. The hydride transfer is irreversible,14,15 but the proton abstraction is fast and reversible. This difference in kinetic behavior of the two H-transfers may suggests different physical nature of bond activations within the same enzymatic active site.16
Scheme 1.

The principle mechanism of TSase.
Quantum mechanic/molecular mechanic (QM/MM) calculations have recently suggested that the traditional mechanism illustrated in Scheme 1 is missing some key features.17–21 Calculations on the proton abstraction (step 4),17,21 suggested that the covalent bond between the enzymatic nucleophile C146 and the pyrimidine dUMP (SC146-C6U) cleaves with the abstraction of the proton from the C5 of the dUMP, resulting in a Cys-thiol anion elimination from the C6 of the pyrimidine base, leading to the formation of a new and unexpected reaction intermediate that comprised of the nucleotide and the folate, and is not covalently bound to the enzyme (Scheme 2, compound I). Existing chemotherapeutic drugs targeting TSase are either derivatives of the pyrimidine (e.g., 5F-dUMP) or the folate (e.g., Raltitrexed); the proposed new nucleotide-folate intermediate presents a potential target for a new class of antibiotics and chemotherapeutics. Calculations18,19 on the subsequent hydride transfer (step 5) predicted a concerted hydride transfer and C146 elimination to form the final product dTMP, while the traditional mechanism proposes a step-wise mechanism with the enolate as an intermediate (Scheme 2, compound E).22 Key to both calculations was a highly conserved residue arginine (R166) that seems to stabilize the transition states for both the proton abstraction and the hydride transfer. The outcome of the QM/MM calculations indicates that R166 alternately fluctuates towards and away from the nucleophile thiol on C146 to stabilize it as a leaving group for each H-transfer and to prepare it for the following nucleophilic attack, respectively (Scheme 2). In contrast to the traditional TSase mechanism,11,13,22,23 these calculations predicted that the covalent bond between the substrate and the enzyme is quite labile due to the fluctuations of R166. The calculations also predicted that the coordinated motion between R166 and C146 and the resulting charge stabilizations at different transition states make R166 an inextricable part of the reaction coordinate for the both H-transfers catalyzed by this enzyme.
Scheme 2.

Traditional (A) and calculated17,18 (B) mechanisms for the proton abstraction and the hydride transfer in TSase. In the QM/MM calculations, the abstraction of C5U proton induces the cleavage of SC146-C6U, leading to the formation of the nucleotide-folate intermediate. Following the protonation of the N5 of folate, the departed C146 then re-attacks the nucleotide-folate intermediate, assisting in the succeeding β-elimination of the cofactor, generating the exocyclic methylene intermediate. Then, the concerted hydride transfer and elimination of C146 leads to the product dTMP.
Temperature dependence of intrinsic kinetic isotope effects (KIEs) has emerged as a powerful tool to probe the nature of C-H bond activations within complex cascades of chemical events.24,25 This tool has been utilized to investigate C-H bond activations in the wild-type (WT) TSase.15,16 It was found that the intrinsic KIEs on the hydride transfer (step 5 in Scheme 1) are temperature independent, while those on the proton abstraction (step 4) are steeply temperature dependent.15,16 Phenomenological models offer an interpretation of these kinetic results.24–33 In these models, as the transferring particle (hydrogen) is rather light, quantum mechanical tunneling occurs between the H-donor and the H-acceptor when the wave functions for the hydrogen atom in the reactant and product wells overlap with each other. The space along the reaction coordinates where the energy of the donor and acceptor well are degenerate and tunneling probability is more than zero is called “Tunneling Ready State” (TRS), which is merely the quantum mechanically delocalized transition state (TS).24,34,35 Motions of the protein, reactants, and solvent bring the system to the TRS and modulate tunneling by altering the height and width of the energy barrier between the reactant and the product states. The expression of KIE in the framework of these activated tunneling models is given by equation 1 and is illustrated in Figure 1:24,36
Figure 1.

Phenomenological models for activated H-tunneling. Left column represents three stages of the reaction along the H-transfer coordinate. Motion of the protein, solvent, and reactants modulate the potential energy surfaces for the H-transfer. At the reactant state (A), the H-wavefunction is localized in the donor well. The motion of the heavy atoms transiently brings the donor and the acceptor wells into the tunneling ready state (TRS, B), where isotopically sensitive H-transfer from donor to acceptor occurs. Further rearrangement in the heavy atoms interrupts the TRS, resulting in entrapment of the hydrogen in the product well (C). The right column demonstrates the contributing factors that modulate H-transfer probability at the TRS. The panel B1 shows the transmission probabilities of light (l, green) and heavy (h, purple) hydrogen isotopes as a function of DAD. Panel B2 represents a potential energy surface (PES) for the DAD fluctuations. Panel B3 shows the product of DADs and transmission probabilities.
| (1) |
where and are the transfer probabilities for the light and heavy atoms, which depend on their respective masses; and the exponential term is the Boltzmann factor representing a distribution of H-donor and acceptor distances (DADs) at the TRS. From the Eq. 1, the DADs and their distributions at TRS determine both the magnitude and the temperature dependence of KIEs. In the framework of this model, temperature independent KIEs stem from a narrow distribution of DADs at the TRS (well reorganized TS) while the temperature dependent KIEs result from a broader distribution of DADs at the TRS (poorly reorganized dividing line between donor and acceptor with broad ensemble of TSs). In E. coli TSase, the hydride transfer (step 5 in Scheme 1) has a narrow distribution of DADs as reflected by temperature independent KIEs, while the fast proton abstraction (step 4) has a broader distribution of DADs as reflected by temperature dependent intrinsic KIEs.16 Mutation of a residue(s) that are part of the reaction coordinate of a certain H-transfer changes the geometry of the TRS, and thereby the distribution of DADs, which is commonly manifested as a change in the temperature dependence of KIEs for that mutant.24,37 Thus a mutation that results in a change in the temperature dependence of intrinsic KIEs serves as an indication that the residue in the wild-type (WT) enzyme was likely part of the reaction coordinate under study.
While the role of R166 in activating C146 to initiate the catalytic cycle by Michael addition of C146 to the nucleotide was proposed in the traditional mechanism,11,13 the catalytic role of R166 in the subsequent H-transfers was delineated only in the calculated mechanisms.17,18,21 Here, we report experimental examination of these QM/MM predictions, by comparing intrinsic KIEs and their temperature dependence for the proton and the hydride transfers for the WT and R166K mutant of E. coli TSase. The experimental findings confirm and characterize the role of R166 in both C-H bond activations, and consequently provide a critical experimental support for the calculated mechanisms. The findings thus reinforce the existence of an unexpected non-covalently bound intermediate that combines the folate and nucleotide moieties.
Materials and Methods
Chemicals
All chemicals, including deoxyuridine monophosphate (dUMP), 5-fluorodeoxyuridine monophosphate (5-FdUMP), and tris(2-carboxyethyl)phosphine (TCEP), were purchased from Sigma-Aldrich, St. Louis, MO, unless otherwise mentioned. [6-3H] dUMP and [2-14C] dUMP were purchased from Moravek Biochemicals. Unlabeled 5,10-methylene-5,6,7,8-tetrahydrofolate (CH2H4folate) was generously donated by EPROVA (Switzerland). [3H] NaBH4 and [2H] NaBH4 were purchased from American Radiolabeled Chemicals and Cambridge Isotopes respectively. [2-xH] iPrOH was synthesized by reduction of acetone with [xH] NaBH4. Dihydrofolate was prepared according to the Blakley Method.38 Ultima Gold liquid scintillation cocktail and scintillation vials were purchased from Packard Biosciences and Research Products International respectively. Wild-type (WT) and mutant R166K E. coli TSase were expressed and purified according to a previously described procedure.39 Steady-state initial velocities were determined using a Hewlett-Packard Model 8452A diode-array spectrophotometer equipped with a temperature-controlled cuvette assembly. All separations were accomplished using an Agilent Technologies 1100 HPLC system with Supelco Discovery C18 reverse phase analytical or semi-preparative columns. Radioactive samples were analyzed using a liquid scintillation counter (LSC).
Synthesis of [2-14C, 5-2H] dUMP and for Proton Transfer KIE experiments
[2-14C, 5-2H] dUMP was prepared using methodology developed by Wataya and Hayatsu.40 Briefly, 1 mM [2-14C] dUMP is incubated with 1 M L-Cysteine in (>99.96% D) D2O, pD 8.8, at 37°C. Deuteration of C5U was followed by 1H-NMR and verified as completed after 7 days of incubation. Aliquots of the reaction mixture were then stored at −80 °C until used in later proton abstraction D/T KIE experiments. Interestingly, one should note that TSase also activates the C6U position of dUMP by nucleophilic attack of active site residue C146 on C5U of dUMP.
Competitive Primary KIE experiments
The competitive primary (1°) KIE experiments for the proton transfer step in R166K followed the same conditions as used previously to measure the proton transfer KIEs for the WT enzyme.16 Briefly, the reaction mixture contained 2 mM TCEP, 1 mM EDTA, 5 mM HCHO, 50 mM MgCl2 in addition to the substrates and the enzymes. CH2H4folate is taken in ~25–30% excess to dUMP (1.35 mM) and the mixture buffered in 100 mM Tris. Additionally, trace amounts of [5-3H] dUMP, with [2-14C] dUMP or [2-14C, 5-2H] dUMP for H/T and D/T KIE experiments, respectively, are added such that the ratio of 3H to 14C in the reaction mixture was kept above 6.0 for higher accuracies in the LSC analyses. Aliquots of the reaction mixture were equilibrated to 5, 15, 25, or 35 °C, pH adjusted to 7.5 at the respective temperature, and the reaction was initiated with ca. 100–300 μM R166K TSase. Throughout the course of the reactions, 6 aliquots were removed at different time points (t), where fraction conversion was between 25–80%. These aliquots were immediately quenched with excess 5-FdUMP, a tight binding competitive inhibitor of TSase, and stored at −80°C until later analysis. In addition to these reaction mixtures, 3 aliquots at 0% conversion (t0) controls, and 3 aliquots at 100% conversion (tinf), were collected by incubating the reaction mixture overnight with WT TSase at 35 °C. Reactants and products of each aliquot were separated by RP HPLC and radioactivity measured by LSC. From the 14C-radioactivity of reactants and products, fraction conversions (f) can be determined according to equation 2. In addition, Ratio of 3H to 14C in the products of the time points (Rt) and time infinity (Rinf) was used to determine the observed KIE using equation 3 whose derivation is described elsewhere.41
| (2) |
| (3) |
Since in contrast to the hydride transfer step the proton abstraction is reversible, further analysis is needed to avoid artifacts. While tritium and deuterium abstractions are practically irreversible (due to dilution into H2O), the formed intermediate could be followed by reversible protonation of the C5-dUMP. When this reversible step effectively competes with the forward reaction an up-going trend for the D/T KIEobs is observed, ranging from the actual D/T KIEobs value at early fraction conversion (f→0) to H/T KIEobs at late fraction conversion (f→1). While this phenomenon has been observed for other mutants (data not shown), a plot of KIEobs as a function of f for both R166K and the WT TSase (Figure S1) shows no such trend. This finding suggests that the forward H-transfer rate and the subsequent irreversible formation of the product were dominant compared to the rate of reverse reaction. Furthermore, for KIEs on the proton abstraction both the reversibility and the fact that CH2H4folate inflates the commitment,43 deflate the KIEobs. However, the Northrop method used to calculate the intrinsic KIEs (Eq. 4 below) drops the commitment factor out of the final outcome as discussed in the following section (Intrinsic KIEs).
The experimental details for the KIEobs on the hydride transfer were reported in ref 42. In brief, [6R-xH]CH2H4folate (xH=D and T) was synthesized according to the procedure described in ref 15. The competitive H/T and D/T KIEobs on the hydride transfer were measured using H/T and D/T labeled CH2H4folate and the reaction was carried out under the same condition as for the proton abstraction measurements described above.
Intrinsic KIEs
Intrinsic KIEs were calculated from their observed values using Northrop method as described in refs 16,41. Briefly, in this method, a combination of KIEs (H/T and D/T in the current case) is measured, where the hydrogen isotope common to both KIE measurements (T) serves as the reference isotope.43 For the H/T and D/T KIE measurements, Northrop method uses Eq. 4 that allows extracting the intrinsic KIEs by eliminating the commitment to catalysis on the reference isotope T.35,41
| (4) |
Here T(V/K)Hobs, T(V/K)Dobs, and (kH/kT) are the observed H/T, D/T and Intrinsic KIEs, respectively. To calculate the intrinsic KIEs, a program freely available at http://chem.uiowa.edu/kohen-research-group/calculation-intrinsic-isotope-effects, numerically solved Eq. 4 and extracted intrinsic KIEs (KIEint) with every combination of the observed values at a particular temperature. The intrinsic KIEs were then exponentially fitted to a modified Arrhenius equation where weighted root means square exponential regression (KaleidaGraph version 4.03) yields the KIE on Arrhenius pre-exponential factor (AL/AH) as well as the isotope effect on activation energies (ΔEa) for respective isotope comparisons.
Results and Discussion
Activation of two sequential C-H bonds
Observed KIEs on the proton abstraction for the WT TSase depend on the concentration of the second-binding ligand CH2H4folate and reaches unity at high CH2H4folate concentration, indicating an ordered-binding mechanism.16,43,44 However, for the mutant R166K, a high concentration of CH2H4folate yields an observed KIE larger than unity, suggesting the mutation caused a more random binding mechanism, which was also observed for other mutants.45,43 This observation accords with steady-state kinetics with R166K that demonstrated inflated KMs for both the substrate and cofactor.42 This can be rationalized by examination of WT crystal structures, where the residue R166 forms two hydrogen bonds with the phosphate of the dUMP. The mutation to lysine thereby is likely to destabilize those tie-ups, reducing the affinity for dUMP and causing an overall disruption of the original order of binding preferences. Steady-state kinetic measurements indicate that this mutation reduced the turnover rate (kcat) by about a 90-fold, and increased KM for dUMP and CH2H4folate by more than 300-fold and 18-fold, respectively.42
In order to investigate how R166 modulates the C-H bond activation, KIEs and their temperature dependence were measured on both the hydride transfer and the proton abstraction for its only active variant, R166K. Competitive KIEs (H/T and D/T) that report on the second order rate constant (kcat/Km)41 were measured at temperatures ranging from 5 to 35 °C (Table S1). The intrinsic KIEs, which are often masked by kinetic complexity, were extracted from the observed KIEs using the Northrop method.35,41,46,47 Figure 2 shows an Arrhenius plot of intrinsic KIEs on the proton abstraction (red) and the hydride transfer (blue) for both the WT and R166K TSase. Isotope effects on the activation energy (ΔEa (T-H)) and on the pre-exponential factor (AH/AT) were obtained from the fittings of KIE data into the following Arrhenius equation:
| (5) |
where H and T denotes hydrogen and tritium respectively.
Figure 2.
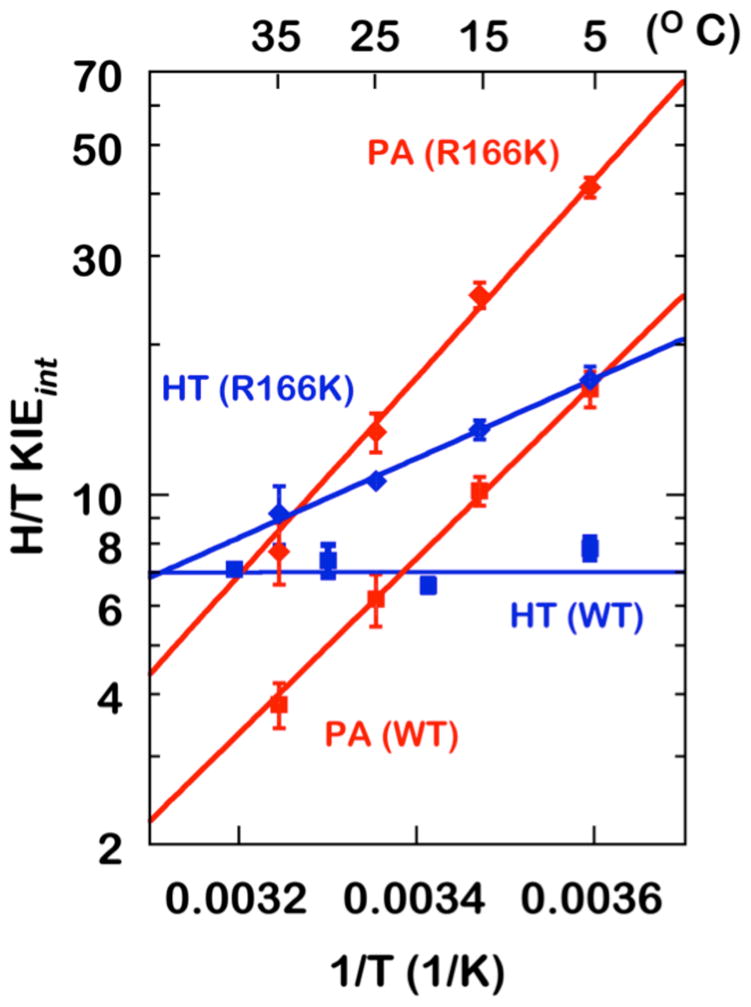
Arrhenius plots of intrinsic H/T KIEs on the proton abstraction (PA, red) and the hydride transfer (HT, blue)15,42 for the WT (Squares)16 and R166K (Diamonds) TSase.
As evident from Figure 2, Table 1, the mutation of R166 increases the temperature dependence of KIEs for both the hydride transfer and the proton abstraction. In R166K, the intrinsic KIEs on the hydride transfer become temperature dependent while, for the proton abstraction, the temperature dependency becomes steeper (higher ΔEa,T/H) compared to that for the WT. The mutation also caused a significant increase in the magnitude of intrinsic KIEs for the both H-transfers across the examined temperatures. This observation is consistent with R166 in the WT TSase being a component of the reaction coordinate and the TS for both hydride and proton transfers, as predicted by the QM/MM calculations discussed above.
Table 1.
Isotope effects on the activation energy (ΔEa,T/H) and the Arrhenius pre-exponential factor (AH/AT) for the proton abstraction (PA) and the hydride transfer (HT) in the WT and R166K.
In the framework of phenomenological models discussed above,24,25,34,37 the larger magnitudes and the greater temperature dependences of KIEs indicate a larger average DAD and a broader DAD distribution, respectively. Figure 3 shows schematic illustrations of DAD distributions in terms of potential energy surfaces (PES) along the DAD coordinate at the TRS for the hydride transfer (left) and the proton abstraction (right) in both the WT and R166K TSases. A stiff PES (large force constant and high DAD sampling frequency) represents a narrowly distributed ensemble of DADs, suggesting a well-defined TRS as in the case for the hydride transfer for the WT TSase. A wide PES, on the other hand, corresponds to a less-organized TRS (small force constant and low DAD sampling frequency), as in the case for the proton abstraction. These DAD’s PESs are wider in the mutant R166K (larger ΔEa,T/H), demonstrating perturbations to the TRSs caused by the mutation. The minima of these PESs representing the average DADs at the TRS shift towards larger DADs in the mutant R166K. These combined features, the larger average DAD and the broad distribution of DADs, would explain both the inflated magnitude and temperature dependence of the intrinsic KIEs in the mutant. An interpretation consistent with the QM/MM calculations would be that the mutation caused a disruption in the coordinated motion between R166 and C146 and in the following charge stabilizations, affecting both the hydride transfer and the proton abstraction reaction coordinate. It is important to note that alternative explanations, such as coincidental similar remote effect of R166 on the TS of both C-H activations, cannot be excluded based on the experimental observations alone. However Ockham’s razor should prefer one mechanism that explains both similar effects, as offered by the R166 role in activating C146 for both steps, which also accords with the calculations. It is also important to note that several mutations closer to the reactions’ center had no or much smaller effect on the intrinsic KIEs or their temperature dependence relative to R166K (e.g., W80M or Y209F)43,45,48,49 In contrast to R166, residues W80 and Y209 were not predicted by the QM/MM calculations to be part of the reaction coordinate for either H-transfers, thus their lack of effect on these transfers may serve as important control further supporting the validity of the mechanism proposed in Scheme 2B.
Figure 3.

Illustration of potential energy surfaces (PESs) along the donor-acceptor distance (DAD) coordinate in TRS for WT (red) and R166K (blue) TSase catalyzed proton abstraction (PA) and hydride transfer (HT) reactions. The relative changes in the distribution of DADs are demonstrated.
To graphically present the effect of R166K on both C-H activations, the isotope effects on the activation parameters from Table 1 are illustrated in Figure 4, which shows a plot of the isotope effects on the activation enthalpy (ΔEa (T-H)) against the isotope effects on Arrhenius pre-exponential factors (AH/AT). Within TS theory, these two parameters are independent of each other, and no theoretical model predicts a trend between them. From Figure 4 it appears that the isotope effects on ΔEa (T-H) and AH/AT for the both H-transfers are oppositely affected by the mutation, indicating similar perturbations of their respective reaction coordinates in this case. Additionally, it seems that the effect of the mutation on the hydride transfer is greater than for the proton abstraction as evident from the higher magnitude of ΔEa (T-H) in R166K for the former. This can be rationalized by the hydride transfer being slower in the WT,14,15 thus requiring more assistance by R166 for the activation of the SC146-C6U bond than that for the faster proton abstraction. That rationale is similar to the one that explains why the proton abstraction has a broad DAD-distribution in the WT, i.e., proton abstraction can happen without the enzyme catalysis and thus the enzyme need not force accurate TS and narrow DAD-distribution. In stark contrast, the slower hydride transfer requires the enzyme to provide strict control over the TS, creating a narrow DAD distribution for that step.
Figure 4.
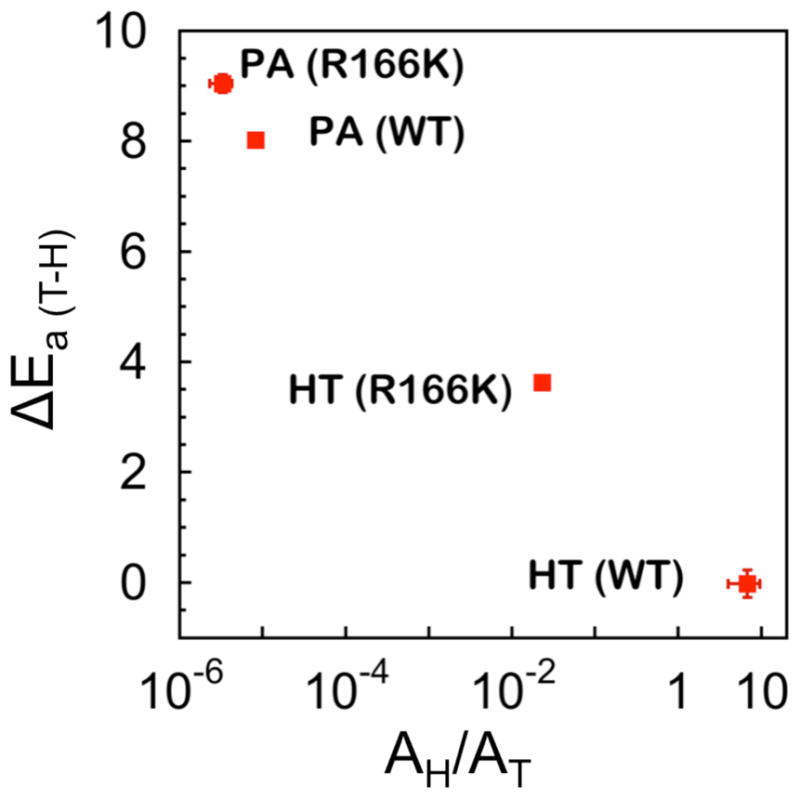
Presentation of the interrelation between isotope effects on activation parameters in the two H-transfers catalyzed by the WT and R166K TSases.
Role of R166 in the reversal of C146-nucleotide covalent adduct
C146 is the active-site nucleophile that initiates the catalysis by Michael addition to C6 of dUMP. In the traditional mechanism of TSase, C146 remains covalently bonded to the substrate dUMP until the very last step of the reaction (Step1–5, Scheme 1).11,13 However, in the QM/MM calculated mechanisms, the SC146-C6U bond was found to be very labile, and it alternates between a full and no covalent bond at different stages of the reaction.17,18,21,50 Montfort et al51 also proposed an unstable nucleotide-C146 conjugate following an observation of a diffused electron density between C6 in 5FdUMP and C146 in a crystal structure of TSase-5FdUMP-CH2H4folate complex (1TSN) even though a more stable SC146-C6U bond (relative to the one with dUMP ligand) would be expected due to the strong electron-withdrawing potential of fluorine in the 5FdUMP. The diffused electron density around SC146-C6U was also observed in several other ternary complexes (e.g. 3BHL, 1KZI, 2G8O), supporting a labile SC146-C6U conjugate.
Another key feature in the traditional mechanism includes the formation of enolates in the binary (compound B in Scheme 1 and compound E in Scheme 2) and ternary complexes (compound C′ in Scheme 2).11–13,22,23 In the crystal structure of TSase, the side of chain of N177 is found to be situated within the hydrogen bonding distance to the C4=O4 of dUMP (Figure 5), presumably stabilizing the charge on the oxyanion. Nevertheless, in the calculations,17,18 both H-transfers were found to occur through mechanisms that bypass complete enolate formations, and only suggested some negative charge accumulation at that carbonyl.17,18 A recently reported normal secondary KIE (2°) on C6 of dUMP for the conversion of the exocyclic methylene intermediate to dTMP in the WT TSase indeed supported a concerted hydride transfer.42 Importantly, this experimental 2° KIE was recently reproduced by a QM/MM calculation.50 Another recent high level QM/MM calculation carried out by an independent group52 on the formation of the covalently-bonded ternary complex from the non-covalent one (from step 1 to 3 in Scheme 1) supported a previous report21 suggesting that the initial Michael addition and the covalent ternary complex formation (compound C′ in Scheme 2) are also concerted. In this recent and independent calculation,52 R166 was also found to promote the concerted process that bypasses the initial enolate in the binary TSase-dUMP complex (compound B in Scheme 1).
Figure 5.
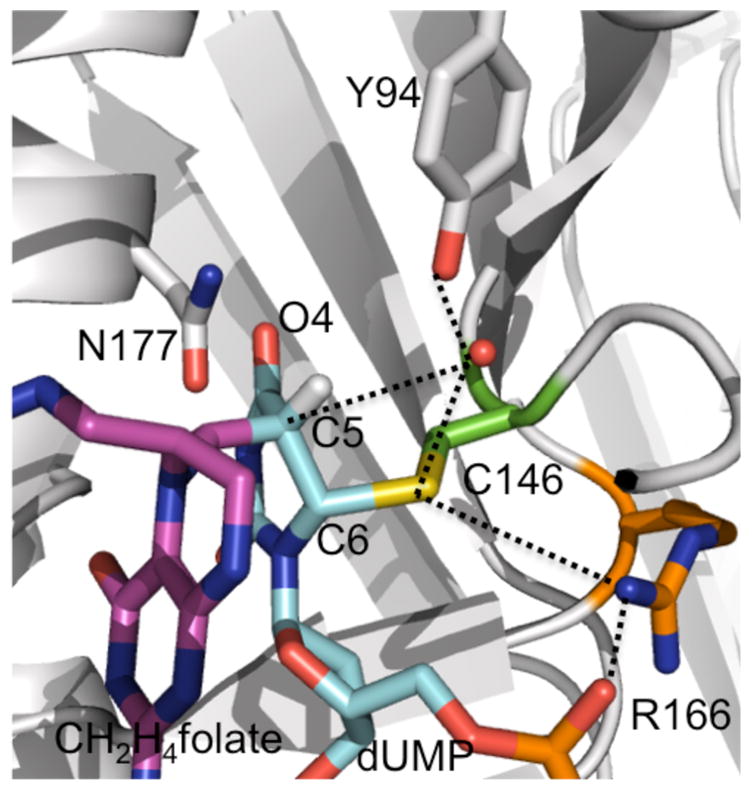
Active site structure of TSase covalently bound with 5-fluoro dUMP and CH2H4folate (PDB ID 1TLS). The ligands (dUMP, CH2H4folate) and residues close to H-transfers’ site are shown in sticks, and atoms that were predicted to interact during the catalytic cycle are connected by dashed lines.
Conclusions
Understanding the role of protein motions in the chemical bond activation, particularly for C-H bonds, is of contemporary interest. TSase catalyzes a series of cleavages and formations of chemical bonds including two C-H bond activations. The studies presented above tested predictions by the QM/MM calculations17,18 that proposed alternative mechanisms for the two different C-H bond activations catalyzed by the same active site. By these calculations, residue R166 is central for both hydride and proton transfers. It was suggested that TSase exploits the reversals of C146-nucleotide adduct coordinated by R166 motions to catalyze the breaking of several covalent bonds. Indeed, R166 has been ranked as one of the five most essential residues for the catalysis as the enzyme cannot tolerate any substitution at this position other than lysine.13 The intrinsic KIEs and their temperature dependence on the proton abstraction and the hydride transfer presented above implicate R166 as an important component of both H-transfers’ coordinate, and thus support the QM/MM calculation-predicted role of R166 in these H-transfers. It seems that R166 plays three possible roles in catalysis: (i) activating C146 to initiate the Michael addition (step 2 in Scheme 1) as proposed in the traditional mechanism,13 which was also supported by the QM/MM calculations,21,52 (ii) facilitating the proton abstraction by promoting the cleavage of SC146 –C6U, forming a newly predicted nucleotide-folate intermediate and (iii) assisting the concerted hydride transfer. The last two roles were first predicted by the QM/MM calculations and are now corroborated by the experiments presented above. The alternative mechanism for the proton abstraction has an important significance as it proposed a new nucleotide-folate intermediate that is not covalently bound to the enzyme. Such an intermediate, if it exists, could represent a new target for designing TSase inhibitors as leads toward a new class of antibiotics and chemotherapeutics. The current kinetic data support the steps before (the proton abstraction, 4B in Scheme 2) and after (the hydride transfer, 5B in Scheme 2) the formation of intermediate, increasing the confidence in the existence of that putative intermediate.
Supplementary Material
Acknowledgments
This work was supported by NIH (R01GM65368) and NSF (CHE-1149023) to AK, and the Iowa Center of Biocatalysis and Bioprocessing associated with NIH T32 GM008365 to ZI.
Footnotes
Authors’ Contributions
AK led and oversaw the studies and ZI, TS and AG conducted the experiments. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information Available
Tables presenting the observed and intrinsic KIEs for both proton abstraction and hydride transfer in R166K are provided. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Carosati E, Tochowicz A, Marverti G, Guaitoli G, Benedetti P, Ferrari S, Stroud RM, Finer-Moore J, Luciani R, Farina D, Cruciani G, Costi MP. J Med Chem. 2012;55:10272–10276. doi: 10.1021/jm300850v. [DOI] [PubMed] [Google Scholar]
- 2.Sergeeva OA, Khambatta HG, Cathers BE, Sergeeva MV. Biochem Biophys Res Commun. 2003;307:297–300. doi: 10.1016/s0006-291x(03)01173-2. [DOI] [PubMed] [Google Scholar]
- 3.Costi MP, Tondi D, Rinaldi M, Barlocco D, Pecorari P, Soragni F, Venturelli A, Stroud RM. Biochim Biophys Acta, Mol Basis Dis. 2002;1587:206–214. doi: 10.1016/s0925-4439(02)00083-2. [DOI] [PubMed] [Google Scholar]
- 4.Popat S, Matakidou A, Houlston RS. J Clin Oncol. 2004;22:529–536. doi: 10.1200/JCO.2004.05.064. [DOI] [PubMed] [Google Scholar]
- 5.Salonga D, Danenberg KD, Johnson M, Metzger R, Groshen S, Tsao-Wei DD, Lenz HJ, Leichman CG, Leichman L, Diasio RB, Danenberg PV. Clin Cancer Res. 2000;6:1322–1327. [PubMed] [Google Scholar]
- 6.Copur S, Aiba K, Drake JC, Allegra CJ, Chu E. Biochem Pharmacol. 1995;49:1419–1426. doi: 10.1016/0006-2952(95)00067-a. [DOI] [PubMed] [Google Scholar]
- 7.Wilson PM, Danenberg PV, Johnston PG, Lenz HJ, Ladner RD. Nat Rev Clin Oncol. 2014;11:282–298. doi: 10.1038/nrclinonc.2014.51. [DOI] [PubMed] [Google Scholar]
- 8.Calascibetta A, Contino F, Feo S, Gulotta G, Cajozzo M, Antona A, Sanguedolce G, Sanguedolce R. J Nucleic Acids. 2010;2010:306754. doi: 10.4061/2010/306754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phan J, Steadman DJ, Koli S, Ding WC, Minor W, Dunlap RB, Berger SH, Lebioda L. J Biol Chem. 2001;276:14170–14177. doi: 10.1074/jbc.M009493200. [DOI] [PubMed] [Google Scholar]
- 10.Johnston PG, Lenz HJ, Leichman CG, Danenberg KD, Allegra CJ, Danenberg PV, Leichman L. Cancer Res. 1995;55:1407–1412. [PubMed] [Google Scholar]
- 11.Carreras CW, Santi DV. Annu Rev Biochem. 1995;64:721–762. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 12.Newby Z, Lee TT, Morse RJ, Liu Y, Liu L, Venkatraman P, Santi DV, Finer-Moore JS, Stroud RM. Biochemistry. 2006;45:7415–7428. doi: 10.1021/bi060152s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finer-Moore JS, Santi DV, Stroud RM. Biochemistry. 2003;42:248–256. doi: 10.1021/bi020599a. [DOI] [PubMed] [Google Scholar]
- 14.Spencer HT, Villafranca JE, Appleman JR. Biochemistry. 1997;36:4212–4222. doi: 10.1021/bi961794q. [DOI] [PubMed] [Google Scholar]
- 15.Agrawal N, Hong B, Mihai C, Kohen A. Biochemistry. 2004;43:1998–2006. doi: 10.1021/bi036124g. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Kohen A. J Am Chem Soc. 2010;132:9820–9825. doi: 10.1021/ja103010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Ferrer S, Moliner V, Kohen A. Biochemistry. 2013;52:2348–2358. doi: 10.1021/bi400267q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanaan N, Ferrer S, Marti S, Garcia-Viloca M, Kohen A, Moliner V. J Am Chem Soc. 2011;133:6692–6702. doi: 10.1021/ja1114369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanaan N, Roca M, Tunon I, Marti S, Moliner V. J Phys Chem B. 2010;114:13593–13600. doi: 10.1021/jp1072457. [DOI] [PubMed] [Google Scholar]
- 20.Kanaan N, Marti S, Moliner V, Kohen A. J Phys Chem A. 2009;113:2176–2182. doi: 10.1021/jp810548d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanaan N, Marti S, Moliner V, Kohen A. Biochemistry. 2007;46:3704–3713. doi: 10.1021/bi061953y. [DOI] [PubMed] [Google Scholar]
- 22.Huang W, Santi DV. Biochemistry. 1997;36:1869–1873. doi: 10.1021/bi961982g. [DOI] [PubMed] [Google Scholar]
- 23.Bruice TW, Santi DV. Enzyme mechanism from isotope effects. CRC Press; Boca Raton: 1991. pp. 457–479. [Google Scholar]
- 24.Kohen A. Acc Chem Res. 2015;48:466–473. doi: 10.1021/ar500322s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klinman JP, Kohen A. Annu Rev Biochem. 2013;82:5543–5567. doi: 10.1146/annurev-biochem-051710-133623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanoian P, Liu CT, Hammes-Schiffer S, Benkovic S. Acc Chem Res. 2015;48:482–489. doi: 10.1021/ar500390e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Layfield JP, Hammes-Schiffer S. Chem Rev. 2013;114:3466–3494. doi: 10.1021/cr400400p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagel ZD, Klinman JP. Chem Rev. 2010;110:PR41–PR67. doi: 10.1021/cr1001035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maglia G, Allemann RK. J Am Chem Soc. 2003;125:13372–13373. doi: 10.1021/ja035692g. [DOI] [PubMed] [Google Scholar]
- 30.Hay S, Scrutton NS. Nat Chem. 2012;4:161–168. doi: 10.1038/nchem.1223. [DOI] [PubMed] [Google Scholar]
- 31.Pudney CR, Hay S, Levy C, Pang J, Sutcliffe MJ, Leys D, Scrutton NS. J Am Chem Soc. 2009;131:17072–17073. doi: 10.1021/ja908469m. [DOI] [PubMed] [Google Scholar]
- 32.Marcus RA. J Phys Chem B. 2007;111:6643–6654. doi: 10.1021/jp071589s. [DOI] [PubMed] [Google Scholar]
- 33.Antoniou D, Basner J, Nunez S, Schwartz SD. Chem Rev. 2006;106:3170–3187. doi: 10.1021/cr0503052. [DOI] [PubMed] [Google Scholar]
- 34.Roston D, Islam Z, Kohen A. Arch Biochem Biophys. 2014;544:96–104. doi: 10.1016/j.abb.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roston D, Islam Z, Kohen A. Molecules. 2013;18:5543–5567. doi: 10.3390/molecules18055543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roston D, Cheatum CM, Kohen A. Biochemistry. 2012;51:6860–6870. doi: 10.1021/bi300613e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klinman JP. Acc Chem Res. 2015;48:449–456. doi: 10.1021/ar5003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blakley RL. Nature (London, U K) 1960;188:231–232. [Google Scholar]
- 39.Changchien LM, Garibian A, Frasca V, Lobo A, Maley GF, Maley F. Protein Expr Purif. 2000;19:265–270. doi: 10.1006/prep.2000.1245. [DOI] [PubMed] [Google Scholar]
- 40.Wataya Y, Hayatsu H. J Am Chem Soc. 1972;94:8927–8928. doi: 10.1021/ja00780a059. [DOI] [PubMed] [Google Scholar]
- 41.Cook PF, Cleland WW. Enzyme kinetics and mechanism. Garland Science; London; New York: 2007. p. 253. [Google Scholar]
- 42.Islam Z, Strutzenberg TS, Gurevic I, Kohen A. J Am Chem Soc. 2014;136:9850–9853. doi: 10.1021/ja504341g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong B, Maley F, Kohen A. Biochemistry. 2007;46:14188–14197. doi: 10.1021/bi701363s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghosh AK, Islam Z, Krueger JD, Abeysinghe T, Kohen A. Phys Chem Chem Phys. 2015 doi: 10.1039/c5cp01246e. http://dx.doi.org/10.1039/C5CP01246E. [DOI] [PMC free article] [PubMed]
- 45.Abeysinghe T, Kohen A. Int J Mol Sci. 2015;16:7304–7319. doi: 10.3390/ijms16047304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Northrop DB. Enzyme mechanism from isotope effects. CRC Press; Boca Raton, FL: 1991. pp. 181–202. [Google Scholar]
- 47.Parkin DW. In: Enzyme mechanism from isotope effects. Cook PF, editor. CRC Press; Boca Raton: 1991. pp. 269–290. [Google Scholar]
- 48.Hong B, Haddad M, Maley F, Jensen JH, Kohen A. J Am Chem Soc. 2006;128:5636–5637. doi: 10.1021/ja060196o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Z, Abeysinghe T, Finer-Moore JS, Stroud RM, Kohen A. J Am Chem Soc. 2012;134:17722–17730. doi: 10.1021/ja307859m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swiderek K, Kohen A, Moliner V. Phys Chem Chem Phys. 2015 doi: 10.1039/c5cp01239b. http://dx.doi.org/10.1039/C5CP01239B. [DOI] [PMC free article] [PubMed]
- 51.Hyatt DC, Maley F, Montfort WR. Biochemistry. 1997;36:4585–4594. doi: 10.1021/bi962936j. [DOI] [PubMed] [Google Scholar]
- 52.Kaiyawet N, Lonsdale R, Rungrotmongkol T, Mulholland AJ, Hannongbua S. J Chem Theory Comput. 2015;11:713–722. doi: 10.1021/ct5005033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.