

Non-coding DNA elements differentially control stem and progenitor cell transitions required for development.
Keywords: hematopoiesis, differentiation, stem cell, progenitor, GATA factor, GATA-2, cis-element, myeloid, transcriptome
Abstract
Cis-element encyclopedias provide information on phenotypic diversity and disease mechanisms. Although cis-element polymorphisms and mutations are instructive, deciphering function remains challenging. Mutation of an intronic GATA motif (+9.5) in GATA2, encoding a master regulator of hematopoiesis, underlies an immunodeficiency associated with myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). Whereas an inversion relocalizes another GATA2 cis-element (−77) to the proto-oncogene EVI1, inducing EVI1 expression and AML, whether this reflects ectopic or physiological activity is unknown. We describe a mouse strain that decouples −77 function from proto-oncogene deregulation. The −77−/− mice exhibited a novel phenotypic constellation including late embryonic lethality and anemia. The −77 established a vital sector of the myeloid progenitor transcriptome, conferring multipotentiality. Unlike the +9.5−/− embryos, hematopoietic stem cell genesis was unaffected in −77−/− embryos. These results illustrate a paradigm in which cis-elements in a locus differentially control stem and progenitor cell transitions, and therefore the individual cis-element alterations cause unique and overlapping disease phenotypes.
INTRODUCTION
Ascribing function to the vast ensemble of genomic cis-regulatory elements emerging from in silico and experimental analyses represents a formidable problem. Whereas strategies exist to predict enhancers (1, 2), gauging endogenous enhancer mechanisms at specific anatomical sites in vivo poses considerable challenges. It is instructive to consider this problem with respect to how diverse inputs control the production of master regulators in developmentally dynamic contexts, which themselves drive enhancer function to establish or maintain complex genetic networks.
By controlling hematopoietic stem cell (HSC) genesis and differentiation and progenitor proliferation/survival, the transcription factor GATA-2 mediates the development of the hematopoietic system (3–6). Whether common or distinct enhancers regulate Gata2 function in diverse settings is unclear. Deletions of prospective Gata2 enhancers, termed GATA switch sites based on GATA-1 replacement of GATA-2 at these sites during erythropoiesis (7, 8), revealed results inconsistent with enhancer predictions (9–12). Whereas certain GATA switch sites have little to no importance for regulating Gata2, the +9.5 intronic site is crucial during embryogenesis when the hemogenic endothelium of the aorta-gonad-mesonephros (AGM) produce HSCs (11, 13). Homeostasis is exquisitely sensitive to the GATA-2 level because ectopically high and low GATA-2 affect disease onset and severity (8, 14–16). GATA2 haploinsufficiency underlies primary immunodeficiency characterized by a panoply of phenotypes, including hematologic deficits, malignancy, lymphedema, and deafness (17–20). Patients with monocytopenia and disseminated Mycobacterium avium complex infection (MonoMAC), linked to myelodysplastic syndrome (MDS) and progression to acute myeloid leukemia (AML), exhibit heterozygous GATA2 coding region or +9.5 enhancer mutations (11, 16, 21).
The most distal upstream GATA switch site (−77) (22) resides within a region of AML-linked chromosomal rearrangements [inv(3)(q21q26) and t(3:3)(q21q26)] (23). The 3q21:q26 inversion repositions −77 and flanking sequences to the distant proto-oncogene EVI1, up-regulating EVI1 expression (24, 25) and down-regulating GATA2 expression in bone marrow cells from affected AML patients (25). Unresolved issues include the questions such as, does the inversion down-regulate GATA2 owing to the loss of −77 or flanking sequences? Does EVI1 up-regulation affect GATA2 expression? Does −77 control GATA2 during development? Additionally, does −77 control unique aspects of hematopoiesis not predictable from existing knowledge? We generated a −77−/− mouse strain that revealed a severe hematopoietic progenitor defect and a phenotypic constellation distinct from other models. HSC genesis was unaffected, contrasting with Gata2−/− (26) and +9.5−/− (11, 13) mouse phenotypes. These studies reveal a paradigm in which a single gene can harbor multiple cis-elements, each being essential for embryonic development but differentially important for controlling stem and progenitor cell transitions. The amalgamated cis-element mechanisms ensure the seamless execution of a complex developmental process.
RESULTS
Essential leukemogenic long-range enhancer
The −77 GATA switch site resides upstream of Gata2 near Rpn1 in a region well conserved among vertebrates (Fig. 1A) (22). Chromosome conformation capture analysis demonstrated close proximity of −77 to Gata2 in murine GATA-1–null G1E cells (22) and human bone marrow cells (25). Whereas relocalization of −77-containing sequences to EVI1, almost 40 Mb away, up-regulates EVI1 in 3q21:q26 AML, it was unclear whether −77 is critical, modulatory, or of no consequence for GATA2 expression and hematopoiesis in a normal context. Rpn1 is not GATA factor–regulated (22) or dysregulated by the −77 alteration in AML (25).
Fig. 1. −77 leukemogenic, long-range enhancer is essential for embryogenesis and hematopoiesis.

(A) Vertebrate conservation plot of −77 showing positions of GATA motifs (G; WGATAR) and E-box (E; CANNTG) within the deleted region. (B) Genotypes of −77+/+, −77+/−, and −77−/− NeoR- embryos at timed developmental stages and genotypes of NeoR− and NeoR+ pups at time of weaning. (C) Representative E13.5 +9.5−/− embryo exhibiting anemia, hemorrhage, and edema. (D) Representative E15.5 −77+/+, −77+/−, and −77−/− littermates. (E) Representative E15.5 fetal livers from −77+/+ and −77−/− littermates. (F and G) E15.5 peripheral blood quantitation [−77+/+ (n = 3) and −77−/− (n = 3)] and representative Wright-Giemsa staining. (H) Relative expression of fetal and adult β-globin mRNA in peripheral blood at E15.5. (I) Total cells [−77+/+ (n = 10), −77+/− (n = 24), and −77−/− (n = 13)] and Ter119+ cells [−77+/+ (n = 7), −77+/− (n = 17), and −77−/− (n = 5)] in E13.5 fetal livers. (J) Representative flow cytometric analysis and quantitation of E13.5 fetal livers for CD71+Ter119− R1 and R2 erythroid progenitors [−77+/+ (n = 7), −77+/− (n = 17), and −77−/− (n = 5)]. Graphs show means ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
To test −77 physiological function, we generated a mouse strain with a targeted deletion of 257 base pairs (bp) spanning conserved GATA motifs. Although E-boxes reside near the GATA motifs, their spacing and orientation deviate from the canonical E-box–GATA motif composite element (27) characteristic of the +9.5 site (28). Mouse embryos homozygous for the −77 deletion (−77−/−) survived beyond embryonic day (E) 15.5, but the vast majority died before weaning, regardless of whether they retained NeoR at the targeted site or if NeoR was excised (Fig. 1B). Whereas +9.5−/− embryos exhibited severe anemia and hemorrhaging by E12.5 and died by ~E14 (Fig. 1C) (11), −77−/− embryos exhibited little to no hemorrhaging (Fig. 1D), though they had reduced fetal liver size (Fig. 1E) and were anemic with drastically lower circulating blood cells (Fig. 1F). A greater proportion of cells in −77−/− embryo circulation were nucleated (Fig. 1G), and the cells expressed higher levels of embryonic Hbb-y and Hbb-bh1 globin RNA versus −77+/+ littermates (Fig. 1H), indicating a severe definitive erythrocyte production deficit and a predominance of primitive erythroid cells. Flow cytometric analysis revealed that reduced −77−/− fetal liver size at E13.5 was due, in large part, to Ter119+ cell loss (Fig. 1I). CD71+Ter119− erythroid precursor populations (R1 and R2) were also reduced, indicating an early erythroid differentiation blockade (Fig. 1J). The late embryonic lethality, coupled with the definitive hematopoiesis defect described above, suggested a −77 role in hematopoietic stem/progenitor cell (HSPC) genesis and/or function.
Stem and progenitor cell transitions controlled by distinct enhancers at a genetic locus
The +9.5−/− embryos lack fetal liver HSPCs because of an HSC genesis defect in the AGM region (13). Since the −77 resides at Gata2, we asked whether it also controls HSC genesis. We enumerated c-Kit+ HSC clusters in E10.5 AGM of −77+/+ and −77−/− embryos and detected no difference in c-Kit+ cell numbers in dorsal aortas (Fig. 2A). By contrast, the percentage of immunophenotypic HSCs and multipotent progenitors (MPPs) in E13.5 fetal livers significantly increased in −77−/− embryos (Fig. 2B), as did the HSC number per liver. Consistent with elevated HSCs, the percentage of −77−/− HSCs in G0 was significantly lower compared with wild-type HSCs (fig. S1). HSC function was quantitated using a competitive transplantation assay. CD45.1+ recipient mice were lethally irradiated and transplanted with CD45.2+ fetal liver cells from −77+/+, −77+/−, or −77−/− E13.5 embryos and an equal number of wild-type CD45.1+ bone marrow cells. At 4-week intervals, recipient mice were bled, and hematopoietic parameters were quantitated by flow cytometry. Consistent with increased HSCs and MPPs in −77−/− fetal livers, −77−/− donor–derived circulating CD45.2+ cells in recipient mice were higher than those in −77+/+ and −77+/− donor–derived recipient mice (Fig. 2C). By 16 weeks, the −77−/− donor cells contributed significantly more to the myeloid lineage versus the −77+/+ cells; the −77−/− contribution to T cell and B cell lineages was reduced. The functionality of −77−/− donor–derived HSCs was further analyzed by serial transplantation. At all times after secondary transplantation, recipient mice that received −77−/−-derived (CD45.2+) bone marrow HSPCs exhibited higher levels of monocytes versus controls.
Fig. 2. Long-term repopulating HSC generation and function do not require −77.
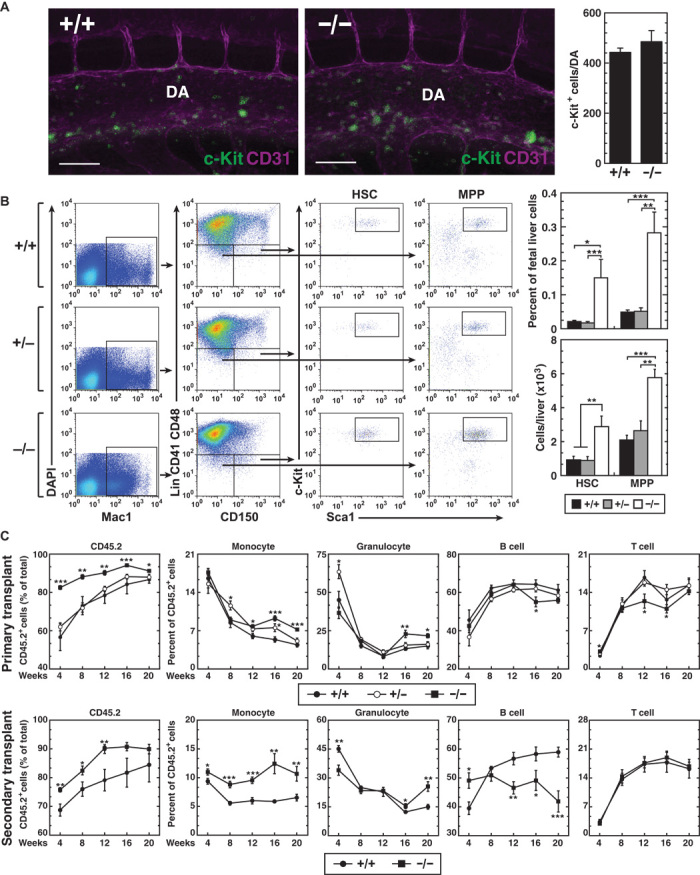
(A) Whole-mount immunostaining of E10.5 −77+/+ and −77−/− embryos showing CD31+ cells (magenta) and c-Kit+ cells (green) within the dorsal aorta (DA). Scale bars, 100 μm. Quantitation of c-Kit+ cells within the whole dorsal aorta [−77+/+ (n = 3) and −77−/− (n = 3)] is shown on the right. (B) Representative flow cytometric analysis of E13.5 fetal livers for HSCs (Lin−Mac1+CD41−CD48−CD150+Sca1+Kit+) and MPPs (Lin−Mac1+CD41−CD48−CD150−Sca1+Kit+). Quantitation of HSCs and MPPs is presented both as a percentage of total fetal liver cells (top) and as the number of cells per liver (bottom) [−77+/+ (n = 5), −77+/− (n = 6), and −77−/− (n = 4)]. (C) Contribution of −77+/− and −77−/− versus −77+/+ fetal liver cells in a competitive transplantation assay [−77+/+ (4 livers; 10 recipients), −77+/− (6 livers; 12 recipients), and −77−/− (3 livers; 11 recipients)]. After 20 weeks, secondary transplants were performed with bone marrow from fetal liver–transplanted −77−/− versus −77+/+ mice (2 bone marrow donors for each genotype; 8 recipient mice). The peripheral blood of recipient mice was analyzed for CD45.2 expression by flow cytometry at 4-week intervals after transplantation. Flow cytometric analysis of the proportions of CD45.2+ monocytes (Mac1+Gr−), granulocytes (Mac1+Gr+), B cells (CD19+), and T cells (Thy1.2+) in the peripheral blood of recipient mice. Graphs show means ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
The percentages of HSPCs (Lin−Sca1+Kit+) and myeloid progenitors (Lin−Sca1−Kit+) increased (3.6- and 2.0-fold, respectively) in −77−/− fetal livers (Fig. 3, A and B). However, when considering the reduced −77−/− fetal liver size, only HSPCs increased significantly (fig. S2). Lin−Sca1−Kit+ cells were analyzed for CD34 and Fc receptor expression to define common myeloid progenitor (CMP) (CD34+FcγRlow), granulocyte-macrophage progenitor (GMP) (CD34+FcγRhigh), and megakaryocyte-erythrocyte progenitor (MEP) (CD34−FcγRlow) populations as described (29) (Fig. 3A). Compared to wild-type littermates, the percentage of MEPs in −77−/− fetal livers was reduced 8.3-fold, whereas the percentages of CMPs and GMPs were increased 7.8- and 2.5-fold, respectively (Fig. 3B). Gata2 expression was quantitated in HSPCs and myeloid progenitor cells. Intriguingly, whereas Gata2 expression was unchanged in −77−/− versus −77+/+ HSPCs, its expression was 4.8-fold lower in the myeloid progenitor population, with similar reductions in CMPs and GMPs (Fig. 3C). Accordingly, chromatin attributes of active enhancers (accessibility and monomethylation of H3K4) are lower at −77 versus +9.5 in HSCs, but are enriched in myeloid progenitors in which Gata2 is active (Fig. 3D). Another mark of active enhancers, H3K27 acetylation, was uniquely developmentally regulated at +9.5 (fig. S3). That −77 activity selectively confers Gata2 expression in myeloid progenitors provides a molecular explanation for the divergent +9.5 and −77 mutant phenotypes.
Fig. 3. Selective loss of Gata2 expression in −77−/− myeloid progenitors disrupts homeostasis.

(A) Representative flow cytometric analysis of E13.5 fetal livers for Lin−Sca1+Kit+ and Lin−Sca1−Kit+ cells, CMPs (Lin−CD34+FcγRlowKit+Sca1−), GMPs (Lin−CD34+FcγRhighKit+Sca1−), and MEPs (Lin−CD34−FcγRlowKit+Sca1−). (B) Percentages of Lin−Sca1+Kit+ and Lin−Sca1−Kit+ cells [top; −77+/+ (n = 7), −77+/− (n = 17), and −77−/− (n = 6)] and CMPs, GMPs, and MEPs [bottom; −77+/+ (n = 8), −77+/− (n = 6), and −77−/− (n = 9)]. (C) Quantitation of Gata2 mRNA levels in Lin−Sca1+Kit+ and Lin−Sca1−Kit+ cells [−77+/+ (n = 5) and −77−/− (n = 7)] and CMPs, GMPs, and MEPs [−77+/+ (n = 4) and −77−/− (n = 4)]. (D) Profiles for chromatin accessibility via the assay for transposase-accessible chromatin (ATAC) and histone H3K4 monomethylation mined from existing data (53). Graphs show means ± SEM; **P < 0.01, ***P < 0.001.
Acquiring myeloid differentiation potential
The Gata2 expression defect in myeloid progenitors suggested that −77 selectively controls myeloid progenitor cell function, and therefore, our mouse model may provide a unique window into myeloid cell biology/pathology. Colony assays were conducted to quantitate myeloerythroid differentiation potential of −77−/− fetal liver progenitors. The −77−/− fetal livers were greatly impaired in their capacity to form CFU-GEMM (colony-forming unit–granulocyte, erythroid, macrophage, megakaryocyte) and BFU-E (burst-forming unit–erythroid) colonies, whereas myeloid colonies [CFU-GM (colony-forming unit–granulocyte, macrophage)] decreased about two-fold (Fig. 4A). The differentiation potential of flow-sorted immunophenotypic CMPs from −77−/− fetal livers was also enumerated by colony assay. Whereas −77+/+ CMPs generated the full repertoire of colony types (CFU-GEMM, CFU-GM, and BFU-E) (Fig. 4B), −77−/− CMP–derived colonies were smaller, less abundant (Fig. 4C), and strikingly composed almost exclusively of macrophages (Fig. 4D). This gross macrophage bias was also detected in colony assays with −77−/− fetal liver and flow-sorted GMPs (fig. S4). The reduced CFU-GEMM and BFU-E colony-forming potential of −77−/− fetal livers (Fig. 4) and the concomitant increase in CMPs and GMPs suggest that Gata2 stimulates myeloid progenitor cells to generate myeloerythroid progeny. Although reduced Gata2 expression upon −77 deletion blocks differentiation, macrophage generation is maintained. The MEP reduction may or may not reflect impaired CMP activity because MEP generation can bypass classically defined CMPs (30–32). The elevated multipotent cells represent an expected compensatory response, considering the severely defective myeloid progenitor cells and gross anemia of the −77−/− mutant embryos. The long-term repopulating activity of the mutant HSCs reinforces the specificity of −77 function to control myeloid cell function.
Fig. 4. Limited differentiation potential of −77−/− fetal liver progenitors.

(A) Colony-forming activity of hematopoietic progenitors from E14.5 fetal livers [−77+/+ (n = 12), −77+/− (n = 17), and −77−/− (n = 3)]. (B) Colony-forming activity of CMPs and GMPs sorted from E13.5 fetal livers [−77+/+ (n = 3) and −77−/− (n = 3)]. (C and D) Representative colonies and Wright-Giemsa–stained cells obtained from plating of CMPs. Scale bars, 2 mm. Mac, macrophage; Ery, erythroblast; Neu, neutrophil; Mye, myeloid. Graphs show means ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
Establishing a progenitor cell transcriptome
As the −77 mutation reduced myeloid progenitor abundance and impaired function, we tested whether the mutation altered the expression of established regulators of myeloerythroid differentiation. Consistent with the MEP loss, Gata1 expression was reduced in CMPs and GMPs, and Zfpm1 (FOG-1) expression was reduced in CMPs (Fig. 5A). In the very few −77−/− MEPs produced, the expression of both genes was unaffected. Among myeloid regulatory factors, −77−/− progenitors expressed normal levels of Sfpi1 (PU.1) and C/EBPα (CCAAT/enhancer binding protein-α), whereas growth factor independence 1 (Gfi1) was reduced 2.6-fold selectively in GMPs. The expression of pre–B cell leukemia homeobox 1 (Pbx1), a proto-oncogene that regulates CMP levels and differentiation potential in the fetal liver (33), was reduced three-fold in CMPs but was increased significantly in GMPs. Although the CMP compartment was substantially reduced in Pbx1−/− embryos but increased in −77−/− fetal livers, both mutations similarly altered the CMP colony-forming potential, leading to reduced erythroid colonies and increased monocytes.
Fig. 5. −77 establishes a functionally vital sector of the myeloid progenitor cell transcriptome.

(A) Quantitative gene expression analysis in CMPs, GMPs, and MEPs from −77+/+ and 77−/− E13.5 fetal livers [−77+/+ (n = 4) and −77−/− (n = 4)]. (B) MA plot of RNA-seq–based comparison of CMP transcriptomes from −77+/+ and −77−/− E13.5 fetal livers [−77+/+ (n = 3) and −77−/− (n = 3)]. Red points indicate down- or up-regulated genes [false discovery rate (FDR) <0.05]. (C) Heatmap depicting statistically significant genes down- or up-regulated by >1.5-fold. (D) Venn diagram depicting the extent of overlap between genes regulated by +9.5 in AGM (13) and −77 in CMPs. Genes shared between data sets are indicated in red in (C). (E) Evidence for direct GATA-2 regulation of −77 target genes. ChIP-seq profiles of GATA-2 and Scl/TAL1 in HPC-7 cells (58) and quantitative gene expression analysis in CMPs from −77+/+ and −77−/− E13.5 fetal livers [−77+/+ (n = 4) and −77−/− (n = 4)]. Graphs show means ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
Presumably, the quantitatively large and selective impact of the −77 mutation on myeloid progenitor numbers and myeloerythroid differentiation potential reflects a unique perturbation of the GATA-2–dependent genetic network, which has not been established in primary myeloid progenitors. In principle, corrupting differentiation by removing −77 may involve gross reconfiguration of the progenitor transcriptome, modest changes in genes encoding essential constituents, or alterations in the spatiotemporal regulation of a very limited number of genes. Because our biased gene expression analysis revealed selective, unpredictable influences of the −77 deletion on the progenitor transcriptome, we used RNA-seq to discriminate between these models and compared −77+/+ and −77−/− CMP transcriptomes. The analysis revealed 105 genes with significantly reduced transcript abundance in −77−/− versus −77+/+ CMPs and 28 genes with increased transcript abundance (FDR <0.05; Fig. 5, B and C).
Most down-regulated genes are highly expressed in myeloid, erythroid, and megakaryocytic lineages (http://biogps.org and http://gexc.stanford.edu), and this expression is consistent with functional clustering of the genes using String (fig. S5). This cohort included the master regulator of erythropoiesis, Gata1 (34, 35). Accordingly, GATA-1 down-regulation decreases erythroid and megakaryocytic target gene transcription; the expression of these genes may reflect lineage priming in the progenitors. Gene Ontology analysis of the down-regulated cohort highlighted links between down-regulated genes and fundamental processes including cell adhesion, inflammatory response, and hematopoiesis. Analysis of the −77-regulated cohort that is not +9.5-regulated in the AGM also revealed cell adhesion and hematopoiesis (fig. S6A). Whereas the most highly down-regulated genes were expressed higher in myeloerythroid cells versus multipotent hematopoietic precursors, the most highly up-regulated genes did not conform to this behavior (fig. S6B). The −77 activated genes encoding established myeloid and/or erythroid cell regulators, including Trib2 encoding Tribbles-2, which inactivates C/EBPα and induces AML (36); IL-5Rα encoding the IL-5 receptor, which induces eosinophil differentiation (37); and Hdc encoding histidine decarboxylase, which controls myeloid differentiation (38).
Additional −77-regulated genes not linked to the control of myeloid cell biology or hematopoiesis include Acss2 encoding acyl-CoA synthetase short-chain family member-2, a metabolic enzyme that generates acetyl-coA (39) and a potential epigenetic regulator, and Mfsd2b encoding major facilitator superfamily domain–containing protein 2b. Mfsd2b is selectively expressed in myeloid progenitor cells (fig. S7) and is related to Mfsd2a encoding an ω-3 fatty acid transporter essential for blood-brain barrier formation (40). The analysis also revealed −77-mediated suppression of nonhematopoietic genes, for example, liver-specific Lpl encoding lipoprotein lipase (41).
Our previous genomic analysis of +9.5-regulated genes in the AGM demonstrated enrichment of HSC and hematopoietic lineage–specific genes (13). Of these 133 genes, only 39 genes were −77 targets in CMPs (Fig. 5D). Furthermore, despite the 2777 GATA-1–regulated genes in erythroid cells (42), only 32 of the 101 −77-regulated genes overlapped with GATA-1–regulated genes (fig. S8). Thus, most −77-regulated genes are not regulated by GATA-1.
We investigated −77 regulation of novel targets that had not been linked to GATA factor mechanisms. Endogenous GATA-2 and Scl/TAL1 occupied Mfsd2b, Samd14, Mylk3, and Il1rl1 in HPC-7 cells, and real-time reverse transcription polymerase chain reaction (RT-PCR) analysis revealed a −77 requirement for their expression (Fig. 5E). In aggregate, the transcriptomic analysis uncovered a vital −77 activity to establish a critical sector of the myeloid progenitor transcriptome, distinct from that described in other mouse models, thereby endowing myeloid progenitors with potential to generate the full repertoire of myeloerythroid cell progeny.
DISCUSSION
Of the vast cis-element entourage, very few have been reported to be essential for embryonic development and for critical processes. The work described herein establishes −77 as the sole cis-element essential for embryogenesis and conferring progenitor cell multipotentiality. Whereas −77 does not regulate Gata2 transcription in multipotent LSK cells, it up-regulates Gata2 expression in myeloid progenitors. The Gata2 +9.5 site is the sole cis-element essential for embryogenesis and stem cell genesis (11, 13). Because +9.5 and −77 exert qualitatively different activities, distinct cis-elements in a locus differentially control stem and progenitor cell transitions within a common developmental blueprint.
The −77−/− mice revealed a novel GATA-2–dependent mechanism to control myeloid progenitor cell function. GATA-2–null yolk sac primitive hematopoietic progenitors are defective in generating mast cells, but not erythroid cells, megakaryocytes, and neutrophils (4). In an IL-3–dependent hematopoietic progenitor cell line, PU.1 and GATA-2 cooperate to generate mast cells, whereas PU.1 antagonizes GATA-2 to generate macrophages (43). Given the gross macrophage bias of −77−/− myeloid progenitors, leveraging this mechanism may represent an innovative strategy to produce macrophages for experimental and/or therapeutic goals. Mechanistic analysis revealed that the −77 deletion dysregulated genes in CMPs that did not include most established myeloid-specific genes, including the master transcriptional regulators of myelopoiesis, C/EBPα, PU.1, and the myeloid signaling factor granulocyte-macrophage colony-stimulating factor (GM-CSF). Thus, −77 establishes a novel and functionally critical sector of the myeloid progenitor cell transcriptome.
An assemblage of essential cis-elements with distinct activities in a locus has important implications for disease mechanisms. GATA2 haploinsufficiency underlies syndromes with unique and overlapping phenotypes (16, 44). Mutational disruption of +9.5 and repositioning of −77 yield MonoMAC immunodeficiency (11, 21) and AML (24, 25), respectively, in humans. As MonoMAC is often associated with MDS, which progresses to AML (16, 44), the integrity of the sites ensures normal hematopoiesis while suppressing leukemogenesis. It is instructive to relate the unique features of −77-linked AML and +9.5-linked MonoMAC to their differential physiological functions to orchestrate progenitor and stem cell transitions (Fig. 6). The deleterious impact of +9.5 deletion on stem cell generation/function (even apparent with +9.5+/− mice) disrupts hematopoiesis in qualitatively and quantitatively different ways versus inversions and translocations that displace −77 from the Gata2 locus. As −77 loss greatly reduces Gata2 expression in CMPs and GMPs but not in LSK cells, mutant myeloid progenitors with an intrinsic differentiation blockade accumulate, and both CMPs and GMPs initiate AML (45–48), the integrity of the −77-dependent genetic network in myeloid progenitors must be exquisitely maintained. The severe phenotypic defects of the −77−/− mice described herein provide mechanistic insights into pathologies, as human GATA2 heterozygous mutations are frequently associated with epigenetic silencing of the wild-type allele to yield, in effect, a nullizygous mutation (49). An additional aspect of the differential mechanisms relevant to 3q21:q26 AML relates to −77-mediated up-regulation of EVI1 expression, which is not relevant to +9.5-linked pathologies. Because EVI1 up-regulation is not an attribute of our −77−/− model, we dissociated phenotypes resulting from −77 loss and EVI1 up-regulation.
Fig. 6.
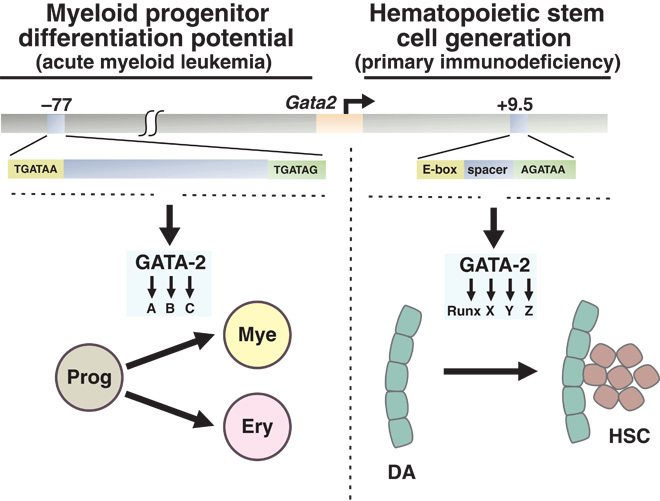
Selective control of stem and progenitor cell transitions by distinct cis-elements within a genetic locus. The model depicts differential +9.5 and −77 functions to control critical stages of hematopoiesis. Disruption of −77 and +9.5 in humans causes acute myeloid leukemia and primary immunodeficiency, respectively. Whereas both enhancers are essential for GATA-2 regulation, they operate in distinct cellular contexts, and genomic analyses indicate major differences in their target gene ensembles. Prog, myeloerythroid progenitor; Mye, myeloid progenitor; Ery, erythroid; DA, dorsal aorta.
In summary, our study revealed a mechanism for conferring multipotentiality to a progenitor cell population that yields erythroid and myeloid cell progeny. The gene controlled by the −77 element, GATA2, regulates the development of non–small cell lung cancer (50), aggressive prostate cancer (51), primary immunodeficiency, MDS, and myeloid leukemia (16, 44). GATA2 is also linked to the development/function of the pituitary (52) and important components of the nervous system (53). Thus, the underlying biology and mechanisms affect a wide swath of biology/medicine. Our results establish a paradigm in which multiple cis-elements in a single locus function nonredundantly to differentially control stem and progenitor cell transitions, and the aggregate mechanisms orchestrate a complex developmental process. We focused on our discovery of two essential Gata2 cis-elements causally linked to GATA-2–dependent pathologies. For disease loci containing more complex cis-element assemblages, one might predict a relationship between the sheer number of qualitatively distinct, essential cis-elements, and disease phenotype complexity, in the absence of coding region mutations.
MATERIALS AND METHODS
Gata2 −77 mutant mouse generation
A 257-bp region (mm10; chr6: 88116954–88117210) upstream of Gata2 was replaced with a LoxP-PGKneo-LoxP cassette via homologous recombination as previously described (11). Gene-targeted CJ9 embryonic stem cell clones were screened by PCR, and targeting was confirmed by Southern blotting. Chimeric mice were generated by blastocyst injection, and F1 pups were screened for germline transmission by PCR. NeoR was excised by mating to cytomegalovirus (CMV) Cre-expressing strain B6.C-Tg(CMV-cre)1Cgn/J (The Jackson Laboratory). Cre-mediated excision of NeoR in the progeny was confirmed by PCR using primers flanking the targeted sequence. All animal experiments were performed with the ethical approval of the AAALAC International (Association for the Assessment and Accreditation of Laboratory Animal Care) at the University of Wisconsin–Madison.
Whole-embryo confocal microscopy
Embryos were fixed, stained, and analyzed as previously described (12, 13, 54). Embryos were stained with anti–PECAM-1 (platelet endothelial cell adhesion molecule 1) (553371, BD Biosciences) and anti–c-Kit (553352, BD Biosciences) antibodies. The samples were mounted in a 1:2 mix of benzyl alcohol and benzyl benzoate to increase the transparency of tissues and visualized with a Nikon A1R confocal microscope. Three-dimensional reconstructions were generated from z-stacks (50 to 150 optical sections) using Fiji software.
Fetal liver cell transplantation
Adult C57BL/6 recipient mice (CD45.1+, 6 to 8 weeks old; National Cancer Institute) were lethally irradiated with a cesium source for a single dose of 11 Gy. Fetal liver cells were harvested from individual E13.5 embryos (CD45.2+). A total of 250,000 live nucleated fetal liver cells were mixed with the same number of CD45.1+ bone marrow cells and injected into individual irradiated CD45.1+ recipients. The transplanted recipient mice were maintained on trimethoprim/sulfamethoxazole-treated water for 2 weeks. Blood was regularly obtained from the retro-orbital venous sinus after transplantation and analyzed using flow cytometry for donor-derived hematopoiesis as previously described (55). Secondary transplants were performed after 20 weeks, using bone marrow pooled from two fetal liver recipients. Bone marrow cells (5 × 106) were injected into irradiated CD45.1+ recipients.
Flow cytometry
Fetal liver cells from E13.5 individual embryos were dissociated and resuspended in phosphate-buffered saline with 2% fetal bovine serum (FBS) and passed through 25-μm cell strainers to obtain single-cell suspensions before antibody staining. The early hematopoietic populations analyzed were fetal liver HSCs (Lin−CD41−CD48−Mac1+CD150+Kit+Sca1+), MPPs (Lin−CD41−CD48−Mac1+CD150−Kit+Sca1+), Lin−Kit+Sca1+, and Lin−Kit+Sca1−. All antibodies were purchased from eBioscience unless otherwise stated. Lineage markers for these populations were stained with fluorescein isothiocyanate (FITC)–conjugated B220 (11-0452), CD3 (11-0031), CD4 (11-0041), CD5 (11-0051), CD8 (11-0081), CD41 (11-0411), CD48 (no. 11-0481), Gr-1 (11-5931), and TER-119 (11-5921) antibodies. Other surface proteins were detected with phycoerythrin (PE)–conjugated CD71 (R17217), CD150 (115904, BioLegend), and Sca1 (E13-161.7); PE-Cy7–conjugated Mac1 (25-0112); peridinin chlorophyll protein (PerCP)–Cy5.5–conjugated Sca1 (45-5981); and allophycocyanin-conjugated c-Kit (2B8) antibodies. Analysis of fetal liver CMPs (Lin−CD34+FcγRlowKit+Sca1−), GMPs (Lin−CD34+FcγRhighKit+Sca1−), and MEPs (Lin−CD34−FcγRlowKit+Sca1−) was performed as previously described (29). Lineage markers were stained with FITC-conjugated B220, CD3, CD4, CD5, CD8, CD19 (11-0193), immunoglobulin M (11-5890), Il7Ra (11-1271), AA4.1 (561990, BD Biosciences), and TER-119 antibodies. Other surface proteins were detected with PE-conjugated Fc receptor (12-0161), Alexa Fluor 647–conjugated CD34 (BDB560230, BD Biosciences), and PerCP-Cy5.5–conjugated Sca1 (no. 45-5981) antibodies. For c-Kit detection, we used biotin-conjugated CD117 (13-1171) with PE-Cy7–conjugated streptavidin (24-4317) antibodies. The stained cells were collected on a FACSCalibur flow cytometer (BD Biosciences), except HSCs, which were analyzed on a LSR II flow cytometer (BD Biosciences). The data were analyzed using FlowJo v9.0.2 software (TreeStar).
Colony assays
For fetal liver colony assays, dissociated cells from E14.5 embryos were plated in duplicate in Methocult M03434 complete medium (StemCell Technologies) at a density of 2 × 104 cells per 35-mm plate. Plates were incubated for 7 to 8 days according to the manufacturer’s recommendation, and colonies were identified and counted using an Olympus Szx16 stereomicroscope. For purified CMPs and GMPs, cells were sorted into IMDM/2% FBS, and a specified number of cells were plated in duplicate in Methocult M03434 complete medium.
Gene expression analysis
Total RNA was purified from fetal livers or sorted cell populations using TRIzol (Invitrogen). Complementary DNA (cDNA) was synthesized by Moloney murine leukemia virus reverse transcription (MMLV RT). Real-time PCR was performed with SYBR Green Master Mix. Control reactions lacking MMLV RT yielded little to no signal. Relative expression was determined from a standard curve of serial dilutions of cDNA samples, and values were normalized to 18S RNA expression. The specific primers used for real-time PCR are given in table S1. RNA-seq was conducted with an Illumina HiSeq 2000 system. Transcript quantification and differential expression were conducted using the software packages RSEM (56) and DESeq2 (57), respectively. RSEM v1.2.18 was provided with a reference transcript set consisting of all protein-coding and large intergenic noncoding transcripts from the Ensembl release 67 annotation of the National Center for Biotechnology Information (NCBI) Build 38 mouse genome assembly. Default parameters and Bowtie v1.1.1 were used for transcript quantification with RSEM. Transcript read counts were summed to produce counts at the level of gene symbols. These counts were then given as inputs to DESeq2 v1.5.90 for differential expression analysis. DESeq2 was run with default parameters except for betaprior = F and alpha = 0.05. Gene symbols with Benjamini-Hochberg FDR values <0.05 were deemed to be differentially expressed between mutant and wild type. The accession number for the RNA-seq data is GEO: GSE69786.
Statistics
For quantitative measurement of cells, cell colonies, or mRNA, the results are presented as means ± SEM. Determination of significance was conducted using two-tailed unpaired Student’s t test. A P value <0.05 was considered significant.
Funding
This work was supported by NIH grants DK68634 and DK50107 (to E.H.B.), CA152108 and HL113066 (to J.Z.), and U01 HG007019 (to C.N.D. and E.H.B.) and Cancer Center Support Grant P30 CA014520. K.J.H. is supported by an American Heart Association Postdoctoral Fellowship. Author contributions: K.D.J., G.K., J.Z., and E.H.B designed the research; K.D.J., G.K., X.G., Y.-I.C., R.S., and R.P. performed experiments; K.D.J., G.K., K.J.H., E.A.R., C.N.D., J.Z., and E.H.B. analyzed the results; and K.D.J. and E.H.B. wrote the manuscript. Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/8/e1500503/DC1
Table S1. Sequences of primers used for qPCR.
Fig. S1. Reduced quiescence of −77−/− HSCs.
Fig. S2. Expansion of HSPC compartment in −77−/− fetal livers.
Fig. S3. Distinct dynamic chromatin signatures at the −77 and +9.5 during hematopoiesis.
Fig. S4. Macrophage bias of −77−/− myeloid progenitors.
Fig. S5. −77 target genes define myeloerythroid, megakaryocyte, and nonhematopoietic protein interaction networks.
Fig. S6. −77 controls a cohort of genes with hematopoietic function.
Fig. S7. Selective myeloerythroid expression patterns of −77 target genes.
Fig. S8. A minority of −77-regulated genes are GATA-1–regulated.
REFERENCES AND NOTES
- 1.Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B. D., Shen Y., Pervouchine D. D., Djebali S., Thurman R. E., Kaul R., Rynes E., Kirilusha A., Marinov G. K., Williams B. A., Trout D., Amrhein H., Fisher-Aylor K., Antoshechkin I., DeSalvo G., See L.-H., Fastuca M., Drenkow J., Zaleski C., Dobin A., Prieto P., Lagarde J., Bussotti G., Tanzer A., Denas O., Li K., Bender M. A., Zhang M., Byron R., Groudine M. T., McCleary D., Pham L., Ye Z., Kuan S., Edsall L., Wu Y.-C., Rasmussen M. D., Bansal M. S., Kellis M., Keller C. A., Morrissey C. S., Mishra T., Jain D., Dogan N., Harris R. S., Cayting P., Kawli T., Boyle A. P., Euskirchen G., Kundaje A., Lin S., Lin Y., Jansen C., Malladi V. S., Cline M. S., Erickson D. T., Kirkup V. M., Learned K., Sloan C. A., Rosenbloom K. R., Lacerda de Sousa B., Beal K., Pignatelli M., Flicek P., Lian J., Kahveci T., Lee D., Kent W. J., Santos M. R., Herrero J., Notredame C., Johnson A., Vong S., Lee K., Bates D., Neri F., Diegel M., Canfield T., Sabo P. J., Wilken M. S., Reh T. A., Giste E., Shafer A., Kutyavin T., Haugen E., Dunn D., Reynolds A. P., Neph S., Humbert R., Scott Hansen R., De Bruijn M., Selleri L., Rudensky A., Josefowicz S., Samstein R., Eichler E. E., Orkin S. H., Levasseur D., Papayannopoulou T., Chang K.-H., Skoultchi A., Gosh S., Disteche C., Treuting P., Wang Y., Weiss M. J., Blobel G. A., Cao X., Zhong S., Wang T., Good P. J., Lowdon R. F., Adams L. B., Zhou X.-Q., Pazin M. J., Feingold E. A., Wold B., Taylor J., Mortazavi A., Weissman S. M., Stamatoyannopoulos J. A., Snyder M. P., Guigo R., Gingeras T. R., Gilbert D. M., Hardison R. C., Beer M. A., Ren, B. The Mouse ENCODE Consortium , A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kellis M., Wold B., Snyder M. P., Bernstein B. E., Kundaje A., Marinov G. K., Ward L. D., Birney E., Crawford G. E., Dekker J., Dunham I., Elnitski L. L., Farnham P. J., Feingold E. A., Gerstein M., Giddings M. C., Gilbert D. M., Gingeras T. R., Green E. D., Guigo R., Hubbard T., Kent J., Lieb J. D., Myers R. M., Pazin M. J., Ren B., Stamatoyannopoulos J. A., Weng Z., White K. P., Hardison R. C., Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. U.S.A. 111, 6131–6138 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai F. Y., Keller G., Kuo F. C., Weiss M., Chen J., Rosenblatt M., Alt F. W., Orkin S. H., An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature 371, 221–226 (1994). [DOI] [PubMed] [Google Scholar]
- 4.Tsai F.-Y., Orkin S. H., Transcription factor GATA-2 is required for proliferation/survival of early hematopoietic cells and mast cell formation, but not for erythroid and myeloid terminal differentiation. Blood 89, 3636–3643 (1997). [PubMed] [Google Scholar]
- 5.de Pater E., Kaimakis P., Vink C. S., Yokomizo T., Yamada-Inagawa T., van der Liden R., Kartalaei P. S., Camper S. A., Speck N., Dzierzak E., Gata2 is required for HSC generation and survival. J. Exp. Med. 210, 2843–2850 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim K. C., Hosoya T., Brandt W., Ku C. J., Hosoya-Ohmura S., Camper S. A., Yamamoto M., Engel J. D., Conditional Gata2 inactivation results in HSC loss and lymphatic mispatterning. J. Clin. Invest. 122, 3705–3717 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bresnick E. H., Lee H. Y., Fujiwara T., Johnson K. D., Keles S., GATA switches as developmental drivers. J. Biol. Chem. 285, 31087–31093 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeVilbiss A. W., Sanalkumar R., Johnson K. D., Keles S., Bresnick E. H., Hematopoietic transcriptional mechanisms: From locus-specific to genome-wide vantage points. Exp. Hematol. 42, 618–629 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snow J. W., Trowbridge J. J., Fujiwara T., Emambokus N. E., Grass J. A., Orkin S. H., Bresnick E. H., A single cis element maintains repression of the key developmental regulator Gata2. PLOS Genet. 6, e1001103 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snow J. W., Trowbridge J. J., Johnson K. D., Fujiwara T., Emambokus N. E., Grass J. A., Orkin S. H., Bresnick E. H., Context-dependent function of "GATA switch" sites in vivo. Blood 117, 4769–4772 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson K. D., Hsu A. P., Ryu M. J., Wang J., Gao X., Boyer M. E., Liu Y., Lee Y., Calvo K. R., Keles S., Zhang J., Holland S. M., Bresnick E. H., Cis-element mutated in GATA2-dependent immunodeficiency governs hematopoiesis and vascular integrity. J. Clin. Invest. 122, 3692–3704 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanalkumar R., Johnson K. D., Gao X., Boyer M. E., Chang Y. I., Hewitt K. J., Zhang J., Bresnick E. H., Mechanism governing a stem cell-generating cis-regulatory element. Proc. Natl. Acad. Sci. U. S. A. 111, E1091–E1100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao X., Johnson K. D., Chang Y. I., Boyer M. E., Dewey C. N., Zhang J., Bresnick E. H., Gata2 cis-element is required for hematopoietic stem cell generation in the mammalian embryo. J. Exp. Med. 210, 2833–2842 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luesink M., Hollink I. H., van der Velden V. H., Knops R. H., Boezeman J. B., de Haas V., Trka J., Baruchel A., Reinhardt D., van der Reijden B. A., van den Heuvel-Eibrink M. M., Zwaan C. M., Jansen J. H., High GATA2 expression is a poor prognostic marker in pediatric acute myeloid leukemia. Blood 120, 2064–2075 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Vicente C., Vazquez I., Conchillo A., García-Sánchez M. A., Marcotegui N., Fuster O., González M., Calasanz M. J., Lahortiga I., Odero M. D., Overexpression of GATA2 predicts an adverse prognosis for patients with acute myeloid leukemia and it is associated with distinct molecular abnormalities. Leukemia 26, 550–554 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Spinner M. A., Sanchez L. A., Hsu A. P., Shaw P. A., Zerbe C. S., Calvo K. R., Arthur D. C., Gu W., Gould C. M., Brewer C. C., Cowen E. W., Freeman A. F., Olivier K. N., Uzel G., Zelazny A. M., Daub J. R., Spalding C. D., Claypool R. J., Giri N. K., Alter B. P., Mace E. M., Orange J. S., Cuellar-Rodriguez J., Hickstein D. D., Holland S. M., GATA2 deficiency: A protean disorder of hematopoiesis, lymphatics, and immunity. Blood 123, 809–821 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu A. P., Sampaio E. P., Khan J., Calvo K. R., Lemieux J. E., Patel S. Y., Frucht D. M., Vinh D. C., Auth R. D., Freeman A. F., Olivier K. N., Uzel G., Zerbe C. S., Spalding C., Pittaluga S., Raffeld M., Kuhns D. B., Ding L., Paulson M. L., Marciano B. E., Gea-Banacloche J. C., Orange J. S., Cuellar-Rodriguez J., Hickstein D. D., Holland S. M., Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 118, 2653–2655 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dickinson R. E., Griffin H., Bigley V., Reynard L. N., Hussain R., Haniffa M., Lakey J. H., Rahman T., Wang X. N., McGovern N., Pagan S., Cookson S., McDonald D., Chua I., Wallis J., Cant A., Wright M., Keavney B., Chinnery P. F., Loughlin J., Hambleton S., Santibanez-Koref M., Collin M., Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood 118, 2656–2658 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahn C. N., Chong C. E., Carmichael C. L., Wilkins E. J., Brautigan P. J., Li X. C., Babic M., Lin M., Carmagnac A., Lee Y. K., Kok C. H., Gagliardi L., Friend K. L., Ekert P. G., Butcher C. M., Brown A. L., Lewis I. D., LB To, Timms A. E., Storek J., Moore S., Altree M., Escher R., Bardy P. G., Suthers G. K., D’Andrea R. J., Horwitz M. S., Scott H. S., Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat. Genet. 43, 1012–1017 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ostergaard P., Simpson M. A., Connell F. C., Steward C. G., Brice G., Woollard W. J., Dafou D., Kilo T., Smithson S., Lunt P., Murday V. A., Hodgson S., Keenan R., Pilz D. T., Martinez-Corral I., Makinen T., Mortimer P. S., Jeffery S., Trembath R. C., Mansour S., Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat. Genet. 43, 929–931 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Hsu A. P., Johnson K. D., Falcone E. L., Sanalkumar R., Sanchez L., Hickstein D. D., Cuellar-Rodriguez J., Lemieux J. E., Zerbe C. S., Bresnick E. H., Holland S. M., GATA2 haploinsufficiency caused by mutations in a conserved intronic element leads to MonoMAC syndrome. Blood 121, 3830–3837, S3831–S3837 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grass J. A., Jing H., Kim S. I., Martowicz M. L., Pal S., Blobel G. A., Bresnick E. H., Distinct functions of dispersed GATA factor complexes at an endogenous gene locus. Mol. Cell. Biol. 26, 7056–7067 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vicente C., Conchillo A., Garcia-Sanchez M. A., Odero M. D., The role of the GATA2 transcription factor in normal and malignant hematopoiesis. Crit. Rev. Oncol. Hematol. 82, 1–17 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Yamazaki H., Suzuki M., Otsuki A., Shimizu R., Bresnick E. H., Engel J. D., Yamamoto M., A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 25, 415–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gröschel S., Sanders M. A., Hoogenboezem R., de Wit E., Bouwman B. A., Erpelinck C., van der Velden V. H., Havermans M., Avellino R., van Lom K., Rombouts E. J., van Duin M., Döhner K., Beverloo H. B., Bradner J. E., Döhner H., Löwenberg B., Valk P. J., Bindels E. M., de Laat W., Delwel R., A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157, 369–381 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Fujiwara Y., Browne C. P., Cunniff K., Goff S. C., Orkin S. H., Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc. Natl. Acad. Sci. U.S.A. 93, 12355–12358 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wadman I. A., Osada H., Grütz G. G., Agulnick A. D., Westphal H., Forster A., Rabbitts T. H., The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J. 16, 3145–3157 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wozniak R. J., Keles S., Lugus J. J., Young K. H., Boyer M. E., Tran T. M., Choi K., Bresnick E. H., Molecular hallmarks of endogenous chromatin complexes containing master regulators of hematopoiesis. Mol. Cell. Biol. 28, 6681–6694 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Traver D., Miyamoto T., Christensen J., Iwasaki-Arai J., Akashi K., Weissman I. L., Fetal liver myelopoiesis occurs through distinct, prospectively isolatable progenitor subsets. Blood 98, 627–635 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Miyawaki K., Arinobu Y., Iwasaki H., Kohno K., Tsuzuki H., Iino T., Shima T., Kikushige Y., Takenaka K., Miyamoto T., Akashi K., CD41 marks the initial myelo-erythroid lineage specification in adult mouse hematopoiesis: Redefinition of murine common myeloid progenitor. Stem Cells 33, 976–987 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto R., Morita Y., Ooehara J., Hamanaka S., Onodera M., Rudolph K. L., Ema H., Nakauchi H., Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 154, 1112–1126 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Sun J., Ramos A., Chapman B., Johnnidis J. B., Le L., Ho Y. J., Klein A., Hofmann O., Camargo F. D., Clonal dynamics of native haematopoiesis. Nature 514, 322–327 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DiMartino J. F., Selleri L., Traver D., Firpo M. T., Rhee J., Warnke R., O’Gorman S., Weissman I. L., Cleary M. L., The Hox cofactor and proto-oncogene Pbx1 is required for maintenance of definitive hematopoiesis in the fetal liver. Blood 98, 618–626 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Evans T., Felsenfeld G., The erythroid-specific transcription factor eryf1: A new finger protein. Cell 58, 877–885 (1989). [DOI] [PubMed] [Google Scholar]
- 35.Tsai S. F., Martin D. I., Zon L. I., D’Andrea A. D., Wong G. G., Orkin S. H., Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through expression in mammalian cells. Nature 339, 446–451 (1989). [DOI] [PubMed] [Google Scholar]
- 36.Keeshan K., He Y., Wouters B. J., Shestova O., Xu L., Sai H., Rodriguez C. G., Maillard I., Tobias J. W., Valk P., Carroll M., Aster J. C., Delwel R., Pear W. S., Tribbles homolog 2 inactivates C/EBPα and causes acute myelogenous leukemia. Cancer Cell 10, 401–411 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beekman J. M., Verhagen L. P., Geijsen N., Coffer P. J., Regulation of myelopoiesis through syntenin-mediated modulation of IL-5 receptor output. Blood 114, 3917–3927 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Yang X. D., Ai W., Asfaha S., Bhagat G., Friedman R. A., Jin G., Park H., Shykind B., Diacovo T. G., Falus A., Wang T. C., Histamine deficiency promotes inflammation-associated carcinogenesis through reduced myeloid maturation and accumulation of CD11b+Ly6G+ immature myeloid cells. Nat. Med. 17, 87–95 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fujino T., Kondo J., Ishikawa M., Morikawa K., Yamamoto T. T., Acetyl-CoA synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J. Biol. Chem. 276, 11420–11426 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Betsholtz C., Physiology: Double function at the blood-brain barrier. Nature 509, 432–433 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Weinstock P. H., Levak-Frank S., Hudgins L. C., Radner H., Friedman J. M., Zechner R., Breslow J. L., Lipoprotein lipase controls fatty acid entry into adipose tissue, but fat mass is preserved by endogenous synthesis in mice deficient in adipose tissue lipoprotein lipase. Proc. Natl. Acad. Sci. U.S.A. 94, 10261–10266 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeVilbiss A. W., Boyer M. E., Bresnick E. H., Establishing a hematopoietic genetic network through locus-specific integration of chromatin regulators. Proc. Natl. Acad. Sci. U.S.A. 110, E3398–E3407 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walsh J. C., DeKoter R. P., Lee H. J., Smith E. D., Lancki D. W., Gurish M. F., Friend D. S., Stevens R. L., Anastasi J., Singh H., Cooperative and antagonistic interplay between PU.1 and GATA-2 in the specification of myeloid cell fates. Immunity 17, 665–676 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Dickinson R. E., Milne P., Jardine L., Zandi S., Swierczek S. I., McGovern N., Cookson S., Ferozepurwalla Z., Langridge A., Pagan S., Gennery A., Heiskanen-Kosma T., Hämäläinen S., Seppänen M., Helbert M., Tholouli E., Gambineri E., Reykdal S., Gottfreðsson M., Thaventhiran J. E., Morris E., Hirschfield G., Richter A. G., Jolles S., Bacon C. M., Hambleton S., Haniffa M., Bryceson Y., Allen C., Prchal J. T., Dick J. E., Bigley V., Collin M., The evolution of cellular deficiency in GATA2 mutation. Blood 123, 863–874 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goardon N., Marchi E., Atzberger A., Quek L., Schuh A., Soneji S., Woll P., Mead A., Alford K. A., Rout R., Chaudhury S., Gilkes A., Knapper S., Beldjord K., Begum S., Rose S., Geddes N., Griffiths M., Standen G., Sternberg A., Cavenagh J., Hunter H., Bowen D., Killick S., Robinson L., Price A., Macintyre E., Virgo P., Burnett A., Craddock C., Enver T., Jacobsen S. E., Porcher C., Vyas P., Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 19, 138–152 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Huntly B. J., Shigematsu H., Deguchi K., Lee B. H., Mizuno S., Duclos N., Rowan R., Amaral S., Curley D., Williams I. R., Akashi K., Gilliland D. G., MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 6, 587–596 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Krivtsov A. V., Twomey D., Feng Z., Stubbs M. C., Wang Y., Faber J., Levine J. E., Wang J., Hahn W. C., Gilliland D. G., Golub T. R., Armstrong S. A., Transformation from committed progenitor to leukaemia stem cell initiated by MLL–AF9. Nature 442, 818–822 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Kirstetter P., Schuster M. B., Bereshchenko O., Moore S., Dvinge H., Kurz E., Theilgaard-Mönch K., Månsson R., Pedersen T. A., Pabst T., Schrock E., Porse B. T., Jacobsen S. E., Bertone P., Tenen D. G., Nerlov C., Modeling of C/EBPα mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell 13, 299–310 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Celton M., Forest A., Gosse G., Lemieux S., Hebert J., Sauvageau G., Wilhelm B. T., Epigenetic regulation of GATA2 and its impact on normal karyotype acute myeloid leukemia. Leukemia 28, 1617–1626 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Kumar M. S., Hancock D. C., Molina-Arcas M., Steckel M., East P., Diefenbacher M., Armenteros-Monterroso E., Lassailly F., Matthews N., Nye E., Stamp G., Behrens A., Downward J., The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell 149, 642–655 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Plymate S. R., Bhatt R. S., Balk S. P., Taxane resistance in prostate cancer mediated by AR-independent GATA2 regulation of IGF2. Cancer Cell 27, 158–159 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Charles M. A., Saunders T. L., Wood W. M., Owens K., Parlow A. F., Camper S. A., Ridgway E. C., Gordon D. F., Pituitary-specific Gata2 knockout: Effects on gonadotrope and thyrotrope function. Mol. Endocrinol. 20, 1366–1377 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Craven S. E., Lim K. C., Ye W., Engel J. D., de Sauvage F., Rosenthal A., Gata2 specifies serotonergic neurons downstream of sonic hedgehog. Development 131, 1165–1173 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Yokomizo T., Yamada-Inagawa T., Yzaguirre A. D., Chen M. J., Speck N. A., Dzierzak E., Whole-mount three-dimensional imaging of internally localized immunostained cells within mouse embryos. Nat. Protoc. 7, 421–431 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang J., Wang J., Liu Y., Sidik H., Young K. H., Lodish H. F., Fleming M. D., Oncogenic Kras-induced leukemogeneis: Hematopoietic stem cells as the initial target and lineage-specific progenitors as the potential targets for final leukemic transformation. Blood 113, 1304–1314 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilson N. K., Foster S. D., Wang X., Knezevic K., Schütte J., Kaimakis P., Chilarska P. M., Kinston S., Ouwehand W. H., Dzierzak E., Pimanda J. E., de Bruijn M. F., Göttgens B., Combinatorial transcriptional control in blood stem/progenitor cells: Genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell 7, 532–544 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/8/e1500503/DC1
Table S1. Sequences of primers used for qPCR.
Fig. S1. Reduced quiescence of −77−/− HSCs.
Fig. S2. Expansion of HSPC compartment in −77−/− fetal livers.
Fig. S3. Distinct dynamic chromatin signatures at the −77 and +9.5 during hematopoiesis.
Fig. S4. Macrophage bias of −77−/− myeloid progenitors.
Fig. S5. −77 target genes define myeloerythroid, megakaryocyte, and nonhematopoietic protein interaction networks.
Fig. S6. −77 controls a cohort of genes with hematopoietic function.
Fig. S7. Selective myeloerythroid expression patterns of −77 target genes.
Fig. S8. A minority of −77-regulated genes are GATA-1–regulated.