

Novel nanoarray for single molecule detection from peptide mixture.
Keywords: plasmonics, self-similar chain device, nano lens, single molecule detection, single point mutation detection, BRCA1 protein
Abstract
Control of the architecture and electromagnetic behavior of nanostructures offers the possibility of designing and fabricating sensors that, owing to their intrinsic behavior, provide solutions to new problems in various fields. We show detection of peptides in multicomponent mixtures derived from human samples for early diagnosis of breast cancer. The architecture of sensors is based on a matrix array where pixels constitute a plasmonic device showing a strong electric field enhancement localized in an area of a few square nanometers. The method allows detection of single point mutations in peptides composing the BRCA1 protein. The sensitivity demonstrated falls in the picomolar (10−12 M) range. The success of this approach is a result of accurate design and fabrication control. The residual roughness introduced by fabrication was taken into account in optical modeling and was a further contributing factor in plasmon localization, increasing the sensitivity and selectivity of the sensors. This methodology developed for breast cancer detection can be considered a general strategy that is applicable to various pathologies and other chemical analytical cases where complex mixtures have to be resolved in their constitutive components.
INTRODUCTION
The control of the top-down organization of matter at nanoscale is a mature field where convergence of design and accurate fabrication can be obtained in a reproducible way (1–8). Therefore, designing devices for applications, especially in areas such as few-molecules detection, is now possible.
Here, we propose the use of organized plasmonic nanostructures, in particular a matrix array of self-similar chains (SSCs) (9–12), to solve the molecular composition of complex mixture solutions. We intend to address early disease detection (13–15), in particular early detection of breast cancer (16, 17). The conceptual scheme of this paper interlaces the design, fabrication, and measurement of the architecture of the chosen device, as well as sample preparation. For measurement, we combine SSC nanostructures with Raman spectroscopy. In fact, these kinds of devices, thanks to strong plasmon localization, have the enhanced sensitivity and specificity needed for solving complex mixtures.
The combination of plasmonic nanostructures and Raman spectroscopy is recognized as a powerful analytical approach (18). Nevertheless, for single-molecule or few-molecules detection, the method has strong limitations when used as a brute force for complex constructs (such as a mixture of different kinds of molecules) (9, 10), as in early disease detection or, in general, in molecular medicine. These limitations are easily explained by the fact that Raman spectra from a mixture of several kinds of macromolecules or from high–molecular weight proteins do not show any specific signatures necessary to solve or distinguish each component or basic molecular group constituting high–molecular weight proteins. That is, Raman bands originating from different components of a mixture tend to overlap one another, and the attribution of Raman peaks and bands to each molecule or to the building block of molecules’ constituents is almost impossible. In particular, the method fails when the number of components is higher than 5 (19) or in proteins with high molecular weight.
We propose, on the basis of human proteins implicated in breast cancer development, a way to detect a mixture of peptides unambiguously representing the signature of a tumor. This method, designed for detecting biomarkers in the early stage of cancer, was applied to human samples. Moreover, in a mixture of peptides, we demonstrated specifically down to the single amino acid level. This result was achieved by retaining the high sensitivity of a single SSC and by achieving high selectivity for SSCs combined in an m × n matrix array. In particular, we designed a matrix where each SSC is the sensitive pixel of the array (Fig. 1) and total pixel interrogation is used to reconstruct the molecular composition of the mixture. By setting an appropriate dilution, each pixel detects a reduced number of molecular species in the mixture, compatible with the solution concentration and diffusion laws. The whole mixture can be reconstructed by reading the Raman spectra of a dried solution on all m × n matrix elements.
Fig. 1. Scheme of the whole process from peptide extraction to Raman detection.
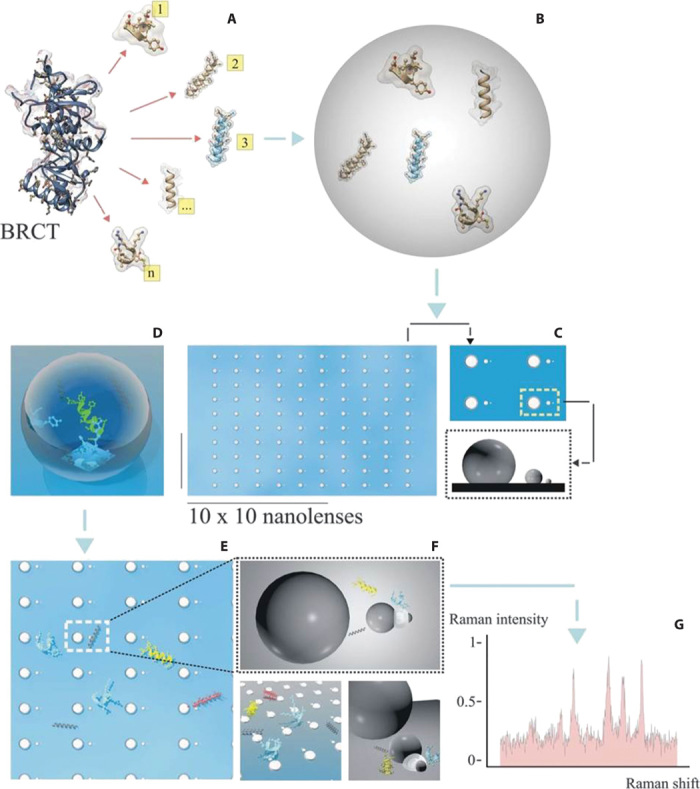
(A) A mixture of peptides is extracted from the BRCT domain derived from the BRCA1 protein. (B) The mixture is collected in aqueous solution at a concentration of 100 pM. (C to E) A drop is deposited and dried on a matrix array of SSCs (each chain is a pixel). (F) The whole content of peptides is spread over the matrix, and some (three different types per pixel, on average) are deposited in the smallest gap. These are the only ensembles per pixel that contribute to the Raman signal. (G) Representative Raman signal from pixel i,j of the matrix array composed of, on average, three different types of peptides.
THE ANALYTICAL APPROACH
The success of this method lies in two peculiar aspects of the SSC: first, the electric field generated in each SSC is strongly spatially confined (hotspot) to a region of a few square nanometers; and, second, Raman enhancement is high enough (about 107) to allow for the detection of a few molecules falling in the hotspot region. With this strategy, our device has the appropriate sensitivity and selectivity necessary to solve complex mixtures. A further element is that the device is compatible with biomolecular methods used to extract peptide content from the protein domain of interest. The peptides extracted from the protein domain differ in the substitution of a single amino acid, which is the cause of missense mutations in the BRCA1 protein. BRCA1 is a tumor suppressor gene whose germline mutations predispose to breast and ovarian cancers (20, 21). Particularly, the literature has shown that a woman carrying a germline mutation of BRCA1 has an 85% risk of developing breast and ovarian cancers, and it is estimated that about 5 to 10% of all breast cancers are caused by inheritance of dominant disease genes (22). BRCA1 encodes a 220-kD nuclear phosphoprotein, which interacts with many proteins and is involved in responses to DNA damage, cell cycle control, transcription regulation, apoptosis, and protein degradation. The protein is functionally organized in domains located at the N terminus and C terminus of the molecule (23, 24). In particular, the C-terminal region of BRCA1 has two amino acid repeats, both named BRCT, characterized by hydrophobic clusters of amino acids, which are thought to stabilize the three-dimensional (3D) structure of the protein.
The C-terminal BRCT domain (Fig. 1A) is involved in the stability of the BRCA1 protein’s conformation and plays a role in transcriptional control and DNA repair by interacting with other cellular factors (25–27). For this reason, the loss of normal functions mediated by the BRCT domain, resulting from inherited mutations, is thought to increase the risk of developing breast and ovarian cancers (see Supplementary Materials, section 1).
It has been shown that one missense mutation in the BRCT domain is a crucial event because it alters the hydrophobic patch and modifies protein folding, stability, and functionality. One of the first cancer-associated missense mutations studied is M1775R peptide, which is characterized by the substitution of a methionine (a highly hydrophobic amino acid) with arginine (a highly hydrophilic amino acid) at the fixed sequential position 1775 (28). This mutation results in a complete loss of the bound protein. Some functional studies have shown that the effects of M1775R missense mutation in BRCA1 are specifically related to the position of the amino acid involved in the mutation and probably depend on the charge switch of the mutated amino acid (29). This mutation could affect different intracellular pathways that may play a key role in the pathogenesis of hereditary breast cancer. Identification of specific alterations in protein profiles associated with the BRCA1 protein offers an opportunity for early diagnosis of breast and ovarian cancers.
The direct detection of a single amino acid point mutation in the BRCA1 protein through Raman or other spectroscopic techniques is an impossible task because the signature in the spectra is washed out by other complex molecular groups in the protein. For this reason, we used molecular methods based on digestive enzyme technology to extract the relative set of peptides contained in the BRCA1-BRCT protein domain. In this way, detection of mutation is transformed into a multicomponent mixture analysis.
In the present work, we obtained both in vitro wild-type and mutated BRCA1-BRCT protein domains. The mutated form is characterized by a missense mutation known as M1775R, which has been described previously. The two protein domains were digested by Glu-C enzyme and fractionated by micro–high-performance liquid chromatography (HPLC). Peptides obtained from the wild-type and mutated domains are referred to as M1775 and M1775R, respectively.
The two fractions containing the M1775 peptide plus 10 other peptides (for a total mix of 11 peptides) and the M1775R peptide plus 13 other peptides (for a total mix of 14 peptides) (Table 1) were analyzed by Raman spectroscopy using a 10 × 10 SSC array (Fig. 1, B to G). In the following, we describe SSC design, fabrication, and electromagnetic behavior, followed by detailed accounts of spectroscopic measurements.
Table 1. Fractional peptide content of M1775 (wild type) and M1775R (mutated).
The point mutation (arginine replaces methionine in peptide 1) is underlined.
|
Number of peptides |
Fractional M1775 (wild type) sequence peptides |
Composition (%) (error, 8%) |
Fractional M1775R (mutated) sequence peptides |
Composition (%) (error, 8%) |
| 1 | ICCYGPFTNMPTDQLE | 11.18 | ICCYGPFTNRPTDQLE | 6.36 |
| 2 | APVVTREWVLD | 7.85 | SDPSEDRAPE | 9.92 |
| 3 | ADALYTNPAQARE | 2.56 | TSYLPRQDLE | 5.91 |
| 4 | TAANLHAPVILAGTPGTFT HAGTE | 14.25 | SARVGNIPSSTSALKVPQLK VAE | 10.25 |
| 5 | NLVQRVPKDVFMGVDE | 7.30 | ASHLPFAQNISRVKE | 7.72 |
| 6 | GAILVVAATDGPMPQTRE | 11.76 | VYILSKDE | 8.76 |
| 7 | GGDALIPMLKE | 9.26 | TFNVGSFASGKE | 5.76 |
| 8 | KFMKIISLAPE | 11.34 | RYLGAKFPGAKRFSLE | 7.75 |
| 9 | VIAHLVNWE | 10.22 | AAKAKGAMALFGEKYDE | 6.98 |
| 10 | RINKALDFIAE | 8.25 | GRQGGTLQLFRTE | 4.22 |
| 11 | LRAKNQITLPVILKNE | 5.03 | KFTALTAELTAE | 7.44 |
| 12 | GGRTVGAGVVAKVLS | 6.38 | ||
| 13 | RFQADTLARFE | 6.15 | ||
| 14 | FLKAGGVFTDE | 6.57 |
SSC DESIGN AND FABRICATION
The proposed device consists of three noble metal nanospheres of appropriate diameter and interdistance relation, as explained by Li et al. (9). The device, firstly proposed by Li et al. (9), shows two spatial regions with plasmonic field localization. In particular, an intense hotspot (Fig. 2D) is localized in the smallest gap (between the smallest sphere and the middle sphere). The footprint area of the hotspot is comparable with the smallest gap. Moreover, the relative position between the nanospheres, calculated by Li et al. (9), was further optimized to maximize the ratio of the electric field in the hotspot to the electric field in the middle gap. However, from the point of view of fabrication, the SSC device presents a challenge because it is necessary to control the smallest nanosphere down to a few tens of nanometers and the smallest gap below the 5-nm range. To meet these fabrication requirements, we combined two techniques: top-down and bottom-up (electron beam lithography and metal electroless deposition) (30–32) (for process details, see Supplementary Materials, section 2). Use of high-resolution electron beam lithography allows the best control of structure definition and positioning, rendering the overall process controllable and reproducible, whereas site-selective and self-assembling silver nanoparticle electroless deposition (31, 33) is used for creating real 3D nanostructures of appropriate size and shape. Figure 2 (A to C) reports the steps of the process and shows representative scanning electron microscopy (SEM) images of SSCs in four different configurations: monomer, dimer, trimer, and tetramer (Fig. 2, E to H).
Fig. 2. Fabrication process of silver SSC.
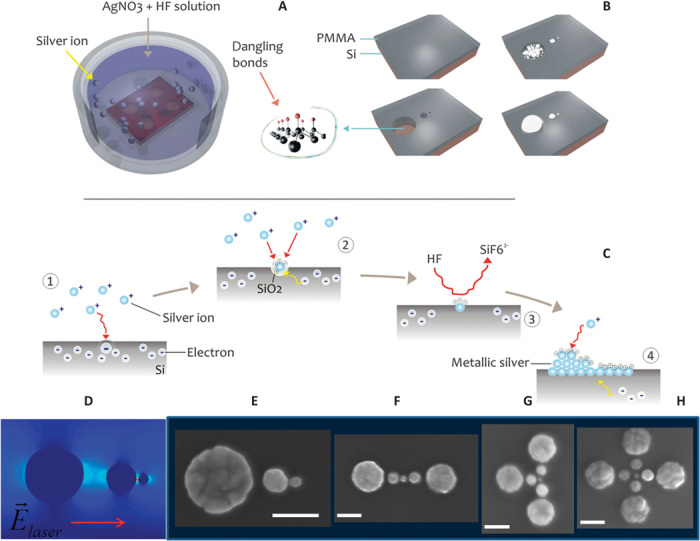
(A) After electron beam lithography and surface treatment with 2 M HF, the sample is immersed in HF/AgNO3 aqueous solution, where Ag+ is reduced to silver metal through a redox reaction chain. (B) In nanowells (reduction surface), silver growth follows a spherical symmetry and generates three spheres of appropriate diameter and interdistance. (C) Redox reactions inside a nanowell starting from the silicon surface. (D) SSC architecture and 2D map of electric field. Evidence of external laser polarization along the chain axis. The electric hotspot is localized in the smallest gap. (E to H) SEM images of silver SSCs and possible combinations in monomer, dimer, trimer, and tetramer. Scale bars, 50 nm. PMMA, polymethyl methacrylate.
The fabrication process illustrated in Fig. 2 shows that, after the lithography process leading to the formation of nanoholes, a metal site-selective growth (Fig. 2, A and B) forms the SSC. Figure 2C reports the redox reactions responsible for silver nanosphere growth. Figure 2 (E to H) presents SEM images of SSCs and higher architectural combinations showing full fabrication control. The combination of SSCs, from dimers to tetramers, can improve sensitivity detection below picomolar concentrations. Here, we used only SSC monomers. This technology can easily be scaled up for mass production by using imprinting as a parallel and low-cost lithography technique.
LOCAL FIELD ENHANCEMENT OF SSC AND ROLE OF DISORDER
Structural surface roughness or disorder is present on a scale much smaller than the laser wavelength as a result of an intrinsic fabrication mechanism. By using finite-difference time-domain (FDTD) analysis, we investigated the role of disorder in sustaining energy localization effects (34–40) (for details, see Supplementary Materials, section 3). Figure 3 (A to C) shows a comparison between the ideal geometry and our final disordered enhanced geometry (41). SSC metrology values, such as nanosphere sizes and gaps, were taken from actual fabricated devices. Figure 3 shows a map of an electric field for two configurations of SSC. In the first case, the SSCs are ideal, without surface roughness. In the second case, an average roughness of 2 nm (measured by SEM on fabricated SSCs) was introduced in the calculation. Furthermore, in both cases, the smallest gap was 5 nm. From the comparison between the ideal case and the fabricated SSC, we discovered that spatial localization and field enhancement are further improved in the final devices.
Fig. 3. Effects of disorder and sphere positioning on hotspot localization and enhancement.
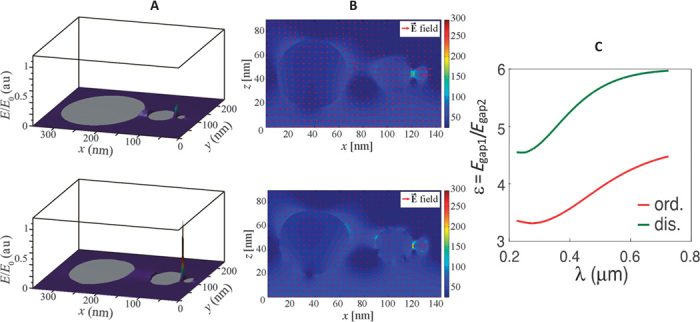
(A) Three-dimensional field enhancement and localization of (top) ideal SSC with zero sphere roughness and (bottom) fabricated SSC with 2-nm surface roughness. (B) Two-dimensional field map with zero roughness (top) and average surface disorder of 2 nm (bottom). (C) Comparison of enhancement between ideal SSC and fabricated SSC. A systematic electric field enhancement of a factor of 2 (as a function of wavelength) is expected of a 2-nm disorder. E0 is the maximal field value used as normalization factor in the 3D plot. In all simulations, laser polarization occurred along the SSC axis. au, arbitrary units; dis., disorder; ord., order.
In both situations, the results show an enhancement factor in the electric field of around 100 (based on our fabricated geometry). The results of Fig. 3C were based on a statistical ensemble of more than 200 different samples, which means that the effect of disorder is reproducible and stable. Compared to surface-enhanced Raman scattering (SERS) measurements obtained after depositing a monolayer of benzene-thiol molecules, this enhancement value is close to the experimental value (about 107) (2, 42). Moreover, from the point of view of localization field, it is important to consider another parameter (that is, ratio ε = Egap1/Egap2) between the fields Egap1 and Egap2, located in the smallest gap and in the middle gap. As from Fig. 3C, the ratio ε at λ = 514 nm is around 4 for the ideal SSC and 6 for the fabricated SSC (Fig. 3A). Under uniform deposition of molecules over the whole SSC device, the dominant Raman signal will originate from the molecules located in the smallest gap. In fact, Raman intensity is proportional to the fourth power (E4) of the local electric field. In the case of fabricated SSCs, ε4disordered = 64 = 1296; instead, ε4ordered = 44 = 256.
Raman intensity is dominated by the scattering of molecules sitting in the smallest hotspot. In a Raman experiment where SSC devices were used as SERS building block, the intensity signal coming from the molecules in the smallest gap was three orders of magnitude higher than the signal coming from the intermediate gap (the intermediate gap is the only region where a residual electric field is different from zero). As a consequence, we can exploit this behavior of the SSC to assume that the detected signal came from the smallest gap, without the need for any aligning procedure to locate the molecules specifically in the hotspot. Therefore, if we randomly cover the SSCs with molecules, the Raman signal detected is dominated by the signal of the molecules sitting in the area defined by the smallest gap.
DETECTION METHOD FOR ANALYSIS OF BRCA1 PEPTIDE MIXTURE
When the construct under examination is composed of a mixture of different kinds of molecules, the number of molecules that can fit into the hotspot is directly related to their concentration in the solution and to the volume of the hotspot. If we want to analyze a complex mixture, we need to calibrate the concentration in such a way that the mutual distance of the molecules after deposition is statistically in the range of the linear size of the hotspot. In this way, only a few and a subset of them will sit in the smallest gap after the solution has dried. In reconstructing the whole content of the mixture, an m × n matrix array of SSCs, whose m × n element is high enough to have all types of molecules statistically represented in the overall m × n pixel ensemble, is necessary and sufficient.
The biomolecular procedure followed for extracting the peptides of interest from BRCA1 led, after purification, to a mixture of two fractions of 11 and 14 peptides, respectively. The experimental dilution used fell in the range of 100 pM. At this concentration, few types of molecules were deposited in each hotspot area, as indicated in the analysis of Raman spectra (Figs. 1 and 4).
Fig. 4. Linear combination fitting procedure of peptide spectra.
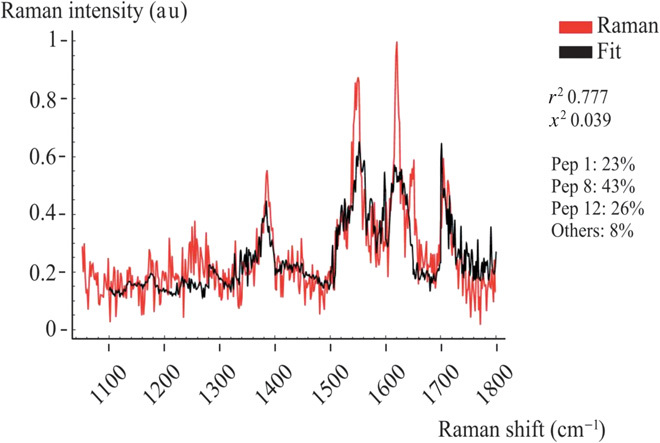
Raman spectra were collected in a representative pixel of the array. The fit gives a weighted composition of peptides 1, 8, and 12.
Detection of mutated peptides in BRCA1 after protein digestion becomes an analytical problem of finding the elemental composition of a multicomponent mixture. In this specific case, which is devoted to detection of point mutation, we need a database where the Raman spectra of each peptide component are known. In our procedure, this database was created by acquiring each single peptide Raman spectrum (base element), and the multicomponent spectrum at each pixel of the matrix was recovered by fitting it with a linear combination of base elements.
Formally, a Raman spectrum, sM, can be fitted by the function:
| (1) |
where the index i runs through the number of peptides n (i = 1,…,m), whereas j runs through the number of intervals n (j = 1,…,n). m is the wave vector range in the Raman spectra. In Eq. 1, the coefficient xi,j is the best combination that minimizes the sum of the squares of deviations of Υ from the experimental values sM. From Eq. 1, the mass fraction ξi of the component i in the total solution is derived and described in Supplementary Materials, section 4. The larger is ξi, the higher is the content of the peptide i in the mixture. By repeating the fitting procedure for all m × n matrix elements, all peptides extracted from BRCA1 are recovered. With this procedure and because of high SERS sensitivity, we could detect both wild-type and mutated species in the mixtures. In Supplementary Materials, section 4, we report all data obtained from the database and all data for the two mixtures, including the relative χ2 and coefficient of determination (r2) values. Figure 4 reports a representative fit of the mixture at a specific pixel element of the matrix. In this case, the Raman spectra contain three peptides.
The amount of peptide mixture derived from human samples was 0.2 ng/ml, with an average molarity of about 100 pM. The peptide mixture was deposited on the plasmonic matrix, where it was air-dried in a few tens of minutes.
As expected, any linear combination contains only a subset of the 25-peptide mixture (25 = 11 + 14) for the in vitro sample and a subset of the 26-peptide mixture (11 + 15) for the synthetic sample.
The typical number of peptides in each pixel of the matrix is between 3 and 5. Therefore, collection of all pixel spectra allowed us to fully reconstruct the mixture composition and, hence, to find the mutated peptide in a patient carrying the disease.
Figure 5 reports additional details explaining SSC code arrays. On the left, a SEM image of the 10 × 10 SSC array is shown (at this magnification, only the overall matrix can be seen). Each pixel (a single SSC) is represented by a color code depicting the linear combination (the fit) of peptides found in that specific pixel. A 10 × 10 matrix is sufficient to fully reconstruct the original peptide mixture.
Fig. 5. Matrix array and data acquisition.
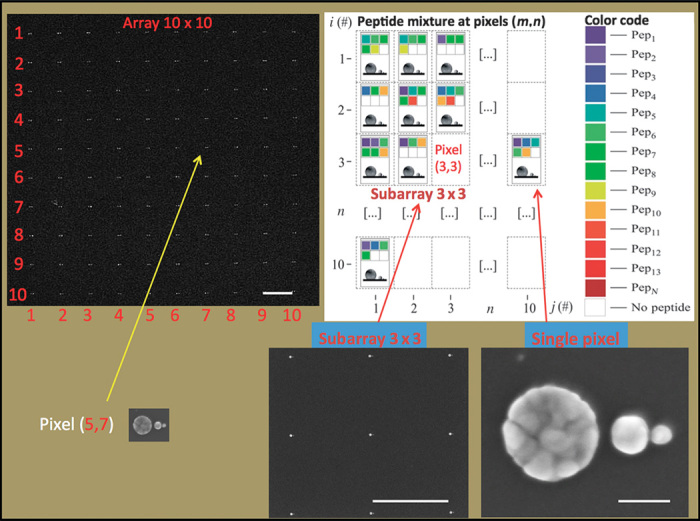
(Top left) SEM image of a 10 × 10 SSC array. Scale bar, 2 μm. Each SSC represents a pixel element i,j of the matrix. (Top right) In each pixel, color code is associated with a specific peptide. (Bottom left) A submatrix of 3 × 3 pixels is evidenced. Scale bar, 2 μm. (Bottom right) A detailed SEM image of the SSC representing the pixel. Scale bar, 50 nm. Empty pixels represent specific positions in the array where no peptides are detected.
Figure 6 reports the Raman spectra of the peptide M1775 (wild type and mutated from pure samples). The noticeable difference in the spectra gives the distinction between wild-type species and mutated species in the linear combination procedure. Furthermore, principal components analysis (PCA) is performed on the Raman data set constituting 2D maps of single pixels in the low-frequency range (43). PCA is performed on the whole data set at once; consequently, the computed principal components (PCs) are exactly the same for both pixels, thus allowing a quantitative comparison of the Raman signatures recorded over them. In particular, although the first PC (not shown) is directly associated with the average signal over the two pixels, the second PC is instead related to the differences between the two Raman signals. The 2D map of PC2 coefficients (Fig. 6C) shows two different pixels: one dominated by the wild-type Raman signature (blue) and the other dominated by the mutated signature (red). Figure 6D shows the PC2 load curve, which takes into account the main differences between wild-type signals and mutated signals at once.
Fig. 6. Raman spectra of pure wild-type and mutated peptides.
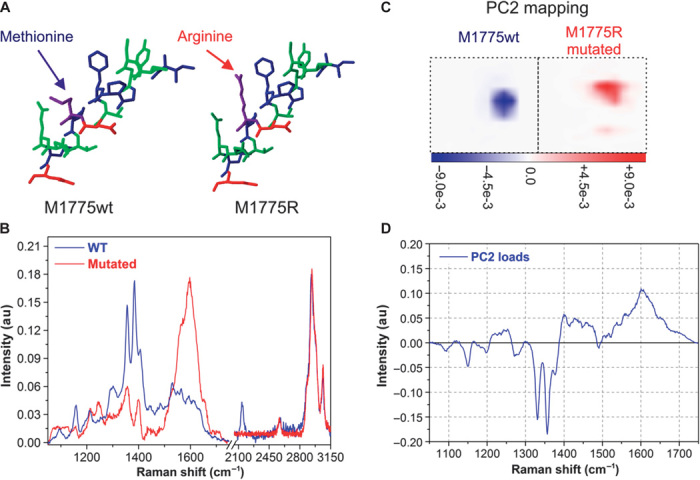
(A and B) Raman spectra (B) show a net difference between two peptides differentiated by only the exchange of one amino acid (A; a methionine with an arginine). These spectra constitute the base set for the fitting procedure. Their net difference allows identification of mutated peptides in the mixture. (C and D) The results of PCA are also shown: a 2D map of the PC2 coefficients of two pixels (C), one pixel for each peptide, where color code is proportional to the significance of the PC2 parameter over the map; the PC2 parameter load curve (D) takes into account spectral differences between the two peptides. The combination of PC2 mapping and PC2 load curve allows identification of pixels dominated by wild-type or mutated species.
In this approach, we looked for peptide components, instead of Raman signatures, of the whole BRCA1 protein. This analytical strategy allows detection of even a single point mutation, namely, the exchange of a single amino acid in the peptide (in this case the exchange of a methionine with an arginine). The strategy of breaking a big protein into a mixture of peptides can be considered a general approach that is applicable to other cases and disease typologies.
Details of the whole procedure are reported in Supplementary Materials, section 4, where we describe how the database (needed for Eq. 1 and eqs. S4.1 to S4.4) was obtained. In Fig. 6D, we add a PCA calculation to show quantitative discrimination between the Raman spectra of wild-type peptides and the Raman spectra of mutated peptides. PCA quality confirms the net difference in the two Raman spectra.
The analytical results of the present procedure are reported in Table 1, where the analytical compositions of both mixtures, containing wild-type and mutated peptides, are indicated (see Supplementary Materials, section 5, complete list of peptides and corresponding IDs used for analysis). These results were also compared with mass spectrometry results for benchmarking. A full qualitative agreement between the two methods was obtained despite the concentration and quantity of the analytes being three orders of magnitude lower than those used in liquid chromatography–tandem mass spectrometry (LC-MS/MS). The present method is fully compatible with superhydrophobic devices, whose sensitivity was proven to fall in the attomolar concentration range (44).
From the point of view of molecular medicine, detection of mutated proteins (or their degradation products) in circulating plasma is a challenging task. On the other hand, its demonstration that an altered gene is transcribed and translated into an abnormal protein is very crucial to establishing a clear correlation between genotype and phenotype. There is no linear relation between transcriptome and proteome that is attributable to messenger RNA instability, microRNA regulation of gene expression, and related modulatory mechanisms. Therefore, it is likely that a specific gene bearing a mutation in its coding sequence might never produce an abnormal protein.
The experimental strategy described in this work can overcome limitations in detecting and identifying very low concentration proteins or peptides in body fluid and may therefore represent a tremendous advancement in the early diagnosis of cancer and other degenerative diseases. Moreover, this approach can be successfully applied to evaluate drug response and prognosis in patients undergoing complex and often debilitating treatments such as chemotherapy.
To the best of our knowledge, this is the first study to use plasmonics to analyze in vitro human cancer samples with a high level of complexity, such as a 25-component mixture at picomolar concentration. The present method can be applied to a wider category of analytical problems in which both sensitivity and specificity are required.
MATERIALS AND METHODS
Sample preparation
In vitro wild-type and mutated BRCA1-BRCT protein domains were obtained from plasmid construction and expression of recombinant proteins. The two protein domains were digested by Glu-C enzyme and analyzed by matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI-TOF) performed on a Voyager-DE STR (MALDI-TOF) (Applied Biosystems Inc.) equipped with a nitrogen laser emitting at 337 nm. The two samples were fractionated by a micro-HPLC system (Agilent Technologies), attaining wild-type and mutated BRCA1-BRCT protein domains. Both fractionated samples were injected by the QSTAR XL hybrid LC-MS/MS system (Applied Biosystems Inc.). Finally, the data were searched on Mascot search engine (www.matrixscience.com) version 2.2 against the International Protein Index database version 3.58 (56,619 sequences). Detailed descriptions of the extraction of BRCA1-BRCT protein domains are available in Supplementary Materials, section 6. Synthetic BRCA1-BRCT peptides (Table 1) were purchased from Sigma-Aldrich Corp. and used without any further processing.
Samples (synthetic and in vitro) were dissolved in water. For SERS measurements, plasmonic devices were immersed in the solution for 30 min, rinsed with water to wash out excess molecules, and dried under nitrogen flow.
SSC fabrication
SSCs of silver nanospheres were realized by a combination of top-down and bottom-up approaches. We combined high-resolution electron beam lithography and electroless deposition of silver, obtaining metal nanostructures with spherical symmetry.
A high-resolution positive electron beam resist (PMMA A2; MicroChem) was spin-coated on a clean silicon(100) substrate for 60 s at 5000 rpm to obtain a 30-nm-thick layer and was prebaked at 170°C for 2 min to remove the solvent from the resist. A pattern of 10 × 10 SSCs with a 2-nm electron beam diameter was exposed by using an electron beam lithography system (CABL-9000C; CRESTEC) working at 50 keV acceleration voltage and 50 pA current. A dose of 1250 μC/cm2 was set for exposure in a field of 60 μm by 60 μm. The sample was developed for immersion in isopropanol at 4°C to selectively remove the exposed resist, rinsed with water, and dried under nitrogen flux.
Metallic silver was deposited on the patterned sample using an electroless method involving silicon–silicon native oxide, hydrofluoric acid (HF), water, and silver nitrate (AgNO3) (30, 31). The process was performed in a few tens of seconds (30 to 40 s) by directly immersing the silicon sample in an appropriate mixture of reactants (0.15 M HF and 0.05 mM AgNO3) at room temperature. After silver electroless deposition, the wider gap became 20 to 25 nm and the smaller gap was below 5 nm.
Characterization technique
SEM images of nanostructures were recorded using the FEI Nova 600 NanoLab system. The acceleration voltage was kept between 5 and 15 keV in mode 2 configuration. In mode 2 configuration, the immersion lens was turned on, and a through lens detector (TLD) in secondary electron operation was used.
Raman scattering measurements were performed using the Renishaw InVia Raman spectrometer. Raman spectra were excited by a 514-nm laser line (Ar+ laser) in backscattering configuration (150× objective) and polarization along the axis of the SSC. Laser power and accumulation time were kept at about 100 μW and 5 s, respectively. All Raman data were analyzed using WiRE 3.2 built-in software.
Theoretical simulation
FDTD simulations were carried out using our home-made parallel code NANOCPP (36), which scales up to hundreds of thousands of processors. Dispersion of metals was carefully taken into account by using a multipolar expansion that covers material response at visible and infrared frequencies. The code for linear combination algorithm was written in Matematica software and used for analyzing Raman spectra.
Funding
We acknowledge financial support from the King Abdullah University of Science and Technology start-up fund and the Italian Minister of Health (projects nos. GR-2010-2320665 and GR-2010-2311677). Author contributions: M.L.C. contributed to the fabrication process. F.G. and P.C. designed the nanolens and performed data analysis. G.D., M.L.C., A.N., and A.M.P. carried out Raman measurements. R.P.Z. performed plasmonic numerical simulation. G.P. contributed to the fabrication process. J.S.T.G., S.A., and A.F. performed numerical simulations and analysis of disorder. T.L. performed biological analysis. G.C. carried out all the molecular biology and human sample management. E.D.F. conceived the idea and participated in all aspects of the project: design, fabrication, data analysis, and algorithm and Raman measurements. All authors discussed the results and finalized the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Authors will make data available on request.
SUPPLEMENTARY MATERIALS
Supplementary materials for this article are available at http://advances.sciencemag.org/cgi/content/full/1/8/1500487/DC1
Crystallographic model of human BRCA1-BRCT protein
Fig. S1. Three-dimensional crystal structure of the BRCA1-BRCT protein.
Design and fabrication of device
Fig. S2. Top-down and bottom-up process fabrication.
Fig. S3. SEM images of different SSCs.
Theoretical comparison of ordered and disordered SSC device
Fig. S4. FDTD analysis of the role of disorder.
Complete peptide content of all the solutions resolved using the nanolens SERS device
Fig. S5. Nanolens matrix position of measurement points for the synthetic peptide solution.
Fig. S6. Nanolens matrix position of measurement points for the wild-type peptide solution.
Fig. S7. Nanolens matrix position of measurement points for the mutated peptide solution.
Table S1. Peptide fraction calculated for each measurement point in the grid of nanolens devices and χ2 and r2 statistics for the synthetic peptide solution.
Table S2. Relative content of each peptide in the synthetic peptide solution.
Table S3. Peptide fraction calculated for each measurement point in the grid of nanolens devices and χ2 and r2 statistics for the wild-type peptide solution.
Table S4. Relative content of each peptide in the wild-type peptide solution.
Table S5. Peptide fraction calculated for each measurement point in the grid of nanolens devices and χ2 and r2 statistics for the mutated peptide solution.
Table S6. Relative content of each peptide in the mutated peptide solution.
List of synthetic peptides utilized in the work and corresponding IDs
Extraction of wild-type and mutated BRCA1-BRCT protein domain
REFERENCES AND NOTES
- 1.Chirumamilla M., Toma A., Gopalakrishnan A., Das G., Zaccaria R. P., Krahne R., Rondanina E., Leoncini M., Liberale C., De Angelis F., Di Fabrizio E., 3D nanostar dimers with a sub-10-nm gap for single-/few-molecule surface-enhanced Raman scattering. Adv. Mater. 26, 2353–2358 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Im H., Bantz K. C., Lindquist N. C., Haynes C. L., Oh S.-H., Vertically oriented sub-10-nm plasmonic nanogap arrays. Nano Lett. 10, 2231–2236 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Fleischer M., Zhang D., Braun K., Jäger S., Ehlich R., Häffner M., Stanciu C., Hörber J. K., Meixner A. J., Kern D. P., Tailoring gold nanostructures for near-field optical applications. Nanotechnology 21, 065301 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Kessentini S., Barchiesi D., D’Andrea C., Toma A., Guillot N., Di Fabrizio E., Fazio B., Maragó O. M., Gucciardi P. G., Lamy de la Chapelle M., Gold dimer nanoantenna with slanted gap for tunable LSPR and improved SERS. J. Phys. Chem. C 118, 3209–3219 (2014). [Google Scholar]
- 5.Schäfer C., Gollmer D. A., Horrer A., Fulmes J., Weber-Bargioni A., Cabrini S., Schuck P. J., Kern D. P., Fleischer M., A single particle plasmon resonance study of 3D conical nanoantennas. Nanoscale 5, 7861–7866 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Guillot N., Shen H., Frémaux B., Péron O., Rinnert E., Toury T., Lamy de la Chapelle M., Surface enhanced Raman scattering optimization of gold nanocylinder arrays: Influence of the localized surface plasmon resonance and excitation wavelength. Appl. Phys. Lett. 97, 023113 (2010). [Google Scholar]
- 7.Day J. K., Neumann O., Grady N. K., Halas N. J., Nanostructure-mediated launching and detection of 2D surface plasmons. ACS Nano 4, 7566–7572 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Yu A. A., Savas T., Cabrini S., Difabrizio E., Smith H. I., Stellacci F., High resolution printing of DNA feature on poly(methyl methacrylate) substrates using supramolecular nano-stamping. J. Am. Chem. Soc. 127, 16774–16775 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Li K., Stockman M. I., Bergman D. J., Self-similar chain of metal nanospheres as an efficient nanolens. Phys. Rev. Lett. 91, 227402 (2003). [DOI] [PubMed] [Google Scholar]
- 10.De Angelis F., Liberale C., Coluccio M. L., Cojoc G., Di Fabrizio E., Emerging fabrication techniques for 3D nano-structuring in plasmonics and single molecule studies. Nanoscale 3, 2689–2696 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Mezeme M. E., Brosseau C., Are scaling laws of sub-optical wavelength electric field confinement in arrays of metal nanoparticles related to plasmonics or to geometry? Opt. Express 20, 17591–17599 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Höppener C., Lapin Z. J., Bharadwaj P., Novotny L., Self-similar gold-nanoparticle antennas for a cascaded enhancement of the optical field. Phys. Rev. Lett. 109, 017402 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Anker J. N., Hall W. P., Lyandres O., Shah N. C., Zhao J., Van Duyne R. P., Biosensing with plasmonic nanosensors. Nat. Mater. 7, 442–453 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Garai-Ibabe G., Grinyte R., Golub E. I., Canaan A., de la Chapelle M. L., Marks R. S., Pavlov V., Label free and amplified detection of cancer marker EBNA-1 by DNA probe based biosensors. Biosens. Bioelectron. 30, 272–275 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Rakovich T. Y., Mahfoud O. K., Mohamed B. M., Prina-Mello A., Crosbie-Staunton K., Van Den Broeck T., De Kimpe L., Sukhanova A., Baty D., Rakovich A., Maier S. A., Alves F., Nauwelaers F., Nabiev I., Chames P., Volkov Y., Highly sensitive single domain antibody–quantum dot conjugates for detection of HER2 biomarker in lung and breast cancer cells. ACS Nano 8, 5682–5695 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Das G., Chirumamilla M., Toma A., Gopalakrishnan A., Zaccaria R. P., Alabastri A., Leoncini M., Di Fabrizio E., Plasmon based biosensor for distinguishing different peptides mutation states. Sci. Rep. 3, 1792 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vendrell M., Maiti K. K., Dhaliwal K., Chang Y. T., Surface-enhanced Raman scattering in cancer detection and imaging. Trends Biotechnol. 31, 249–257 (2013). [DOI] [PubMed] [Google Scholar]
- 18.D’Andrea C., Bochterle J., Toma A., Huck C., Neubrech F., Messina E., Fazio B., Maragò O. M., Di Fabrizio E., Lamy de La Chapelle M., Gucciardi P. G., Pucci A., Optical nanoantennas for multiband surface-enhanced infrared and Raman spectroscopy. ACS Nano 7, 3522–3531 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Goulet P. J., Aroca R. F., Distinguishing individual vibrational fingerprints: Single-molecule surface-enhanced resonance Raman scattering from one-to-one binary mixtures in Langmuir–Blodgett monolayers. Anal. Chem. 79, 2728–2734 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Duncan J. A., Reeves J. R., Cooke T. G., BRCA1 and BRCA2 proteins: Roles in health and disease. Mol. Pathol. 51, 237–247 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshida K., Miki Y., Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 95, 866–871 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teng L. S., Zheng Y., Wang H. H., BRCA1/2 associated hereditary breast cancer. J. Zhejiang Univ. Sci. B 9, 85–89 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruffner H., Verma I. M., BRCA1 is a cell cycle–regulated nuclear phosphoprotein. Proc. Natl. Acad. Sci. U.S.A. 94, 7138–7143 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shakya R., Reid L. J., Reczek C. R., Cole F., Egli D., Lin C. S., deRooij D. G., Hirsch S., Ravi K., Hicks J. B., Szabolcs M., Jasin M., Baer R., Ludwig T., BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 334, 525–528 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bork P., Hofmann K., Bucher P., Neuwald A. F., Altschul S. F., Koonin E. V., A superfamily of conserved domains in DNA damage–responsive cell cycle checkpoint proteins. FASEB J. 11, 68–76 (1997). [PubMed] [Google Scholar]
- 26.Huyton T., Bates P. A., Zhang X., Sternberg M. J., Freemont P. S., The BRCA1 C-terminal domain: Structure and function. Mutat. Res. 460, 319–332 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Callebaut I., Mornon J. P., From BRCA1 to RAP1: A widespread BRCT module closely associated with DNA repair. FEBS Lett. 400, 25–30 (1997). [DOI] [PubMed] [Google Scholar]
- 28.Scully R., Ganesan S., Vlasakova K., Chen J., Socolovsky M., Livingston D. M., Genetic analysis of BRCA1 function in a defined tumor cell line. Mol. Cell 4, 1093–1099 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Williams R. S., Glover J. N., Structural consequences of a cancer-causing BRCA1-BRCT missense mutation. J. Biol. Chem. 278, 2630–2635 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Yae S., Nasu N., Matsumoto K., Hagihara T., Fukumuro N., Matsuda H., Nucleation behavior in electroless displacement deposition of metals on silicon from hydrofluoric acid solutions. Electrochim. Acta 53, 35–41 (2007). [Google Scholar]
- 31.Qiu T., Chu P. K., Self-selective electroless plating: An approach for fabrication of functional 1D nanomaterials. Mater. Sci. Eng. R 61, 59–77 (2008). [Google Scholar]
- 32.Goia D. V., Matijević E., Preparation of monodispersed metal particles. New J. Chem. 22, 1203–1215 (1998). [Google Scholar]
- 33.Coluccio M. L., Das G., Mecarini F., Gentile F., Pujia A., Bava L., Tallerico R., Candeloro P., Liberale C., De Angelis F., Di Fabrizio E., Silver-based surface enhanced Raman scattering (SERS) substrate fabrication using nanolithography and site selective electroless deposition. Microelectron. Eng. 86, 1085–1088 (2009). [Google Scholar]
- 34.Alabastri A., Tuccio S., Giugni A., Toma A., Liberale C., Das G., De Angelis F., Di Fabrizio E., Zaccaria R. P., Molding of plasmonic resonances in metallic nanostructures: Dependence of the non-linear electric permittivity on system size and temperature. Materials 6, 4879–4910 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.V. M. Shalaev, Nonlinear Optics of Random Media: Fractal Composites and Metal-Dielectric Films (Springer, New York, 2000). [Google Scholar]
- 36.Dai J., Čajko F., Tsukerman I., Stockman M. I., Electrodynamic effects in plasmonic nanolenses. Phys. Rev. B 77, 115419 (2008). [Google Scholar]
- 37.Liu C., Di Falco A., Molinari D., Khan Y., Ooi B. S., Krauss T. F., Fratalocchi A., Enhanced energy storage in chaotic optical resonators. Nat. Photonics 7, 473–478 (2013). [Google Scholar]
- 38.A. Taflove, S. C. Hagness, Computational Electrodynamics: The Finite-Difference Time-Domain Method (Artech House, Norwood, MA, ed. 3, 2005). [Google Scholar]
- 39.Conti C., Fratalocchi A., Dynamic light diffusion, three-dimensional Anderson localization and lasing in inverted opals. Nat. Phys. 4, 794–798 (2008). [Google Scholar]
- 40.Garcia N., Stoll E., Monte Carlo calculation for electromagnetic-wave scattering from random rough surfaces. Phys. Rev. Lett. 52, 1798–1801 (1984). [Google Scholar]
- 41.E. M. Di Fabrizio, A. Fratalocchi, J. S. T. Gongora, M. L. Coluccio, M. P. Candeloro, G. Cuda, Analytic device including nanostructures, U.S. Patent 61/920,725 (2013).
- 42.Reilly T. H. III, Chang S. H., Corbman J. D., Schatz G. C., Rowlen K. L., Quantitative evaluation of plasmon enhanced Raman scattering from nanoaperture arrays. J. Phys. Chem. C 111, 1689–1694 (2007). [Google Scholar]
- 43.Chan J. W., Taylor D. S., Zwerdling T., Lane S. M., Ihara K., Huser T., Micro-Raman spectroscopy detects individual neoplastic and normal hematopoietic cells. Biophys. J. 90, 648–656 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Angelis F., Gentile F., Mecarini F., Das G., Moretti M., Candeloro P., Coluccio M. L., Cojoc G., Accardo A., Liberale C., Zaccaria R. P., Perozziello G., Tirinato L., Toma A., Cuda G., Cingolani R., Di Fabrizio E., Breaking the diffusion limit with super-hydrophobic delivery of molecules to plasmonic nanofocusing SERS structures. Nat. Photonics 5, 682–687 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials for this article are available at http://advances.sciencemag.org/cgi/content/full/1/8/1500487/DC1
Crystallographic model of human BRCA1-BRCT protein
Fig. S1. Three-dimensional crystal structure of the BRCA1-BRCT protein.
Design and fabrication of device
Fig. S2. Top-down and bottom-up process fabrication.
Fig. S3. SEM images of different SSCs.
Theoretical comparison of ordered and disordered SSC device
Fig. S4. FDTD analysis of the role of disorder.
Complete peptide content of all the solutions resolved using the nanolens SERS device
Fig. S5. Nanolens matrix position of measurement points for the synthetic peptide solution.
Fig. S6. Nanolens matrix position of measurement points for the wild-type peptide solution.
Fig. S7. Nanolens matrix position of measurement points for the mutated peptide solution.
Table S1. Peptide fraction calculated for each measurement point in the grid of nanolens devices and χ2 and r2 statistics for the synthetic peptide solution.
Table S2. Relative content of each peptide in the synthetic peptide solution.
Table S3. Peptide fraction calculated for each measurement point in the grid of nanolens devices and χ2 and r2 statistics for the wild-type peptide solution.
Table S4. Relative content of each peptide in the wild-type peptide solution.
Table S5. Peptide fraction calculated for each measurement point in the grid of nanolens devices and χ2 and r2 statistics for the mutated peptide solution.
Table S6. Relative content of each peptide in the mutated peptide solution.
List of synthetic peptides utilized in the work and corresponding IDs
Extraction of wild-type and mutated BRCA1-BRCT protein domain