

Abstract
A deposition process has been developed to fabricate a complete-monolayer Pt coating on a large-surface-area three-dimensional (3D) Ni foam substrate using a buffer layer (Ag or Au) strategy. The quartz crystal microbalance, current density analysis, cyclic voltammetry integration, and X-ray photoelectron spectroscopy results show that the monolayer deposition process accomplishes full coverage on the substrate and the deposition can be controlled to a single atomic layer thickness. To our knowledge, this is the first report on a complete-monolayer Pt coating on a 3D bulk substrate with complex fine structures; all prior literature reported on submonolayer or incomplete-monolayer coating. A thin underlayer of Ag or Au is found to be necessary to cover a very reactive Ni substrate to ensure complete-monolayer Pt coverage; otherwise, only an incomplete monolayer is formed. Moreover, the Pt monolayer is found to work as well as a thick Pt film for catalytic reactions. This development may pave a way to fabricating a high-activity Pt catalyst with minimal Pt usage.
Keywords: Catalyst, complete monolayer, monolayer, Ni foam, platinum
Platinum is not only the most precious metal for jewelry but also the best material for electrical contacts, particularly in wet chemical processing. It is also the best catalyst for many commercial applications, including fuel cells, autocatalytic converters, and the petroleum industry (1). Platinum has high catalytic activity, and it is the most common catalyst for almost all major industrial reactions, including oxidation, oxygen reduction reaction (2–6), hydrogen evolution reaction (HER) (7–10), and fuel cells (11–13). Platinum is in high demand but of low abundance, making Pt material too expensive to be used for many industrial applications (14). The U.S. Department of Energy has established its 2017 target for the U.S. DRIVE Fuel Cell Tech Team to reduce its total Pt group metal loading to 0.125 mg cm−2 from its current level of 0.4 to 1.0 mg cm−2 (15, 16). It is for this very reason that there have been numerous attempts to fabricate Pt monolayer (ML) films and even to develop single-atom alloy (17) catalysts. For example, Brankovic et al. (18) demonstrated the preparation of a submonolayer of Pt film using galvanic displacement of a Cu monolayer obtained by underpotential deposition (UPD) on an Au(111) surface. Using the same galvanic displacement, Li et al. found that there is a correlation between substrate-induced lateral strain in the Pt submonolayer and its activity/selectivity and when the expanded Pt layer on Au(111) exhibits a sevenfold activity increase in catalyzing methanol electrooxidation relative to Pt(111) (19). Kye et al. (20) fabricated a Pt(shell)/Au(core) to demonstrate a similar catalytic activity as a Pt nanoparticle for photoelectrochemical HER. The new concept facilitates the formation of catalysts with several unique features including high Pt utilization, enhanced activity, and stability.
Although there has been a tremendous amount of research conducted in minimizing the Pt usage, it is nontrivial to fabricate a complete-monolayer Pt coating on a large-area substrate, particularly on a 3D substrate with complex structures. For example, Ding et al. demonstrated a successful epitaxial casting of Pt films on 3D nanoporous gold membrane substrates when the Pt thickness reaches more than 1 nm (21).
Indeed, there are reports on Pt monolayer deposition on different single-crystalline surfaces and nanoparticles such as Au (20), Rh (22), Pd (23), and Ir (24). So far, successful Pt monolayer deposition has been realized only recently on relatively small-area single-crystalline facets (25). There are also reports on galvanic displacement of a Cu monolayer obtained by UPD on single-crystalline surfaces or nanoparticles (18, 20, 22, 23, 26, 27). Because the Cu UPD layer uniformly covers the underlying substrate, the probability of Pt-Cu interaction is similar over the entire surface, leading to uniform nucleation for evenly distributed Pt nanoclusters or incomplete-monolayer Pt (3, 18). To our knowledge, there has been no report on successfully fabricating complete-monolayer Pt on large-piece 3D bulk substrates with complex geometries.
Here, we describe the successful synthesis of Pt monolayers on a large-surface-area 3D Ni foam network using a buffer layer technique. It is the most effective way to minimize Pt usage (1 ML is equivalent to ~550 ng cm−2), allowing the use of Pt in commercial applications such as fuel cells at minimal cost. Surprisingly, it works as well as a thick Pt film for catalyzing HER.
Because we intended to develop a complete-monolayer Pt film on a large-surface-area carrier for broad applications, we selected as substrate a commercially available nickel foam for its high electrical conductivity, high porosity, large surface area, high-temperature stability, resistance to most corrosive and oxidizing environments, and its large dimension allowing for feasible recycling. Unfortunately, we found that direct Pt deposition on Ni resulted in nanoparticles or incomplete-monolayer coating.
Inspired by a recent development by Liu et al. (25), who successfully controlled Pt coating to monolayer thickness on single-crystalline Au(111) surfaces using underpotential deposited hydrogen to protect the Pt surface from further deposition, we implemented a buffer layer of thin Ag (or Au) film on the Ni foam before the Pt deposition. We found that the monolayer Pt worked best when it was deposited on a thin buffer layer or nanofilm (NF) of Ag or Au, as shown in Fig. 1. We successfully used this method for the complete-monolayer Pt coating on a complex 3D Ni foam substrate. For the deposition, a piece of commercial grade Ni foam of 10 × 10 × 1.5 mm was used as the substrate, and a thin layer of Ag or Au NF was deposited onto the Ni foam as a buffer layer. To minimize the material usage, the buffer layer coating was controlled to be just thick enough to ensure that the underlying Ni surface is covered by the Ag or Au buffer. The Pt monolayer was then deposited using an electrochemical workstation by first stepping the potential to more positive values to activate the substrate surface and then lowering the potential to deposit the Pt monolayer.
Fig. 1. Schematic illustration of the Pt monolayer fabrication process on Au-buffered Ni foam.

Figure 2 demonstrates the effectiveness of monolayer deposition. Figure 2A shows the current density and potential changes for five electroplating cycles on a complex 3D Ni foam substrate with an Au NF buffer (see the Supplementary Materials for experimental details). The solid curve is for current density (left axis) and the dashed line (right axis) is for plating voltage. When the voltage was set at −0.4 VSCE (volts versus a saturated calomel electrode), the current density increased from −4.4 to ~0 mA cm−2 in 0.2 s, signifying that the deposition cycle was completed. The current density then stabilized even though the plating voltage was maintained at −0.4 VSCE for an extended period of time, demonstrating that the deposition was completely terminated. When the voltage was set to 0 VSCE, the current density first jumped to ~4.4 mA cm−2 and then quickly dropped to 0, signifying that the adsorbed hydrogen layer was instantly desorbed to release a fresh Pt surface. Afterward, as soon as the potential was set to −0.4 VSCE, another cycle of deposition was started, and the previous cycle was repeated.
Fig. 2. Sequential deposition of Pt monoatomic layers by pulsed electrodeposition in a pH 4 solution.
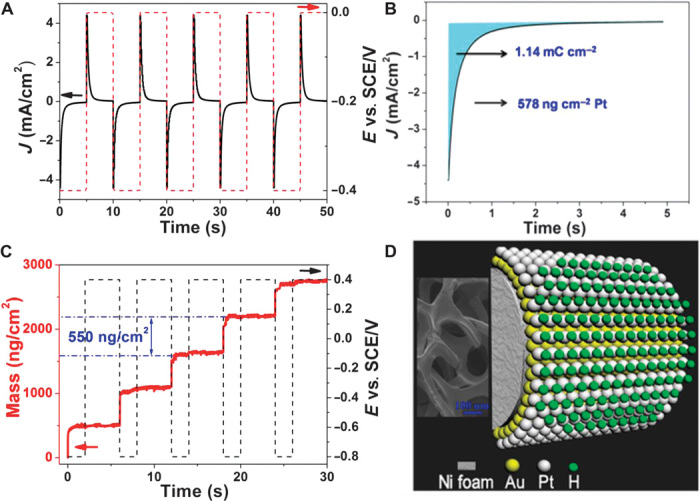
(A) Current density and potential versus time plot for Pt monolayer deposition on 3D Ni foam substrate using the sequential technique. (B) Mass and potential change during the sequential deposition of Pt monolayers using QCM on an Au-coated quartz crystal substrate. (C) Voltammetry curve for the deposition of Pt on the Au NF/Ni foam surface by using a pulsed potential waveform in 0.5 M NaCl and 3 mM K2PtCl4 solution. Sweep rate, 50 mV s−1. (D) Illustration of self-terminating Pt deposition. Inset: Scanning electron microscopy image of Ni foam.
To further confirm that the deposition process of the Pt monolayer by this electrochemical technique is indeed self-terminating to a single complete-monolayer Pt each time, we used a quartz crystal microbalance (QCM) to monitor mass change as a function of deposition time. For this experiment, we used a thin layer of gold-coated quartz crystal substrate to grow Pt monolayer films from a PtCl42− solution at pH 4 (see the Supplementary Materials for experimental details). Figure 2B shows the mass and voltage changes for five electroplating cycles. At the beginning of each cycle, the plating voltage was set at −0.8 VSCE. A sudden increase in the mass was observed as soon as the voltage pulse was started. The mass increased by a stable value of 550 ng cm−2 for a monolayer of Pt and remained essentially unchanged afterward, although the plating voltage was maintained at −0.8 VSCE for an extended period. For the next cycle, the potential was first set at 0.4 VSCE to allow the adsorbed hydrogen layer to be desorbed, releasing a fresh Pt surface. Afterward, as soon as the potential was set at −0.8 VSCE, the mass rapidly increased by the same amount of 550 ng cm−2 to another steady value again and remained unchanged even though the potential was maintained for an extended period. Because monolayer deposition conserves surface area, each monolayer deposition cycle results in the same mass gain of 550 ng cm−2, as shown in Fig. 2B. Assuming that the Pt atoms are uniformly distributed on the surface, this value would imply that each Pt atom occupies an area of 59,469 pm2, or a Pt radius of 137 pm, in excellent agreement with the covalent radius of 136 pm for Pt (28). This in turn proves that the coating process is indeed based on a complete-monolayer mechanism.
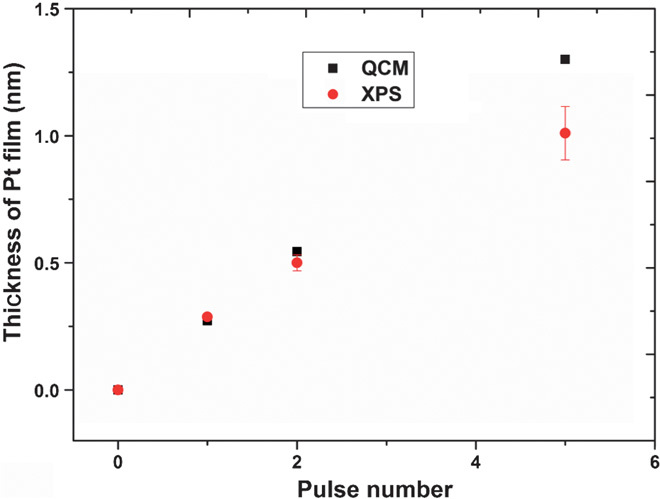
X-ray photoelectron spectroscopy (XPS) is a powerful technique for surface-specific composition measurement. Figure 3 shows the Pt monolayer film thickness, as quantified by the XPS analysis, as a function of the number of electrochemical deposition cycles. For comparison, the QCM measurement on a planner quartz crystal is used as a reference. Surprisingly, the XPS results for the monolayer deposition on nickel foam agree well with the QCM measurement on the planner substrate for the 1 and 2 ML samples, and a significant deviation is found for thicker deposition for its surface-specific nature (29).
Fig. 3. QCM mass increase converted to thickness and compared with XPS measurements.
Hydrogen adsorption-desorption is a powerful technique to determine the electroactive surface area for the Pt catalyst on the Ni foam (18). Figure S1 shows a cyclic voltammogram for the underpotential deposition of hydrogen (HUPD) on a Pt monolayer/Au NF/Ni foam electrode in 0.5 M H2SO4 at 50 mV s−1.
Figure 2C shows more detailed changes in current density (J) as a function of deposition time for the Pt monolayer coating on the Au NF/Ni foam surface, using a pulsed potential waveform. During the process, the electrode was maintained at a potential of −0.4 VSCE. By integrating the current density against deposition time as shown by the shaded area in Fig. 2C, we found that 1.14 mC cm−2 was used for each cycle to reach saturation at J = 0 mA cm−2. Using the Faraday equation, we concluded that 578 ng cm−2 Pt was deposited for each monolayer. Again, assuming that all Pt atoms are uniformly dispersed on surface, each Pt atom would take 56,014 pm2, or a Pt radius of 134 pm, in agreement with the covalent radius of 136 pm (3) and with that calculated from the QCM measurement of Pt monolayer deposited on the Au(111) surface of an Au coating on the quartz crystal substrate.
The above observation is very similar to what was observed by Liu et al. on the Au(111) surface (25). Because the proton reduction and the diffusion-limited PtCl42− reduction occur at essentially the same potential, whenever a Pt atom is deposited a hydrogen atom also forms on top of it, leading to a Pt/H double layer that shields the newly formed Pt surface from further deposition. As long as the H shield layer remains, the Pt deposition is completely quenched, and a well-defined monolayer Pt coating is produced (Fig. 2D), as evidenced by the mass plateau in Fig. 2B and the zero current sections in Fig. 2 (A and C). Because the Pt monolayer is deposited on a large piece of Ni foam with the dimension in inches, the precious Pt coating can be easily recovered with the substrate, treated if needed, and reused.
We evaluated the Pt monolayer/metal NF/Ni foam as a catalyst for HER in acidic conditions using a three-electrode voltammetry configuration (Pt monolayer/metal NF/Ni foam as the working electrode). We found that these catalysts are very effective for water splitting. Figure 4A shows the cyclic voltammograms in 0.5 M H2SO4 solution using a series of catalysts: bare Ni foam without noble metal coating, Pt layer on Ni foam, Au NF/Ni foam, and Pt monolayer films on Ag and Au NF-coated Ni foam. The onset potential for the bare Ni foam is −0.16 V. When a metal NF coating is applied, the onset potential slightly changes to −0.13 V for the Au NF and −0.14 V for the Ag NF. However, when a Pt monolayer was applied onto the metal NFs and the Ni foam, the onset potential jumps to −0.04 V, signifying that the Pt atoms function as HER catalysts that effectively lower the onset potential. However, different catalysts show significantly altered kinetics as characterized by the J-E behavior. In the scan range from 0 to −0.4 V, when potential changed to −0.4 V, the current density J changed to −19, −35, and −30 mA cm−2 for the bare Ni foam, Ag NF/Ni foam, and Au NF/Ni foam, respectively. The Pt monolayers on Ag and Au NFs gave identical current densities (−69 and −70 mA cm−2), significantly larger than the bare Ni form and NFs without the Pt monolayer.
Fig. 4. High-performance Pt monolayer/metal NF/Ni foam electrocatalysts with high stability.
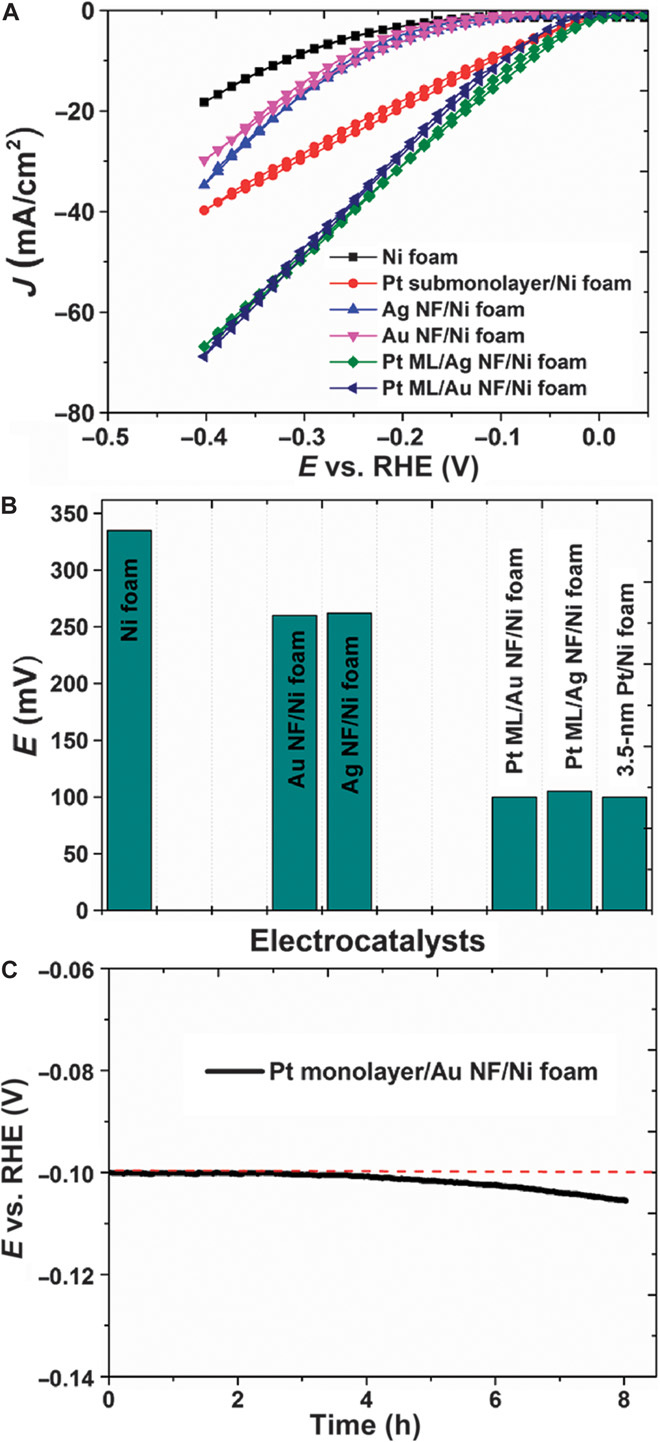
(A) Cyclic voltammograms of various electrocatalysts in 0.5 M H2SO4 solution. RHE, reversible hydrogen electrode. (B) Overpotential (EHER) of the different electrocatalysts to achieve a current density of 10 mA cm−2 for the HER in 0.5 M H2SO4. Note that the 3.5-nm Pt/Ni foam result is from Luo et al. (30). (C) Chronopotentiometry curve of Pt monolayer/Au NF/Ni foam catalyst in 0.5 M H2SO4 solution at a constant current density of 10 mA cm−2. A Pt sheet was used as the counter electrode and a calomel electrode was used as the reference.
To evaluate the catalytic performance for the water-splitting reaction, the electrodes were swept at 50 mV s−1 from negative to positive potential on the reversible hydrogen electrode scale. The overpotential of the present Pt monolayer catalyst on both Ag and Au NFs is much lower than that of its counterpart on the bare Ni foam. For the current density of 10 mA cm−2, the overpotential of the Pt monolayer on the Au NF electrode is only 100 mV, which is the same as that achieved by Luo (30) using a much thicker (3.5 nm) Pt coating on the Ni foam electrode, as shown in Fig. 4B.
It is surprising to see that a Pt monolayer appears to work as well as the much thicker (3.5 nm) Pt coating on the same Ni foam electrode (30), whereas the buffer layer Au or Ag makes little difference. However, for Pt monolayer directly deposited on the Ni foam electrode, although its onset potential is equal to that for the Pt monolayer on Au and Ag NFs, its current density is smaller (Fig. 4, A and B), indicating that only a submonolayer Pt was formed on the Ni foam.
To study the stability of the catalyst system, we measured the chronopotentiometry curves of Pt ML/Au NF/Ni foam catalysts in 0.5 M H2SO4 solution at a constant current density of 10 mA cm−2. For the experiment, a 1.5 × 1.5–cm2 Pt sheet (~0.3 mm thick) was used as the counter electrode along with a calomel reference electrode. Figure 4C shows the catalyst degradation data for the time period. It is remarkable that the current density for the Pt monolayer catalyst decayed from 100 to 105 mV, for only ~5% in a very acidic solution for ~8 hours of the experiment. Because Ni shows far inferior activity (Fig. 4), it is expected that Ni diffusion to the catalyst surface would lead to considerable degradation in catalytic activity. It is commonly believed that there is no significant Ni diffusion driven by the Kirkendalle effect in this system.
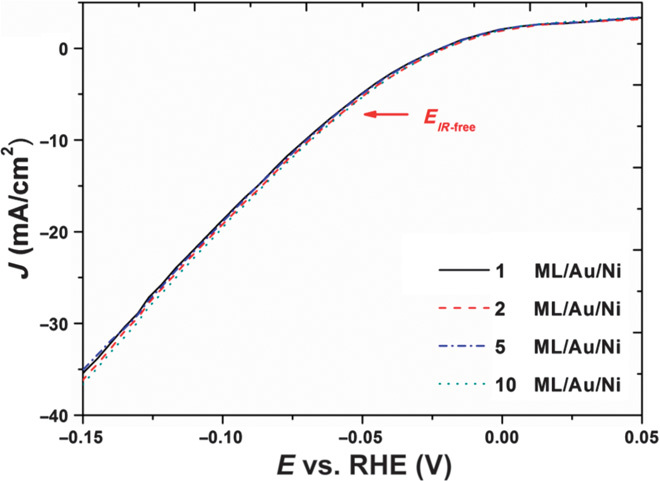
Figure 5 shows the J-E plots of the Au NF/Ni foam electrode coated with 1, 2, 5, and 10 ML Pt in 0.5 M H2SO4 solution. To avoid interference caused by electrodes, electrolytes, and interfaces, the measured J-E curves are iR–corrected (31) to determine the net contribution of the catalyst [details of the correction are provided in the Supplementary Materials (fig. S2)]. All four catalysts showed essentially identical J-E curves in view of measurement error. The additional Pt monolayer coating (up to 10 ML) had only a small effect on the J-E behavior of HER. More specifically, all samples had basically the same onset value at E = −0.01 V for the HER. When |E| < 0.01 V, J is essentially 0, meaning that there is no current flow and therefore no electrochemical reaction. When E increases above −0.01 V, HER starts and |J| linearly increases with |E|.
Fig. 5. The J-E plots of Au NF/Ni foam coated with 1, 2, 5, and 10 ML Pt in 0.5 M H2SO4 solution after iR correction.
In comparison, Ni has a much smaller lattice constant of 352.4 pm, meaning that Ni is less favorable compared to Au for direct Pt monolayer formation. In addition, because Ni has a very negative electrochemical potential, the following galvanic replacement reaction is spontaneous:
In other words, atomic Pt particles can be directly formed on the Ni foam surface without the need for electric potential, leading to island formation of Pt nanoparticles with incomplete surface coverage.
This is analogous to the prior art Pt deposition (32) in which an atomic layer of a less noble metal (for example, copper) is applied first using the UPD deposition technique, with the less noble metal subsequently replaced by Pt atoms. By repeating the above combination cycle, a thicker Pt film is collected. Using a combination of electrochemistry, in situ scanning tunneling microscopy equipped with an electrochemical flow cell, and ultrahigh vacuum surface studies combined with electrochemistry, Kim et al. discovered that a single deposition cycle yielded an “incomplete monolayer” and partially distorted “Pt nanoislands” (33). This explains why the Pt monolayer/Ni foam shows significantly lower catalytic activity.
The Tafel plot η versus log(J), where η is the overpotential and J is the current density, provides insight into the HER mechanism on catalyst surface. The overpotential (η) is related to the reaction rate by the Tafel equation:
where J0 is the exchange current density and b is the Tafel slope. A reduction in overpotential leads to increased catalytic activity for the HER. The Tafel slope b is an intrinsic quantity that is independent of the surface area of the catalyst but is dependent on the electronic factors or reaction mechanism (18). Figure 6 shows the Tafel plots for four different catalysts, namely, Ni foam, Au NF/Ni foam, Pt ML/Au NF/Ni foam, and Pt monolayer/Ag NF/Ni foam. Using the least-squares analysis, the Tafel slopes obtained were −77, −67, and −53 mV dec−1 for Ni foam (34, 35), Au NF/Ni foam, and Pt monolayer/Au NF/ Ni foam, respectively. The Tafel slope of the Pt monolayer/Au NF/Ni foam increased compared to Au NF/Ni foam and Ni foam, indicating a significant enhancement of electronic effect by the Pt monolayer on Au NF (18). In addition, Pt monolayers on Au NF/Ni foam and Ag NF/Ni foam gave very similar Tafel slopes of −53 and −55 mV dec−1, respectively, because they seemed to be independent of the buffer layer, indicating that only the top monolayer surface is responsible for the catalytic activity.
Fig. 6. Tafel plots of the Pt monolayer on different metal NFs recorded at 50 mV s−1 in 0.5 M H2SO4 after iR correction.
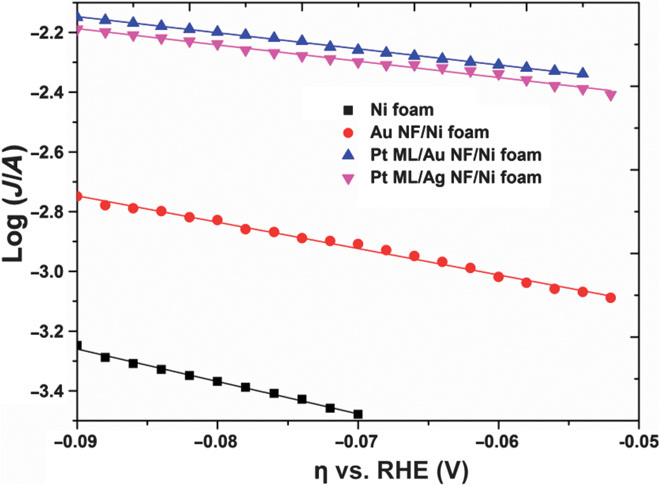
A comparison before and after the iR correction is shown in fig. S3.
Exchange current densities reflect intrinsic rates of electron transfer between the electrolyte and electrodes (36, 37). As shown in table S1, the exchange current densities for platinum sheet and Pt monolayers for reduction of protons are very similar, indicating that thePt monolayers work as well as the Pt sheet as the catalysts for the HER.
The Pt monolayer–coated catalysts reached catalytic activity as high as that of the thick Pt film, indicating that the Ni foam is completely covered by the Pt monolayer, meaning that there is a complete-monolayer Pt coating generated on the large–surface area Ni foam substrate. Assuming that all metal sites are involved in the electrochemical reaction, the turnover frequencies (TOFs) are calculated and listed in table S2. It shows that the TOF values for the Pt monolayer catalysts are far greater than those for metal NF/Ni foam catalysts. Specifically, the TOF for the Ag NF/Ni foam catalyst is 24, whereas the Pt monolayer/Ag NF/Ni foam catalyst shows a much higher TOF value of 1839, about 55 times higher. The Pt monolayer/Au NF/Ni foam catalyst shows an even higher TOF value of 1655, in line with what was achieved using the state-of-the-art catalysts (38).
To further confirm the above conclusion, we conducted an XPS analysis on three individual areas separated by >1 cm from a Pt monolayer coating. Figure S4 displays that all three areas, each measuring 0.3 × 0.7 mm in the x-ray beam spot, have very similar XPS peak intensities. Note that the element mapping analysis on the Au NF/Ni foam shows uniform Au coating on the Ni substrate (fig. S5). Table S3 shows relative Pt concentrations at three different areas on the catalyst surface. The Pt percentages on all three different areas obtained from the XPS are essentially the same, demonstrating that the catalyst surface is indeed completely covered by Pt.
CONCLUSIONS
In conclusion, we demonstrated a technique to prepare a complete-monolayer Pt coating on large-surface-area 3D Ni foam–based substrates using a buffer layer (Ag or Au) strategy. The QCM shows that each monolayer deposition cycle results in the same mass gain of 550 ng cm−2, corresponding to a complete surface coverage by Pt atoms. A detailed analysis on the current density change in each deposition cycle achieved the same conclusion. The XPS analysis confirms the result. On the basis of the analyses of the J-E curve, Tafel plot, and surface area from HUPD, the catalyst performance of the Pt monolayer/metal NF/Ni foam electrode is tantamount to that of the thick Pt film and outperformed Ni foam. Because Pt is the most important catalyst for many industrial applications, and this development provides a way to fabricate a high-activity Pt catalyst with minimal Pt usage, it may pave a way for Pt to be used in fuel cells, petroleum processing, and other commercial applications at minimal cost.
MATERIALS AND METHODS
Materials
Sodium chloride, potassium chloride, potassium hydroxide, ammonium hydroxide, sulfuric acid, silver nitrate (AgNO3), potassium tetrachloroplatinate (K2PtCl4), potassium hexachloroplatinate (K2PtCl6), and potassium chloroaurate (K2AuCl4) are from Alfa Aesar. Perchloric acid is from Fisher Scientific. Organic reagents including AR (analytical reagent) grade acetone, ethanol, and ethylenediamine tetraacetic acid disodium are from J&K Chemicals. The substrate Ni foam (purity, 99.9%; thickness, 1.5 mm; pore density, 100 ppi) is from Zhuoer Advanced Metallic Materials. The substrate was cleaned using ultrasonication with ethanol (>99.7%), hydrochloric acid (2%), and deionized (DI) water (18.3 megohms·cm, Merck Millipore).
Characterization
A Zennium Zahner electrochemistry workstation with a QCM was used for electrochemical deposition and analysis. The Pt deposition was done using a constant voltage mode. For the Pt monolayer deposition, a pulse duration of 5 s was set for each deposition cycle, whereas on the basis of the J-t curve (Fig. 2), the effective deposition lasted for ~0.2 s. The cyclic voltammetry (CV) and Tafel analysis were conducted in a nitrogen-purged H2SO4 solution with a scan rate of 50 mV s−1. The catalysts were cycled many times by CV until a stable CV curve developed. Chronopotentiometry was carried out at a constant current density of 10 mA cm−2 for Pt monolayer/Au NF/Ni foam. A Pt sheet was used as the counter electrode, and a calomel electrode was used as the reference (sweep rate, 50 mV s−1). A Kratos Axis Ultra XPS system was used with a monochromatic Al-Kα source.
Preparation of the working electrode on Ni foam
Au and Ag NF/Ni foam: An Au buffer layer on Ni foam was produced by displacement of Ni using gold ions in 0.5 M NaCl and 1 mM KAuCl4 aqueous solution at pH 4. The spontaneous deposition involves the following reaction:
The Ag buffer layer was synthesized in a constant-voltage mode at 2 V, using a solution of 10 mM AgNO3 and 5 mM ethylenediamine tetraacetic acid disodium. During deposition, the pH was maintained at 9 to 10 with ammonium hydroxide.
Growth of Pt monolayer
Deposition on the Ni foam substrate with the buffer layer
A pulsed potential method was used to grow Pt monolayer and multilayer films from a solution consisting of 0.5 M NaCl and 3 mM K2PtCl4; the pH was maintained at 4 with perchloric acid and sodium hydroxide solution. The Pt monolayer films deposited one monolayer at a time by cycling the electrode potential between 0 and −0.4 VSCE. By repeating the process, 2, 5, and 10 ML Pt films were produced. Upon electrodeposition, the films were immediately rinsed with DI water and dried in a stream of N2.
The QCM experiment
The QCM experiment was performed using Ti/Au-coated sensor crystals. The electrode was cleaned by soaking in a solution [3:1 volume ratio of concentrated H2SO4 (70%) to H2O2 (30%)] before being placed into the electrolyte solution [50 ml of N2-saturated 0.5 M NaCl and 3 mM K2PtCl4 (pH 4)]. The Pt monolayer was deposited by first setting the potential to 0.4 VSCE to activate the substrate surface and then lowering potential to −0.8 VSCE for deposition.
Pt monolayer/Ni foam
The specific surface area of the nickel foam was measured using an optical 3D video microscope (78 cm2 cm−2) (HIROX, KH-7700). The total surface area was used to calculate K2PtCl6 needed to achieve single atomic layer platinum deposition. The Pt monolayer on Ni foam was achieved by the galvanic exchange of Ni with a calculated amount of PtCl62−.
Acknowledgments
We acknowledge support from Thousand Talents Program, Chinese National University Research Fund (GK261001009), Program for Changjiang Scholars and Innovative Research Team in University (IRT_14R33), the Overseas Talent Recruitment Project (B14041) and Shaanxi Normal University, Xi’an, China. Author contributions: M.L., W.Z., X.Z., and S.L. conceived the experiments and contributed to the design of the experiments. M.L. and Q.M. carried out the fabrication of the samples and performed the experiments. M.L., X.L., and S.L. contributed to the data analysis, interpretation of the results, and preparation of the manuscript. M.L. and S.L. wrote the manuscript. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/8/1400268/DC1
Fig. S1. Cyclic voltammogram for HUPD on Pt monolayer/Au NF/Ni foam electrode in Ar-saturated 0.5 M H2SO4 at 50 mV s−1 with two different positive potential limits. The current density is referenced to the actual Pt surface area.
Fig. S2. HER polarization curve on Pt monolayer/Au NF/Ni foam in 0.5 M H2SO4 solution at 50 mV s−1 (solid black lines) and after iR correction (EIR-free, dotted red lines).
Fig. S3. Tafel curve on Pt monolayer/Au NF/Ni foam in 0.5 M H2SO4 solution at 50 mV s−1 (solid black lines) and after iR correction (EIR-free, dotted red lines).
Fig. S4. The XPS spectra of the different points on a Pt monolayer/Au NF/Ni foam electrode.
Fig. S5. EDX and the image of element mapping of the Au NF/Ni foam.
Table S1. Comparison of exchange current density for proton reduction reaction in 1 M H2SO4.
Table S2. TOFs (s−1) for electrocatalytic hydrogen generation by various catalysts in 0.5 M H2SO4 solution.
Table S3. Relative metal concentrations at different areas of a Pt monolayer/Au NF/Ni foam sample measured using XPS.
REFERENCES AND NOTES
- 1.http://en.wikipedia.org/wiki/Platinum.
- 2.Xie J., Yang X., Han B., Shao-Horn Y., Wang D., Site-selective deposition of twinned platinum nanoparticles on TiSi2 nanonets by atomic layer deposition and their oxygen reduction activities. ACS Nano 7, 6337–6345 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Adzic R. R., Zhang J., Sasaki K., Vukmirovic M. B., Shao M., Wang J. X., Nilekar A. U., Mavrikakis M., Valerio J. A., Uribe F., Platinum monolayer fuel cell electrocatalysts. Top. Catal. 46, 249–262 (2007). [Google Scholar]
- 4.Zhang J., Lima F. H. B., Shao M. H., Sasaki K., Wang J. X., Hanson J., Adzic R. R., Platinum monolayer on nonnoble metal–noble metal core–shell nanoparticle electrocatalysts for O2 reduction. J. Phys. Chem. B 109, 22701–22704 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Zhang J., Sasaki K., Sutter E., Adzic R. R., Stabilization of platinum oxygen-reduction electrocatalysts using gold clusters. Science 315, 220–222 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Kim C., Oh J.-G., Kim Y.-T., Kim H., Lee H., Platinum dendrites with controlled sizes for oxygen reduction reaction. Electrochem. Commun. 12, 1596–1599 (2010). [Google Scholar]
- 7.Hsu I. J., Kimmel Y. C., Jiang X., Willis B. G., Chen J. G., Atomic layer deposition synthesis of platinum–tungsten carbide core–shell catalysts for the hydrogen evolution reaction. Chem. Commun. 48, 1063–1065 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Liu Y., Mustain W. E., Evaluation of tungsten carbide as the electrocatalyst support for platinum hydrogen evolution/oxidation catalysts. Int. J. Hydrogen Energy 37, 8929–8938 (2012). [Google Scholar]
- 9.Lombardi I., Marchionna S., Zangari G., Pizzini S., Effect of Pt particle size and distribution on photoelectrochemical hydrogen evolution by p-Si photocathodes. Langmuir 23, 12413–12420 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Dai P., Xie J., Mayer M. T., Zhan J., Wang D., Solar hydrogen generation by silicon nanowires modified with platinum nanoparticle catalysts by atomic layer deposition. Angew. Chem. Int. Ed. 52, 11119–11123 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Tang J. M., Jensen K., Waje M., Li W., Larsen P., Pauley K., Chen Z., Ramesh P., Itkis M. E., Yan Y., Haddon R. C., High performance hydrogen Fuel Cells with ultralow Pt loading carbon nanotube thin film catalysts. J. Phys. Chem. C 111, 17901–17904 (2007). [Google Scholar]
- 12.Wang G., Huang B., Xiao L., Ren Z., Chen H., Wang D., Abruña H. D., Lu J., Zhuang L., Pt skin on AuCu intermetallic substrate: A strategy to maximize Pt utilization for fuel cells. J. Am. Chem. Soc. 136, 9643–9649 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Yan X., Ge X., Cui S., Pt-decorated nanoporous gold for glucose electrooxidation in neutral and alkaline solutions. Nanoscale Res. Lett. 6, 313 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen A., Holt-Hindle P., Platinum-based nanostructured materials: Synthesis, properties, and applications. Chem. Rev. 110, 3767–3804 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Jha N., Ramesh P., Bekyarova E., Tian X., Wang F., Itkis M. E., Haddon R. C., Functionalized single-walled carbon nanotube-based fuel cell benchmarked against US DOE 2017 technical targets. Sci. Rep. 3, 2257 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.US-DRIVE, 27 July 2011: Fuel Cell Technical Team Technology Roadmap: Target Tables, www.uscar.org/commands/files_download.php?files_id=279.
- 17.Kyriakou G., Boucher M. B., Jewell A. D., Lewis E. A., Lawton T. J., Baber A. E., Tierney H. L., Flytzani-Stephanopoulos M., Sykes E. C. H., Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations. Science 335, 1209–1212 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Brankovic S. R., Wang J. X., Adžić R. R., Metal monolayer deposition by replacement of metal adlayers on electrode surfaces. Surf. Sci. 474, L173–L179 (2001). [Google Scholar]
- 19.Li M., Liu P., Adzic R. R., Platinum monolayer electrocatalysts for anodic oxidation of alcohols. J. Phys. Chem. Lett. 3, 3480–3485 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Kye J., Shin M., Lim B., Jang J.-W., Oh I., Hwang S., Platinum monolayer electrocatalyst on gold nanostructures on silicon for photoelectrochemical hydrogen evolution. ACS Nano 7, 6017–6023 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Ding Y., Mathur A., Chen M., Erlebacher J., Epitaxial casting of nanotubular mesoporous platinum. Angew. Chem. Int. Ed. 44, 4002–4006 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Karan H. I., Sasaki K., Kuttiyiel K., Farberow C. A., Mavrikakis M., Adzic R. R., Catalytic activity of platinum monolayer on iridium and rhenium alloy nanoparticles for the oxygen reduction reaction. ACS Catal. 2, 817–824 (2012). [Google Scholar]
- 23.Zhang J., Mo Y., Vukmirovic M. B., Klie R., Sasaki K., Adzic R. R., Platinum monolayer electrocatalysts for O2 reduction: Pt monolayer on Pd(111) and on carbon-supported Pd nanoparticles. J. Phys. Chem. B 108, 10955–10964 (2004). [Google Scholar]
- 24.Lee K.-S., Yoo S. J., Ahn D., Jeon T.-Y., Choi K. H., Park I.-S., Sung Y.-E., Surface structures and electrochemical activities of Pt overlayers on Ir nanoparticles. Langmuir 27, 3128–3137 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Liu Y., Gokcen D., Bertocci U., Moffat T. P., Self-terminating growth of platinum films by electrochemical deposition.. Science 338, 1327–1330 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Loukrakpam R., Brankovic S. R., Strasser P., A study of Au/C nanoparticles with Pt monolayer and sub-monolayer electrocatalysts for ethanol oxidation reaction. ECS Trans. 58, 1733–1736 (2013). [Google Scholar]
- 27.Sasaki K., Naohara H., Choi Y. M., Cai Y., Chen W.-F., Liu P., Adzic R. R., Highly stable Pt monolayer on PdAu nanoparticle electrocatalysts for the oxygen reduction reaction. Nat. Commun. 3, 1115 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Cordero B., Gómez V., Platero-Prats A. E., Revés M., Echeverría J., Cremades E., Barragán F., Alvarez S., Covalent radii revisited. Dalton Trans., 2832–2838 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Jablonski A., Powell C. J., Relationships between electron inelastic mean free paths, effective attenuation lengths, and mean escape depths. J, Electron Spectros. Relat. Phenomena 100, 137–160 (1999). [Google Scholar]
- 30.Luo J., Im J.-H., Mayer M. T., Schreier M., Nazeeruddin M. K., Park N.-G., Tilley S. D., Fan H. J., Grätzel M., Water photolysis at 12.3% efficiency via perovskite photovoltaics and Earth-abundant catalysts. Science 345, 1593–1596 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Wu R., Zhang J., Shi Y., Liu D., Zhang B., Metallic WO2–carbon mesoporous nanowires as highly efficient electrocatalysts for hydrogen evolution reaction. J. Am. Chem. Soc. 137, 6983–6986 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Gokcen D., Bae S.-E., Brankovic S. R., Stoichiometry of Pt submonolayer deposition via surface-limited redox replacement reaction. J. Electrochem. Soc. 157, D582–D587 (2010). [Google Scholar]
- 33.Kim Y. G., Kim J. Y., Vairavapandian D., Stickney J. L., Platinum nanofilm formation by EC-ALE via redox replacement of UPD copper: Studies using in-situ scanning tunneling microscopy. J. Phys. Chem. B 110, 17998–18006 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Reece S. Y., Hamel J. A., Sung K., Jarvi T. D., Esswein A. J., Pijpers J. J. H., Nocera D. G., Wireless solar water splitting using silicon-based semiconductors and earth-abundant catalysts. Science 334, 645–648 (2011). [DOI] [PubMed] [Google Scholar]
- 35.McKone J. R., Sadtler B. F., Werlang C. A., Lewis N. S., Gray H. B., Ni–Mo nanopowders for efficient electrochemical hydrogen evolution. ACS Catal. 3, 166–169 (2013). [Google Scholar]
- 36.Sun Y., Lu J., Zhuang L., Rational determination of exchange current density for hydrogen electrode reactions at carbon-supported Pt catalysts. Electrochim. Acta 55, 844–850 (2010). [Google Scholar]
- 37.Neyerlin K. C., Gu W., Jorne J., Gasteiger H. A., Study of the exchange current density for the hydrogen oxidation and evolution reactions. J. Electrochem. Soc. 154, B631–B635 (2007). [Google Scholar]
- 38.Hou Y., Abrams B. L., Vesborg P. C. K., Björketun M. E., Herbst K., Bech L., Setti A. M., Damsgaard C. D., Pedersen T., Hansen O., Rossmeisl J., Dahl S., Nørskov J. K., Chorkendorff I., Bioinspired molecular co-catalysts bonded to a silicon photocathode for solar hydrogen evolution. Nat. Mater. 10, 434–438 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Herrero E., Buller L. J., Abruña H. D., Underpotential deposition at single crystal surfaces of Au, Pt, Ag and other materials. Chem. Rev. 101, 1897–1930 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Esswein A. J., McMurdo M. J., Ross P. N., Bell A. T., Tilley T. D., Size-dependent activity of Co3O4 nanoparticle anodes for alkaline water electrolysis. J. Phys. Chem. C 113, 15068–15072 (2009). [Google Scholar]
- 41.D. T. Sawyer, A. Sobkowiak, J. L. Roberts, Electrochemistry for Chemists (John Wiley & Sons, New York, ed. 2, 1995). [Google Scholar]
- 42.Popczun E. J., Read C. G., Roske C. W., Lewis N. S., Schaak R. E., Highly active electrocatalysis of the hydrogen evolution reaction by cobalt phosphide nanoparticles. Angew. Chem. Int. Ed. 53, 5427–5430 (2014). [DOI] [PubMed] [Google Scholar]
- 43.N. Fairley, CasaXPS Manual 2.3.15, Rev 1.2 (Casa Software Ltd., Teignmouth, UK, 2009). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/8/1400268/DC1
Fig. S1. Cyclic voltammogram for HUPD on Pt monolayer/Au NF/Ni foam electrode in Ar-saturated 0.5 M H2SO4 at 50 mV s−1 with two different positive potential limits. The current density is referenced to the actual Pt surface area.
Fig. S2. HER polarization curve on Pt monolayer/Au NF/Ni foam in 0.5 M H2SO4 solution at 50 mV s−1 (solid black lines) and after iR correction (EIR-free, dotted red lines).
Fig. S3. Tafel curve on Pt monolayer/Au NF/Ni foam in 0.5 M H2SO4 solution at 50 mV s−1 (solid black lines) and after iR correction (EIR-free, dotted red lines).
Fig. S4. The XPS spectra of the different points on a Pt monolayer/Au NF/Ni foam electrode.
Fig. S5. EDX and the image of element mapping of the Au NF/Ni foam.
Table S1. Comparison of exchange current density for proton reduction reaction in 1 M H2SO4.
Table S2. TOFs (s−1) for electrocatalytic hydrogen generation by various catalysts in 0.5 M H2SO4 solution.
Table S3. Relative metal concentrations at different areas of a Pt monolayer/Au NF/Ni foam sample measured using XPS.