

Virus-free gene editing possible at chromosomes through stretching cells in a micropost array, even for hard-to-transfect cells.
Keywords: CRISPR-Cas9, Delivery, Mechanical deformation, Hard-to-transfect cells
Abstract
The CRISPR (clustered regularly interspaced short palindromic repeats)–Cas (CRISPR-associated) nuclease system represents an efficient tool for genome editing and gene function analysis. It consists of two components: single-guide RNA (sgRNA) and the enzyme Cas9. Typical sgRNA and Cas9 intracellular delivery techniques are limited by their reliance on cell type and exogenous materials as well as their toxic effects on cells (for example, electroporation). We introduce and optimize a microfluidic membrane deformation method to deliver sgRNA and Cas9 into different cell types and achieve successful genome editing. This approach uses rapid cell mechanical deformation to generate transient membrane holes to enable delivery of biomaterials in the medium. We achieved high delivery efficiency of different macromolecules into different cell types, including hard-to-transfect lymphoma cells and embryonic stem cells, while maintaining high cell viability. With the advantages of broad applicability across different cell types, particularly hard-to-transfect cells, and flexibility of application, this method could potentially enable new avenues of biomedical research and gene targeting therapy such as mutation correction of disease genes through combination of the CRISPR-Cas9–mediated knockin system.
INTRODUCTION
The CRISPR (clustered regularly interspaced short palindromic repeats)–Cas (CRISPR-associated) nuclease system is an easy-to-use, highly specific, efficient, and multiplexable genome editing tool that has been used in various organisms, including human and mouse cell lines (1–3). In the two-component system, a single-guide RNA (sgRNA) directs Cas9 nuclease to introduce sequence-specific targeted loss-of-function mutations into the genome (3, 4). Cas9 can be easily programmed to induce DNA double-strand breaks through RNA guides, which can generate insertions and deletions (indels) and stimulate genome editing at specific target genomic loci (5, 6). The ability to perturb the genome in a precise and targeted manner is crucial to understanding genetic contributions to biology and disease (3, 7).
Successful delivery of sgRNA and Cas9 into cells guarantees efficient genome editing. Typical intracellular delivery techniques use liposomes or polymeric nanoparticles to induce cell membrane poration or endocytosis (8–11), and recently, cell-penetrating peptide-mediated delivery of sgRNA and Cas9 has been used for gene disruption (12). In these methods, delivery efficiency is often dependent on cell type and the structure of the target molecule. Electroporation is an attractive alternative for many applications and allows highly efficient RNA-guided genome editing via delivery of purified Cas9 ribonucleoprotein (13–15). However, this method can cause cell damage and generate a high cell death rate. Moreover, commonly used virus (adeno-associated virus, retrovirus, or lentivirus)–mediated delivery of sgRNA and Cas9 is often associated with uncontrolled chromosomal integration (16, 17), limiting its clinical potential.
Rapid mechanical deformation of cells can produce transient membrane disruptions that facilitate passive diffusion of material into the cytosol. Using physical constriction to deform and shear cells for delivery has achieved high efficiency with low cell death rate. This method has the advantage of high-throughput delivery of almost any macromolecule into almost any cell type (18). Membrane deformation–based microfluidic devices have been used in the delivery of a range of materials such as carbon nanotubes, proteins, and short interfering RNAs (siRNAs). They have been used for delivering transcription factors for cell reprogramming (18). Microfluidic membrane deformation has the potential to serve as a broad-based universal delivery platform and boasts the advantages of precise control over treatment conditions at the single-cell level, with macroscale throughput. Here, we optimized the physical constriction in a microfluidic setup, considering both delivery efficiency and cell viability. Through this, we successfully delivered single-stranded DNA (ssDNA), siRNAs, and large-sized plasmids into different cell types, including adherent and non-adherent cells, hard-to-transfect lymphoma, and embryonic stem cells. Sequence analysis, together with biochemical and functional analyses, demonstrated highly efficient genome editing and successful generation of gene-knockout cell lines, using our delivery device in different cell types. To the best of our knowledge, this is the first demonstration of membrane deformation for CRISPR/Cas9 gene editing. Thus, we expect that our new microfluidic delivery method will facilitate RNA-guided genome editing and gene loss-of-function analysis across different cell types, especially difficult-to-transfect cells. Achievement of high genome editing efficiency in non-adherent lymphoma cells suggests that the approach also has potential for clinical use.
RESULTS
Delivery principle and chip design
When a cell passes through a constriction smaller than the cell diameter, it undergoes rapid mechanical deformation, causing transient membrane disruption or holes. The shear and compressive forces imposed on the cell during passage through the constriction determine the degree of disruption and the size and frequency of the holes. Macromolecules small enough to pass through the holes can diffuse into the cytosol from the surrounding medium and may remain and function in the cell after the membrane recovers from the deformation (Fig. 1, A and B). To apply this principle, we designed a family of microfluidic devices with a series of constrictions of different dimensions formed by structures of different shapes (fig. S1A).
Fig. 1. Delivery mechanism and device design.
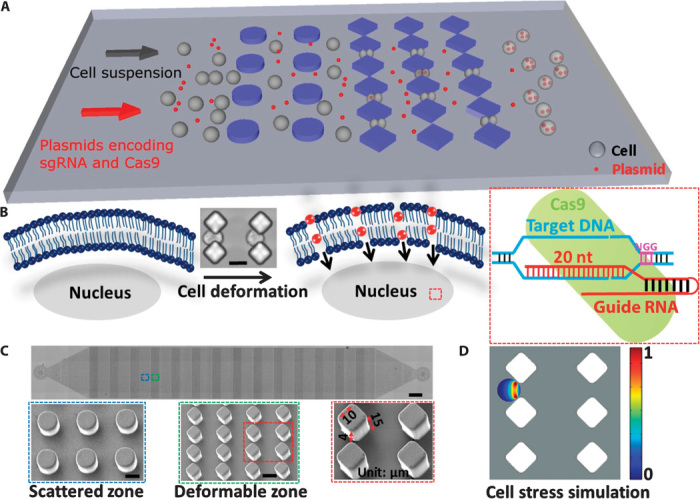
(A) Illustration of the delivery process wherein cells pass through the microconstriction and experience deformability. Plasmids encoding sgRNA and Cas9 protein are mixed with the cells to flow through the chip. (B) Illustration of the delivery mechanism whereby transient membrane holes are generated when cells pass through the microconstriction and specific genome editing is conducted after plasmids encoding sgRNA and Cas9 protein are delivered into the cell. Cell deformation was shown by microscopy when cells passed through the microconstriction. Scale bar, 15 μm. nt, nucleotide. (C) Microscopy of the whole device structure. Scale bar, 0.5 mm. Scanning electron microscopy (SEM) of scattered and deformable zones in the device is also shown. Scale bar, 15 μm. One diamonded microconstriction of 15-μm depth and 4-μm width is indicated by the red arrow. The length of the diamond edge is 10 μm. (D) Cell stress simulation was applied on the diamonded microconstriction design with 15-μm depth and 4-μm width when a cell began to penetrate the constriction. A graphical representation of the cell stress gradient that forms across the membrane is shown.
Devices were fabricated with standard polydimethylsiloxane (PDMS) microfluidics technology. Each chip consists of 14 identical cell-scattering and deformation zones, and each zone contains 10 arrays of structures forming microconstrictions (Fig. 1C). The scatter zone is designed to prevent device collapse and also disperse or “scatter” the cell suspension. The deformation zone is where cells pass through microconstrictions, becoming deformed and generating transient membrane holes that ensure delivery of the macromolecule(s) of interest. Interconnected channels enable high throughput of treated cells by preventing clogging. To optimize the microconstriction design, we first prepared constrictions using structures of several different shapes, including circle, ellipse, and diamond (fig. S1A). Suspended cells were applied to the chip through a Tygon tube connected to the inlet, and fluid flow was controlled by a syringe pump. To optimize the design, we did a series of test deliveries of fluorescein isothiocyanate (FITC)–labeled ssDNA into human embryonic kidney 293T (HEK293T) cells. The smallest constriction width of the three designs, 4 μm, was chosen for further experiments. Of the three designs, the diamond pattern showed nearly identical delivery efficiency at a range of flow rates from 50 to 250 μl/min, with much higher cell viability than the circle or ellipse patterns (fig. S1, B and C), and so this pattern was chosen for further experiments. To maximize the functional area, we minimized the length of the diamond edge to 10 μm (Fig. 1C). The parallel chip design (fig. S1D) was generated by arranging multiple devices side by side to demonstrate that delivery can be multiplexed. The cell recovery rate after delivery for both HEK293T and SUM159 cell lines was close to 100% (fig. S1E). Movie S1 shows cells passing through microconstrictions formed by the diamond pattern at a flow rate of 30 μl/min. Cell stress simulation (Fig. 1D and fig. S2) and flow velocity simulation (fig. S3) were applied to the diamond pattern design at the time point when a cell began to penetrate the microconstriction. Movie S2 shows the flow velocity simulation. With this chip design, we expect to successfully deliver plasmids encoding different sgRNAs and Cas9 into different types of cells and achieve precise genome editing and perform specific gene loss-of-function analysis, as depicted in Fig. 1B.
Optimization of the delivery chip specifications
To optimize the delivery performance of the chip, we took into consideration constriction dimensions, fluid flow rates, and duration of cell passage through the chip as three key parameters. In the diamond design, the constriction depth was 15 μm, and the width varied from 4 to 5 μm (Fig. 1C). In pursuit of high delivery efficiency coupled with high cell viability, we did a series of testing deliveries of FITC-labeled ssDNA into HEK293T cells (Fig. 2A). Our data showed that delivery efficiency increased with increasing flow rate across design patterns (Fig. 2B). The 4-μm constriction width presented higher delivery efficiency than the 5-μm width at all flow rates, with minimal effect on cell viability. Increasing the number of operational cycles with the same chip allowed multiple cell passaging times, which would also enhance the delivery efficiency; however, the operation clearly decreased cell viability (Fig. 2, B and C). The data for the 0 μl/min flow rate represents a control whereby the cells were treated exactly as the other samples but were not applied with the membrane deformation, thus ruling out the possibility that cell FITC positivity was the result of any endocytotic or surface binding events.
Fig. 2. Governing parameters and broad applicability.
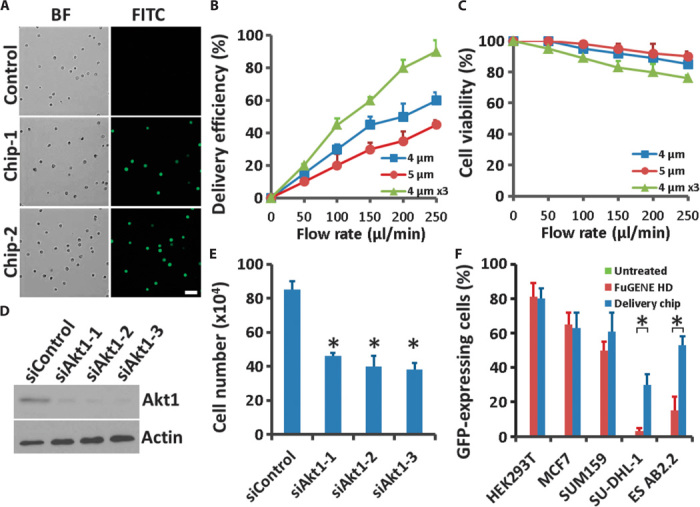
(A) Microscopy of HEK293T cells into which FITC-labeled ssDNA was delivered through our chip. Results shown are from two independent chips. Control indicated all the same treatments for the cells except passing through the chip. Scale bar, 50 μm. BF, bright field. (B and C) Delivery efficiency (B) and cell viability (C) 16 hours after treatment were calculated for (A) as a function of fluid speed at different parameter designs; 4 or 5 μm indicates the constriction width, and 4 μm ×3 indicates cells passing through the same device three times. Error bars indicate SEM (n = 3). (D) Western blotting of PC-3 cells 48 hours after delivery with three different siRNA oligos targeting Akt1. Actin is showed as a loading control. (E) Cells from (D) were seeded in complete medium and, after 6 days, were recovered and trypsinized to count the numbers with a Countess II FL Automated Cell Counter (Life Technologies). Error bars indicate SEM (n = 3). *P < 0.005 determined by Student’s t test. (F) Delivery efficiency in different cell lines. HEK293T cells, human luminal-like MCF7 and basal-like SUM159 breast cancer cells, human SU-DHL-1 anaplastic large cell lymphoma cells, and mouse AB2.2 embryonic stem cells were delivered with plasmids encoding GFP. Untreated serves as a negative control and FuGENE HD serves as a positive control. Error bars indicate SEM (n = 3). *P < 0.005 determined by Student’s t test.
Broad applicability
To investigate the adaptability of this technique, we first tried siRNA delivery for gene knockdown. Considering both delivery efficiency and cell viability, we chose a microconstriction width of 4 μm, a fluid flow rate of 250 μl/min, and single passage of the cells through the chip for all subsequent experiments. When we delivered three siRNAs specific for Akt1 into PC-3 cells, all of the oligos achieved >70% knockdown efficiency in 48 hours after delivery (Fig. 2D). Moreover, depletion of Akt1 by all three siRNAs suppressed cell growth, which is consistent with previous research (Fig. 2E), indicating that our technique is reliable for cell phenotype analysis and gene function study (19).
To further assess the delivery ability of the chip across different cell types, we used plasmids encoding green fluorescent protein (GFP) to measure the delivery efficiency. We successfully delivered plasmids encoding GFP with high efficiency into HEK293T cells, human luminal-like MCF7 and basal-like SUM159 breast cancer cells, human SU-DHL-1 anaplastic large cell lymphoma cells, and mouse AB2.2 embryonic stem cells (Fig. 2F), all with minimal cell death. Using our method, we achieved nearly the same percentage of GFP-expressing cells as obtained with traditional FuGENE HD transfection (Fig. 2F and fig. S4, A to E). Our delivery method achieved even higher efficiency than FuGENE HD transfection in human anaplastic large cell lymphoma cells and mouse embryonic stem cells without inducing stem cell differentiation (Fig. 2F and fig. S4F), suggesting potential application in difficult-to-transfect cells. Device designs have not been optimized for different cell types, indicating that we can expect further improvement in delivery efficiency, with the goal of cell-specific delivery protocols, in future applications.
EGFP knockout via chip
We used cells stably expressing enhanced GFP (EGFP) to illustrate the potential application of this method in CRISPR-Cas9–mediated genome editing. EGFP was introduced into cells with lentivirus, and the EGFP encoding sequences were integrated into chromosomal DNA. Plasmids encoding Cas9 only or sgRNAs targeting EGFP (sgEGFP-1 and sgEGFP-2) and Cas9 were delivered into adherent MDA-MB-231 cells and non-adherent SU-DHL-1 lymphoma cells. To enhance delivery efficiency, cells were passed through the same chip three times. After delivery, cells were allowed to recover in culture for 7 days. Bright-field and fluorescence microscopic (Fig. 3A) and flow cytometric analyses (Fig. 3B and fig. S5A) showed that plasmid delivery was efficient and genome editing was successful in MDA-MB-231 cells, achieving >90% EGFP knockout efficiency with both sgRNAs targeting different EGFP coding sequences. In SU-DHL-1 lymphoma cells, bright-field and fluorescence microscopic analyses (Fig. 3C) and flow cytometric analyses (Fig. 3D and fig. S5B) showed >70% EGFP knockout efficiency, which was satisfactory for this difficult-to-transfect lymphoma cell line and could not be achieved by current transfection methods. As expected, EGFP expression was not affected in the negative control cells, which were delivered with plasmids encoding Cas9 only.
Fig. 3. EGFP knockout via a microfluidic method.
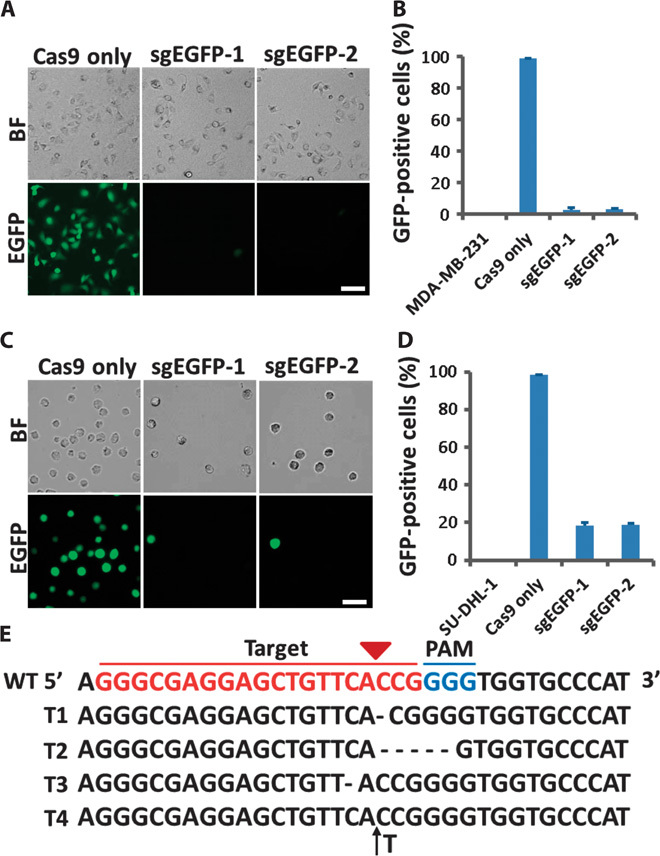
(A) Microscopy of MDA-MB-231 cells stably expressing EGFP 7 days after being delivered with plasmids encoding only Cas9 protein or both sgEGFP and Cas9 protein. Scale bar, 20 μm. (B) Percentage of cells displaying EGFP fluorescence from (A) was quantified by flow cytometry. MDA-MB-231 serves as a negative control for EGFP fluorescence signal. Error bars indicate SE (n = 3). (C) Microscopy of SU-DHL-1 lymphoma cells stably expressing EGFP 7 days after being delivered with plasmids encoding only Cas9 protein or both sgEGFP and Cas9 protein. Scale bar, 20 μm. (D) Percentage of cells displaying EGFP fluorescence from (C) was quantified by flow cytometry. SU-DHL-1 serves as a negative control for EGFP fluorescence signal. Error bars indicate SE (n = 3). (E) PCR product sequencing data for the sgEGFP-1 targeting region in SU-DHL-1 lymphoma cells. The 20–base pair (bp) target sequence is shown in red; the PAM sequence is shown in blue. Representative sequences for indels are shown. Short black lines denote different deletions. Black arrow denotes an insertion. WT, wild type.
To analyze the indels at the EGFP locus generated by CRISPR-Cas9–mediated genome editing, we amplified the specific sgEGFP-1 target regions by polymerase chain reaction (PCR) and conducted TA cloning of the products in SU-DHL-1 lymphoma cells (fig. S5C). The results of sequence analysis showed that delivery of plasmids encoding sgRNA targeting EGFP and Cas9 via our chip caused different types of mutations in the EGFP locus (Fig. 3E). These data indicate that we successfully delivered plasmids encoding sgRNAs and Cas9 into different human cell lines using our chip and achieved highly efficient genome editing.
Gene disruption platform
To determine whether our delivery platform could be used for gene disruption and function analysis, we carried out further delivery of plasmids encoding Cas9 and sgRNAs targeting different genes in different types of cell lines. Plasmids encoding sgRNA targeting the endogenous AAVS1 locus and Cas9 were delivered into MCF7 cells. The cells were allowed to recover in culture for 7 days, followed by PCR amplification of the specific sgRNA target region. The results of TA cloning and sequence analysis showed that the delivery of plasmids encoding Cas9 and sgRNA targeting AAVS1 resulted in mutations, including indels, at the specific genomic loci (Fig. 4A). Surveyor mutation detection assay revealed substantial cleavage at the AAVS1 locus, with indels occurring at a frequency of about 18 to 46% when delivery was optimized by passage of the cells through the chip three times (Fig. 4B).
Fig. 4. Gene disruption via chip.
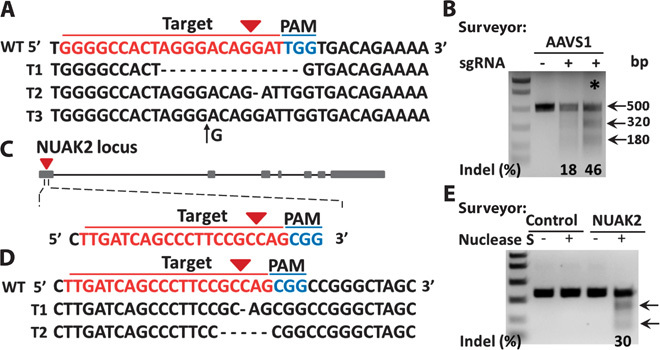
Plasmids encoding both sgRNA targeting AAVS1 locus or NUAK2 and Cas9 protein were delivered into MCF7 and HeLa cells, respectively. After 7 days of cell culture, genomic DNA was extracted. PCR product sequencing for specific targeting regions was performed. (A) PCR product sequencing data for the sgAAVS1 targeting region. The 20-bp target sequence is shown in red; the PAM sequence is shown in blue. Representative sequences for indels are shown. Short black lines denote different deletions. Black arrow denotes an insertion. (B) Surveyor mutation detection assay for sgAAVS1- and Cas9 protein–mediated indels via chip. Arrows indicate the expected positions of DNA bands cleaved by Surveyor Nuclease S. The symbol * indicates the cleavage lane of DNA bands after cells went through the same chip three times. (C) Illustration of sgNUAK2 targeting region at the first exon. The 20-bp target sequence is shown in red; the PAM sequence is shown in blue. (D) PCR product sequencing data for the sgNUAK2 targeting region. Representative sequences for deletions are shown. Short black lines denote different deletions. (E) Surveyor mutation detection assay for sgNUAK2- and Cas9 protein–mediated indels via chip. Arrows indicate the expected positions of DNA bands cleaved by Surveyor Nuclease S.
We designed an sgRNA targeting the first exon of the NUAK2 gene and cloned it into a vector for coexpression with sgRNA and Cas9 (Fig. 4C). Plasmids encoding Cas9 and sgRNA targeting NUAK2 were delivered into HeLa cells via our membrane deformation method, and the cells were allowed to recover in culture for 7 days. PCR amplification of the sgRNA target region followed by TA cloning and sequence analysis showed deletion mutations at the specific genomic loci (Fig. 4D). Mutation detection assay revealed substantial cleavage at the NUAK2 gene locus, with indels occurring at a frequency of about 30% (Fig. 4E). The indel mutation frequencies could be optimized in a few ways such as passaging cells multiple times through the deformation chip, increasing the concentration of the plasmids, and using a selective drug to kill the nontransfected cells.
Next, we explored gene function and cell phenotype via our delivery chip. Plasmids encoding Cas9 and sgRNA targeting phosphatase and tensin homolog (Pten) (fig. S6A) were delivered into MCF7 cells, followed by culture for 48 hours and puromycin selection. More than 80% of the cells survived the selection process, indicating the high delivery efficiency of our method. Cells were allowed to recover for 7 days and then analyzed by Western blotting. The results of Western blotting analysis showed that endogenous Pten expression was abolished compared with expression in control cells transfected only with plasmid encoding Cas9. Moreover, the level of Akt phosphorylation increased with Pten depletion, consistent with activation of Akt by loss of Pten (Fig. 5A). Cells were immunostained to further confirm successful knockout of Pten and Akt activation (fig. S6B). Cell proliferation was also increased in MCF7 cells after Pten knockout (Fig. 5B), which is consistent with a previous study (20).
Fig. 5. Microfluidic platform for cell phenotype and gene function analysis.
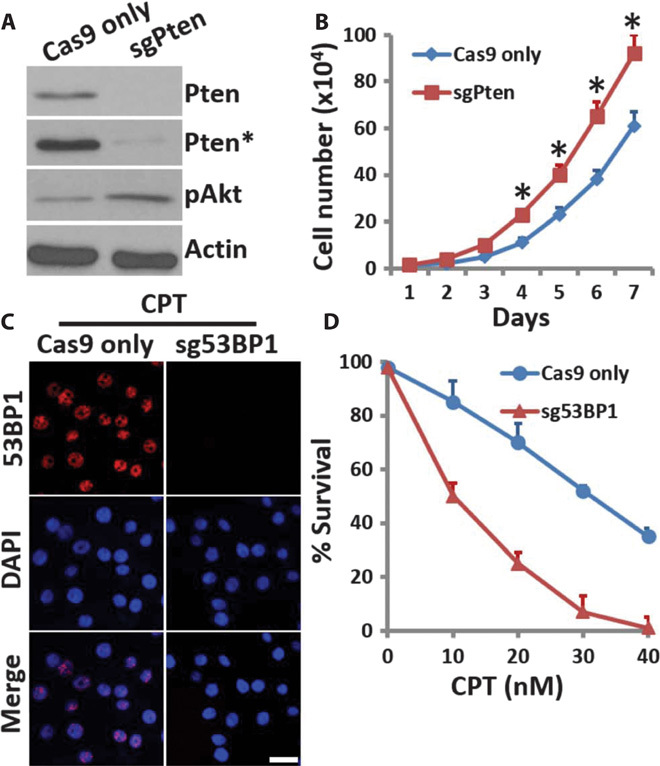
(A) MCF7 cells delivered with plasmids encoding only Cas9 protein or both sgPten and Cas9 protein were cultured for 7 days and then analyzed by Western blotting with the indicated antibodies. Actin was used as a loading control. The symbol * indicates long exposure. (B) Cells (5 × 104) from (A) were seeded in 60-mm dishes in complete medium and cultured for 7 days. Cells were trypsinized and collected for cell count in a Countess II FL Automated Cell Counter (Life Technologies) daily for 7 days. Error bars indicate SEM (n = 3). *P < 0.005 determined by Student’s t test. (C) HeLa cells delivered with plasmids encoding only Cas9 protein or both sg53BP1 and Cas9 protein were cultured 7 days. Then, the cells were treated with 1 μM CPT for 2 hours and then examined by immunostaining with anti-53BP1 antibodies (red). Scale bar, 10 μm. DAPI, 4′,6-diamidino-2-phenylindole. (D) Survival rate of HeLa cells from (C) after control or CPT treatment was assessed by colony survival assay. Error bars indicate SEM (n = 3).
Tumor suppressor p53 binding protein 1 (53BP1) is required for DNA damage response and tumor suppression (21–23). We designed an sgRNA targeting a 53BP1 gene locus and delivered plasmids encoding both sg53BP1 and Cas9 via our chip into HeLa cells (fig. S6C). Cells were cultured for 48 hours and then selected with puromycin. Similar to Pten knockout, more than 80% of 53BP1 knockout cells survived the selection process. Western blotting analysis showed the clear absence of 53BP1 expression compared with control cells (fig. S6D). Camptothecin (CPT) causes DNA strand breaks mediated by transcription and induces clear 53BP1 foci in the nuclei. Here, we showed that CPT treatment resulted in clear 53BP1 foci formation in the nuclei of control cells, but not in the cells treated with plasmids encoding both sg53BP1 and Cas9 (Fig. 5C). Consistent with this, cell survival was also greatly decreased in the cells delivered with plasmids encoding both sg53BP1 and Cas9 after CPT treatment (Fig. 5D). Together, these data show that our chip-mediated delivery is a rapid, efficient, and high-throughput method for CRISPR-Cas9–mediated genome editing and gene knockout analysis and may provide a multiplexable and integrated platform for gene phenotype and functional analysis.
DISCUSSION
Our delivery method uses the mechanical deformability of cells to generate transient holes in the cell membrane, permitting diffusion of biomaterials in the extracellular milieu into the cytoplasm. We achieved high delivery efficiency and high cell viability with delivery of siRNAs and plasmids. On the basis of the delivery principle, this method also has the potential to deliver other materials, such as proteins and nanoparticles. Moreover, the delivery method can be applied across different types of cells, including hard-to-transfect cells, such as immune cells and stem cells, to address clinical needs. In the future, with a better understanding of the nature of the deformation experienced by cells passing through a microconstriction and optimization of device parameters, one can expect to achieve better performance in a range of cell types and applications.
The mechanical deformability–based principle provides a new solution for delivery and has advantages over some existing methods. To our knowledge, this is the first application of this microfluidic deformation method to the delivery of the CRISPR-Cas9 system to achieve genome editing and gene disruption. Similar to microinjection, the method does not rely on cell type or the structure of the target molecule (24, 25); however, it is easier to use with higher throughput than microinjection. Electroporation has been successfully applied to CRISPR-Cas9 delivery and allows highly efficient RNA-guided genome editing. However, unlike our microfluidic method of delivery, electroporation damages cells and often affects cell viability. The high delivery efficiency and associated high cell viability of our method guarantee efficient genome editing and precise gene functional analysis. To increase genome editing activity, we may apply the cells multiple times through the deformation chip times, increase the concentration of the plasmids, and/or use a selective drug to kill the nontransfected cells. Using stable Cas9-expressing cells for sgRNA delivery or Cas9 protein/sgRNA co-complexes may also be helpful to increase the indel frequencies. Given our achieved capability of the deformation-based CRISPR/Cas9 gene editing, we expect to expand the work to many other cells and model systems.
Microfluidics as a basic research tool has the advantage that it is capable of integration and incorporation into a larger system including multiple posttreatment modules. This enables potential integration of our CRISPR-Cas9 system delivery and gene loss-of-function or mutation correlation analysis. For example, the device could be integrated with the single-cell protrusion microfluidic chip developed in our laboratory for screening genes potentially involved in cell protrusion mechanics (26). Use of our device would generate large quantities of CRISPR-Cas9–mediated knockout or knockin cells for high-throughput cell phenotypic screening.
CRISPR-Cas9–mediated delivery for gene therapy has been reported recently for correction of some mutations associated with disease (7, 27–30). Our technique enables novel approaches to this type of gene therapy. We have achieved high delivery efficiency compared with traditional liposome-mediated delivery in SU-DHL-1 lymphoma cells, and successful application in anaplastic large cell lymphoma cells provides the possibility of delivery in primary patient cells. For example, a patient’s target cells could be isolated from blood or other tissue, treated with the device to deliver the CRISPR-Cas9 knockin system with wild-type template to correct the disease gene mutation, and then reintroduced into the patient. The enhanced delivery efficiency of our method would increase the likelihood of correcting disease mutation genes by gene targeting therapy.
MATERIALS AND METHODS
Materials and reagents
SPR 220-7 photoresist was purchased from Rohm and Haas Electronic Materials. PDMS (GE 615 RTV) was purchased from Fisher Scientific. Tygon tubing was purchased from Saint-Gobain. Flat steel pins were purchased from New England Small Tube. Fetal bovine serum (FBS), trypsin, and penicillin-streptomycin were purchased from Fisher Scientific. Dulbecco’s modified Eagle’s medium (DMEM), Ham’s F-12 medium, RPMI 1640 and F-12K medium, insulin, hydrocortisone, and phosphate-buffered saline (PBS) were purchased from Life Technologies. FITC-labeled ssDNA DNA was purchased from Integrated DNA Technologies. SiRNAs targeting Akt1 (siAkt1-1 SASI_Hs01_00105952, siAkt1-2 SASI_Hs01_00105953, and siAkt1-3 SASI_Hs01_00105954) were used previously and purchased from Sigma-Aldrich (31). Plasmids encoding sgRNA and Cas9 were purchased from Addgene, and specific sgRNA target sequences were cloned into the CRISPR v2 vector (Addgene plasmid #52961). The 20-bp target sequences of sgRNAs targeting EGFP, AAVS1, and Pten were used previously (4–6). The 20-bp target sequences of the indicated sgRNAs were as follows: sgEGFP-1, GGGCGAGGAGCTGTTCACCG; sgEGFP-2, GAGCTGGACGGCGACGTAAA; sgAAVS1, GGGGCCACTAGGGACAGGAT; sgNUAK2, TTGATCAGCCCTTCCGCCAG; sgPten, AGATCGTTAGCAGAAACAAA; sg53BP1, CATAATTTATCATCCACGTC. The primers used for PCR amplification of sgRNA target regions were as follows: EGFP-FP, ATGGTGAGCAAGGGCGAGGA; EGFP-RP, TTACTTGTACAGCTCGTCCA; AAVS1-FP, CCCCGTTCTCCTGTGGATTC; AAVS1-RP, ATCCTCTCTGGCTCCATCGT; NUAK2-FP, GCTTTACTGCGCGCTCTGGTACTGC; NUAK2-RP, CAGGCGCCCCGAGCTCTCCC.
Chip design and fabrication
The microchip pattern was designed with AutoCAD (Autodesk). Each chip consists of 14 identical cell-scattering and deformation zones, and each zone contains 10 arrays of constrictions. The constriction depth is 15 μm, and the width varies from 4 to 5 μm. The parallel chip design was generated by arranging multiple devices side by side. The microfluidic chip was fabricated using standard photolithography and soft lithography procedures. The negative photoresist SU8-3025 (MicroChem) was used to fabricate patterns on a silicon wafer. The silicon wafer was then silanized using trimethylchlorosilane (Thermo Scientific) for 30 min to facilitate PDMS mold release. PDMS prepolymer (10A:1B, Sylgard 184 silicone elastomer kit, Dow Corning) was poured onto the silicon wafer and cured at 80°C for 1 hour. Holes were then punched in the PDMS for the inlets and outlets, and oxygen plasma treatment was used to chemically bond the PDMS mold to a glass slide.
Finite element method
The flow velocity distribution, cell trajectory, and stress on the cell were simulated using the finite element method. To perform the temporal simulation, the fluidic dynamics equation (incompressible Navier-Stokes equations) and solid mechanics equation (Newton’s second law of motion) were coupled and implemented by fluid-solid interactions. This combined the spatial frame interface for fluid flow and the material frame for the cell. The mesh geometry was continuously moved and deformed by applying the arbitrary Lagrangian-Eulerian method. The dimensions of model geometries and mechanical properties were identical to the actual experiment. The stress on the cell was computed as the von Mises stress, which is a scalar value determined from the stress tensor of a particle under the pressure in fluid flow.
Cell culture
HEK293T, MCF7, MDA-MB-231, and HeLa cells were grown in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin in a humidified atmosphere of 5% CO2/95% air at 37°C. PC-3 cells were grown in F-12K medium supplemented with 10% FBS and 1% penicillin-streptomycin. SUM159 cells were grown in Ham’s F-12 medium supplemented with 5% FBS, 1% penicillin-streptomycin, insulin (5 μg/ml), and hydrocortisone (1 μg/ml). Human SU-DHL-1 anaplastic large cell lymphoma cells were cultured in RPMI 1640 supplemented with 10% FBS and 1% penicillin-streptomycin. Mouse AB2.2 embryonic stem cells were maintained on a 0.1% gelatin (Sigma-Aldrich)–coated tissue culture dish in high-glucose DMEM, supplemented with 15% FBS, 55 μM β-mercaptoethanol (Life Technologies), and 0.01% mouse leukemia inhibitory factor (Millipore) under feeder-free conditions.
Delivery procedure and puromycin selection
The channels in the device were wetted with PBS and blocked with 1% bovine serum albumin in PBS for 10 min. Cells were first suspended in the desired volume of Opti-MEM medium (Life Technologies) and then mixed with the desired amount of delivery material (ssDNA, siRNA, or plasmid) and loaded into plastic Tygon tubing with a 5-ml syringe. The tubing was then connected to the device inlet reservoir by a flat steel pin. During the flow experiments, a syringe pump controlled the fluid flow through the device. Treated cells were incubated in a 37°C incubator for 20 min to recover before further treatment.
Plasmids encoding both Cas9 and sgRNA targeting Pten or 53BP1 were delivered into MCF7 or HeLa cells, respectively, via our chip. After 48 hours of culture, the cells were grown in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, and puromycin (2 μg/ml; Sigma) for 2 to 3 days to kill the undelivered cells.
Immunostaining, Western blotting, and flow cytometry
Cells grown overnight on coverslips were fixed in 4% paraformaldehyde and then permeabilized with 0.5% Triton X-100 plus 300 mM sucrose. Cells were then immunostained and visualized under an Olympus FV1000 confocal microscope. The primary antibodies used were anti-53BP1 (NB100-304, Novus Biologicals), anti-Oct4 (ab18976, Abcam), anti-Pten (ab130224, Abcam), and anti–phospho-Akt (Ser473) (ab81283, Abcam). The secondary antibodies used were Alexa Fluor 488–conjugated goat anti-mouse (A-11001, Life Technologies) and Texas red–conjugated goat anti-rabbit (T-2767, Life Technologies).
For Western blotting after siRNA-mediated knockdown or sgRNA-Cas9–mediated knockout, cells were allowed to recover in culture for 2 or 7 days, respectively. The primary antibodies used were anti-Akt1 (ab32505, Abcam), anti-53BP1 (ab21083, Abcam), and anti-actin (A3853, Sigma-Aldrich). For flow cytometric analysis after sgEGFP-mediated knockout, cells were allowed to recover in culture for 7 days followed by analysis of EGFP fluorescence with a BD LSRFortessa cell analyzer.
Mutation detection assay, TA cloning, and sequencing
Genomic DNA was extracted using the PureLink Genomic DNA Mini Kit (K1820-00, Life Technologies) according to the manufacturer’s instructions. PCR amplicons of nuclease target sites were generated and analyzed for the presence of mismatch mutations using the Transgenomic Surveyor Mutation Detection Kit (Integrated DNA Technologies) according to the manufacturer’s instructions. Briefly, PCR amplicons of sgRNA target regions were denatured by heating for 10 min at 95°C, annealed to form heteroduplex DNA using a thermocycler from 95° to 25°C at −0.3°C/s, digested with Surveyor Nuclease S for 2 hours at 42°C, and separated by 1% agarose gel electrophoresis. For sequence analysis, PCR products corresponding to genomic modifications were cloned into pCR4-TOPO vector using the TOPO TA Cloning Kit (Life Technologies). Cloned products were sequenced using the M13 primer.
Cell proliferation assay, CPT treatment, and sensitivity assay
After chip-mediated delivery and recovery in culture for siRNA knockdown or sgRNA-Cas9–mediated knockout, cells (5 × 104) were seeded in 60-mm dishes in complete medium and cultured for 7 days. Cells were harvested by trypsinization daily and counted in a Countess II FL Automated Cell Counter (Life Technologies).
To assess CPT sensitivity, cells were treated with 1 μM CPT for 2 hours and immunostained with anti-53BP1 or treated with 10, 20, 30, or 40 nM CPT for sensitivity assay. CPT sensitivity was assessed by colony survival assay. Briefly, CPT-treated cells (500 to 1000) were plated in 60-mm dishes in complete medium and incubated for 2 to 3 weeks to form clones. Clones were stained with Coomassie blue, and survival rate was calculated.
Supplementary Material
Funding
We acknowledge funding support from the Cancer Prevention and Research Institute of Texas (CPRIT-R1007), NIH-CA180083, NIH-U54CA143837, and Golfers Against Cancer Foundation. Author contributions: X.H. and L.Q. designed the experiments and developed the method. X.H. and Z.L. performed the experiments. M.c.J. assisted with flow velocity simulation. K.Z. and Y.L. assisted with device optimization. N.L. assisted with the CPT sensitivity assay. Z.Z. and Y.Z. provided SU-DHL-1 lymphoma cells and helpful suggestions for improved user-friendliness. X.H. and L.Q. wrote the paper. All co-authors reviewed and approved the manuscript. Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/7/e1500454/DC1
Fig. S1. Performance of chip with different designs.
Fig. S2. Cell stress simulation.
Fig. S3. Flow velocity simulation.
Fig. S4. Comparison of FuGENE HD transfection and delivery via chip.
Fig. S5. Flow cytometric analysis of EGFP knockout cells.
Fig. S6. Pten and 53BP1 knockout mediated by delivery via chip.
Movie S1. Cells passing through the diamonded microconstrictions.
Movie S2. Flow velocity simulation in the diamonded microconstriction chip.
REFERENCES AND NOTES
- 1.Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., Church G. M., RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F., Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsu P. D., Lander E. S., Zhang F., Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xue W., Chen S., Yin H., Tammela T., Papagiannakopoulos T., Joshi N. S., Cai W., Yang G., Bronson R., Crowley D. G., Zhang F., Anderson D. G., Sharp P. A., Jacks T., CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514, 380–384 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang T., Wei J. J., Sabatini D. M., Lander E. S., Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shalem O., Sanjana N. E., Hartenian E., Shi X., Scott D. A., Mikkelsen T. S., Heckl D., Ebert B. L., Root D. E., Doench J. G., Zhang F., Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.High K., Gregory P. D., Gersbach C., CRISPR technology for gene therapy. Nat. Med. 20, 476–477 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Heitz F., Morris M. C., Divita G., Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol. 157, 195–206 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verma A., Uzun O., Hu Y., Hu Y., Han H. S., Watson N., Chen S., Irvine D. J., Stellacci F., Surface-structure-regulated cell-membrane penetration by monolayer-protected nanoparticles. Nat. Mater. 7, 588–595 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zabner J., Cationic lipids used in gene transfer. Adv. Drug Deliv. Rev. 27, 17–28 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Duan H., Nie S., Cell-penetrating quantum dots based on multivalent and endosome-disrupting surface coatings. J. Am. Chem. Soc. 129, 3333–3338 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Ramakrishna S., Kwaku Dad A. B., Beloor J., Gopalappa R., Lee S. K., Kim H., Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 24, 1020–1027 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fox M. B., Esveld D. C., Valero A., Luttge R., Mastwijk H. C., Bartels P. V., van den Berg A., Boom R. M., Electroporation of cells in microfluidic devices: A review. Anal. Bioanal. Chem. 385, 474–485 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Li S., Electroporation gene therapy: New developments in vivo and in vitro. Curr. Gene Ther. 4, 309–316 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Kim S., Kim D., Cho S. W., Kim J., Kim J. S., Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 24, 1012–1019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu Y. C., Baculoviral vectors for gene delivery: A review. Curr. Gene Ther. 8, 54–65 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Waehler R., Russell S. J., Curiel D. T., Engineering targeted viral vectors for gene therapy. Nat. Rev. Genet. 8, 573–587 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharei A., Zoldan J., Adamo A., Sim W. Y., Cho N., Jackson E., Mao S., Schneider S., Han M. J., Lytton-Jean A., Basto P. A., Jhunjhunwala S., Lee J., Heller D. A., Kang J. W., Hartoularos G. C., Kim K. S., Anderson D. G., Langer R., Jensen K. F., A vector-free microfluidic platform for intracellular delivery. Proc. Natl. Acad. Sci. U.S.A. 110, 2082–2087 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasaki T., Nakashiro K., Tanaka H., Azuma K., Goda H., Hara S., Onodera J., Fujimoto I., Tanji N., Yokoyama M., Hamakawa H., Knockdown of Akt isoforms by RNA silencing suppresses the growth of human prostate cancer cells in vitro and in vivo. Biochem. Biophys. Res. Commun. 399, 79–83 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Zhang J., Zhang P., Wei Y., Piao H. L., Wang W., Maddika S., Wang M., Chen D., Sun Y., Hung M. C., Chen J., Ma L., Deubiquitylation and stabilization of PTEN by USP13. Nat. Cell Biol. 15, 1486–1494 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panier S., Boulton S. J., Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 15, 7–18 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Chapman J. R., Barral P., Vannier J. B., Borel V., Steger M., Tomas-Loba A., Sartori A. A., Adams I. R., Batista F. D., Boulton S. J., RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 49, 858–871 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rappold I., Iwabuchi K., Date T., Chen J., Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage–signaling pathways. J. Cell Biol. 153, 613–620 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y., Yu L. C., Microinjection as a tool of mechanical delivery. Curr. Opin. Biotechnol. 19, 506–510 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Luo D., Saltzman W. M., Synthetic DNA delivery systems. Nat. Biotechnol. 18, 33–37 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Zhang K., Chou C. K., Xia X., Hung M. C., Qin L., Block-Cell-Printing for live single-cell printing. Proc. Natl. Acad. Sci. U.S.A. 111, 2948–2953 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Y., Zhou H., Fan X., Zhang Y., Zhang M., Wang Y., Xie Z., Bai M., Yin Q., Liang D., Tang W., Liao J., Zhou C., Liu W., Zhu P., Guo H., Pan H., Wu C., Shi H., Wu L., Tang F., Li J., Correction of a genetic disease by CRISPR-Cas9-mediated gene editing in mouse spermatogonial stem cells. Cell Res. 25, 67–79 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yin H., Xue W., Chen S., Bogorad R. L., Benedetti E., Grompe M., Koteliansky V., Sharp P. A., Jacks T., Anderson D. G., Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 32, 551–553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu Y., Liang D., Wang Y., Bai M., Tang W., Bao S., Yan Z., Li D., Li J., Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 13, 659–662 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Schwank G., Koo B. K., Sasselli V., Dekkers J. F., Heo I., Demircan T., Sasaki N., Boymans S., Cuppen E., C. K. van der Ent, Nieuwenhuis E. E., Beekman J. M., Clevers H., Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 13, 653–658 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Han X., Liu D., Zhang Y., Li Y., Lu W., Chen J., Songyang Z., Akt regulates TPP1 homodimerization and telomere protection. Aging Cell 12, 1091–1099 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/7/e1500454/DC1
Fig. S1. Performance of chip with different designs.
Fig. S2. Cell stress simulation.
Fig. S3. Flow velocity simulation.
Fig. S4. Comparison of FuGENE HD transfection and delivery via chip.
Fig. S5. Flow cytometric analysis of EGFP knockout cells.
Fig. S6. Pten and 53BP1 knockout mediated by delivery via chip.
Movie S1. Cells passing through the diamonded microconstrictions.
Movie S2. Flow velocity simulation in the diamonded microconstriction chip.