

Abstract
BACKGROUND & AIMS
Human immunodeficiency virus (HIV) protease inhibitor (PI)-induced adverse effects have become a serious clinical problem. In addition to their metabolic and cardiovascular complications, these drugs also frequently cause severe gastrointestinal disorders, including mucosal erosions, epithelial barrier dysfunction, and diarrhea. However, the exact mechanisms underlying gastrointestinal adverse effects of HIV PIs remain unknown. This study investigated whether HIV PIs disrupt intestinal epithelial barrier integrity by activating endoplasmic reticulum (ER) stress.
METHODS
The most commonly used HIV PIs (lopinavir, ritonavir, and amprenavir) were used; their effects on ER stress activation and epithelial paracellular permeability were examined in vitro as well as in vivo using wild-type and CHOP−/− mice.
RESULTS
Treatment with lopinavir and ritonavir, but not amprenavir, induced ER stress, as indicated by a decrease in secreted alkaline phosphatase activities and an increase in the unfolded protein response. This activated ER stress partially impaired the epithelial barrier integrity by promoting intestinal epithelial cell apoptosis. CHOP silencing by specific small hairpin RNA prevented lopinavir-and ritonavir-induced barrier dysfunction in cultured intestinal epithelial cells, whereas CHOP−/− mice exhibited decreased mucosal injury after exposure to lopinavir and ritonavir.
CONCLUSIONS
HIV PIs induce ER stress and activate the unfolded protein response in intestinal epithelial cells, thus resulting in disruption of the epithelial barrier integrity.
Human immunodeficiency virus (HIV) protease inhibitors (PIs) are major components of highly active antiretroviral therapy and have been successfully used to suppress HIV replication. However, the benefits of HIV PIs are compromised by various undesirable side effects.1 HIV PI-induced gastrointestinal dysfunction represents a major adverse effect of HIV PIs. Drug-induced diarrhea occurs in 16%–62% of patients on highly active antiretroviral therapy.2,3
Diarrhea may be acute or chronic. In addition to abnormal intestinal motility and malabsorption, diarrhea can be caused either by increased active ion secretion (secretory diarrhea) or by a leaky epithelial barrier (leak-flux diarrhea). Recent studies by Bode et al showed that HIV PIs have no effect on epithelial ion secretion but induce apoptosis and decrease barrier function in human intestinal epithelial cells (IECs).2–4 However, the underlying mechanism of HIV PI-induced disruption of barrier function in IECs remains unclear.
The endoplasmic reticulum (ER) is a principal site for protein synthesis and folding, calcium storage and signaling, and biosynthesis of corticosteroids, cholesterol, and other lipids, and it is highly sensitive to alterations in calcium homeostasis and perturbations in its environment.5,6 A number of biochemical and physiologic stimuli can change ER homeostasis, impose stress on the ER, and subsequently lead to accumulation of unfolded protein or misfolded proteins in the ER lumen.7 The ER has evolved highly specific signaling pathways, collectively termed the unfolded protein response (UPR), to ensure that its protein-folding capacity is not overwhelmed.8 However, persistent activation of the UPR can induce cell apoptosis. ER stress-induced apoptosis is directly implicated in the pathology of various human diseases, including gastrointestinal diseases.9
Our previous studies have shown that HIV PIs induce ER stress, activate the UPR, and induce cell apoptosis in both macrophages and hepatocytes.1–4,10 Recent studies by Djedaini et al also found that lopinavir (LOPV)-induced ER stress is correlated to insulin resistance in human adipocytes.11 However, whether HIV PIs also induce ER stress in IECs has not been investigated. In the present study, we examined the effects of different HIV PIs on UPR activation and apoptosis in normal IECs. Furthermore, we examined whether HIV PI-induced ER stress is responsible for HIV PI-associated cell apoptosis and disruption of barrier function both in vitro and in vivo.
Materials and Methods
Cell Culture and HIV PI Treatment
The IEC-6 cell line at passage 13 was purchased from American Type Culture Collection (Manassas, VA) and used at passages 15–20 for the current experiments. The cell line was derived from normal rat intestine and was developed and characterized by Quaroni et al.12 It is nontumorigenic and retains the undifferentiated character of epithelial stem cells. The stable IEC-Cdx2L1 cells were developed and characterized by Suh and Traber13 and were kind gifts from Dr Peter G. Traber (Baylor College of Medicine, Houston, TX). Before experiments, cells were grown in Dulbecco’s modified Eagle medium containing 4 mmol/L isopropyl-β-D-thiogalactopyranoside for 16 days to induce cell differentiation. The differentiated cells have multiple morphologic characteristics of villus-type enterocytes with few goblet cells, and sucrase-isomaltase is highly expressed (Supplementary Figure 1).
Amprenavir (AMPV), LOPV, and ritonavir (RITV) were dissolved in dimethyl sulfoxide and directly added to culture medium (final concentrations, 5–25 μmol/L) and incubated for 0.5–24 hours. For each result, a minimum of 3 independent experiments was performed.
Western Blot Analysis
Total cell lysates, cytoplasmic proteins, and nuclear proteins were prepared, and the protein levels of CHOP, ATF-4, XBP-1, GRP78, lamin B, or β-actin were detected and analyzed as previously described.1
Construction of IEC-6 Stable Cell Line Expressing Secreted Alkaline Phosphatase and Secreted Alkaline Phosphatase Enzyme Activity Assay
Activity of secreted alkaline phosphatase (SEAP) is rapidly down-regulated by ER stress independent of transcriptional regulation.14 IEC-6 cells were cotransfected with pSEAP2-control and pEGFP-N1 using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA). Stable clones containing both pSEAP2 and pEGFP-N1 were selected by G418 and purified by colonial cloning and flow cytometry cell sorting. Expression of SEAP and green fluorescent protein were confirmed by Western blot analysis, chemiluminescent assays, and fluorescent microscopy.
Enzyme activity of SEAP was measured by chemiluminescent assays using the Great EscAPe SEAP Detection Kit according to the manufacturer’s instructions (Promega, Madison, WI). The viability of cells was detected with CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega).
Analysis of Apoptosis by Annexin V and Propidium Iodide Staining
IEC-6 cells were treated with various concentrations of HIV PIs for 24 hours and stained with Annexin V/fluorescein isothiocyanate (FITC) and propidium iodide using the BD ApoAlert Annexin V Kit (Clontech, Mountain View, CA) according to the protocol recommended by the manufacturer. Annexin V/propidium iodide–stained cells were visualized under an Olympus inverted fluorescence microscope (Olympus, Center Valley, PA) with a 63 × oil immersion objective using FITC and rhodamine filters or analyzed by flow cytometry using the Beckman Coulter Elite XL-MCL single-laser flow system (Beckman Coulter, Inc, Fullerton, CA).1
RNA Isolation and Real-Time Quantitative Polymerase Chain Reaction
Total cellular RNA was isolated using the Ambion RNAqueous Kit (Austin, TX). Total RNA (5 μg) was used for first-strand complementary DNA synthesis using the High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). The messenger RNA (mRNA) levels of CHOP, GRP78, and β-actin were quantified using the following primers: CHOP, forward:5′-GGAGCAGGAGAATGAGAG-3′, reverse: 5′-GACAGACAGGAGGTGATG-3′; GRP78, forward: 5′-TCCTGCGTCGGTGTATTC-3′, reverse:5′-TCCTGCGTCGGTGTATTCCGT-3′ β-actin, forward: 5′-TATCGGCAATGAGCGGTTCC-3′, reverse: 5′-AGCACTGTGTTGGCATAGAGG-3′. iQ SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA) was used as a fluorescent dye to detect the presence of double-stranded DNA. The mRNA values for each gene were normalized to internal control β-actin mRNA using the ΔΔ Ct method.15
Construction of Lentiviral Small Hairpin RNA for CHOP
The small hairpin RNA (shRNA) specifically targeting the nucleotides of CHOP was designed through siRNA Target Finder (Ambion). The lentiviral expression vectors containing CHOP shRNA were constructed as previously described.15 The sequences of 3 CHOP shRNAs were as follows: shRNA1 is 5′-CTGGAAGCCTGGTATGAGGA-3′, shRNA2 is GGAAACGGAAACAGAGTGGTC-3′, and shRNA3 is 5′-GCAGGAAATCGAGCGCCTGAC-3′ The recombinant lentiviruses were produced and titered as described previously.15
IEC-6 cells were incubated with lentivirus at a multiplicity of infection of 100 for 48 hours. The CHOP RNA silencing effect of the lentiviral shRNA was confirmed by real-time reverse-transcription polymerase chain reaction (RT-PCR) and Western blot analysis.
Paracellular Tracer Flux Assay
Flux assays were performed with the 12-mm Transwell (Corning Life Sciences, Corning, NY) as described by Wong and Gumbiner16 with minor modification. Briefly, cells were plated at a confluent density of 4 × 105 cells/cm2 on the insert and maintained under the same culture conditions for an additional 48 hours to establish tight monolayers. The membrane-impermeable molecule, [14C]-mannitol (MW 184), served as the paracellular tracer. At the beginning of the flux assay, both sides of the bathing wells of Transwell filters were replaced with fresh medium containing individual HIV PI (5–25 μmol/L) and 0.5 mmol/L unlabeled mannitol and incubated at 37°C for 24 hours. [14C]-Mannitol was added to a final concentration of 3.6 nmol/L to the apical bathing wells that contained 0.5 mL of medium. The basal bathing well had no added tracers and contained 1.5 mL of the same flux assay medium as in the apical compartment. All flux assays were performed at 37°C, and the basal medium was collected 2 hours after addition of [14C]-mannitol for counting in a Beckman liquid scintillation counter. The results were expressed as a percentage of total count values of each tracer.17
In Vivo Studies
C57BL/6 wild-type and CHOP−/− mice with C56BL/6 background (male, 6–8 weeks old; Jackson Laboratories, Bar Harbor, ME) were used in this study. Mice were housed and fed standard mouse chow and tap water ad libitum throughout the study following protocols approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University.
Mice were randomly assigned to the following 4 groups: control group, AMPV, RITV, and LOPV. Mice were gavaged daily with individual HIV PI (50 mg/kg) or control solution for 2 or 4 weeks. Animals were monitored daily for appearance of diarrhea, body weight loss, and other distress. At the end of treatment, mice were killed. Blood was collected to measure drug concentration. The intestines and colons were removed, and sections were taken for formalin fixation and histologic examination.
Intestinal Permeability In Vivo
Mice were gavaged with FITC/dextran (4.4 kilodaltons; Sigma–Aldrich, St Louis, MO) at a dose of 600 mg/kg body wt 4 hours before harvest. Blood was collected from the hepatic portal vein. The serum concentration of the FITC-dextran was determined using a fluorescence plate reader with an excitation wavelength at 490 nm and an emission wavelength of 530 nm.18,19
Histologic Assessment and Microscopic Scoring
Small intestine tissues were fixed in 10% neutral buffered formalin overnight and then washed with phosphate-buffered saline and transferred to 70% ethanol. Formalin-fixed tissues were then embedded in paraffin, sectioned at 5 μm, and stained with H&E using standard procedures. Images were analyzed using a Motic BA200 microscope (Motic Instruments, Inc, Baltimore, MD). Inflammation and tissue damage were assessed microscopically and histologically based on epithelial tissue damage (0, none; 1, destruction of villous tips; 2, destruction of distal half of villi; 3, complete destruction of villi) and neutrophil infiltration (0, none; 1, rare inflammation cells; 2, extravagated inflammation cells; 3, inflammation cells throughout lamina propria and epithelium). Microscopic and histologic damage were recorded and scored for each mouse by 2 different investigators who were blind to the treatment conditions.
Terminal Deoxynucleotidyl Transferase–Mediated Deoxyuridine Triphosphate Nick-End Labeling Assay
To detect the apoptosis in intestine tissue, 5-μm sections were deparaffinized and rehydrated through washes with graded concentrations of ethanol. Tissue was pretreated with proteinase K (20 μg/mL) for 15 minutes at room temperature, followed by incubation in 3% H2O2 in phosphate-buffered saline for 5 minutes at room temperature to quench endogenous peroxidase activity. Apoptotic cells were detected using ApoAlert DNA Fragmentation Assay Kit following the manufacturer’s protocol (BD Biosciences, Palo Alto, CA). Control stains were obtained by processing in parallel duplicate sections, omitting only the TnT enzyme. The apoptotic cells were counted.20
Statistical Methods
Values are means ± SD from 3–6 samples. Western blot results were repeated at least 3 times. One-way analysis of variance was used to analyze the data. Statistics were performed using Prism 5 (GraphPad, San Diego, CA).
Results
HIV PIs Disrupt Paracellular Permeability in IECs
Maintenance of mammalian intestinal epithelial integrity is critical for normal gastrointestinal function. We first examined whether HIV PIs affect intestinal epithelial paracellular barrier function by assessing the paracellular flux of a membrane-impermeable tracer, [14C]-mannitol, across the confluent monolayer after treatment with individual HIV PIs in IEC-6 cells. To verify the system used for paracellular permeability assays, the effect of decreased cytosolic Ca2+ on the paracellular flux of [14C]-mannitol was tested and used as a positive control. As expected, exposure to Ca2+-free medium for 2 hours significantly increased paracellular permeability in IEC-6 cells (data not shown). Both LOPV and RITV markedly increased paracellular permeability in IEC-6 cells, even at concentrations as low as 5 μmol/L (Figure 1B and C). At a concentration of 15 μmol/L, LOPV and RITV increased paracellular permeability by 4.6-and 1.9-fold, respectively (Figure 1B and C), but AMPV had no significant effect even at a concentration of 25 μmol/L (Figure 1A).
Figure 1.
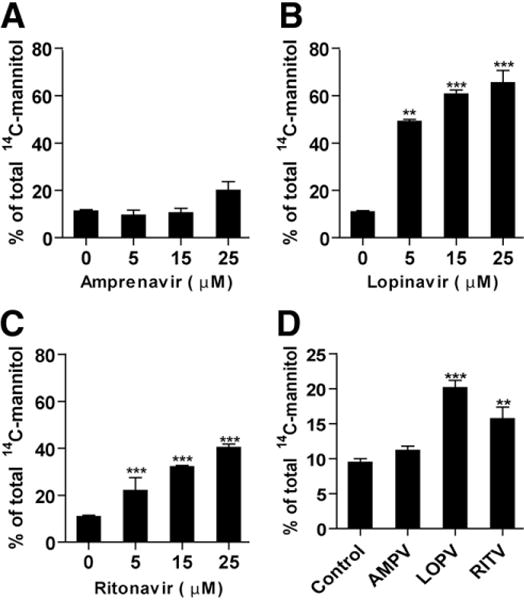
Effect of HIV PIs on paracellular permeability in IEC cells. (A–C) IEC-6 cells were treated with individual HIV PIs (0–25 μmol/L) for 24 hours. (D) IEC-cdx2L1 cells were cultured on the filter in Dulbecco’s modified Eagle medium containing 4 mmol/L isopropyl- β - D-thiogalac-topyranoside for 16 days to induce differentiation and treated with individual HIV PIs(25 μmol/L) for 24 hours. The paracellular permeability was measured as described in Materials and Methods. Values are mean ± SD of 3 independent experiments and analyzed using one-way analysis of variance. **P < .01, ***P < .001.
To further determine whether individual HIV PIs have a similar effect on paracellular permeability in differentiated IECs, we examined the effect of HIV PIs (15 μmol/L) on paracellular permeability in differentiated IEC-Cdx2L1 cells. As shown in Figure 1D, LOPV and RITV also increased paracellular permeability in differentiated IEC-Cdx2L1 cells. Paracellular permeability was increased 2.2-and 1.9-fold in IEC-Cdx2L1 cells exposed to LOPV and RITV (15 μmol/L) for 24 hours, respectively. AMPV had no significant effect on paracellular permeability in differentiated IEC-Cdx2L1 cells.
HIV PIs Induce Apoptosis in IECs
Dysregulation of tight junction and induction of apoptosis or necrosis in IECs are responsible for epithelial damage. Our previous studies have shown that HIV PIs induced apoptosis in macrophages and hepatocytes.1,10 We further examined whether HIV PIs also induce apoptosis in IECs. IEC-6 cells were treated with an individual HIV PI (15 or 25 μmol/L) for 24 hours. As shown in Figure 2, both LOPV and RITV dose-dependently induced apoptosis and necrosis in IEC-6 cells, but AMPV had no significant effect. At a concentration of 15 μmol/L, LOPV-and RITV-induced apoptotic cells were 14% and 19%, respectively, which is similar to that induced by 10 nmol/L of thapsigargin (Figure 2C). Similar results were obtained in IEC-6 Cdx2L1-cells (data not shown). Immunofluorescent staining also indicated that LOPV, RITV, and thapsigargin (a known ER stress inducer) inhibited cell differentiation, but AMPV had less effect (Supplementary Figure 2).
Figure 2.
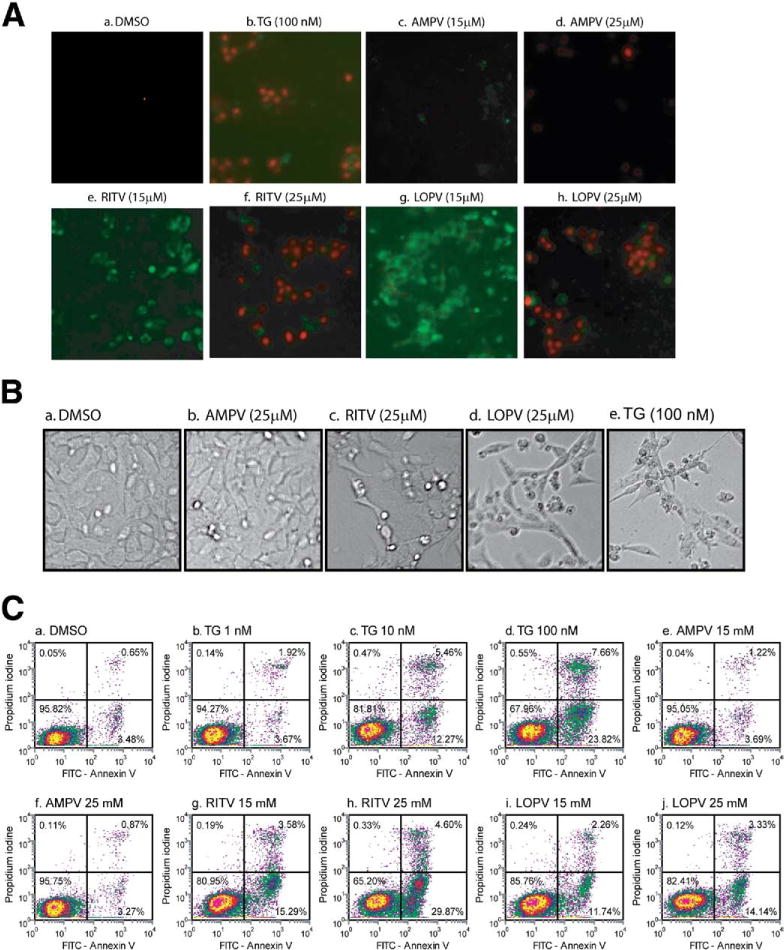
HIV PIs induce apoptosis in IECs. IEC-6 cells were treated with dimethyl sulfoxide (DMSO), individual HIV PIs (AMPV, LOPV, and RITV; 15 or 25 μmol/L), or thapsigargin (TG; 100 nmol/L) for 24 hours and then stained with Annexin V-FITC/propidium iodide and analyzed as described in Materials and Methods. (A) The representative merged images of IEC-6 cells treated with individual HIV PIs or TG from 3 independent experiments are shown. (B) Representative phase-contrast images of IEC-6 cells treated with individual HIV PI (25 μmol/L) or TG (100 nmol/L) for 24 hours. (C) The representative flow cytometry diagrams of 3 independent experiments.
HIV PIs Induce ER Stress and Activate the UPR in IECs
ER stress is implicated in a wide range of pathologies.1,5 It has been found that HIV PIs induce apoptosis and decrease barrier function in human HT-29/B6 cells (a human colon carcinoma cell line).2 To delineate the cellular and molecular mechanisms responsible for HIV PI-induced apoptosis, we first examined the HIV PI-induced ER stress response in IEC-6 cells using the SEAP reporter system. IEC-6 cells, which were stably transfected with SEAP, were treated with individual HIV PIs for 24 hours. As shown in Figure 3, both RITV and LOPV dose-dependently induced ER stress, which was indicated by a decrease of SEAP activity. However, AMPV had no significant effect. Thapsigargin was used as the positive control.
Figure 3.
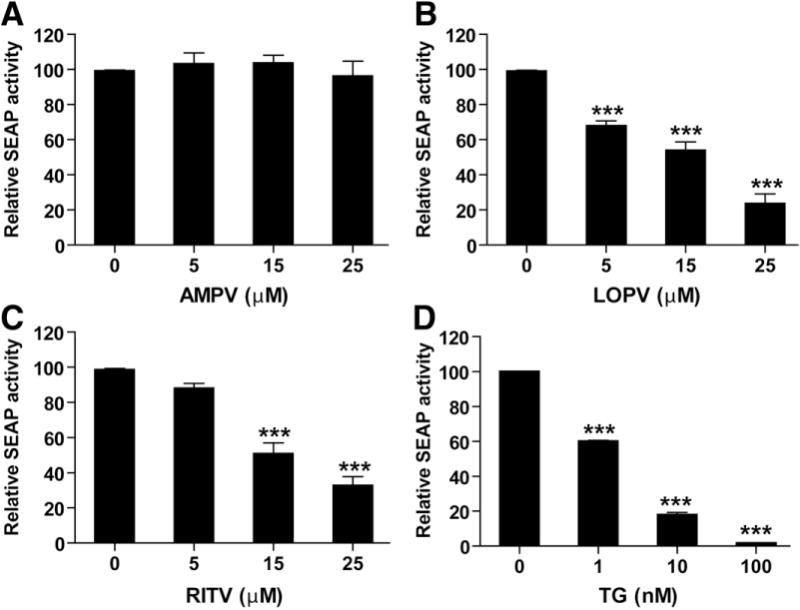
HIV PIs induce ER stress in IEC-6 cells. IEC-6 cells stably transfected with pSEAP plasmid were treated with HIV PIs (AMPV, LOPV, and RITV; 0–25 μmol/L) or thapsigargin (TG; 100 nmol/L) for 24 hours. Activity of SEAP was measured as described in Materials and Methods and expressed as percent of control. Values are mean ± SD of 5 independent experiments. Statistical significance relative to vehicle control: ***P < .001.
To further determine whether HIV PI-induced ER stress was correlated with the activation of the UPR in IEC-6 cells, we measured the expression of the ER stress master regulator, GRP78/Bip, and downstream transcriptional factors CHOP, XBP-1s (spliced form of XBP-1), and ATF-4. As shown in Supplementary Figure 3, both LOPV and RITV significantly induced GRP78, CHOP, XBP-1s, and ATF-4 expression. Up-regulation of XBP-1s and ATF-4 could be observed as early as 1 hour after treatment. Both LOPV-and RITV-induced UPR activation were dose dependent (Figure 4), but AMPV had no effect on UPR activation even at a concentration of 50 μmol/L.
Figure 4.
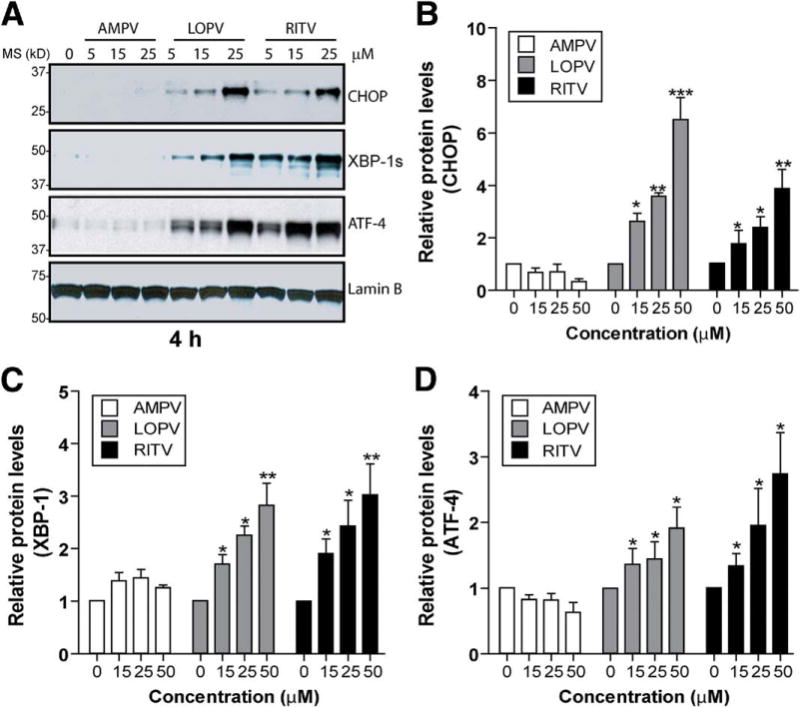
Activation of the UPR by HIV PIs. (A) Representative immunoblots against CHOP, ATF-4, XBP-1, and lamin B from the nuclear extracts of IEC-6 cells treated with individual HIV PIs (0–25 μmol/L) for 4 hours. Lamin B was used as a loading control of nuclear protein. (B–D) Relative protein levels of CHOP, XBP-1, and ATF-4. Statistical significance relative to vehicle control: *P < .05; **P < .01; ***P < .001.
To determine whether HIV PIs also activate the UPR in differentiated IECs, we measured UPR activation in differentiated IEC-Cdx2L1 cells.17 As shown in Figure 5A and B, LOPV and RITV significantly increased CHOP and GRP78 mRNA levels both in IEC-Cdx2L1 cells and IEC-6 cells. AMPV also increased GRP78 mRNA level in IEC-Cdx2L1 cells but not in IEC-6 cells. Similarly, LOPV and RITV increased CHOP, XBP-1s, and ATF-4 protein levels both in IEC-Cdx2L1 cells and IEC-6 cells (Figure 5C and E). Consistent with the real-time RT-PCR data, AMPV also up-regulated the expression of the UPR genes (XBP-1s and ATF-4) in IEC-Cdx2L1 cells but not in parental IEC-6 cells. These results indicate that individual HIV PIs have different effects on UPR activation in IECs, which is consistent with our previous observations in macrophages and hepatocytes.1,10
Figure 5.
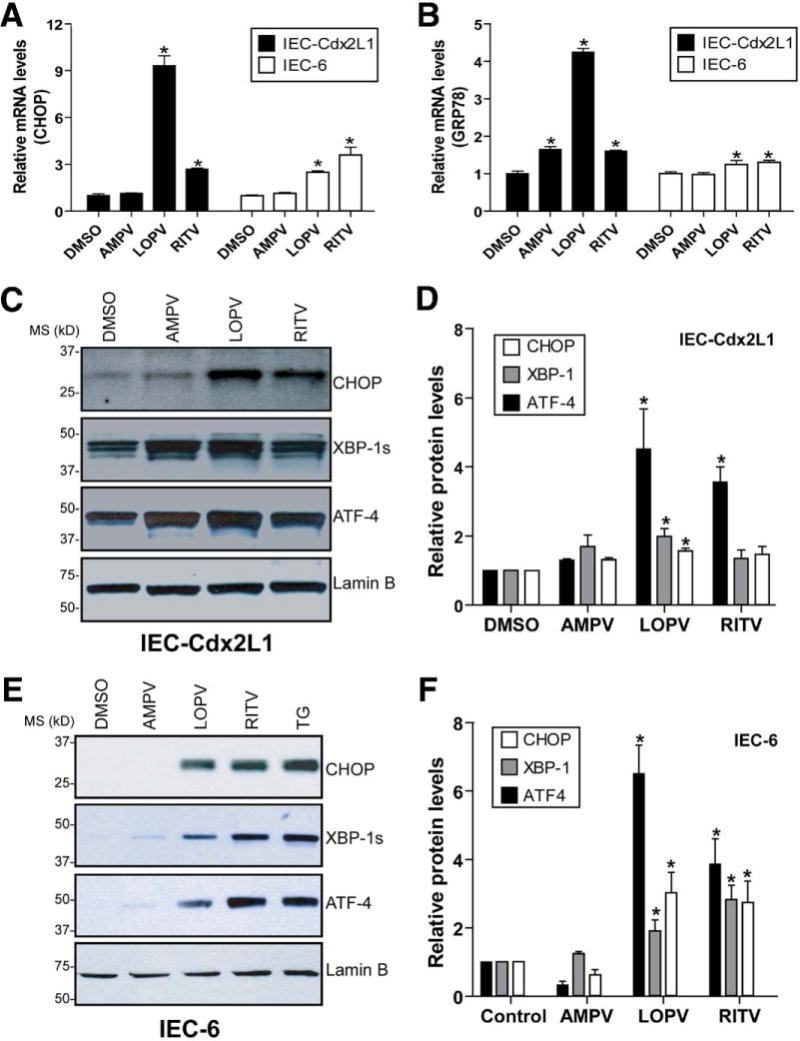
HIV PIs activate the UPR in differentiated IECs. (A and B) Real-time RT-PCR analysis of mRNA levels of CHOP and GRP78 in IEC-Cdx2L1 and IEC-6 cells treated with AMPV, LOPV, and RITV (25 μmol/L) for 24 hours. The relative mRNA levels for each gene were normalized to internal control β-actin mRNA using the ΔΔ Ct method. DMSO, dimethyl sulfoxide. Statistical significance relative to vehicle control, *P < .05. (C and E) Representative immunoblots against CHOP, ATF-4, XBP-1, and lamin B from the nuclear extracts of IEC-Cdx2L1 and IEC-6 cells treated with AMPV, LOPV, RITV (25 μmol/L), or TG (100 nmol/L) for 4 hours. Lamin B was used as a loading control. (D and F) Relative protein levels of CHOP, XBP-1, and ATF-4. Statistical significance relative to vehicle control: *P < .05.
Effect of HIV PI-Induced ER Stress on Paracellular Permeability in IECs
Although it has been shown that HIV PIs induce apoptosis and disrupt intestinal barrier function,2 the exact cellular mechanisms remain to be identified. Our previous studies1,10 and others21 suggest that HIV PI-induced ER stress and activation of the UPR play important roles in HIV PI-associated adverse side effects. To determine whether HIV PI-induced UPR activation in IEC-6 cells is responsible for HIV PI-induced disruption of intestinal barrier integrity, we constructed 3 lentiviral shRNA to knock down the expression of CHOP in IEC-6 cells. As shown in Figure 6, shRNA1 and shRNA3 significantly inhibited CHOP mRNA and protein expression by 48% and 56%, respectively, in IEC-6 cells, but shRNA2 had no inhibitory effect and was used as a negative control in functional studies. The transfection efficiency of traditional lipid-based reagents in IEC-6 is relatively low (~30%), but the infection rate of lentivirus in IEC-6 could reach as high as 90% (data not shown).
Figure 6.
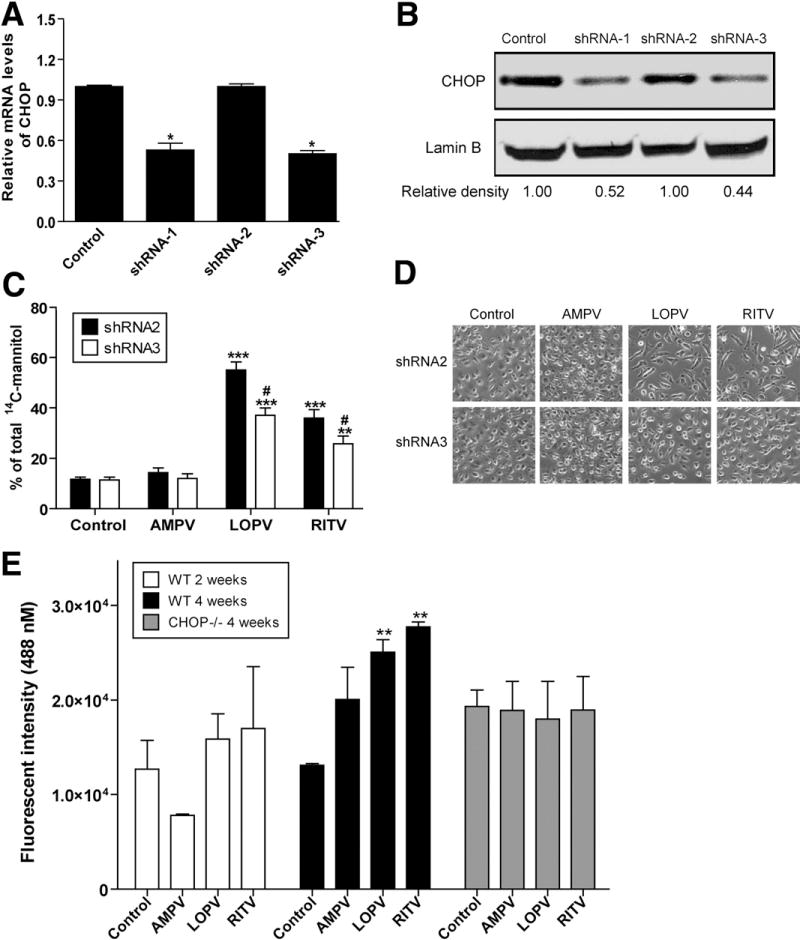
Effect of CHOP knockdown on HIV PI-induced increase of paracellular permeability in IEC-6 cells. (A) Real-time RT-PCR analysis of CHOP mRNA levels in IEC-6 cells infected with individual lentiviral-CHOP-shRNA for 48 hours. The mRNA levels of CHOP were normalized to internal control β-actin mRNA using the ΔΔ Ct method. Statistical significance relative to vehicle control: *P < .001. (B) Representative immunoblots against CHOP and lamin B from the nuclear extracts of IEC-6 cells treated with the individual lentiviral CHOP shRNA for 48 hours. Lamin B was used as a loading control. (C) IEC-6 cells were infected with lentiviral-CHOP shRNA2 or shRNA3 for 48 hours and then treated with individual HIV PIs (15 μmol/L) for 24 hours. The paracellular permeability was measured as described in Materials and Methods. Values are mean ± SD of 3 independent experiments. Statistical significance relative to vehicle control: ***P < .001, **P < .01. Statistical significance relative to shRNA2: #P < .05. (D) Representative phase-contrast images of IEC-6 cells infected with CHOP shRNAs and treated with individual HIV PIs for 24 hours. (E) Effect of HIV PIs on intestinal permeability in vivo. Wild-type and CHOP−/− mice were treated with individual HIV PIs (50 mg/kg) for 2 or 4 weeks. The intestinal permeability was measured using FITC-dextran as described in Materials and Methods. Statistical significance relative to vehicle control: **P < .01.
To determine whether down-regulation of CHOP expression will prevent HIV PI-induced disruption of paracellular permeability in IEC-6 cells, we infected the cells with lentiviral-CHOP shRNA2 and shRNA3 at a multiplicity of infection of 100 for 48 hours and measured the paracellular permeability as described previously. As shown in Figure 6C, knockdown of CHOP expression inhibited a LOPV-and RITV-induced increase of paracellular permeability by 37% and 29%, respectively, in IEC-6 cells. Consistently, LOPV-and RITV-induced apoptosis was also reduced after knockdown of CHOP gene expression (Figure 6D). These results indicate that HIV PI-induced ER stress and subsequent activation of CHOP expression play a critical role in HIV PI-induced disruption of paracellular permeability.
HIV PIs Induce ER Stress and Increase Intestinal Permeability In Vivo
To determine whether HIV PIs also induce ER stress in intestine in vivo, we examined the effect of individual HIV PIs on UPR activation in vivo using C57BL/6 wild-type male mice. As shown in Figure 7A, both RITV and LOPV significantly increased CHOP and XBP-1s expression in intestine after 4 weeks of treatment, whereas AMPV had no significant effect. We further examined the effect of HIV PIs on intestinal permeability in vivo using FITC-dextran as probe. As shown in Figure 6E, 2-week treatment of HIV PIs had no significant effect on intestinal permeability, but 4-week treatment with LOPV and RITV resulted in a significant increase of the intestinal permeability.
Figure 7.
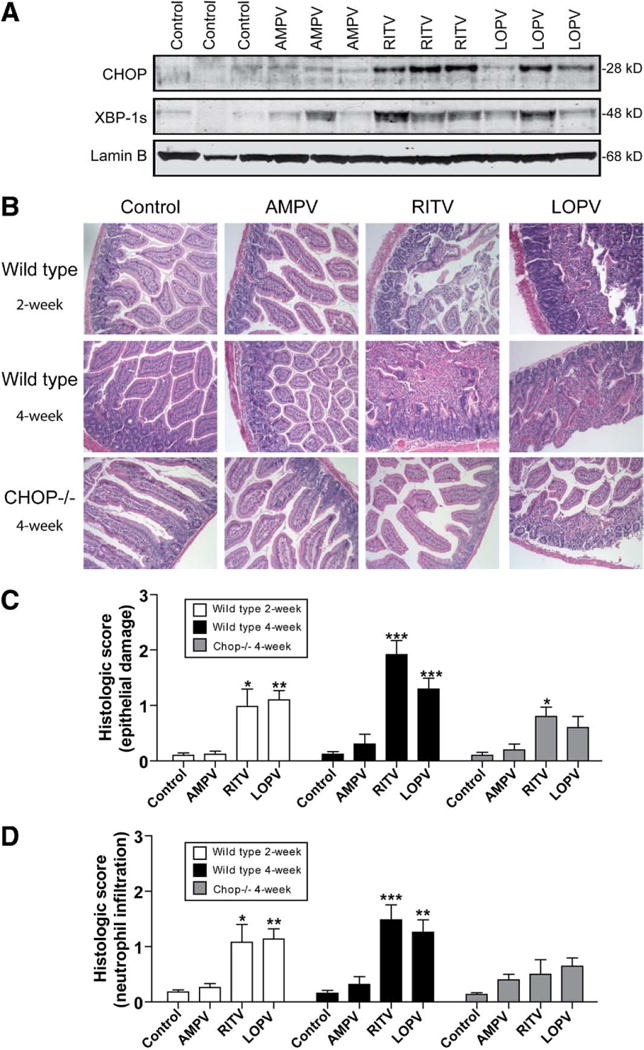
HIV PIs activate the UPR and induce damage in intestine. Mice were treated with individual HIV PIs (50 mg/kg) for 2–4 weeks. (A) Representative immunoblots of nuclear extracts isolated from intestine tissues against CHOP, XBP-1, and lamin B are shown. (B) Representative images of H&E staining for each treatment group is shown. (C and D) Histologic score of HIV PI-induced epithelial tissue damage and neutrophil infiltration. Statistical significance relative to vehicle control: *P < .05, **P < .01, ***P < .001.
HIV PIs Induce Apoptosis and Pathologic Changes in Intestine In Vivo
Histologic examination revealed that HIV PIs had no significant effect on the colon (Supplementary Figure 4) and AMPV caused minor pathologic changes in intestine. However, both LOPV and RITV started to cause significant pathologic changes in intestine at 2 weeks of treatment, inducing a loss of villi, edema, neutrophil infiltration, and necrosis. The 4-week treatment with LOPV and RITV induced more severe damage in intestinal epithelium and lamina propria (Figure 7B). In control and AMPV-treated animals, less tissue damage and few terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling assay (TUNEL)-positive cells were detected, whereas in LOPV-and RITV-treated animals, many more TUNEL-positive cells were observed in the intestine (Figure 8).
Figure 8.
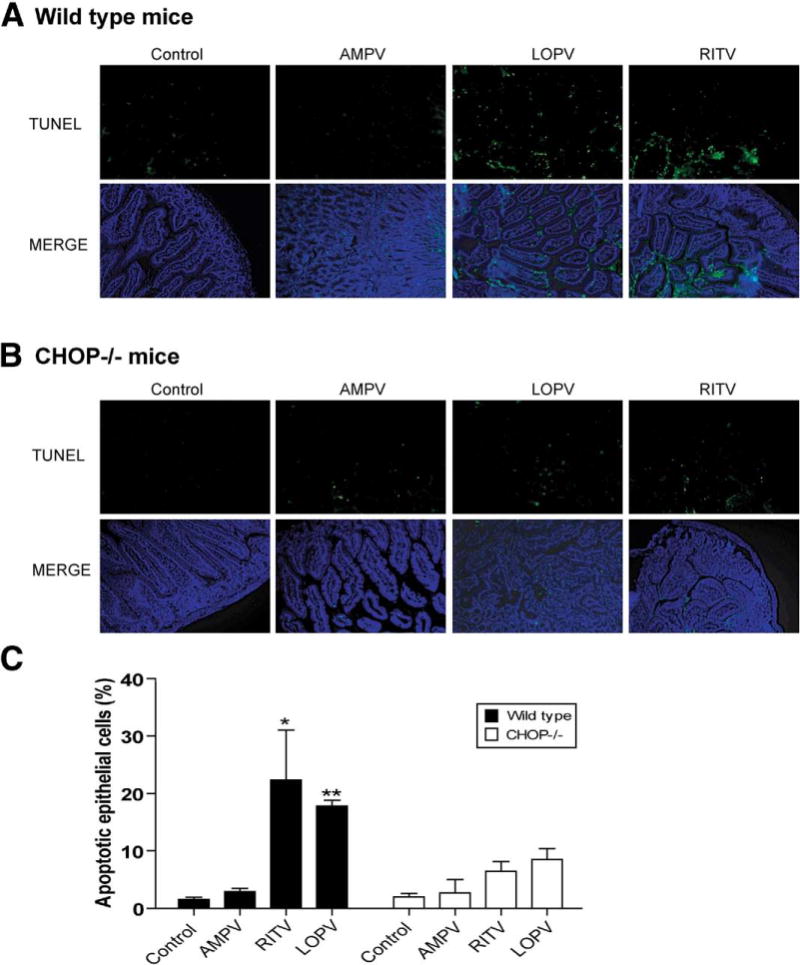
HIV PIs induce apoptosis in intestine in vivo. The representative TUNEL staining images of intestine tissue sections from (A) wild-type and (B) CHOP−/− mice treated with individual HIV PIs as described in Materials and Methods. (C) The apoptotic cells were counted. Statistical significance relative to vehicle control: *P < .05, **P < .01.
Deletion of CHOP Ameliorates HIV PI-Induced Pathologic Changes in Intestine and Prevents HIV PI-Induced Increase of Intestinal Permeability
Histologic assessment showed that wild-type and CHOP−/− mice appeared similar under basal conditions. However, HIV PI-induced apoptosis and pathologic changes in wild-type mice were diminished in CHOP−/− mice, with significantly reduced edema and tissue damage, and fewer TUNEL-positive cells were detected (Figures 7B–D and 8). Similarly, LOPV and RITV had less effect on intestinal permeability in CHOP−/− mice (Figure 6E). Thus, deletion of the ER stress responsive gene, CHOP, dramatically prevents HIV PI-induced apoptosis and intestinal damage.
Discussion
HIV PIs have been successfully incorporated into highly active antiretroviral therapy to control virus replication in HIV-infected patients. However, the benefits of HIV PIs in reducing morbidity and mortality of patients with HIV require high compliance of the patients, which is seriously hampered by HIV PI-associated gastrointestinal adverse effects. The most frequently reported gastrointestinal side effects of HIV PIs include abdominal pain, abnormal stools, and diarrhea.3 However, the exact cellular mechanisms underlying HIV PI-induced gastrointestinal dysfunction remain largely unknown.
Recent studies by Bode et al suggest that HIV PIs induce apoptosis and impair the barrier function in human HT-29/B6 cells, which suggests a leak-flux type of diarrhea in patients receiving HIV PIs.2 Whether HIV PIs have similar effects on normal IECs has not been examined. In the present study, we found that the 2 most commonly used HIV PIs, RITV and LOPV, significantly induced apoptosis and disrupted intestinal epithelial barrier integrity both in vitro and in vivo, whereas AMPV had less effect.
The mammalian intestinal epithelium is a rapidly self-renewing tissue in the body, and its integrity is maintained by a strictly regulated process of cell proliferation, growth arrest, and apoptosis.22 Our previous studies have shown that HIV PI-induced ER stress response and activation of the UPR are responsible for HIV PI-induced cell apoptosis both in macrophages and hepatocytes.1,10 It also has been reported that HIV PI-induced ER stress response is linked to insulin resistance and dysregulation of lipid metabolism in adipocytes.11,23 Numerous studies have suggested that ER stress signaling pathways are implicated in various diseases, including atherosclerosis, metabolic disease, liver disease, and inflammatory bowel disease.6,24–27 However, whether HIV PI-induced apoptosis is also associated with the activation of the ER stress response in IECs has not been explored. Herein, we describe our investigation of whether HIV PI-induced activation of the ER stress response is involved in the induction of apoptosis and disruption of normal intestinal barrier function both in in vitro cell culture models and in in vivo mice models.
Several recent studies have shown that ER stress responses contribute to the pathogenesis of chronic intestinal inflammation.9,24,27 Disruption of ER homeostasis by adverse environmental and/or metabolic conditions triggers cellular stress responses including the UPR, the ER specific stress response. The signaling pathways of the UPR are important for maintenance of normal cellular homeostasis. However, inappropriate activation of the UPR can lead to cellular dysfunction and ultimately cell death. The results from our functional studies showed that HIV PIs not only activated the UPR but also disrupted the barrier integrity both in undifferentiated and differentiated IECs (Figure 1). In vivo animal studies further showed that HIV PIs, LOPV and RITV, induced severe damage to intestinal tissue (Figure 7). However, AMPV, which had less effect on UPR activation, had little effect on intestinal barrier function. Both in vitro and in vivo studies suggest that HIV PI-induced activation of the UPR is correlated to its effect on intestinal barrier function.
To further demonstrate our notion that HIV PI-induced ER stress response is critical to HIV PI-induced damage in intestinal epithelia, we examined the effect of knocking down CHOP expression on HIV PI-induced dysfunction of IECs. CHOP is a UPR-induced transcription factor that mediates apoptosis.6 It has been reported that macrophages from CHOP−/− mice are highly protected from free cholesterol-induced cell death.28 The results from our current study also clearly showed that down-regulation of CHOP expression significantly blocked LOPV-and RITV-induced paracellular permeability in IECs (Figure 6). In vivo studies with CHOP−/− mice further demonstrated that expression of CHOP plays a critical role in HIV PI-induced intestinal tissue damage and barrier function (Figures 6–8). Therefore, HIV PI-induced ER stress and subsequent activation of the UPR represent an important cellular mechanism underlying HIV PI-induced dysfunction of IECs. However, how HIV PIs induce ER stress in IECs remains to be further elucidated.
It has been well established that the UPR can be activated by many stress signals that cause the accumulation of unfolded or misfolded proteins in the ER lumen, such as accumulation of free cholesterol, depletion of ER calcium stores, and inhibition of N-glycosylation.6 Our previous studies have shown that HIV PIs induced the accumulation of intracellular free cholesterol and depleted the ER calcium store in macrophages and primary hepatocytes.1,10 Our preliminary studies also found that LOPV and RITV, but not AMPV, depleted the ER calcium store in IEC-6 cells (Supplementary Figure 5).
It has been reported that unresolved ER stress activates the c-Jun-N-terminal kinase (JNK) signaling pathway and induces inflammatory response.9,24 We also examined the effect of HIV PIs on activation of mitogen-activated protein kinases both in vitro and in vivo. As shown in Supplementary Figure 6A, LOPV and RITV not only activated the JNK but also inhibited the extracellular signal–regulated kinase (ERK) and AKT signaling pathways in differentiated intestinal cells. It has been shown that activation of ERK and AKT signaling pathways prevents tumor necrosis factor α–induced apoptosis in IECs.29 Furthermore, RITV also activated JNK in wild-type mice intestine but not CHOP−/− mice intestine.
Recent studies by Heazlewood et al show that activation of ER stress response in goblet cells leads to spontaneous colitis.18 It also has been shown that disruption of Xbp1 depletes Paneth cells and reduces goblet cells in small intestine but has no effect on epithelial barrier.24 In the present study, we did not observe significant changes in goblet cells. Both in vitro and in vivo studies suggest that HIV PIs induce apoptosis of epithelial cells.
In summary, our present studies provide the first evidence suggesting that ER stress response is implicated in the HIV PI-induced apoptosis and disruption of barrier integrity both in in vitro cultured IECs and in in vivo intestinal tissue. Together with our previous findings in macrophages and hepatocytes, our studies suggest that the HIV PI-induced ER stress response represents a critical cellular mechanism underlying HIV PI-associated various adverse effects in HIV PI-treated patients. Understanding of the molecular mechanisms of HIV PI-induced ER stress will provide important information for developing more effective anti-HIV therapeutics that could avoid some of the adverse effects associated with current HIV PIs.
Supplementary Material
Acknowledgments
The authors thank the AIDS Research and Reference Reagent Program, National Institutes of Health, and GlaxoSmithKline (amprenavir) for providing the compounds used in this research.
Funding: Supported by grants from the National Institutes of Health (R21AI068432, R01AT004148, R01AI057189, P01DK38030, P30CA16059, and R01DK064240), A.D. Williams fund, and Jeffress Memorial Trust.
Abbreviations
- AMPV
amprenavir
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinase
- FITC
fluorescein isothiocyanate
- HIV
human immunodeficiency virus
- IEC
intestinal epithelial cell
- JNK
c-Jun-N-terminal kinase
- LOPV
lopinavir
- PI
protease inhibitor
- RITV
ritonavir
- RT-PCR
reverse-transcription polymerase chain reaction
- SEAP
secreted alkaline phosphatase
- shRNA
short hairpin RNA
- TUNEL
terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling assay
- UPR
unfolded protein response
Footnotes
Supplementary Data
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi: 10.1053/j.gastro.2009.08.054.
Conflicts of interest
The authors disclose no conflicts.
References
- 1.Zhou H, Pandak WM, Jr, Lyall V, et al. HIV protease inhibitors activate the unfolded protein response in macrophages: implication for atherosclerosis and cardiovascular disease. Mol Pharmacol. 2005;68:690–700. doi: 10.1124/mol.105.012898. [DOI] [PubMed] [Google Scholar]
- 2.Bode H, Schmidt W, Schulzke JD, et al. The HIV protease inhibitors saquinavir, ritonavir, and nelfinavir but not indinavir impair the epithelial barrier in the human intestinal cell line HT-29/B6. AIDS. 1999;13:2595–2597. doi: 10.1097/00002030-199912240-00016. [DOI] [PubMed] [Google Scholar]
- 3.Bode H, Schmidt W, Schulzke JD, et al. Effects of HIV protease inhibitors on barrier function in the human intestinal cell line HT-29/B6. Ann N Y Acad Sci. 2000;915:117–122. doi: 10.1111/j.1749-6632.2000.tb05233.x. [DOI] [PubMed] [Google Scholar]
- 4.Bode H, Lenzner L, Kraemer OH, et al. The HIV protease inhibitors saquinavir, ritonavir, and nelfinavir induce apoptosis and decrease barrier function in human intestinal epithelial cells. Antivir Ther. 2005;10:645–655. [PubMed] [Google Scholar]
- 5.Lin JH, Walter P, Yen TSB. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen X, Zhang K, Kaufman RJ. The unfolded protein response—a stress signaling pathway of the endoplasmic reticulum. J Chem Neuroanat. 2004;28:79–92. doi: 10.1016/j.jchemneu.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaser A, Blumberg RS. Endoplasmic reticulum stress in the intestinal epithelium and inflammatory bowel disease. Semin Immunol. 2009;21:156–163. doi: 10.1016/j.smim.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou H, Gurley EC, Jarujaron S, et al. HIV protease inhibitors activate the unfolded protein response and disrupt lipid metabolism in primary hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2006;291:G1071–G1080. doi: 10.1152/ajpgi.00182.2006. [DOI] [PubMed] [Google Scholar]
- 11.Djedaini M, Peraldi P, Drici MD, et al. Lopinavir co-induces insulin resistance and ER stress in human adipocytes. Biochem Biophys Res Commun. 2009;386:96–100. doi: 10.1016/j.bbrc.2009.05.148. [DOI] [PubMed] [Google Scholar]
- 12.Quaroni A, Wands J, Trelstad RL, et al. Epithelioid cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria. J Cell Biol. 1979;80:248–265. doi: 10.1083/jcb.80.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suh E, Traber PG. An intestine-specific homeobox gene regulates proliferation and differentiation. Mol Cell Biol. 1996;16:619–625. doi: 10.1128/mcb.16.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hiramatsu N, Kasai A, Hayakawa K, et al. Real-time detection and continuous monitoring of ER stress in vitro and in vivo by ES-TRAP: evidence for systemic, transient ER stress during endotoxemia. Nucleic Acids Res. 2006;34:e93. doi: 10.1093/nar/gkl515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou H, Jarujaron S, Gurley EC, et al. HIV protease inhibitors increase TNF-alpha and IL-6 expression in macrophages: involvement of the RNA-binding protein HuR. Atherosclerosis. 2007;195:e134–e143. doi: 10.1016/j.atherosclerosis.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 16.Wong V, Gumbiner BM. A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J Cell Biol. 1997;136:399–409. doi: 10.1083/jcb.136.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo X, Rao JN, Liu L, et al. Regulation of adherens junctions and epithelial paracellular permeability: a novel function for polyamines. Am J Physiol Cell Physiol. 2003;285:C1174–C1187. doi: 10.1152/ajpcell.00015.2003. [DOI] [PubMed] [Google Scholar]
- 18.Heazlewood CK, Cook MC, Eri R, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008;5:e54. doi: 10.1371/journal.pmed.0050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laukoetter MG, Nava P, Lee WY, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204:3067–3076. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukata M, Chen A, Klepper A, et al. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: role in proliferation and apoptosis in the intestine. Gastroenterology. 2006;131:862–877. doi: 10.1053/j.gastro.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hruz PW, Murata H, Mueckler M. Adverse metabolic consequences of HIV protease inhibitor therapy: the search for a central mechanism. Am J Physiol Endocrinol Metab. 2001;280:E549–E553. doi: 10.1152/ajpendo.2001.280.4.E549. [DOI] [PubMed] [Google Scholar]
- 22.Johnson LR. Regulation of gastrointestinal mucosal growth. Physiol Rev. 1988;68:456–502. doi: 10.1152/physrev.1988.68.2.456. [DOI] [PubMed] [Google Scholar]
- 23.Parker RA, Flint OP, Mulvey R, et al. Endoplasmic reticulum stress links dyslipidemia to inhibition of proteasome activity and glucose transport by HIV protease inhibitors. Mol Pharmacol. 2005;67:1909–1919. doi: 10.1124/mol.104.010165. [DOI] [PubMed] [Google Scholar]
- 24.Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocrinol Rev. 2008;29:317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shkoda A, Ruiz PA, Daniel H, et al. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology. 2007;132:190–207. doi: 10.1053/j.gastro.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 28.Feng B, Yao PM, Li Y, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–792. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 29.Bhattacharya S, Ray RM, Johnson LR. Prevention of TNF-alphainduced apoptosis in polyamine-depleted IEC-6 cells is mediated through the activation of ERK1/2. Am J Physiol Gastrointest Liver Physiol. 2004;286:G479–G490. doi: 10.1152/ajpgi.00342.2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.