

Summary
IκB kinase/nuclear factor κB (IKK/NF-κB) signaling exhibits important yet opposing functions in hepatocarcinogenesis. Mice lacking NEMO in liver parenchymal cells (LPC) spontaneously develop steatohepatitis and hepatocellular carcinoma (HCC) suggesting that NF-κB prevents liver disease and cancer. Here, we show that complete NF-κB inhibition by combined LPC-specific ablation of RelA, c-Rel, and RelB did not phenocopy NEMO deficiency, but constitutively active IKK2-mediated NF-κB activation prevented hepatocellular damage and HCC in NEMOLPC-KO mice. Knock-in expression of kinase inactive receptor-interacting protein kinase 1 (RIPK1) prevented hepatocyte apoptosis and HCC, while RIPK1 ablation induced TNFR1-associated death domain protein (TRADD)-dependent hepatocyte apoptosis and liver tumors in NEMOLPC-KO mice, revealing distinct kinase-dependent and scaffolding functions of RIPK1. Collectively, these results show that NEMO prevents hepatocarcinogenesis by inhibiting RIPK1 kinase activity-driven hepatocyte apoptosis through NF-κB-dependent and -independent functions.
Graphical Abstract
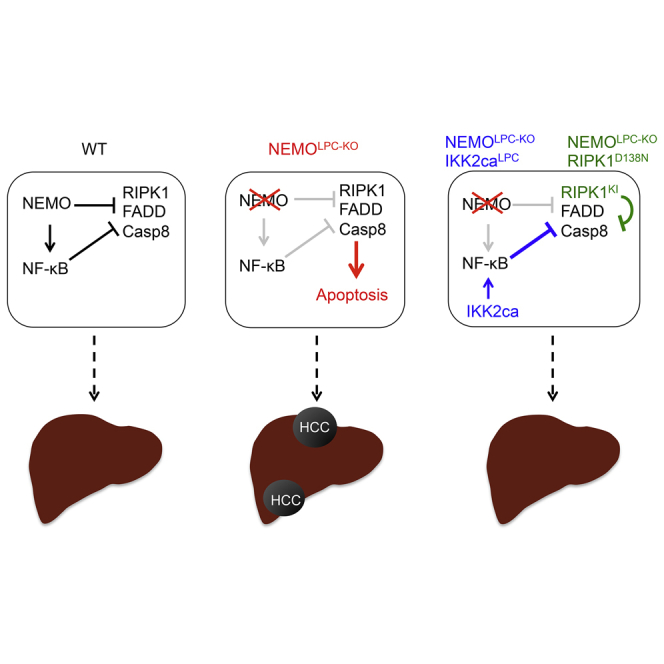
Highlights
-
•
NF-κB inhibition in LPCs does not cause spontaneous HCC in contrast to NEMO ablation
-
•
Constitutive IKK2/NF-κB activation prevents liver damage and HCC in NEMOLPC-KO mice
-
•
RIPK1 kinase activity-mediated hepatocyte apoptosis drives HCC in NEMOLPC-KO mice
-
•
NEMO prevents formation of a RIPK1/FADD/Casp8 complex inducing hepatocyte apoptosis
Kondylis et al. identify an NF-κB-independent function of NEMO in controlling RIPK1 kinase activity-dependent apoptosis of hepatocytes and thus hepatocarcinogenesis. NEMO-mediated NF-κB activity and kinase activity-independent scaffold function of RIPK1 also regulate hepatocyte apoptosis.
Significance
Pathways regulating cell survival, cell death, and inflammation play a key role in the maintenance of liver homeostasis and the pathogenesis of chronic liver disease and cancer. Caspase-dependent apoptosis and receptor-interacting protein (RIP) kinase-dependent necroptosis are key pathways of programmed cell death and have been implicated in disease pathogenesis. Here, we reveal a RIP kinase 3 (RIPK3)-independent function of RIP kinase 1 (RIPK1) in mediating hepatocyte apoptosis and triggering chronic hepatitis and HCC development. This function of RIPK1 depends on its kinase activity and is controlled by nuclear factor κB (NF-κB)-dependent and NF-κB-independent NEMO functions. These results identify the interplay between NEMO and RIPK1 as a key mechanism that is essential for hepatocyte survival and the prevention of chronic liver damage leading to hepatocarcinogenesis.
Introduction
Liver cancer is the fifth most common cancer in men and the seventh in women (El-Serag, 2011, Shariff et al., 2009). Hepatocellular carcinoma (HCC), the most prominent primary liver cancer, usually develops in the presence of cirrhosis and chronic inflammatory conditions resulting from hepatitis B virus (HBV) or hepatitis C virus (HCV) infections, alcoholic and non-alcoholic steatohepatitis, and aflatoxin-mediated toxicity (Shariff et al., 2009). Due to the limited treatment options, HCC is the third most common cause for cancer-related deaths worldwide (El-Serag, 2011, Shariff et al., 2009). Therefore, there is an urgent need to better understand the mechanisms contributing to the pathogenesis of chronic liver inflammation and HCC in order to identify possible preventive strategies and therapeutic targets.
Death receptors, such as tumor necrosis factor receptor 1 (TNFR1), regulate cell death and inflammation and are implicated in the pathogenesis of acute and chronic liver diseases and in liver cancer (Schattenberg et al., 2011, Wang, 2014). Upon stimulation, TNFR1 activates proinflammatory nuclear factor κB (NF-κB) and mitogen-activated protein (MAP) kinase cascades through a receptor-proximal signaling complex (complex I), composed of TNFR1-associated death domain protein (TRADD), receptor-interacting protein kinase 1 (RIPK1), as well as the E3 ubiquitin ligases TNF receptor-associated factor 2 (TRAF2), cellular inhibitors of apoptosis 1 (cIAP1) and cellular inhibitors of apoptosis 2 (cIAP2), and the linear ubiquitin chain assembly complex (LUBAC) (Micheau and Tschopp, 2003, Walczak et al., 2012). In sensitized cells (e.g., upon NF-κB inhibition), complex I dissociates from the receptor and recruits Fas-associated via death domain protein (FADD) and caspase-8 to induce apoptosis (Micheau and Tschopp, 2003). When caspase-8 is inhibited, TNFR1 triggers necroptosis via receptor-interacting protein kinase 3 (RIPK3) and mixed lineage kinase domain-like protein (MLKL) (Vanden Berghe et al., 2014, Weinlich et al., 2011). RIPK1 determines the outcome of TNFR1 signaling by inducing pro-survival and proinflammatory signaling via kinase-independent scaffolding functions, but also apoptosis or necroptosis through its kinase activity (Christofferson et al., 2014, Pasparakis and Vandenabeele, 2015).
The IκB kinase (IKK)/NF-κB pathway regulates cell survival, inflammation, and cancer (Ben-Neriah and Karin, 2011, Hayden and Ghosh, 2012) and is implicated in the pathogenesis of hepatitis and HCC (Luedde and Schwabe, 2011, Sun and Karin, 2008). Mammalian cells express five NF-κB transcription factors, RelA/p65, RelB, c-Rel, p50/p105, and p52/p100. RelA, c-Rel, and RelB contain transactivation domains and are capable to activate gene transcription. In resting cells, NF-κB dimers are kept inactive by association with inhibitory IκB proteins. Upon cell stimulation, the IKK complex, consisting of the IKK1/IKKα and IKK2/IKKβ kinases and the NEMO/IKKγ regulatory subunit, phosphorylates IκB proteins targeting them for proteasomal degradation. Consequently, NF-κB dimers accumulate in the nucleus where they regulate the transcription of many genes involved in inflammation, cell growth, cell survival, and proliferation (Hayden and Ghosh, 2012, Pasparakis, 2009, Perkins, 2012).
Mouse model studies revealed important but contradictory functions of IKK/NF-κB signaling in liver carcinogenesis. IκBα super-repressor-mediated NF-κB inhibition in hepatocytes delayed HCC progression but not tumor initiation in the Mdr2−/− mouse model of inflammation-driven liver carcinogenesis by counteracting TNF-mediated tumor promotion in hepatocytes (Pikarsky et al., 2004). In addition, hepatocyte-specific ablation of IKK2 prevented hepatitis and liver tumorigenesis induced by transgenic overexpression of lymphotoxin α/β by inhibiting LTβ receptor-mediated expression of chemokines and cytokines (Haybaeck et al., 2009). These reports support a predominantly tumor-promoting function of NF-κB in hepatocytes. In contrast, other studies revealed tumor-suppressing functions of IKK/NF-κB signaling in the liver. In a mouse model of diethylnitrosamine (DEN)-induced hepatocarcinogenesis, hepatocyte-specific ablation of IKK2 resulted in increased tumor load due to enhanced DEN-induced liver damage and compensatory proliferation of IKK2-deficient hepatocytes (Maeda et al., 2005). Mice with liver parenchymal cell (LPC)-specific ablation of NEMO (NEMOLPC-KO) spontaneously developed chronic steatohepatitis and HCC, further supporting a tumor suppressing function of IKK/NF-κB signaling in hepatocytes (Luedde et al., 2007). The development of hepatitis and HCC in NEMOLPC-KO mice is triggered by FADD- and caspase-8-dependent apoptosis of NEMO-deficient hepatocytes (Ehlken et al., 2014, Liedtke et al., 2011, Luedde et al., 2007). However, the molecular mechanisms by which NEMO inhibits hepatocyte apoptosis, hepatitis and HCC remain poorly understood.
Here, we addressed the role of NF-κB and RIPK1 signaling in the pathogenesis of liver disease and cancer caused by NEMO ablation in LPCs.
Results
Inhibition of NF-κB Signaling in LPCs Does Not Phenocopy NEMO Deficiency
To study whether NEMO deficiency triggered hepatitis and HCC by inhibiting NF-κB, we generated and analyzed mice lacking NF-κB subunits in LPCs (Figures 1A and 1B). As reported previously (Geisler et al., 2007, Luedde et al., 2008), RelALPC-KO mice did not develop considerable liver pathology, although they had slightly elevated alanine aminotransferase (ALT) levels compared to wild-type mice at the age of 8 weeks (Figure 1C). To assess possible functional redundancy between NF-κB factors, we generated mice with LPC-specific single or combined deficiency of RelA, RelB, and c-Rel, hereafter referred to as NF-κBLPC-KO.
Figure 1.

LPC-Specific NF-κB Inhibition Does Not Cause Similar Liver Damage as NEMO Deficiency
(A and B) Immunoblot analysis of liver (A) and hepatocyte (B) lysates from 8-week-old mice with the indicated genotypes. GAPDH and α-tubulin were used as loading controls.
(C) Graph depicting basal serum ALT levels in 8-week-old mice with the indicated genotypes. Horizontal lines indicate mean values.
(D) Graph depicting serum ALT levels in mice with the indicated genotypes before and 4 and 10 hr after LPS injection (mean ± SD).
(E) Representative images of liver sections from 8-week-old mice with the indicated genotypes that are stained with H&E or immunostained with the indicated antibodies. Graphs depict quantification of indicated parameters (mean ± SD, n = 3–5 mice per genotype). Scale bars, 200 μm.
See also Figure S1.
To address the role of hepatocyte NF-κB signaling in protecting the liver from endotoxin-induced injury, we compared the response of NF-κBLPC-KO and NEMOLPC-KO mice to a dose of LPS that does not induce considerable liver damage in wild-type mice. RelA/c-RelLPC-KO and triple NF-κBLPC-KO mice showed increased serum ALT levels after LPS injection compared to RelALPC-KO animals, suggesting that c-Rel can partly substitute pro-survival functions of RelA in hepatocytes (Figure 1D). However, NEMOLPC-KO mice were considerably more sensitive to endotoxin-induced liver damage showing ∼8-fold higher ALT levels compared to NF-κBLPC-KO animals.
Surprisingly, 8-week-old RelALPC-KO, RelALPC-KO;c-RelLPC-KO, and NF-κBLPC-KO mice had only mildly elevated serum ALT levels (≤100 U/l) that were ∼10-fold lower compared to NEMOLPC-KO mice at this age (Figure 1C), showing that NF-κB inhibition was not sufficient to cause substantial spontaneous liver damage. Histological analysis of livers from 8-week-old NF-κBLPC-KO mice revealed an overall normal liver architecture, with only a limited number of apoptotic hepatocytes (Figure 1E). Interestingly, in contrast to the NEMO-deficient livers that showed widespread distribution of apoptotic hepatocytes throughout the liver parenchyma, the few apoptotic hepatocytes in the livers of NF-κBLPC-KO mice were localized mostly in the vicinity of focal inflammatory cell infiltrates (Figure 1E). The limited hepatocyte death in NF-κBLPC-KO livers correlated with a moderate increase of Ki-67+ proliferating hepatocytes, which was significantly lower than in NEMOLPC-KO livers and was not accompanied by hepatic stellate cell (HSC) or liver progenitor cell activation, as shown by immunostaining for α-smooth muscle actin (α-SMA) and cytokeratin 19 (CK19), respectively (Figures 1E and S1). Immunohistochemistry revealed that macrophages, granulocytes, T cells, and B cells populated the foci of dying hepatocytes in the livers of NF-κBLPC-KO mice (Figures 1E and S1).
In contrast to NEMOLPC-KO mice, livers from 1-year-old mice with individual or combined LPC-specific deficiency of RelA, c-Rel, and RelB did not display macroscopic signs of tumor development, and only ∼25% (6 out of 25) of NF-κBLPC-KO mice showed a number of very small macroscopically visible steatotic nodules (Figure 2A). Accordingly, liver-to-body weight (LW/BW) ratio in NF-κBLPC-KO mice was similar to controls and significantly lower than that of NEMOLPC-KO mice (Figure 2B). One-year-old NF-κBLPC-KO mice showed significantly elevated serum ALT levels compared to controls but on average lower than NEMOLPC-KO mice (Figure 2C). Notably, ALT levels in NF-κBLPC-KO mice with macroscopically visible liver nodules were statistically higher compared to nodule-free mice (Figure 2C; 332 versus 135, p = 0.003), indicating that the appearance of small steatotic nodules correlated with increased liver damage.
Figure 2.

Mice with LPC-Specific NF-κB Inhibition Do Not Develop HCC
(A) Representative liver images from 52-week-old mice with the indicated genotypes. Scale bar, 1 cm.
(B) Liver to body weight (LW/BW) ratios in 52-week-old mice with the indicated genotypes. Horizontal lines indicate mean values.
(C) Serum ALT levels in 52-week-old mice with the indicated genotypes. The ALT values of NF-κBLPC-KO mice with very small (<2 mm diameter) macroscopically visible nodules are marked in red.
(D) Representative images of liver sections from 52-week-old NF-κBLPC-KO, NEMOLPC-KO and floxed control mice stained with Masson's trichrome (collagen staining in blue) or immunostained with the indicated antibodies. HCC areas are outlined and marked with an asterisk. Graph shows quantification of Ki67+ cells (mean ± SD, n = 3–5 mice per genotype). Scale bars, 200 μm.
See also Figure S2.
Histologically, livers from 1-year-old NF-κBLPC-KO mice showed a relatively normal overall architecture without signs of HCC or preneoplastic lesions, with the macroscopically visible nodules identified as focal fatty changes resembling clear cell foci (Figures 2D, S2A, and S2B). Interestingly, collagen staining by Masson’s trichrome showed that two-thirds of the examined livers (n = 9) displayed periportal or bridging fibrosis (Figures 2D and S2A). The areas of focal apoptosis observed in the liver of 8-week-old mice were also present at 1 year of age (Figures 2D, S2C, and S2D), but with higher frequency reflecting their increased serum ALT values (Figure 2C). Hepatocyte proliferation was mildly elevated in NF-κBLPC-KO mice compared to NF-κBFL littermates but lower than the non-tumor liver tissue of NEMOLPC-KO mice (Figure 2D). Therefore, combined ablation of RelA, RelB, and c-Rel in LPCs was not sufficient to cause spontaneous development of HCC as observed in NEMOLPC-KO mice, suggesting that NEMO prevents liver tumorigenesis by functions independent of NF-κB-mediated gene transcription.
Constitutively Active IKK2 Expression Prevents Hepatocellular Damage and HCC in NEMOLPC-KO Mice by Activating NF-κB
Although NF-κB inhibition alone was not sufficient to cause liver tumors, impairment of NF-κB could contribute to the liver pathology of NEMOLPC-KO mice when combined with the loss of NF-κB-independent NEMO functions. To address this possibility, we examined whether enforced NF-κB activation in NEMO-deficient hepatocytes induced by the overexpression of a constitutively active IKK2 (IKK2ca) transgene (Sasaki et al., 2006) could rescue the liver phenotype of NEMOLPC-KO mice. Heterozygous expression of the R26-StopFL-IKK2ca transgene in NEMOLPC-KO;IKK2caLPC mice resulted in ∼5-fold increased IKK2 protein levels in the liver compared to endogenous IKK2 levels correlating with nuclear accumulation of RelA in hepatocytes (Figures 3A and S3A). Strikingly, serum ALT levels as well as hepatocyte apoptosis and proliferation, myeloid cell and HSC activation, and fibrosis were drastically reduced in 8-week-old NEMOLPC-KO;IKK2caLPC mice compared to NEMOLPC-KO (Figures 3B, 3C, and S3B, compare with Figures 1E and S1). Furthermore, NEMOLPC-KO;IKK2caLPC mice examined at the age of 12–16 months did not show macroscopically visible liver nodules and had markedly lower LW/BW ratio compared to NEMOLPC-KO mice (Figures 4A–4D). Histological analysis confirmed the lack of dysplastic nodules in livers from NEMOLPC-KO;IKK2caLPC mice (Figures 4E and S4A). Therefore, overexpression of IKK2ca prevented chronic hepatocellular damage and the development of HCC in NEMOLPC-KO mice.
Figure 3.

Overexpression of Constitutively Active IKK2 Prevents Spontaneous Liver Damage in NEMOLPC-KO Mice by Activating NF-κB
(A) Liver lysates from mice with the indicated genotypes were analyzed by immunoblot with the indicated antibodies. α-Tubulin was used as loading control.
(B) Graph depicting basal serum ALT levels in 8- and 52-week-old mice with the indicated genotypes. Horizontal lines indicate mean values.
(C) Representative images of liver sections from 8-week-old mice with the indicated genotypes after staining with H&E or immunostaining with the indicated antibodies. Graphs depict quantification of the indicated parameters (mean ± SD, n = 3–5 mice per genotype). Scale bars, 200 μm.
See also Figure S3.
Figure 4.

Persistent NF-κB Activation by IKK2ca Expression Prevents HCC in NEMOLPC-KO Mice
(A) Representative liver images from 52-week-old mice with the indicated genotypes. Only a fraction of NEMOLPC-KO;RelALPC-KO;IKK2caLPC mice (4 out of 15) developed more than two large-sized (>5 mm) tumors per liver. Scale bar, 1 cm.
(B–D) Tumor load in mice with the indicated phenotypes as estimated by quantification of the LW/BW ratios (horizontal lines indicate mean values) (B), the tumor number per liver (horizontal lines indicate mean values) (C), and tumor size distribution (mean ± SEM, n = 11–15 mice per genotype) (D).
(E) Representative images of Masson’s trichrome stained liver sections from 52-week-old mice with the indicated genotypes. HCC/dysplastic nodule areas are marked with an asterisk. Scale bar, 200 μm.
(F) qRT-PCR analysis of anti-apoptotic and anti-oxidant gene expression in primary hepatocytes from the indicated mice. Graphs show relative mRNA expression normalized to Tbp.
See also Figure S4.
To investigate how IKK2ca expression prevented the death of NEMO-deficient hepatocytes, we prepared primary hepatocytes from NEMOLPC-KO, NEMOLPC-KO;IKK2caLPC, and NF-κBLPC-KO mice. NEMO-deficient, but not NF-κB-deficient, primary hepatocytes underwent spontaneous apoptosis in culture that could be prevented by treatment with z-VAD-fmk and was only partially reduced by TNF blockade (Figure S4B). Consistent with its protective effect in vivo, IKK2ca expression prevented the spontaneous death of primary NEMO-deficient hepatocytes in vitro (Figure S4B). Assessment of NF-κB activation in z-VAD-fmk-treated primary hepatocytes confirmed that IKK2ca induced constitutive nuclear accumulation of RelA (Figure S4C), suggesting that RelA-mediated expression of anti-apoptotic genes could prevent the death of NEMO-deficient hepatocytes. To address this possibility, we generated and analyzed NEMOLPC-KO mice that expressed IKK2ca but at the same time lacked RelA specifically in LPCs (NEMOLPC-KO;RelALPC-KO;IKK2caLPC mice) (Figure 3A). NEMOLPC-KO;RelALPC-KO;IKK2caLPC mice showed elevated serum ALT levels and increased hepatocyte apoptosis, immune cell infiltration, compensatory hepatocyte proliferation, activation of HSCs, and fibrosis (Figures 3B, 3C, and S3B), revealing that IKK2ca expression could not prevent spontaneous liver damage in NEMOLPC-KO mice in the absence of RelA. Moreover, most 1-year-old NEMOLPC-KO;RelALPC-KO;IKK2caLPC mice developed tumors with histopathological features of HCCs, demonstrating that IKK2ca-mediated inhibition of HCC development in NEMOLPC-KO mice depended on RelA (Figures 4A, 4E, and S4A). However, NEMOLPC-KO;RelALPC-KO;IKK2caLPC mice showed reduced number and size of tumors compared to NEMOLPC-KO;RelALPC-KO mice (Figures 4C and 4D), indicating that RelA ablation could not completely counteract the protective effect of IKK2ca overexpression. Therefore, IKK2ca overexpression prevents liver damage and HCC in NEMOLPC-KO mice mainly by inducing RelA-dependent canonical NF-κB activation.
To gain mechanistic insight into how NEMO prevents hepatocyte apoptosis, we first analyzed the expression of a number of prosurvival genes in isolated hepatocytes from NEMOLPC-KO, NF-κBLPC-KO and wild-type mice. Surprisingly, we found that NEMO or NF-κB deficiency did not result in markedly reduced mRNA expression of prosurvival genes, with the exception of Sod2 that was downregulated in the liver of both NEMOLPC-KO and NF-κBLPC-KO mice (Figure 4F). These results suggested that apoptosis of NEMO-deficient hepatocytes is not caused by an impaired transcriptional regulation of NF-κB-dependent antiapoptotic genes, consistent with the findings that complete NF-κB inhibition did not phenocopy NEMO deficiency in the liver. However, IKK2ca expression induced elevated mRNA expression of the anti-apoptotic proteins cIAP1, cIAP2, and A20, as well as of the anti-oxidant enzyme SOD2 in NEMO-deficient livers (Figure 4F). Curiously, IKK2ca did not increase the mRNA expression of cFLIP and Bcl-xl that are considered important transcriptional targets of NF-κB (Figure 4F). In addition, IKK2ca reduced the expression of the proapoptotic protein Bax, which is expressed at increased levels in NEMO-deficient livers. These results suggested that IKK2ca expression prevents the death of NEMO-deficient hepatocytes by inducing NF-κB-dependent expression of anti-apoptotic and anti-oxidant genes.
NEMO-deficient hepatocytes did not show TNF-inducible nuclear translocation of RelA but displayed strongly increased constitutive nuclear accumulation of p50 (Figure S4C). Moreover, RelB was constitutively present in the nucleus of NEMO-deficient hepatocytes at levels similar to those induced by TNF in wild-type cells (Figure S4C). This correlated with a decreased expression of cIAP1, whose degradation promotes non-canonical NF-κB activation (Sun, 2012). These findings prompted us to investigate the potential role of non-canonical NF-κB signaling in NEMOLPC-KO mice by generating NEMOLPC-KO;RelBLPC-KO mice. However, RelB ablation did not inhibit spontaneous liver damage and the development of hepatitis and HCC in NEMOLPC-KO mice arguing against a role of non-canonical NF-κB signaling (Figures S4D–S4G).
RIPK1 Kinase Activity-Dependent Apoptosis Drives Hepatocyte Death and HCC Development in NEMOLPC-KO Mice
RIPK1 and RIPK3 emerged recently as critical regulators of cell death and inflammation. We therefore addressed the role of RIP kinase-mediated signaling in the development of hepatitis and HCC in NEMOLPC-KO mice. Eight-week-old NEMOLPC-KO;Ripk3−/− mice showed elevated ALT levels, increased apoptosis, and compensatory proliferation of hepatocytes, as well as immune cell infiltration in the liver similarly to NEMOLPC-KO;Ripk3WT/- mice (Figure S5A and data not shown). In addition, macroscopic and histological analysis of livers from 1-year-old mice revealed that RIPK3 deficiency did not prevent the development of HCC in NEMOLPC-KO mice (Figures S5B–S5E). Therefore, RIPK3-dependent necroptosis does not contribute to the spontaneous liver pathology of NEMOLPC-KO mice.
Inhibition of RIPK1 kinase activity by necrostatin-1 or other small molecule inhibitors prevents RIPK3/MLKL-dependent necroptosis and under certain conditions apoptosis (Christofferson et al., 2014, Pasparakis and Vandenabeele, 2015, Vanden Berghe et al., 2014). To address the role of RIPK1 kinase activity in hepatocyte death and HCC development in NEMOLPC-KO mice, we crossed them to knock-in mice expressing a kinase-inactive mutant RIPK1 (RIPK1D138N) (Polykratis et al., 2014). Strikingly, 8-week-old NEMOLPC-KO;Ripk1D138N/D138N mice showed strongly reduced serum ALT levels compared to NEMOLPC-KO mice (Figure 5A). Immunoblot analysis showed lack of caspase-3 and JNK activation in liver extracts from 8-week-old NEMOLPC-KO;Ripk1D138N/D138N mice (Figure 5B). Accordingly, histological analysis of livers from NEMOLPC-KO;Ripk1D138N/D138N mice showed significantly reduced hepatocyte apoptosis and proliferation and HSC activation compared to NEMOLPC-KO mice (Figure 5C). To exclude the possibility that inhibition of RIPK1 kinase activity in a cell type different from LPCs was responsible for the protective effect, we generated NEMOLPC-KO mice carrying one loxP-flanked Ripk1 allele and one Ripk1D138N allele. In these mice, Alfp-Cre-mediated deletion of the floxed Ripk1 allele results in expression of only the RIPK1D138N protein in LPCs, while all other cells express both wild-type RIPK1 and the mutant RIPK1D138N protein. Similarly to NEMOLPC-KO;Ripk1D138N/D138N mice, these NEMOLPC-KO;RIPK1LPC-KO/D138N mice showed strongly reduced liver damage (Figure 5A and data not shown). Collectively, these results showed that lack of RIPK1 kinase activity strongly protected NEMO-deficient hepatocytes from apoptosis.
Figure 5.

Lack of RIPK1 Kinase Activity, but Not Its Scaffolding Function, Prevents Hepatocyte Apoptosis in NEMOLPC-KO Mice
(A) Serum ALT levels in 8-week-old mice with the indicated genotypes. Horizontal lines indicate mean values.
(B) Immunoblot analysis of liver lysates from 8-week-old mice with the indicated genotypes. Actin was used as loading control.
(C) Representative images of liver sections from 8-week-old mice with the indicated genotypes after staining with H&E or immunostaining with the indicated antibodies. Graphs depict quantification of the indicated parameters (mean ± SD, n = 3–5 mice per genotype). Scale bars, 200 μm.
See also Figure S5.
To further dissect the role of the kinase activity-dependent and scaffolding functions of RIPK1 in hepatocyte death, we generated mice lacking both NEMO and RIPK1 in the liver by crossing NemoFL/FL;Alfp-Cre mice with Ripk1FL/FL mice. Surprisingly, 8-week-old NEMOLPC-KO;RIPK1LPC-KO mice showed increased serum ALT levels compared to NEMOLPC-KO;Ripk1D138N/D138N or NEMOLPC-KO;RIPK1LPC-KO/D138N mice, although their ALT levels were reduced compared to NEMOLPC-KO animals (Figure 5A). Accordingly, liver extracts from NEMOLPC-KO;RIPK1LPC-KO mice showed activation of caspase-3 albeit at reduced levels compared to NEMOLPC-KO mice (Figure 5B). In addition, histological analysis showed substantial hepatocyte apoptosis and proliferation, inflammation and HSC activation in the liver of NEMOLPC-KO;RIPK1LPC-KO mice (Figure 5C). RIPK1LPC-KO mice did not show considerable liver damage and displayed marginally elevated serum ALT levels and only sparse focal hepatocyte apoptosis, suggesting that the liver damage in NEMOLPC-KO;RIPK1LPC-KO is caused by the lack of NEMO and not RIPK1 (Figures 5A and S5F).
In line with the reduced hepatocyte apoptosis in young mice, 1-year-old NEMOLPC-KO;Ripk1D138N/D138N or NEMOLPC-KO;RIPK1LPC-KO/D138N mice did not show macroscopic signs of liver tumor development (Figures 6A–6C). Histological examination confirmed the lack of HCC or pre-neoplastic lesions and only 23% of the livers examined exhibited areas with clear cell foci and anisokaryosis (Figures 6D and 6E). Interestingly, although the livers of NEMOLPC-KO;Ripk1D138N/D138N did not show HSC activation at 8 weeks, they exhibited mild portal fibrosis with septa formation at the age of 1 year (Figure 6D). Analysis of 1-year-old NEMOLPC-KO;RIPK1LPC-KO mice revealed the development of liver tumors with dysplastic foci, dysplastic nodules and HCC, although macroscopically ∼33% of the mice analyzed (7 out of 20) had smaller nodules compared to NEMOLPC-KO mice suggesting that RIPK1 deficiency inhibited tumor growth (Figures 6A–6E). Therefore, lack of RIPK1 kinase activity but not the absence of RIPK1 protein prevented hepatocyte apoptosis and HCC development in NEMOLPC-KO mice.
Figure 6.

Lack of RIPK1 Kinase Activity, but Not Its Scaffolding Function, Prevents HCC in NEMOLPC-KO Mice
(A) Representative liver images from 52-week-old mice with the indicated genotypes. Scale bar, 1 cm.
(B and C) Tumor load in mice with the indicated phenotypes as estimated by quantification of the LW/BW ratios (B), and the tumor size distribution (C). Horizontal lines indicate mean values.
(D) Representative images of Masson’s trichrome stained liver sections from 52-week-old mice with the indicated genotypes. Arrows indicate anisokaryotic hepatocytes. HCC/dysplastic nodule areas are marked with an asterisk. Scale bar, 200 μm.
(E) Histopathological evaluation of HCC development in 1-year-old mice with the indicated genotypes. Each color bar represents the % of livers per genotype in which the indicated stage was identified as the most advanced disease stage.
TRADD Mediates Apoptosis of NEMO-Deficient Hepatocytes in the Absence of RIPK1
The differential effects of RIPK1 kinase activity inhibition versus loss of its scaffolding functions suggested the existence of two distinct pathways triggering hepatocyte death in NEMO-deficient hepatocytes. When RIPK1 is present, its kinase activity is required for hepatocyte apoptosis, but when RIPK1 protein is absent, an alternative RIPK1-independent apoptotic pathway is induced. Since TRADD was shown to induce RIPK1-independent apoptosis (Vanlangenakker et al., 2011, Zheng et al., 2006), we examined the role of TRADD in NEMOLPC-KO mice by generating mice lacking both NEMO and TRADD in LPCs (Figure S6A). Analysis of serum ALT levels at the age of 8 weeks showed that LPC-specific TRADD deficiency did not prevent hepatocyte death and hepatitis in NEMOLPC-KO mice (Figure S6B). Furthermore, macroscopic and histological examination of livers from 1-year-old NEMOLPC-KO;TRADDLPC-KO mice revealed the development of HCC or pre-neoplastic lesions, although the number of macroscopically visible large tumors was decreased compared to littermates that were heterozygous for TRADD (Figures S6B–S6G). Therefore, TRADD deficiency did not prevent hepatocyte apoptosis and liver damage but partly inhibited liver tumor growth in NEMOLPC-KO mice.
To assess whether TRADD triggers apoptosis in NEMO/RIPK1-deficient hepatocytes, we generated NEMOLPC-KO;RIPK1LPC-KO;TRADDLPC-KO mice (Figure 7A). Strikingly, caspase-3 activation in the liver and serum ALT levels were strongly reduced in 8-week-old NEMOLPC-KO;RIPK1LPC-KO;TRADDLPC-KO mice compared to NEMOLPC-KO;RIPK1LPC-KO animals (Figures 7A and 7B). Accordingly, histological analysis of livers from NEMOLPC-KO;RIPK1LPC-KO;TRADDLPC-KO mice showed that apoptosis, compensatory proliferation, HSC activation and hepatitis were all nearly completely restored back to wild-type levels (Figure 7C). Moreover, 1-year-old NEMOLPC-KO;RIPK1LPC-KO;TRADDLPC-KO mice did not develop liver tumors (Figures 6E and 7D–7F). Therefore, in the absence of RIPK1 scaffolding function, TRADD-mediated apoptosis induces liver damage and HCC development in NEMOLPC-KO mice.
Figure 7.

TRADD Mediates Hepatocyte Apoptosis and HCC Development in the Absence of NEMO and RIPK1
(A) Immunoblot analysis of the indicated proteins in liver lysates from 8-week-old mice with the indicated genotypes. α-tubulin was used as loading control.
(B) Serum ALT levels in 8-week-old mice with the indicated genotypes. Horizontal lines indicate mean values.
(C) Representative images of liver sections from 8-week-old NEMOLPC-KO;RIPK1LPC-KO;TRADDLPC-KO mice immunostained with the indicated antibodies. Graphs depict quantification of the indicated parameters (mean ± SD, n = 3–5 mice per genotype). Scale bar, 200 μm.
(D and E) Representative images of livers (D) and Masson’s trichrome stained liver sections (E) from 52-week-old mice with the indicated genotypes. Scale bars, 1 cm (D); 200 μm (E).
(F) Tumor load quantification by LW/BW ratios. Horizontal lines indicate mean values.
See also Figure S6.
RIPK1 and TRADD are known to induce apoptosis downstream of TNFR1 via distinct protein complexes suggesting that TNF might contribute to hepatocyte apoptosis in NEMOLPC-KO mice. However, we showed previously that LPC-specific ablation of TNFR1, Fas or TRAIL alone or in combination did not prevent hepatocyte death and HCC development in NEMOLPC-KO mice, arguing that apoptosis of NEMO-deficient hepatocytes is not triggered by these death receptors (Ehlken et al., 2014). Furthermore, in contrast to a recent report (Cubero et al., 2013), we found that TNFR1 deficiency did not considerably diminish liver damage in 8-week-old NEMOLPC-KO;Tnfr1−/− mice (Figures S6H–S6K). One-year-old NEMOLPC-KO;Tnfr1−/− mice had mildly reduced ALT levels and LW/BW ratio compared to NEMOLPC-KO;Tnfr1WT/-, and they developed a similar number of dysplastic nodules although they lacked large tumors with histological characteristics of HCC (Figures S6I–S6L). Therefore, global TNFR1 deficiency did not prevent hepatocyte death and tumor initiation but delayed tumor progression in NEMOLPC-KO mice. These results show that TNF is not an essential driver of hepatocyte apoptosis and tumor development in NEMOLPC-KO mice and suggest that other, yet unidentified, TNF-independent pathways trigger this liver pathology.
NEMO Prevents the Formation of a RIPK1/FADD/Caspase-8 Complex in Hepatocytes
To gain mechanistic insight on how NEMO regulates RIPK1 kinase-mediated apoptosis, we analyzed the expression of proteins regulating the formation of the FADD/caspase-8 signaling complexes in NEMOLPC-KO mice. Expression of cFLIP, cIAP1, and TRAF2 was considerably diminished in NEMO-deficient livers and hepatocytes (Figures 8A and 8B). Notably, expression of kinase inactive RIPK1D138N, but not RIPK1 ablation, fully restored the expression of cFLIP and partly normalized the expression of TRAF2 and cIAP1 proteins in the NEMO-deficient livers (Figures 8A and 8B). Moreover, IKK2ca expression restored the protein levels of cIAP1 and SOD2 in NEMO-deficient livers (Figures 8C and S4C). Loss of cFLIP, cIAP1, and TRAF2 could trigger apoptosis in NEMO-deficient hepatocytes by promoting the association of RIPK1 with FADD and caspase-8, as shown for ripoptosome formation in response to DNA damage (Tenev et al., 2011). Indeed, immunoprecipitation of RIPK1 and subsequent immunoblotting with antibodies against FADD and caspase-8 revealed that these proteins interacted with RIPK1 in NEMO-deficient hepatocytes (Figure 8D). Immunoprecipitation of FADD confirmed the interaction of RIPK1 with FADD and caspase-8 in NEMO-deficient hepatocytes (Figure 8E). Importantly, expression of kinase inactive RIPK1D138N prevented the association of FADD and caspase-8 with RIPK1 in NEMO-deficient hepatocytes (Figures 8D and 8E), consistent with the restored expression of cFLIP, cIAP1 and TRAF2 in the liver of NEMOLPC-KO;Ripk1D138N/D138N mice. Collectively, these results suggested that NEMO protects hepatocytes from death by preventing the formation of a RIPK1/FADD/caspase-8 apoptosis-inducing complex.
Figure 8.

NEMO Prevents the Degradation of cFLIPL, cIAP1, and TRAF2 and the Formation of a RIPK1/FADD/Caspase-8 Complex
(A and B) Immunoblot analysis of cFLIPL, cIAP1 and TRAF2 in liver lysates (A) and primary hepatocytes cultured in vitro in the presence of z-VAD-fmk (B) from mice with the indicated genotypes. α-Tubulin and actin were used as loading controls.
(C) Immunoblot analysis of SOD2 in primary hepatocytes from mice with the indicated genotypes.
(D and E) Immunoblotting of RIPK1- (D) and FADD-containing (E) complexes immunoprecipitated from primary hepatocytes from mice with the indicated genotypes cultured for 20 hr in the presence of z-VAD-fmk. GAPDH was used as loading control. ns, non-specific band.
(F) Proposed model for the interplay between NEMO, NF-κB, and RIPK1 in the regulation of hepatocyte survival and death.
Discussion
The IKK/NF-κB signaling pathway has emerged as a key regulator of liver homeostasis and disease. In particular, the finding that mice with LPC-specific NEMO deficiency showed spontaneous hepatocyte apoptosis, chronic hepatitis and HCC suggested a key function of NF-κB in preventing hepatocyte death in the adult liver and development of liver cancer (Luedde et al., 2007, Pasparakis, 2009). To overcome the functional redundancy between NF-κB subunits, we studied the role of NF-κB in the adult liver by generating mice with LPC-specific ablation of all three NF-κB proteins capable of activating gene transcription, namely RelA, c-Rel, and RelB, thereby completely inhibiting NF-κB-mediated gene expression. Surprisingly, in contrast to NEMOLPC-KO mice, NF-κBLPC-KO mice showed only limited spontaneous hepatocyte apoptosis at young age and although a fraction of these mice showed increased liver damage, hepatitis, steatosis, and fibrosis at the age of 1 year they did not develop liver tumors. NEMOLPC-KO mice showed much more severe liver damage compared to NF-κBLPC-KO mice both spontaneously and in response to LPS, demonstrating that NEMO exhibits additional NF-κB-independent pro-survival functions in hepatocytes. Considering that NF-κB inhibition did not phenocopy NEMO deficiency, it was unexpected that overexpression of IKK2ca could prevent hepatocyte death and HCC development in NEMOLPC-KO mice. Our results that the IKK2ca protective effect largely depended on RelA suggested that IKK2ca prevented apoptosis of NEMO-deficient hepatocytes, at least in part, by inducing the NF-κB-dependent expression of pro-survival genes such as cIAP1, cIAP2, and A20. Moreover, IKK2ca expression also restored the expression of SOD2, which could protect NEMO-deficient hepatocytes from oxidative stress. Collectively, these results showed that NEMO ablation in LPCs triggers spontaneous hepatocyte death, chronic hepatitis, and HCC by inhibiting both NF-κB-dependent and NF-κB-independent pro-survival functions.
We showed previously that LPC-specific ablation of either IKK2 or IKK1 did not cause spontaneous liver pathology, but mice with combined IKK1/IKK2 or NEMO/IKK1 deficiency in LPCs developed severe jaundice and fatal cholangitis characterized by inflammatory destruction of small portal bile ducts, suggesting that combined inhibition of canonical and non-canonical NF-κB signaling resulted in cholestatic liver disease (Luedde et al., 2008). However, NF-κBLPC-KO and NEMOLPC-KO;RelBLPC--KO mice did not develop a similar biliary phenotype, suggesting that NF-κB-independent functions of IKK subunits control bile duct integrity in the liver.
Our results revealed important but opposing RIPK1 kinase-dependent and -independent functions in regulating liver disease in NEMOLPC-KO mice. Genetic inhibition of RIPK1 kinase activity strongly reduced hepatocyte death, thereby preventing liver damage and HCC development in NEMOLPC-KO mice. RIPK1 kinase activity induces both apoptosis and necroptosis (Pasparakis and Vandenabeele, 2015). However, RIPK3 deficiency did not prevent hepatitis and HCC development in NEMOLPC-KO mice, arguing that RIPK3/MLKL-driven necroptosis does not play an important role in this model. Moreover, most dying hepatocytes in NEMOLPC-KO mice stain positive for active caspase-3 and ablation of FADD or caspase-8 strongly inhibited hepatocyte death and prevented HCC development in these mice (Ehlken et al., 2014, Liedtke et al., 2011, Luedde et al., 2007). Together, these results show that RIPK1-mediated hepatocyte apoptosis causes chronic liver disease and HCC in NEMOLPC-KO mice. However, RIPK1 ablation could not prevent the death of NEMO-deficient hepatocytes showing that kinase-independent scaffolding properties of RIPK1 are required to prevent hepatocyte apoptosis. Remarkably, combined LPC-specific deficiency of RIPK1 and TRADD, but not of TRADD alone, prevented hepatocyte apoptosis and HCC development in NEMOLPC-KO animals. RIPK1 and TRADD have been suggested to trigger TNFR1-mediated apoptosis via distinct signaling complexes, the TRADD-dependent complex IIa and the RIPK1-dependent complex IIb (Christofferson et al., 2014, Vanden Berghe et al., 2014, Wang et al., 2008). Although TNFR1 deficiency did not prevent liver damage in NEMOLPC-KO mice, it remains unclear whether TNFR1 signaling might contribute to TRADD-dependent hepatocyte death in mice lacking both NEMO and RIPK1 in LPCs.
NEMO-deficient as well as NF-κB-deficient livers did not show considerably reduced transcription of anti-apoptotic genes, revealing that NF-κB-dependent transcriptional regulation is not essential to protect the adult liver under physiological conditions. However, NEMO deficiency resulted in reduced protein expression of cFLIP, cIAP1, and TRAF2 suggesting that NEMO inhibits hepatocyte apoptosis, at least in part, by preventing the degradation of these anti-apoptotic proteins. Furthermore, immunoprecipitation experiments revealed that a RIPK1/FADD/Caspase-8 signaling complex formed spontaneously in NEMO-deficient hepatocytes. Remarkably, expression of kinase inactive RIPK1D138N prevented the degradation of cFLIP, cIAP1, and TRAF2 and the formation of the RIPK1/FADD/Caspase-8 complex in NEMO-deficient livers. Single or combined knockout of TNFR1, Fas or TRAILR1 did not prevent liver damage and tumor development in NEMOLPC-KO mice (Ehlken et al., 2014), suggesting that alternative signals drive the formation of the RIPK1/FADD/caspase-8 apoptosis-inducing complex in NEMO-deficient hepatocytes. It is tempting to speculate that loss of cIAPs and cFLIP may lead to the assembly of a complex resembling the ripoptosome, which forms in response to DNA damage or SMAC-mimetic-induced cIAP degradation (Tenev et al., 2011). Ripoptosome formation was negatively regulated by cFLIP and required RIPK1 kinase activity, further supporting that similar mechanisms may trigger the formation of the RIPK1/FADD/caspase-8 complex inducing hepatocyte apoptosis in NEMOLPC-KO mice. However, it remains unclear whether upstream receptors or other stress response pathways are involved in triggering the loss of cIAPs and cFLIP and induction of apoptosis in NEMO-deficient hepatocytes.
NEMO was previously reported to prevent TNF-induced death of Jurkat T cells by inhibiting RIPK1-mediated activation of caspase-8 independently of NF-κB (Legarda-Addison et al., 2009). However, the molecular mechanisms by which NEMO deficiency induces cell death in Jurkat cells and hepatocytes are fundamentally different. In particular, TNF-induced apoptotic death of NEMO-deficient Jurkat cells was prevented by RIPK1 but not by FADD knockdown (Legarda-Addison et al., 2009). In sharp contrast, the death of hepatocytes, as well as the development of hepatitis and HCC in NEMOLPC-KO mice, were rescued by the ablation of FADD (Ehlken et al., 2014, Luedde et al., 2007) but not of RIPK1 (this study). Additionally, inhibition of RIPK1 kinase activity by Nec-1 did not inhibit TNF-induced apoptosis but prevented TNF+z-VAD-fmk-induced necroptosis of NEMO-deficient Jurkat cells (O’Donnell et al., 2012). In contrast, we showed that inhibition of RIPK1 kinase activity prevented hepatocyte apoptosis in NEMOLPC-KO mice, while necroptosis does not play a role in this model. Therefore, different mechanisms regulate the TNF-induced death of NEMO-deficient Jurkat T cells in vitro and apoptosis of NEMO-deficient hepatocytes in vivo.
Collectively, our results suggest that the interplay between NEMO, RIPK1, and NF-κB determines the survival or death of hepatocytes (Figure 8F). In wild-type livers, NEMO prevents hepatocyte apoptosis by inhibiting the formation of the death-inducing RIPK1/FADD/Caspase-8 signaling complex (complex IIb). This function of NEMO is likely mediated, at least partly, by preventing the RIPK1 kinase activity-dependent degradation of cIAPs, TRAF2, and cFLIP. In addition, NEMO-mediated NF-κB-dependent expression of SOD2 could also contribute to the anti-apoptotic effect by reducing oxidative stress, as feeding with the anti-oxidant butylated hydroxyanisole reduced liver damage in NEMOLPC-KO mice (Luedde et al., 2007). In the absence of NEMO, the RIPK1/FADD/Caspase-8 complex forms in a RIPK1 kinase activity-dependent manner and induces apoptosis. Enforced activation of NF-κB prevents apoptosis of NEMO knockout hepatocytes most likely by inducing the expression of pro-survival genes, such as cIAPs, A20, and SOD2. Lack of RIPK1 kinase activity prevents apoptosis of NEMO knockout hepatocytes by inhibiting the formation of the RIPK1/FADD/Casp8 complex. However, lack of RIPK1 protein removes not only its kinase-dependent but also its scaffolding functions, which are important to prevent degradation of cFLIP, cIAPs, and TRAF2, thereby promoting the formation of the TRADD/FADD/Casp8 complex (complex IIa) inducing apoptosis of NEMO-deficient hepatocytes.
Taken together, our results identify a NF-κB-independent function of NEMO in controlling RIPK1 kinase activity-dependent apoptosis of hepatocytes that is critical to prevent chronic liver damage and HCC development in mice. These results suggest that the interplay between NEMO and RIPK1 could be implicated in chronic hepatitis and hepatocarcinogenesis in humans. Interestingly, a recent study reported loss of NEMO immunoreactivity in a significant percentage of human HCC samples, which correlated with a poor 5-year overall survival (Aigelsreiter et al., 2012). These findings suggest that loss of NEMO-dependent cytoprotective functions may be critical for the development of chronic liver damage and HCC in at least a subset of patients. Hepatocyte death is a key feature of many chronic liver diseases, leading to inflammation, fibrosis, cirrhosis, and HCC development (Luedde et al., 2014). Our results identify RIPK1 kinase activity as a potent trigger of hepatocyte apoptosis that causes chronic liver disease culminating in the development of HCC. Although the role of RIPK1 in human liver disease remains unclear, it is possible that RIPK1 kinase activity triggers hepatocyte death in at least a subset of patients with chronic liver diseases leading to HCC, such as viral hepatitis, alcoholic and non-alcoholic steatohepatitis. Our results suggest that RIPK1 kinase inhibitors could provide an effective and likely well-tolerated treatment option for such patients.
Experimental Procedures
Mice
The following mouse lines were used: Rela/p65FL (Luedde et al., 2008), RelbFL (Powolny-Budnicka et al., 2011), RelFL (Heise et al., 2014), NemoFL (Schmidt-Supprian et al., 2000), R26IKK2casFL (Sasaki et al., 2006), Alfp-Cre (Kellendonk et al., 2000), TraddFL (Ermolaeva et al., 2008), Ripk3−/− (Newton et al., 2004), Ripk1D138N (Polykratis et al., 2014), Ripk1FL (Dannappel et al., 2014), and Tnfr1−/− (Pfeffer et al., 1993). All alleles were maintained on a C57BL/6 genetic background. Littermates carrying the floxed alleles but not the Alfp-Cre transgene served as controls. Animals were housed in individually ventilated cages in a specific pathogen-free (SPF) mouse facility at the Institute for Genetics, University of Cologne, kept under a 12-hr light cycle, and given a regular chow diet (Harlan, diet no. 2918) and water ad libitum. All animal procedures were conducted in accordance with European, national and institutional guidelines and protocols and were approved by local government authorities (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, Germany).
Mouse Experiments
LPS injections were performed on mice between 8 and 10 weeks of age. LPS (Sigma) was administered intraperitoneally, at a concentration of 25 μg/10 g of body weight, and blood was obtained prior to and 4 and 10 hr post injection. ALT levels were measured in serum using a Cobas c111 analyzer (Roche).
Histology, Immunohistochemistry, and Quantification
H&E, Masson’s trichrome staining, immunohistochemistry (IHC), and staining quantification was performed by standard procedures. For more detailed information, see the Supplemental Experimental Procedures.
Quantification of Macroscopically Visible Tumors and Histopathological Evaluation
Livers of 1-year-old mice were excised, digitally photographed with their dorsal side exposed, and weighed to calculate the liver/body weight ratio. The tumor number and size were determined by counting the number of visible tumors/nodules and measuring their diameter using ImageJ. Histopathological evaluation was performed as previously described (Ehlken et al., 2014) and explained in detail in the Supplemental Experimental Procedures.
Hepatocyte Isolation and Cell Death Assays
Primary hepatocytes were isolated from ∼4-week-old mice and cultured in DMEM (Sigma-Aldrich) containing 2% FCS, penicillin, and streptomycin. Cell death was estimated using an LDH release-based cytotoxicity assay (Promega) according to the manufacturers’ instructions. For more detailed information, see the Supplemental Experimental Procedures.
Immunoprecipitation, Subcellular Fractionation, and Immunoblotting
Immunoprecipitation (IP) experiments were performed in primary hepatocytes isolated from mice with the indicated genotypes and cultured for 20 hr in the presence of 20 μM zVAD-fmk. Subcellular fractionation, lysate preparation, immunoprecipitation, and immunoblotting was performed using standard protocols. For detailed description, see the Supplemental Experimental Procedures.
Statistical Analysis
Data shown in column graphs represent the mean ± SD, except tumor size distribution graphs that show the mean ± SEM. For those datasets that fulfilled D’Agostino and Pearson normality test criteria an unpaired Student’s t test was performed; otherwise nonparametric Mann-Whitney test was chosen. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.005. The analysis was performed using Prism software.
Author Contributions
V.K., A.P., H.E., L.O.-C., S.K.-S., and T.-M.V. performed genetic crosses, tissue sampling, and data interpretation. H.M.C. carried out some immunoblot assays. V.K. conducted the immunohistochemical evaluation and performed immunoblot, immunoprecipitation, and qRT-PCR assays. A.P. generated the RIPK1D138N and RIPK1 floxed mice in collaboration with M.K. and performed immunoblot analysis. F.W. provided RelB floxed mice. N.H. and U.K. provided c-Rel floxed mice. B.K.S. and P.S. performed histopathological analysis of mouse livers. M.P. conceived and coordinated the project and wrote the manuscript together with V.K.
Acknowledgments
We thank C. Uthoff-Hachenberg, J. Buchholz, E. Mahlberg, B. Kühnel, B. Hülser, D. Beier and E. Stade for technical assistance. We thank Vishva Dixit and Genentech for providing Ripk3−/− mice and anti-RIPK1 antibodies. Research reported in this publication was supported by grants from the European Research Council (ERC) (grant agreement no. 323040), the European Commission (FP7 grant 223151 [InflaCare]), the Deutsche Krebshilfe (grant 110302), Worldwide Cancer Research (grant 15-0228) and the Helmholtz Alliance Preclinical Comprehensive Cancer Center to M.P., and by the National Institute of Allergy and Infectious Diseases (NIAID) division of the NIH under award RO1AI075118 to M.K. V.K. was supported by a Marie Curie Career Development Fellowship (FP7-PEOPLE-2010-IEF; grant agreement no 275767), and B.K.S. by a grant of the DFG (STR 1160/1-2).
Published: November 9, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures and six figures and can be found with this article online at http://dx.doi.org/10.1016/j.ccell.2015.10.001.
Supplemental Information
References
- Aigelsreiter A., Haybaeck J., Schauer S., Kiesslich T., Bettermann K., Griessbacher A., Stojakovic T., Bauernhofer T., Samonigg H., Kornprat P. NEMO expression in human hepatocellular carcinoma and its association with clinical outcome. Hum. Pathol. 2012;43:1012–1019. doi: 10.1016/j.humpath.2011.08.009. [DOI] [PubMed] [Google Scholar]
- Ben-Neriah Y., Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- Christofferson D.E., Li Y., Yuan J. Control of life-or-death decisions by RIP1 kinase. Annu. Rev. Physiol. 2014;76:129–150. doi: 10.1146/annurev-physiol-021113-170259. [DOI] [PubMed] [Google Scholar]
- Cubero F.J., Singh A., Borkham-Kamphorst E., Nevzorova Y.A., Al Masaoudi M., Haas U., Boekschoten M.V., Gassler N., Weiskirchen R., Muller M. TNFR1 determines progression of chronic liver injury in the IKKγ/Nemo genetic model. Cell Death Differ. 2013;20:1580–1592. doi: 10.1038/cdd.2013.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannappel M., Vlantis K., Kumari S., Polykratis A., Kim C., Wachsmuth L., Eftychi C., Lin J., Corona T., Hermance N. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513:90–94. doi: 10.1038/nature13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlken H., Krishna-Subramanian S., Ochoa-Callejero L., Kondylis V., Nadi N.E., Straub B.K., Schirmacher P., Walczak H., Kollias G., Pasparakis M. Death receptor-independent FADD signalling triggers hepatitis and hepatocellular carcinoma in mice with liver parenchymal cell-specific NEMO knockout. Cell Death Differ. 2014;21:1721–1732. doi: 10.1038/cdd.2014.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Serag H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- Ermolaeva M.A., Michallet M.C., Papadopoulou N., Utermöhlen O., Kranidioti K., Kollias G., Tschopp J., Pasparakis M. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat. Immunol. 2008;9:1037–1046. doi: 10.1038/ni.1638. [DOI] [PubMed] [Google Scholar]
- Geisler F., Algül H., Paxian S., Schmid R.M. Genetic inactivation of RelA/p65 sensitizes adult mouse hepatocytes to TNF-induced apoptosis in vivo and in vitro. Gastroenterology. 2007;132:2489–2503. doi: 10.1053/j.gastro.2007.03.033. [DOI] [PubMed] [Google Scholar]
- Haybaeck J., Zeller N., Wolf M.J., Weber A., Wagner U., Kurrer M.O., Bremer J., Iezzi G., Graf R., Clavien P.A. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell. 2009;16:295–308. doi: 10.1016/j.ccr.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden M.S., Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise N., De Silva N.S., Silva K., Carette A., Simonetti G., Pasparakis M., Klein U. Germinal center B cell maintenance and differentiation are controlled by distinct NF-κB transcription factor subunits. J. Exp. Med. 2014;211:2103–2118. doi: 10.1084/jem.20132613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellendonk C., Opherk C., Anlag K., Schütz G., Tronche F. Hepatocyte-specific expression of Cre recombinase. Genesis. 2000;26:151–153. doi: 10.1002/(sici)1526-968x(200002)26:2<151::aid-gene17>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Legarda-Addison D., Hase H., O’Donnell M.A., Ting A.T. NEMO/IKKgamma regulates an early NF-kappaB-independent cell-death checkpoint during TNF signaling. Cell Death Differ. 2009;16:1279–1288. doi: 10.1038/cdd.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke C., Bangen J.M., Freimuth J., Beraza N., Lambertz D., Cubero F.J., Hatting M., Karlmark K.R., Streetz K.L., Krombach G.A. Loss of caspase-8 protects mice against inflammation-related hepatocarcinogenesis but induces non-apoptotic liver injury. Gastroenterology. 2011;141:2176–2187. doi: 10.1053/j.gastro.2011.08.037. [DOI] [PubMed] [Google Scholar]
- Luedde T., Schwabe R.F. NF-κB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011;8:108–118. doi: 10.1038/nrgastro.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedde T., Beraza N., Kotsikoris V., van Loo G., Nenci A., De Vos R., Roskams T., Trautwein C., Pasparakis M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Luedde T., Heinrichsdorff J., de Lorenzi R., De Vos R., Roskams T., Pasparakis M. IKK1 and IKK2 cooperate to maintain bile duct integrity in the liver. Proc. Natl. Acad. Sci. USA. 2008;105:9733–9738. doi: 10.1073/pnas.0800198105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedde T., Kaplowitz N., Schwabe R.F. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology. 2014;147:765–783. doi: 10.1053/j.gastro.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S., Kamata H., Luo J.L., Leffert H., Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Micheau O., Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- Newton K., Sun X., Dixit V.M. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol. Cell. Biol. 2004;24:1464–1469. doi: 10.1128/MCB.24.4.1464-1469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell M.A., Hase H., Legarda D., Ting A.T. NEMO inhibits programmed necrosis in an NFκB-independent manner by restraining RIP1. PLoS ONE. 2012;7:e41238. doi: 10.1371/journal.pone.0041238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat. Rev. Immunol. 2009;9:778–788. doi: 10.1038/nri2655. [DOI] [PubMed] [Google Scholar]
- Pasparakis M., Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- Perkins N.D. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer. 2012;12:121–132. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- Pfeffer K., Matsuyama T., Kündig T.M., Wakeham A., Kishihara K., Shahinian A., Wiegmann K., Ohashi P.S., Krönke M., Mak T.W. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- Pikarsky E., Porat R.M., Stein I., Abramovitch R., Amit S., Kasem S., Gutkovich-Pyest E., Urieli-Shoval S., Galun E., Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- Polykratis A., Hermance N., Zelic M., Roderick J., Kim C., Van T.M., Lee T.H., Chan F.K., Pasparakis M., Kelliher M.A. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol. 2014;193:1539–1543. doi: 10.4049/jimmunol.1400590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powolny-Budnicka I., Riemann M., Tänzer S., Schmid R.M., Hehlgans T., Weih F. RelA and RelB transcription factors in distinct thymocyte populations control lymphotoxin-dependent interleukin-17 production in γδ T cells. Immunity. 2011;34:364–374. doi: 10.1016/j.immuni.2011.02.019. [DOI] [PubMed] [Google Scholar]
- Sasaki Y., Derudder E., Hobeika E., Pelanda R., Reth M., Rajewsky K., Schmidt-Supprian M. Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity. 2006;24:729–739. doi: 10.1016/j.immuni.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Schattenberg J.M., Schuchmann M., Galle P.R. Cell death and hepatocarcinogenesis: Dysregulation of apoptosis signaling pathways. J. Gastroenterol. Hepatol. 2011;26(Suppl 1):213–219. doi: 10.1111/j.1440-1746.2010.06582.x. [DOI] [PubMed] [Google Scholar]
- Schmidt-Supprian M., Bloch W., Courtois G., Addicks K., Israël A., Rajewsky K., Pasparakis M. NEMO/IKK gamma-deficient mice model incontinentia pigmenti. Mol. Cell. 2000;5:981–992. doi: 10.1016/s1097-2765(00)80263-4. [DOI] [PubMed] [Google Scholar]
- Shariff M.I., Cox I.J., Gomaa A.I., Khan S.A., Gedroyc W., Taylor-Robinson S.D. Hepatocellular carcinoma: current trends in worldwide epidemiology, risk factors, diagnosis and therapeutics. Expert Rev. Gastroenterol. Hepatol. 2009;3:353–367. doi: 10.1586/egh.09.35. [DOI] [PubMed] [Google Scholar]
- Sun S.C. The noncanonical NF-κB pathway. Immunol. Rev. 2012;246:125–140. doi: 10.1111/j.1600-065X.2011.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B., Karin M. NF-kappaB signaling, liver disease and hepatoprotective agents. Oncogene. 2008;27:6228–6244. doi: 10.1038/onc.2008.300. [DOI] [PubMed] [Google Scholar]
- Tenev T., Bianchi K., Darding M., Broemer M., Langlais C., Wallberg F., Zachariou A., Lopez J., MacFarlane M., Cain K., Meier P. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell. 2011;43:432–448. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T., Linkermann A., Jouan-Lanhouet S., Walczak H., Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- Vanlangenakker N., Bertrand M.J., Bogaert P., Vandenabeele P., Vanden Berghe T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011;2:e230. doi: 10.1038/cddis.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak H., Iwai K., Dikic I. Generation and physiological roles of linear ubiquitin chains. BMC Biol. 2012;10:23. doi: 10.1186/1741-7007-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. Molecular mechanisms of hepatic apoptosis. Cell Death Dis. 2014;5:e996. doi: 10.1038/cddis.2013.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Du F., Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- Weinlich R., Dillon C.P., Green D.R. Ripped to death. Trends Cell Biol. 2011;21:630–637. doi: 10.1016/j.tcb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L., Bidere N., Staudt D., Cubre A., Orenstein J., Chan F.K., Lenardo M. Competitive control of independent programs of tumor necrosis factor receptor-induced cell death by TRADD and RIP1. Mol. Cell. Biol. 2006;26:3505–3513. doi: 10.1128/MCB.26.9.3505-3513.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.