

Abstract
Strain K22T is the type species of the recently- described genus Pyrinomonas, in subdivision 4 of the phylum Acidobacteria (Int J Syst Evol Micr. 2014; 64(1):220–7). It was isolated from geothermally-heated soil from Mt. Ngauruhoe, New Zealand, using low-nutrient medium. P. methylaliphatogenes K22T has a chemoheterotrophic metabolism; it can hydrolyze a limited range of simple carbohydrates and polypeptides. Its cell membrane is dominated by iso-branching fatty acids, and up to 40 % of its lipid content is membrane-spanning and ether lipids. It is obligately aerobic, thermophilic, moderately acidophilic, and non-spore-forming. The 3,788,560 bp genome of P. methylaliphatogenes K22T has a G + C content of 59.36 % and contains 3,189 protein-encoding and 55 non-coding RNA genes. Genomic analysis was consistent with nutritional requirements; in particular, the identified transporter classes reflect the oligotrophic nature of this strain.
Electronic supplementary material
The online version of this article (doi:10.1186/s40793-015-0099-5) contains supplementary material, which is available to authorized users.
Keywords: Acidobacteria, Pyrinomonas, New Zealand, Thermophile, Soil, Geothermal
Introduction
Phylotypes from the phylum Acidobacteria1 are commonly detected across a range of ecosystems, including marine and freshwater bodies, sediments, geothermal systems, and soils. Despite the apparent ubiquitous distribution acidobacterial phyotypes, particularly in soil environments, only 17 acidobacterial genera (represented by formal description and publication of respective type strains, in accordance with the International Code of Nomenclature of Prokaryotes [1]) have been validly published [2, 3]. Here we present a description of the complete genome sequence and annotation of Pyrinomonas methylaliphatogenes strain K22T (= DSM 25857 = ICMP 18710), the type species of the genus Pyrinomonas within subdivision 4 of Acidobacteria.
Pyrinomonas methylaliphatogenes K22T was isolated from a fumarole on the outer crater rim of the stratovolcano Mt. Ngauruhoe [4]. It exhibits a Gram-negative cell wall, is non-spore-forming, and is catalase- and oxidase-positive (Table 1). It is a thermophilic and moderately acidophilic obligately aerobic chemoorganotroph. Of particular note is its unusual lipid composition that is dominated by odd-numbered saturated iso-branching fatty acids (iso-C15:0, iso-C17:0, iso-C19:0 and iso-C21:0 that total >88.5 % of the total fatty acid extract) [4]. In addition, >40 % of the total membrane lipid content is made up by iso-branching glycerol ether analogues of the cellular fatty acids and membrane-spanning iso-diabolic acids [5]. Membrane-spanning and ether lipids occur ubiquitously in Archaea, but in recent studies have also been commonly detected in cultivated representatives in subdivision groups 1, 3 and 4 of Acidobacteria [5, 6].
Table 1.
Classification and general features of P. methylaliphatogenes K22T
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [35] | |
| Phylum Acidobacteria | TAS [36] | ||
| Class ‘Insertae sedis 99’ | |||
| Order ‘Insertae sedis 100’ | |||
| Family ‘Insertae sedis 101’ | |||
| Genus Pyrinomonas | TAS [4] | ||
| Species Pyrinomonas methylaliphatogenes | TAS [4] | ||
| Type strain K22T (=DSM 25857T =ICMP 18710T). | TAS [4] | ||
| Gram stain | negative | TAS [4] | |
| Cell shape | rod | TAS [4] | |
| Motility | non-motile | TAS [4] | |
| Sporulation | non-sporulating | TAS [4] | |
| Temperature range | thermophilic (50–69 °C) | TAS [4] | |
| Optimum temperature | 65 °C | TAS [4] | |
| pH range | moderately acidophilic (4.1–7.8) | ||
| Optimum pH | 6.5 | ||
| Carbon source | peptides, proteins, carbohydrates | TAS [4] | |
| Terminal electron receptor | oxygen | TAS [4] | |
| Energy metabolism | chemoorganotroph | TAS [4] | |
| MIGS-6 | Habitat | geothermal soil | TAS [37] |
| MIGS-6.3 | Salinity | non-halophile (no growth > 1 % (w/v) NaCl) | TAS [4] |
| MIGS-22 | Oxygen requirement | obligate aerobe | TAS [4] |
| MIGS-15 | Biotic relationship | free-living | TAS [4] |
| MIGS-14 | Pathogenicity | not reported | NAS |
| MIGS-4 | Geographic location | Mt Ngauruhoe, New Zealand | TAS [37] |
| MIGS-5 | Sample collection | 2006 | NAS |
| MIGS-4.1 MIGS-4.2 | Latitude – Longitude | 39° 9’25.31”S - 175° 38’6.74”E | IDA |
| MIGS-4.3 | Depth | not reported | IDA |
| MIGS-4.4 | Altitude | 2,270 m | IDA |
aEvidence codes - IDA inferred from direct assay, TAS traceable author statement (i.e., a direct report exists in the literature), NAS non-traceable author statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [38]
Subdivision 4 of the Acidobacteria has five validly-named species: P. methylaliphatogenes K22T,[4] Chloracidobacterium thermophilum [7, 8], Blastocatella fastidiosa [9], Aridibacter famidurans, and Aridibacter kavangonensis [3]. The latter three species are phylogenetically distant from P. methylaliphatogenes K22T, are mesophilic and have differing pH ranges and substrate utilization profiles from that of P. methylaliphatogenes K22T. Chloracidobacterium thermophilum is a moderately thermophilic facultatively anoxygenic photoheterotroph isolated from a hotspring microbial mat at Yellowstone National Park [7, 8]. An additional strain, Ellin6075 was isolated from an Australian pasture soil, and is a mesophilic heterotroph that derives its energy from complex carbohydrate sources, but has little information available regarding its phenotypic traits [10]. Common features shared by subdivision 4 strains include an aerobic and heterotrophic phenotype [3, 4], and membrane lipid iso-diabolic acids [5].
Organism information
Classification and features
Phylogenetic distances of closest-related phylotypes and cultivated subdivision 4 acidobacterial strains were determined by aligning the representative near full length 16S rRNA gene sequences (all sequences were > 1,400 nucleotides in length) and calculating sequence similarity via a pair-wise alignment within the ARB software environment [11]. Analysis showed that the 16S rRNA gene sequence of P. methylaliphatogenes K22T (AM749787) is 85 % similar to B. fastidiosa strain A2-16T (JQ309130), and is 84 % similar to both A. famidurans strain A22_HD_4HT (KF245634), and A. kavangonensis Ac_23_E3T (KF245633) [3, 4, 9]. In addition, P. methylaliphatogenes K22T shares 85 % 16S rRNA gene sequence similarity with both Ellin6075 (AY234727) [7] and C. thermophilum BT (EF531339) [8]. The most closely-related phylotypes to P. methylaliphatogenes K22T are two sequences from clonal libraries of environmental 16S rRNA genes (EU490264, EU490279) retrieved from geothermal soils on Mt. Erebus, Antarctica [12]; both of these shared 95 % 16S rRNA gene sequence similarity with P. methylaliphatogenes K22T. Phylogenetic comparison (Fig. 1) showed that P. methylaliphatogenes K22T is a taxonomically-distinct genus and species of subdivision 4 in the phylum Acidobacteria.
Fig. 1.
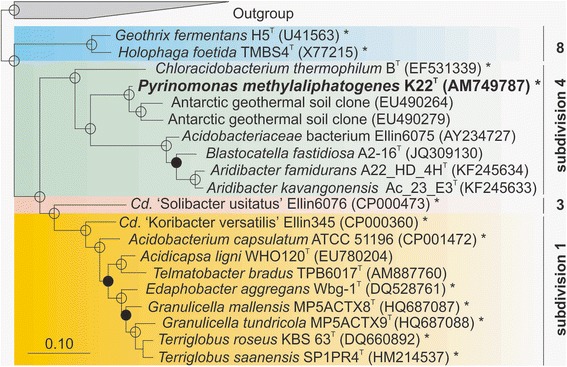
Phylogenetic tree based on 16S rRNA gene sequences of Pyrinomonas methylaliphatogenes K22T (highlighted) and other cultivated strains and clonal phylotypes within the phylum Acidobacteria. Four of the acidobacterial subdivisions are included. The tree was constructed via a Bayesian inference model (MrBayes), using Markov Chain Monte Carlo (MCMC - 2,000,000 resamples, four chains, temperature = 0.5) sampling methods to calculate posterior distributions of trees in the ARB software environment. Posterior probability values ≥ 90 % are indicated by open circles, ≥80 % by filled circles, and ≥70 % by open diamonds. The scale bar represents a 0.1 change per nucleotide position. Strains whose genomes have been sequenced, are marked with an asterisk; G. fermentans H5T (NZ_AUAU00000000), H. foetida TMBS4T (AGSB00000000), C. thermophilum BT (CP002414), P. methylaliphatogenes K22T (CBXV000000000), Candidatus ‘S. usitatus’ Ellin6076 (CP000473), Candidatus ‘K. versatilis’ Ellin345 (CP000360), Acidobacterium capsulatum ATCC 51196T (CP001472), Edaphobacter aggregans Wbg-1T (JQKI00000000), Granulicella mallensis MP5ACTX9T (CP003130), Granulicella tundricola MP5ACTX9T (CP002480), Terriglobus roseus KBS63T (CP003379), and Terriglobus saanensis SP1PR4T (CP002467). The phylotypes strains used as an outgroup included Thermoanaerobaculum aquaticum MP-01T (JX4200244), Dictyoglomus thermophilum H-6-12T (X69194), Caldisericum exile AZM16c01T (AB428365), Hydrogenobacter hydrogenophilus Z-829T (Z30424), Thermodesulfobacterium thermophilum DSM 1276T (AF334601), Deinococcus roseus TDMA-uv51 (AB264136), Truepera radiovicrix RQ-24T (DQ022076), Thermus aquaticus YT-1 (L09663), and Thermus scotoductus SE-1T (AF032127)
Pyrinomonas methylaliphatogenes K22T is non-motile and exhibits straight or bent rod cell morphology (0.3 – 0.6 μm in diameter and 1–4 μm in length) (Fig. 2). It has a temperature range (optimum) for growth of 50–69 °C (65 °C) and a pH range (optimum) of 4.1–7.8 (6.5). The bacterium has an obligately aerobic metabolism and can utilize a small selection of simple carbohydrates including glucose, lactate, alginate, mannose, xanthan, xylan, xylose, arabinose, and sucrose, as well as a limited variety of proteinaceous substrates including casamino acids, peptone, tryptone, yeast extract and nutrient broth (Table 1). It obtains nitrogen via the uptake of NO3−, NH4+, urea, yeast extract and casamino acids but cannot fix dinitrogen gas. The strain is not able to grow via photosynthesis, nor is it able grow autotrophically using CO2 as the sole source of carbon. However, optical density of culture is improved via the provision of additional CO2 in the headspace during heterotrophic growth, suggesting an assistive anapleurotic mechanism [4].
Fig. 2.
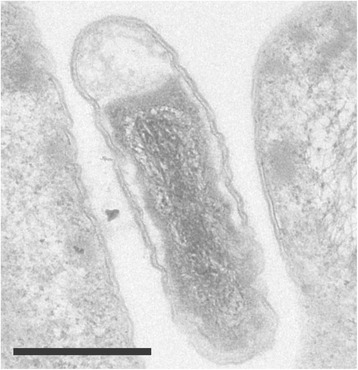
Transmission electron micrograph of P. methylaliphatogenes K22T cultured in R2A liquid medium (60 °C), using a Zeiss LEO 912 Energy-Filtering TEM [34]. The scale bar represents 500 nm
Chemotaxonomic data
The primary cellular fatty acids are iso-C15:0 (40.8 %), iso-C17:0 (30.8 %), iso-C19:0 (12.1 %) and iso-C21:0 (4.8 %). P. methylaliphatogenes K22T also possesses membrane-spanning dicarboxylic acid 13,16-dimethyl octacosanedioic (iso-diabolic) acid and glyceryl ethers of alkyl analogues of iso-C15:0 and iso-C17:0 and iso-diabolic acid. Its primary cellular quinone is MK-8 and its primary cellular lipids are phosphatidylethanolamine and phosphatidylcholine [4].
Genome sequencing information
Genome project history
The genome of P. methylaliphatogenes K22T was selected for sequencing on the basis of its phylogenetic position and phenotypic dissimilarity to other cultured Acidobacteria strains. The quality draft (QD) assembly and annotation was completed in December 2013. The genome project is deposited in the Genomes OnLine Database Gp0050834. A summary of the project information is shown in Table 2. The EMBL-Bank project accession number is CBXV000000000 and consists of 16 scaffolds. Table 2 presents the project information and its association with MIGS version 2.0 compliance [13].
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | High quality draft |
| MIGS-28 | Libraries used | Two libraries used: One 454 library, one Illumina PE library |
| MIGS-29 | Sequencing platforms | 454 GS Junior Titanium, Illumina MiSeq |
| MIGS-31.2 | Fold coverage | 75.0 × |
| MIGS-30 | Assemblers | MIRA 4.0rc2 |
| MIGS-32 | Gene calling method | Prodigal |
| Locus tag | PYK22 | |
| EMBL ID | CBXV000000000 | |
| EMBL Date of Release | 12 January 2015 | |
| GOLD ID | Gp0050834 | |
| BIOPROJECT | PRJEB4906 | |
| MIGS-13 | Source Material Identifier | DSMZ DSM 25857, ICMP ICMP 18710 |
| Project relevance | Microbial diversity of the Taupō Volcanic Zone, Tree of Life |
Growth conditions and genomic DNA preparation
Pyrinomonas methylaliphatogenes K22T was grown in 2 × 500 ml volumes of R2A liquid medium [14] at 60 °C in an air headspace (1 : 1 ratio of headspace to medium). The medium was sterilized at 121 °C (15 min, 15 psi) prior to inoculation. After three days of incubation, cells were collected via centrifugation. Culture purity was confirmed using an RFLP digestion (EcoR1) of the 16S rRNA gene PCR amplification product (amplification used the 9f/1492r primer set) [4]. The restriction digest pattern was identical to known axenic cultures of P. methylaliphatogenes K22T. Genomic DNA was extracted from the wet biomass (200 mg) using the Nucleospin for Tissue extraction kit as per the manufacturer’s instructions (Macherey Nagel). The gDNA extract was purified via electrophoresis on a 0.8 % (w/v) agarose gel. The gel extracts were cleaned using a Gel Purification kit as per the manufacturer’s instructions (Macherey Nagel), giving a final concentration of 595 ng 100 μl−1. The purified gDNA was then frozen at −20 °C until sequenced.
Genome sequencing and assembly
Genomic sequencing was conducted using a combination of the Illumina MiSeq and 454 GS Junior platforms. A single-end 454 library was constructed according to the protocols of 454 GS FLX Titanium Rapid Library kits and GS Junior Titanium emPCR kits (Additional file 1). The sequencing of the 454 library yielded 75,215 reads with an average length of 492 bps. The paired-end Illumina library was constructed using the Nextera XT DNA Sample Preparation kit (Illumina), according to the manufacturer's protocol (Additional file 1), and sequenced on a MiSeq (2 × 150 bp paired-end reads), yielding 1,196,578 reads. The combined 454 (28.9 Mbp) and Illumina (301 Mbp) sequencing data were assembled together using the hybrid assembly capability of MIRA 4.0 rc4 [15] (parameter and methodologies provided in Additional file 1). The resulting contigs were manually curated via the Staden package [16], generating scaffolds with an average 75 × coverage. Scaffolds with average coverage two standard deviations below the aforementioned overall genome average were discarded (i.e. 32.5 × coverage threshold). The resulting 16 scaffolds contained 2,302,690 assembled reads and 3188 protein coding genes. The abundance of clustered regularly interspaced short palindromic repeats (CRISPRs) and other repeating elements (e.g. transposons and RHS repeat-encoded genes) may have contributed to the scaffolds junctions, such as those observed in scaffold CBXV010000001, CBXV010000004, CBXV010000005, and CBXV010000006.
Genome annotation
Genome annotation was processed via the DOE-JGI Integrated Microbial Genome – Expert Review (IMG-ER) annotation pipeline [17] using the following steps/components: Coding sequences (CDSs) were predicted using Prodigal [18]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non-redundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. These data sources were combined to ascribe descriptions of the protein tRNAScan-SE tool [19] was used to find tRNA genes, whereas ribosomal RNAs were found by searching against models of the ribosomal RNA genes built from SILVA. Other non-coding RNA such as the RNA components of the protein secretion complex and the RNaseP were identified by searching the genome for the corresponding Rfam profiles using INFERNAL [20]. Transmembrane helices and signal peptide cleavage sites within the putative proteins were predicted via TMHMM [21], and SignalP [22] respectively. Additional annotation and gene function prediction as well as data visualization was conducted within the IMG-ER system [23].
Genome properties
The QD assembly of the genome consists of 16 scaffolds totaling 3,788,560 bp in length (59.36 % GC content). Of the 3,244 genes predicted, 3,189 were protein-coding genes, and 55 were non-coding RNA genes. A majority (79.0 %) of genes were assigned putative functions, and the remainder were annotated as hypothetical proteins. The properties and the statistics of the P. methylaliphatogenes K22T genome and the distribution of genes into COG functional categories are presented in Table 3, Table 4, and Fig. 3.
Table 3.
Genome statistics
| Attribute | Genome (total) | |
|---|---|---|
| Value | % of totala | |
| Size (bp) | 3,788,560 | 100.0 |
| DNA coding (bp) | 3,353,298 | 88.5 |
| G + C content (bp) | 2,249,198 | 59.36 |
| DNA Scaffolds | 16 | |
| Total genesb | 3,244 | 100.00 |
| Protein-coding genes | 3,189 | 98.3 |
| RNA genes | 55 | 1.7 |
| Pseudo genes | 0 | 0.0 |
| Genes in paralog clusters | 2535 | 78.4 |
| Protein coding genes with function prediction | 2,564 | 79.0 |
| Genes assigned to COGs | 2,023 | 62.3 |
| Genes assigned Pfam domain | 2,605 | 80.3 |
| Genes with signal peptides | 293 | 9.0 |
| Genes with transmembrane helices | 766 | 23.7 |
| CRISPR repeats | 15 | |
aThe percentage total is based on either the size of the genome in base pairs or the total number of protein coding genes in the annotated genome
Table 4.
Number of genes associated with the general COG functional categories
| Code | Value | % of totala | Description |
|---|---|---|---|
| J | 137 | 5.01 | Translation, ribosomal structure and biogenesis |
| A | 1 | 0.03 | RNA processing and modification |
| K | 103 | 3.23 | Transcription |
| L | 77 | 2.41 | Replication, recombination and repair |
| B | 2 | 0.06 | Chromatin structure and dynamics |
| D | 27 | 0.85 | Cell cycle control, cell division, chromosome partitioning |
| V | 65 | 2.04 | Defense mechanisms |
| T | 101 | 3.17 | Signal transduction mechanisms |
| M | 191 | 5.99 | Cell wall/membrane/envelope biogenesis |
| N | 67 | 2.10 | Cell motility |
| U | 32 | 1.00 | Intracellular trafficking and secretion |
| O | 123 | 3.85 | Posttranslational modification, protein turnover, chaperones |
| C | 127 | 3.98 | Energy production and conversion |
| G | 171 | 5.36 | Carbohydrate transport and metabolism |
| E | 202 | 6.33 | Amino acid transport and metabolism |
| F | 65 | 2.04 | Nucleotide transport and metabolism |
| H | 126 | 3.95 | Coenzyme transport and metabolism |
| I | 105 | 3.29 | Lipid transport and metabolism |
| P | 105 | 3.29 | Inorganic ion transport and metabolism |
| Q | 64 | 2.01 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 218 | 6.83 | General function prediction only |
| S | 85 | 2.66 | Function unknown |
| - | 1,223 | 38.33 | Not in COGs |
aThe total is based on the total number of protein coding genes (3180) in the annotated genome
Fig. 3.
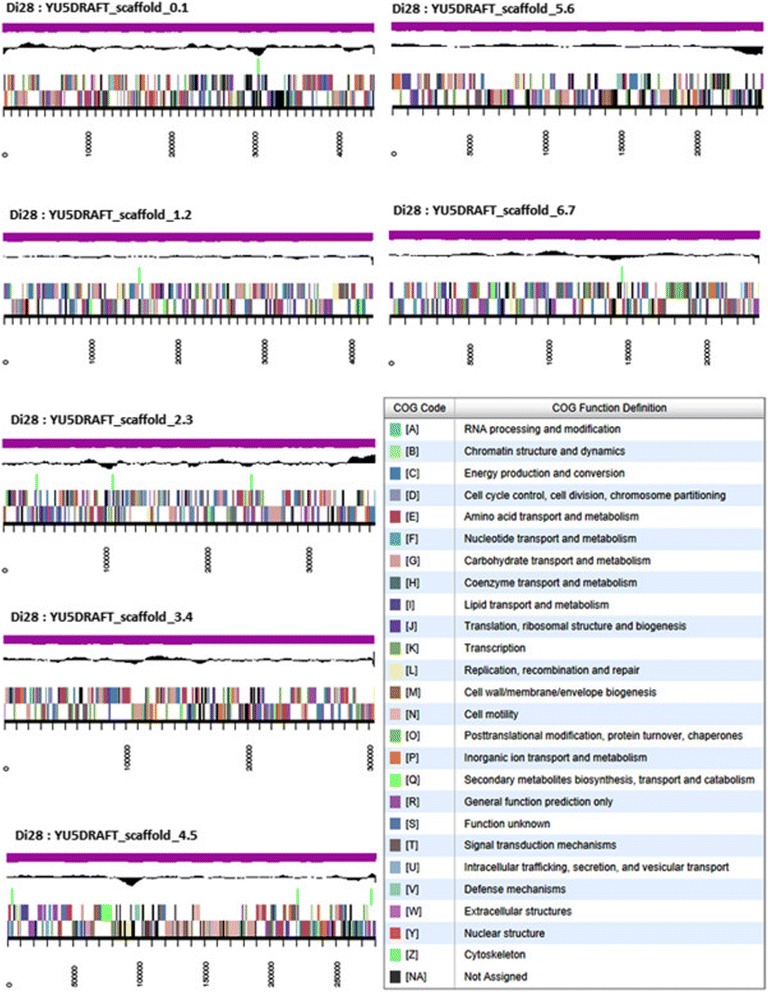
Graphical map of the genome of P. methylaliphatogenes K22T showing the eight largest scaffolds. From bottom to the top of each scaffold: Genes on forward strand (color by COG categories as denoted by the IMG platform), genes on the reverse strand (color by COG categories), RNA genes (tRNAs – green, sRNAs – red, other RNAs – black), GC content, and GC skew
Insights from the genome sequence
The P. methylaliphatogenes K22T genome assembly has a size of 3.79 Mb with a %G + C content of 59.3, both of which are comparable with the genomes of other sequenced Acidobacteria [24]. It possesses complete citric acid and pentose phosphate cycles. A complete electron transport pathway with an F-type ATPase, NADH dehydrogenase and cytochrome C complex, and the presence of genes encoding superoxide dismutase (PYK22_00483-00484) and catalase (PYK22_02691) are consistent with the observed aerobic phenotype. Genes encoding outer membrane secretion (for example, a type II secretion system, PYK22_02507-02511) and protein assembly (Bam complex, PYK22_02371 & 01777) are present, confirming the observed Gram-negative cell wall structure [4]. Interestingly, P. methylaliphatogenes K22T possesses a near-complete complement of flagella encoding-genes (possibly missing the proximal rod flgF gene) despite having no observed motility. Key genes for all autotrophic carbon fixation pathways were absent. However, it was previously noted that while P. methylaliphatogenes K22T was unable to fix carbon, additional CO2 to the headspace while growing heterotrophically improved growth [4]. The presence of phosphoenolpyruvate carboxylase and isocitrate dehydrogenase confirmed the ability of P. methylaliphatogenes K22T to supplement carbon anapleurotically. No genes encoding the ability to fix dinitrogen gas were found, again confirming previous phenotypic observations. Interestingly, the genome contains a gene cluster encoding a group 5-type [NiFe] hydrogenase (PYK22_03058-03084) similar to that found in Mycobacterium smegmatis [25]; this may confer an ability to oxidize tropospheric concentrations of hydrogen for cell maintenance.
Previous phenotypic characterization of P. methylaliphatogenes K22T indicated that it possessed a heterotrophic phenotype with the ability to grow on a range of simple carbohydrates. The P. methylaliphatogenes K22T genome encodes for a large number of beta-glucosidase and exoglucanase-acting glycosyl hydrolases, reflecting its ability to grow on primarily simple oligosaccharides such as cellobiose, sucrose, and maltose. A single C6 endoglucanase-acting glycosyl hydrolase (PYK22_03181) was identified in the genome despite having no reported growth on complex or crystalline cellulose as energy sources [4]. Two endo-1,4-beta-xylanases genes confer an ability to grow on xylan and xanthan gum.
Transporters encoded in the P. methylaliphatogenes K22T genome mainly belong to the ABC-type transporter superfamily and the major facilitator superfamily. This is consistent with previous study of acidobacterial genomes, which suggest these transporters types were adapted for low-nutrient conditions [26]. ABC transporters in P. methylaliphatogenes K22T appear to be involved in the transport of carbohydrates (and derivatives) such as ribose, D-xylose, lipopolysaccharide (rfbAB, e.g. PYK22_01076-77, PYK22_01839-40, PYK22_02287-88), and lipo-oligosaccharide (nodJI, PYK22_00778 and PYK22_00785). These reflect the carbohydrate and polypeptide utilizing phenotype of the bacterium. Pyrinomonas methylaliphatogenes K22T also possesses putative ABC transporters targeting amino acid cysteine, oligopeptides (oppABCDF, e.g. the PYK22_01277-281 cluster), and lipoproteins (lolCDE, PYK22_02373-4). Nitrogen assimilation is facilitated via an ammonia permease (PYK22_02853), the importation of oligopeptides by an oppABCDF ABC transporter system (similar to the system in Salmonella typhimurium [27]), and major facilitator superfamily nitrate/nitrite permeases (PYK22_00018 & PYK22_00946). Additionally, the P. methylaliphatogenes K22T genome contained a cluster of genes tonB-exbB-exbD-exbD (PYK22_00991-94) associated with siderophore transport in some other acidobacterial species [26]. However, genes involved in siderophore synthesis, polyketide synthase, and nonribosomal peptide synthetase were not found, suggesting that it scavenges siderophores produced by other bacteria.
Based upon 16S rRNA gene sequence similarity, the most closely related and cultivated strain to P. methylaliphatogenes K22T is C. thermophilum BT [28] (Fig. 1). The sequence similarity (~86 %) indicates that the two strains may belong to the same subdivision based on taxonomic sequence identity thresholds calculated for other prokaryotic taxa [29]. This phylogenetic dissimilarity between the two strains is also reflected in a comparison of the genomic content and the different metabolic modes of existence (chemoheterotrophic P. methylaliphatogenes K22T vs. photoheterotrophic C. thermophilum BT) of the two strains. For example, the C. thermophilum BT genome encodes for genes for chlorosomes, bacteriochlorophyll pigments a and c and a pigment protein complex for phototrophic growth, whereas no genes encoding for phototrophy were found in K22T. The C. thermophilum BT genome also contained significantly more COGs (15 vs 50) related to signal transduction kinases (COG0515 and COG0642) than were encoded in P. methylaliphatogenes K22T. Conversely, P. methylaliphatogenes K22T contained more genes related to amino acid utilization, such as amino acid transporters (COG0531) and amidohydrolases (COG1228), reflecting its ability to grow using proteinaceous media as the carbon and energy source. While both species possess carbohydrate-related metabolisms, the P. methylaliphatogenes K22T genome encodes a much larger number of glycosyltransferases (COG0438 and COG0463) and beta-glucosidase-related glycosidases (COG1472) than that of C. thermophilum BT.
Conclusions
Acidobacteria is one of the most widely-distributed bacterial phyla, particularly in soils [30–32]. Despite the wide distribution, the number of cultivated and sequenced representatives within most subdivisions within Acidobacteria remains low [33]. The sequencing and annotation of the P. methylaliphatogenes K22T genome presented here links the phenotypic traits of P. methylaliphatogenes K22T [4] with its genetic characteristics, and represents a step that will assist future studies describing the ecological and metabolic capabilities of this widespread phylum.
Acknowledgements
Support for this work was provided by Geothermal Resources of New Zealand (GRN) Programme at GNS Science.
Additional file
Associated MIGS Record and Sequencing and Assembly Methodologies. (DOCX 39 kb)
Footnotes
Editor’s note: Although the name Acidobacteria is in common use at the phylum and class level, readers are advised that it appears on the list of rejected names. By definition, a rejected name must not be used to designate any taxon (Rule 23 a Note Note 4 (i)) at any rank.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KCYL, XCM, PFD, CH and MBS drafted the manuscript. MBS conducted the phylogenetic studies. JFP, PFD and MBS performed the laboratory experiments. KCYL, XCM, PFD, CH and MBS sequenced, assembled and annotated the genome. All authors read and approved the final manuscript.
References
- 1.Tindall BJ, Kampfer P, Euzeby JP, Oren A. Valid publication of names of prokaryotes according to the rules of nomenclature: past history and current practice. Int J Syst Evol Micr. 2006;56(11):2715–20. doi: 10.1099/ijs.0.64780-0. [DOI] [PubMed] [Google Scholar]
- 2.Barns SM, Takala SL, Kuske CR. Wide distribution and diversity of members of the bacterial kingdom Acidobacterium in the environment. Appl Environ Microbiol. 1999;65(4):1731–7. doi: 10.1128/aem.65.4.1731-1737.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huber KJ, Wust PK, Rohde M, Overmann J, Foesel BU. Aridibacter famidurans gen. nov., sp. nov. and Aridibacter kavangonensis sp. nov., two novel members of subdivision 4 of the Acidobacteria isolated from semiarid savannah soil. Int J Syst Evol Micr. 2014;64(6):1866–75. doi: 10.1099/ijs.0.060236-0. [DOI] [PubMed] [Google Scholar]
- 4.Crowe MA, Power JF, Morgan XC, Dunfield PF, Lagutin K, Rijpstra WI, et al. Pyrinomonas methylaliphatogenes gen. nov., sp. nov., a novel group 4 thermophilic member of the phylum Acidobacteria from geothermal soils. Int J Syst Evol Micr. 2014;64(1):220–7. doi: 10.1099/ijs.0.055079-0. [DOI] [PubMed] [Google Scholar]
- 5.Sinninghe Damste JS, Rijpstra WI, Hopmans EC, Foesel BU, Wust PK, Overmann J, et al. Ether- and ester-bound iso-diabolic acid and other lipids in members of Acidobacteria subdivision 4. Appl Environ Microbiol. 2014;80(17):5207–18. doi: 10.1128/AEM.01066-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sinninghe Damste JS, Rijpstra WI, Hopmans EC, Weijers JW, Foesel BU, Overmann J, et al. 13,16-Dimethyl octacosanedioic acid (iso-diabolic acid), a common membrane-spanning lipid of Acidobacteria subdivisions 1 and 3. Appl Environ Microbiol. 2011;77(12):4147–54. doi: 10.1128/AEM.00466-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bryant DA, Costas AM, Maresca JA, Chew AG, Klatt CG, Bateson MM, et al. Candidatus Chloracidobacterium thermophilum: an aerobic phototrophic Acidobacterium. Science. 2007;317(5837):523–6. doi: 10.1126/science.1143236. [DOI] [PubMed] [Google Scholar]
- 8.Tank M, Bryant DA. Chloracidobacterium thermophilum gen. nov., sp. nov.: an anoxygenic microaerophilic chlorophotoheterotrophic acidobacterium. Int J Syst Evol Microbiol. 2015;65(5):1426–30. doi: 10.1099/ijs.0.000113. [DOI] [PubMed] [Google Scholar]
- 9.Foesel BU, Rohde M, Overmann J. Blastocatella fastidiosa gen. nov., sp. nov., isolated from semiarid savanna soil - The first described species of Acidobacteria subdivision 4. Syst Appl Microbiol. 2013;36(2):82–9. doi: 10.1016/j.syapm.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 10.Sait M, Hugenholtz P, Janssen PH. Cultivation of globally distributed soil bacteria from phylogenetic lineages previously only detected in cultivation-independent surveys. Environ Microbiol. 2002;4(11):654–66. doi: 10.1046/j.1462-2920.2002.00352.x. [DOI] [PubMed] [Google Scholar]
- 11.Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004; 32(4):1363–71. [DOI] [PMC free article] [PubMed]
- 12.Soo RM, Wood SA, Grzymski JJ, McDonald IR, Cary SC. Microbial biodiversity of thermophilic communities in hot mineral soils of Tramway Ridge, Mount Erebus Antarctica. Environ Microbiol. 2009;11(3):715–28. doi: 10.1111/j.1462-2920.2009.01859.x. [DOI] [PubMed] [Google Scholar]
- 13.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26(5):541–7. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reasoner DJ, Geldreich EE. A new medium for the enumeration and subculture of bacteria from potable water. Appl Environ Microbiol. 1985;49(1):1–7. doi: 10.1128/aem.49.1.1-7.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chevreux B, Wetter T, Suhai S. Genome sequence assembly using trace signals and additional sequences infromation. Hannover, Germany: German Conference on Bioinformatics; 1999. p. 45–56.
- 16.Bonfield JK, Smith K, Staden R. A new DNA sequence assembly program. Nucleic Acids Res. 1995;23(24):4992–9. doi: 10.1093/nar/23.24.4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mavromatis K, Ivanova NN, Chen IM, Szeto E, Markowitz VM, Kyrpides NC. The DOE-JGI standard operating procedure for the annotations of microbial genomes. Stand Genomic Sci. 2009;1(1):63–7. doi: 10.4056/sigs.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–64. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29(22):2933–5. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol. 2001;305(3):567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 22.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340(4):783–95. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 23.Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25(17):2271–8. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 24.Reddy TB, Thomas AD, Stamatis D, Bertsch J, Isbandi M, Jansson J, et al. The Genomes OnLine Database (GOLD) v. 5: a metadata management system based on a four level (meta)genome project classification. Nucleic Acids Res. 2014;43(D1):D1099–106. doi: 10.1093/nar/gku950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greening C, Berney M, Hards K, Cook GM, Conrad R. A soil actinobacterium scavenges atmospheric H2 using two membrane-associated, oxygen-dependent [NiFe] hydrogenases. Proc Natl Acad Sci U S A. 2014;111(11):4257–61. doi: 10.1073/pnas.1320586111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ward NL, Challacombe JF, Janssen PH, Henrissat B, Coutinho PM, Wu M, et al. Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl Environ Microbiol. 2009;75(7):2046–56. doi: 10.1128/AEM.02294-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guyer CA, Morgan DG, Staros JV. Binding specificity of the periplasmic oligopeptide-binding protein from Escherichia coli. J Bacteriol. 1986;168(2):775–9. doi: 10.1128/jb.168.2.775-779.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia Costas AM, Liu Z, Tomsho LP, Schuster SC, Ward DM, Bryant DA. Complete genome of Candidatus Chloracidobacterium thermophilum, a chlorophyll-based photoheterotroph belonging to the phylum Acidobacteria. Environ Microbiol. 2012;14(1):177–90. doi: 10.1111/j.1462-2920.2011.02592.x. [DOI] [PubMed] [Google Scholar]
- 29.Yarza P, Yilmaz P, Pruesse E, Glockner FO, Ludwig W, Schleifer KH, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014;12(9):635–45. doi: 10.1038/nrmicro3330. [DOI] [PubMed] [Google Scholar]
- 30.Hugenholtz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. 1998;180(18):4765–74. doi: 10.1128/jb.180.18.4765-4774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janssen PH. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol. 2006;72(3):1719–28. doi: 10.1128/AEM.72.3.1719-1728.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tringe SG, von Mering C, Kobayashi A, Salamov AA, Chen K, Chang HW, et al. Comparative metagenomics of microbial communities. Science. 2005;308(5721):554–7. doi: 10.1126/science.1107851. [DOI] [PubMed] [Google Scholar]
- 33.Barns SM, Cain EC, Sommerville L, Kuske CR. Acidobacteria phylum sequences in uranium-contaminated subsurface sediments greatly expand the known diversity within the phylum. Appl Environ Microbiol. 2007;73(9):3113–6. doi: 10.1128/AEM.02012-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weatherby TM, Lenz PH. Mechanoreceptors in calanoid copepods: designed for high sensitivity. Arthropod Struct Dev. 2000;29(4):275–88. doi: 10.1016/S1467-8039(01)00011-1. [DOI] [PubMed] [Google Scholar]
- 35.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms - Proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87(12):4576–9. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thrash JC, Coates JD, et al. Phylum XVII. Acidobacteria phyl. nov. In: Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, et al., editors. The Bacteiodetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes. 2. New York Dordrecht Heidelberg London: Springer; 2011. pp. 725–35. [Google Scholar]
- 37.Stott MB, Crowe MA, Mountain BW, Smirnova AV, Hou S, Alam M, et al. Isolation of novel bacteria, including a candidate division, from geothermal soils in New Zealand. Environ Microbiol. 2008;10(8):2030–41. doi: 10.1111/j.1462-2920.2008.01621.x. [DOI] [PubMed] [Google Scholar]
- 38.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]