

Abstract
Reactive microglia and macrophages are prevalent in damaged retinas. Glucocorticoid signaling is known to suppress inflammation and the reactivity of microglia and macrophages. In the vertebrate retina, the glucocorticoid receptor (GCR) is known to be activated and localized to the nuclei of Müller glia (Gallina et al., 2014). Accordingly, we investigated how signaling through GCR influences the survival of neurons using the chick retina in vivo as a model system. We applied intraocular injections of GCR agonist or antagonist, assessed microglial reactivity, and the survival of retinal neurons following different damage paradigms. Microglial reactivity was increased in retinas from eyes that were injected with vehicle, and this reactivity was decreased by GCR-agonist dexamethasone (Dex) and increased by GCR-antagonist RU486. We found that activation of GCR suppresses the reactivity of microglia and inhibited the loss of retinal neurons resulting from excitotoxicity. We provide evidence that the protection-promoting effects of Dex were maintained when the microglia were selectively ablated. Similarly, intraocular injections of Dex protected ganglion cells from colchicine-treatment and protected photoreceptors from damage caused by retinal detachment. We conclude that activation of GCR promotes the survival of ganglion cells in colchicine-damaged retinas, promotes the survival of amacrine and bipolar cells in excitotoxin-damaged retinas, and promotes the survival of photoreceptors in detached retinas. We propose that suppression of microglial reactivity is secondary to activation of GCR in Müller glia, and this mode of signaling is an effective means to lessen the damage and vision loss resulting from different types of retinal damage.
Keywords: retina, microglia, Müller glia, neuronal survival, retinal detachment
Introduction
Distinct types of glial cells are found within the retina. Müller glia and microglia are found in retinas of all vertebrates. With variability between vertebrate species, retinal glia can include astrocytes and oligodendrocytes. Avascular retinas, including those of chickens, guinea pigs and rabbits, contain oligodendrocytes that myelinate the axons of ganglion cells in the nerve fiber layer (NFL). Vascular retinas contain significant numbers of astrocytes, that are closely associated with the blood vessels (Dorrell and Friedlander 2006; Friedlander et al. 2007). By comparison, the retinas of guinea pigs and birds do not contain conventional types of astrocytes (Fischer and Bongini 2010; Fischer et al. 2010a; Fujita et al. 2001; Won et al. 2000). The chick retina, but not guinea pig retina, contains an atypical glial cell, termed Non-astrocytic Inner Retinal Glia-like (NIRG) cells, that is scattered across inner retinal layer (Fischer et al. 2010a; Fischer et al. 2010b).
Glial reactivity can have a significant impact upon neuronal survival. For example, conditional ablation of the Müller glia compromises photoreceptor survival and vascular integrity in the rodent retina (Shen et al. 2012). Glial cells can profoundly exacerbate neuronal death following excitotoxic injury. For example, N-methyl-D-aspartate (NMDA) -mediated activity of Müller glia is known to activate the NFκB-pathway and stimulate tumor necrosis factor α (TNFα) production by the Müller glia, and the TNFα influences the Ca2+ -permeability of AMPA-receptors in retinal neurons to diminish survival (Lebrun-Julien et al. 2009). Similarly, intraocular injections of insulin stimulate the reactivity of Müller glia, microglia and NIRG cells, and predispose retinal neurons to excitotoxic damage (Fischer et al. 2009a). Conversely, stimulation of glia can provide enhanced neuroprotection. For example, we found that the neuroprotective effects of fibroblast growth factor 2 (FGF2) are manifested through activation of Mitogen-Activated Protein Kinase (MAPK) -signaling in the Müller glia (Fischer et al. 2009a). Similar to the Müller glia, microglia have been implicated in the pathophysiology of different sight-threatening diseases. Microglia were thought to be bystanders cells that do not contribute to retinal degeneration. However, current consensus indicates that activated microglia and/or infiltrating macrophages contribute to the progression of age-related macular degeneration, glaucoma and diabetic retinopathy (reviewed by Buschini et al. 2011; Karlstetter et al. 2010; Langmann 2007; Xu et al. 2009). Taken together, these findings suggest that the activity of retinal glia is coordinated and can have a significant influence upon neuronal survival and the progression of retinal degeneration.
Cytokines can have a significant effect upon glial reactivity and neuronal survival. In IL6-deficient mice, compared to wild-type mice, acute injury results in significantly reduced reactivity in Glial Fibriliary Acidic Protein (GFAP) -positive astrocytes and microglia (Klein et al. 1997). In GFAP-IL6-overexpressing transgenic mice, astrocytes produce elevated levels of IL6 which results in lifelong reactive gliosis of both astrocytes and microglia (Chiang et al. 1994). In response to brain trauma resulting from a cryolesion, neuronal damage was reduced and recovery was accelerated in the GFAP-IL6 mice compared to the injury response seen in wild-type mice, suggesting a neuroprotective role for elevated IL6 in neural tissues (Penkowa et al. 2003). A recent study demonstrated that murine Müller glia exposed to activated microglia have altered cell morphology and gene expression compared to Müller glia undergoing gliosis (Wang et al. 2011). Activated microglia may influence Müller glia directly, and exchange proinflammatory and chemotactic cytokines that mediate adaptive glial activity in the retina in response to injury (Wang et al. 2011). We recently found that reactive microglia stimulate the formation of Müller glia-derived progenitors in the chick retina (Fischer et al. 2014). The current study investigates how activation of GCR signaling in Müller glia influences neuronal survival and retinal folds/detachments in acutely damaged chick retinas in vivo. We have recently reported that activated, nuclear GCR is predominantly found in the Müller glia in the retinas of chicks, mice, guinea pigs, dogs and humans (Gallina et al. 2014).
Methods and Materials
Animals
The use of animals in these experiments was in accordance with the guidelines established by the National Institutes of Health and The Ohio State University. This study was approved by the Ohio State University Institutional Animal Care and Use Committee (IACUC). Newly hatched leghorn chickens (Gallus gallus domesticus) were obtained from the Department of Animal Sciences at the Ohio State University or Meyer Hatchery (Polk, Ohio). Chicks were kept on a cycle of 12 hours light, 12 hours dark (lights on at 7:00 am). Chicks were housed in a stainless steel brooder at about 25°C and received water and Purinatm chick starter ad libitum.
Preparation of clodronate-liposomes
The preparation of clodronate liposomes was based on previous descriptions (Van Rooijen 1989; van Rooijen 1992; Van Rooijen and Sanders 1994). The protocols used to generate clodronate liposome were as described previously (Zelinka et al. 2012). Approximately 1% of the clodronate is encapsulated by the lipsomes (Van Rooijen and Sanders 1994), yielding approximately 7.85 mg/ml. Precise quantitation of the clodronate was difficult, because of the stochastic nature of combination of the clodronate and liposomes. Accordingly, we titrated doses of clodronate liposomes to levels where >99% of the microglia/macrophages were ablated at 1 day after treatment.
Intraocular injections
Chickens were anesthetized via inhalation of 2.5% isoflurane in oxygen and intraocular injections performed as described previously (Fischer et al., 1998; Fischer et al., 1999). For all experiments, the left eyes of chicks were injected with the “test” compound and the contra-lateral right eyes were injected with vehicle as a control. Interleukin-6 (100 or 200ng/dose), NMDA (2μmol), colchicine (200 ng/dose), FGF2 (100ng/dose), and clodronate liposomes (10 to 2000 ng) were injected in 20 μl sterile saline with 0.05 mg/ml bovine serum albumin added as a carrier. Dex (100ng/dose or 200ng/dose) and CpdA (500ng/dose) were injected in 20 μl saline and 25% DMSO. RU486 (1μg/dose) was injected in 20 μl PBS (0.01M) and 50%DMSO. Two μg of BrdU was injected to label proliferating cells. Injection paradigms are included in each figure.
Subretinal injections
P14 chicks were anesthetized via inhalation of 2.5% isoflurane in oxygen and subretinal injections performed as described previously (Cebulla et al. 2012). For all experiments, the left eyes of chicks were injected with vehicle as a control or the “test” compound. Subretinal injections of approximately 25 microliters of sterile saline (n = 4) or saline with 100 ng water-soluble Dex (n = 4) were delivered to the eye to bleb-up the retina. Eyes were enucleated for analysis at day 3. Injection paradigms are included in each figure.
Tissue dissection, fixation, sectioning and immunolabeling
Tissues were fixed, sectioned and immunolabeled as described elsewhere (Fischer et al. 1999; Fischer et al. 1998). Retinal whole-mount preparations were processed as described previously (Fischer and Reh 2002; Fischer et al. 2008; Fischer et al. 2006). Working dilutions and sources of antibodies used in this study included; (1) mouse anti-CD45 was used at 1:200 (HIS-C7; Cedi Diagnostic), (2) rabbit anti-GCR was used at 1:400 (PA1-511A; Thermo Scientific), (3) goat anti-Sox2 was used at 1:1000 (Y-17; Santa Cruz Immunochemicals), (4) mouse anti-Nkx2.2 was used at 1:80 (74.5A5; Developmental Studies Hybridoma Bank (DSHB)), (5) mouse anti-Brn3a (Pouf4a) was used at 1:50 (mab1585; Chemicon), (6) rabbit anti-Sox9 was used at 1:2000 (AB5535; Chemicon), (7) mouse (IgG) anti-transitin was used at 1:50 (EAP3; DSHB), (8) rabbit anti-GFAP was used at 1:2000 (Z0334; DakoCytomation), (9) rabbit anti- L/M opsin was used at 1:400 (AB5405; Chemicon), (10) mouse (IgG) anti rhodopsin was used at 1:200 (Rho 4D2; generous gift Dr. R. Molday, University British Columbia), and (11) rat anti-BrdU was used at 1:200 (OBT00030S; AbD Serotec Raleigh, NC). None of the observed labeling was due to non-specific binding of secondary antibody or auto-fluorescence because sections labeled with secondary antibodies alone were devoid of fluorescence. Secondary antibodies included donkey-anti-goat-Alexa488/568, goat-anti-rabbit-Alexa488/568/647, goat-anti-mouse-Alexa488/568/647, and goat-anti-rat-Alexa488 (Invitrogen) diluted to 1:1000 in PBS plus 0.2% Triton X-100. Some sections were counter-stained with the nuclear label DRAQ5 (Cell Signaling) diluted to 1:2000 and added to the secondary antibody diluent. To permeabilize retinas for whole-mount labeling procedures, samples were frozen (−80°C) and thawed (20°C) three times prior to incubation with the antibody solution. Both primary and secondary antibodies were incubated overnight.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)
To identify dying cells that contained fragmented DNA the TUNEL method was used. We used an In Situ Cell Death Kit (TMR red; Roche Applied Science), as per the manufacturer’s instructions.
qRT-PCR
Tissue dissections, RNA isolation, and reverse transcriptase reactions were performed as described previously ((Fischer et al. 2004; Ghai et al. 2010). PCR primers were designed by using the Primer-BLAST primer design tool at NCBI (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). Primer sequences and predicted product sizes are listed in Table 1. For qPCR, reactions were performed using SYBR Green Master Mix and StepOnePlus Real-Time system (Applied BioSystems). Samples re normalized to GAPDH for each sample and the fold change between control and treated samples was determined using the −ΔΔCt method [=fold change 2(−ΔΔCt)] and represented as a percentage change from the control, which was assigned a value of 100. The significance of any differences in percentage change was determined using a non-parametric Mann-Whitney U-test.
Table 1.
PCR Primer sequences and predicted product sizes
| Gene | Forward primer 5′ߝ3′ | Reverse primer 5′ߝ3′ | Product size (bp) |
|---|---|---|---|
| adam17 | AGC GAG TGC CCT CCTCCT GG | TTG CAG GCA CAC GAG CGG AG | 125 |
| ascl1a | AGG GAA CCA CGT TTA TGC AG | TTA TAC AGG GCC TGG TGA GC | 187 |
| cDelta1 | CAC TGA CAA CCC TGA TGG TG | TGG CAC TGG CAT ATG TAG GA | 152 |
| cDll4 | GGT CTG CAG CGA GAA CTA CT | TGC AGT ATC CAT TCT GTT CG | 181 |
| cHes1 | CGC TGA AGA AGG ATA GTT CG | GTC ACT TCG TTC ATG CAC TC | 175 |
| cHes5 | GGA GAA GGA GTT CCA GAG AC | AAT TGC AGA GCT TCT TTG AG | 143 |
| cJag1 | TGA TAA GTG CAT TCC ACA CC | CAG GTA CCA CCA TTC AAA CA | 149 |
| cNotch1 | GGC TGG TTA TCA TGG AGT TA | CAT CCA CAT TGA TCT CAC AG | 154 |
| gapdh | CAT CCA AGG AGT GAG CCA AG | TGG AGG AAG AAA TTG GAG GA | 161 |
| gli1 | CTA GCG TTG ACC TGC AGA CG | ACA GGG TTT CGT GGG AGC TA | 127 |
| gli3 | ATT TTT GGG GCA ATG GAC AG | TGA ATG CCA TCT CCA ACC AG | 209 |
| il1β | GCA TCA AGG GCT ACA AGC TC | CAG GCG GTA GAA GAT GAA GC | 131 |
| il6 | TTA GTT CGG GCA CAA TCC TC | GGT TCC TGA AAC GGA ACA AC | 72 |
| pax2 | GCC AGG CCT CAT TGT AGG TT | CCA ACT GGA CAA GGC AGC TA | 115 |
| ptch | AAC GCA TGG GCT AGA AGG AA | ATG CTT GCC TAC GCC TGT TT | 221 |
| smo | GGG TGG TTG CTC TTG ATG GA | GAC TCC GTC AGC GGT ATC TG | 152 |
| tnfα | AGC AGC GTT TGG GAG TGG GC | GCA GAT GGG GCA GGA AAG CCA | 133 |
Photography, measurements, and cell counts
Wide-field photomicrographs were obtained by using a Leica DM5000B microscope and Leica DC500 digital camera. Images were optimized for brightness and contrast, multiple-channel images overlaid, and figures constructed by using Adobe Photoshop™6.0. Cell counts were made from at least 4 different animals, and means and standard deviations calculated on data sets. To avoid the possibility of region-specific differences within the retina, cell counts were consistently made from the same region of retina for each data set.
Similar to previous reports (Fischer et al. 2009a; Fischer et al. 2009b; Fischer et al. 2010a; Ghai et al. 2009), immunofluorescence was quantified by using ImagePro 6.2 (Media Cybernetics, Bethesda, MD, USA). Identical illumination, microscope, and camera settings were used to obtain images for quantification. Retinal areas were sampled from 5.4 MP digital images. These areas were randomly sampled across all layers of the retina where microglia where found. Measurements were made for thresholded regions (≥50 contiguous pixels) containing pixels with intensity values of 68 or greater (0 = black and 255 = saturated). The mean density sum was calculated as the average sum of pixel values for all pixels above threshold within fields of view. These calculations were determined for retinal regions sampled from at least 4 different retinas for each experimental condition.
Percentage area of retinal folds and detachments was determined from digital micrographs. The detached areas appeared as opacities that were digitally traced and measured by using ImagePro 6.2. The detached retinal area was calculated as a percentage of total retinal area without compensating for concave shape of the eyecup.
To assess ganglion cell survival, numbers of Brn3a+ nuclei in the GCL (52,000 μm2) per region of the retina (central, temporal, nasal and ventral) were counted per eye. To assess numbers of dying cells in NMDA-damaged retinas, cell counts of TUNEL-positive cells in the INL were made in central regions (14,600 μm2) of retina.
Statistics
Where significance of difference was determined between two treatment groups accounting for inter-individual variability (means of treated-control values) we performed a two-tailed, paired t-test. The significance of differences for the percent change was determined using a two-tailed Mann-Whitney U test. The normality distribution of the data was determined by using the Shapiro-Wilk test. The statistics were calculated using the GraphPad Prism 6 software.
Results
The reactivity of microglia/macrophages is influenced by GCR-signaling
We first examined whether intraocular injections of agonists and antagonists to GCR influenced the reactivity of microglia. We have recently reported that merely puncturing the eye stimulates the reactivity of retinal microglia, and puncture is equally as potent as injections of saline in inducing reactivity (Fischer et al. 2014). Microglial reactivity was assessed by probing for levels of CD45 expression. The CD45-labeling does not distinguish between retinal microglia and infiltrating macrophages. Thus, we use the term “microglia/macrophages” to collectively refer to retinal microglia and infiltrating macrophages. Intraocular injections of Dex, which activates both the GCR dimer and monomer, and does not discriminate between GCR-dependent transcriptional changes and changes in cell signaling pathways, suppressed the reactivity of microglia/macrophages and prevented the accumulation of these cells at the vitread surface of the retina (Figs. 1a–f). We tested whether CpdA, a small molecule, which activates only the monomeric form of GCR that interacts with cytoplasmic proteins (De Bosscher et al. 2010; De Bosscher et al. 2005), and aldosterone which acts at mineralocorticoid receptor (MCR) influenced the reactivity of the microglia/macrophages. We found that CpdA and aldosterone had no significant effect upon the reactivity of microglia/macrophages (Figs. 1g and h), suggesting that neither cytoplasmic GCR nor MCR influence microglial reactivity. By contrast, the GCR antagonist RU486 stimulated the reactivity of microglia/macrophages (Figs. 1i–k). However, RU486 is known to act at progesterone receptors in addition to GCR. Thus, we tested whether Dex, which prefers GCR not the progesterone receptor, overrides the effects of RU486. We found that application of Dex with RU486 suppressed the microglial reactivity that resulted from RU486 alone (Fig. 1l). Consistent with previous reports demonstrating that GCR is not expressed by normal or reactive microglia/macrophages in the chick retina (Fischer et al. 2010a; Gallina et al. 2014; Zelinka et al. 2012), immunoreactivity for nuclear GCR was found only in Sox2+ Müller glia in the INL and in Sox2+/Nkx2.2+ NIRG cells in the IPL and GCL (Fig. 1m). These observations are consistent with the hypothesis that Dex-mediated effects on retinal cells occur via GCR-signaling in Müller glia and/or NIRG cells.
Figure 1.
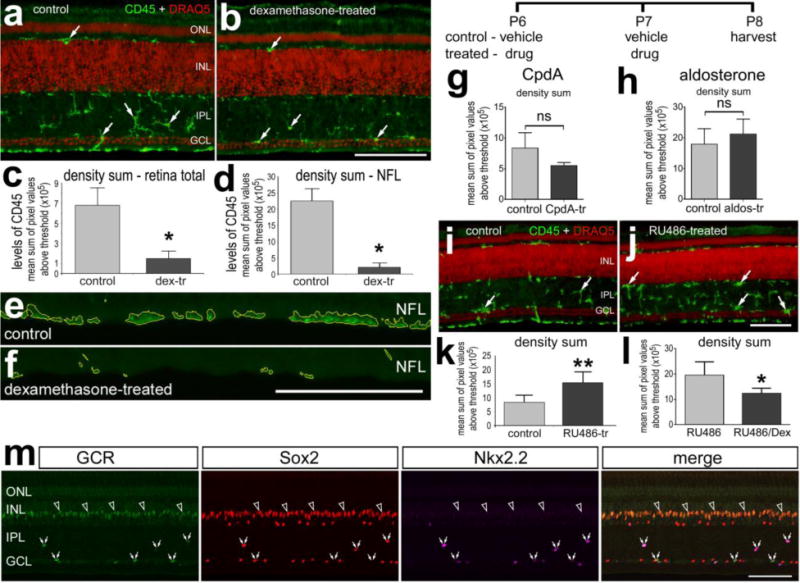
In undamaged retina activation of GCR inhibits, whereas inhibition of GCR activates microglial reactivity. Retinas where obtained from eyes that received 2 consecutive daily injections of Dex (a–f), CpdA (g), aldosterone (h), RU486 (i–k) or vehicle, or the combination of Dex and RU486 or RU486 (l). Tissues were harvested 24 hours after the last injection. Sections of the retina were labeled with DRAQ5 (red; a,b,i,j), antibodies to CD45 (green;a,b,e,f,i,j) or GCR, So×2 and Nk×2.2 (m). In m the immunolabeling was done on normal untreated retinas. e and f; areas about threshold are outlined by yellow. Quantitative immunofluorescence was used to measure the density sum of CD45-immunofluorescence (c,d,g,h,k,l). The histograms illustrate the mean (±SD; n≥5) density (intensity) sum. Arrows indicate reactive microglia (a,b,i,j), hollow arrows indicate the nuclei of GCR+ Müller glia in the INL, and small double-arrows indicate GCR+ NIRG cells in the IPL and GCL (m). All the scale bars are 50 μm. The scale bar in panel b applies to a and b, the bar in f applies to e and f, the bar in j applies to i and j, and the bar in m applies to m alone. Abbreviations: ONL – outer nuclear layer, INL – inner nuclear layer, IPL – inner plexiform layer, GCL – ganglion cell layer, NFL – nerve fiber layer. Significance of difference (*p<0.05; ns – not significant) was determined by using a two-tailed t test.
We next tested whether activation of GCR influences the reactivity of microglia/macrophages in NMDA-damaged retinas. We found that 2 consecutive daily injections of 200 ng Dex significantly reduced the reactivity of microglia/macrophages at 1 and 3 days after NMDA-treatment (Figs. 2a,b,d,e). By comparison, 100 ng doses of Dex had no significant effect upon microglial reactivity in damaged retinas (Figs. 2c and 2f). Similarly, 200ng doses of Dex applied after NMDA-treatment are known to reduce microglial reactivity (Gallina et al. 2014). By comparison, 200ng doses of aldosterone or CpdA before or after NMDA-treatment had no effect upon the reactivity of the microglia/macrophages (Figs. 2g–j).
Figure 2.
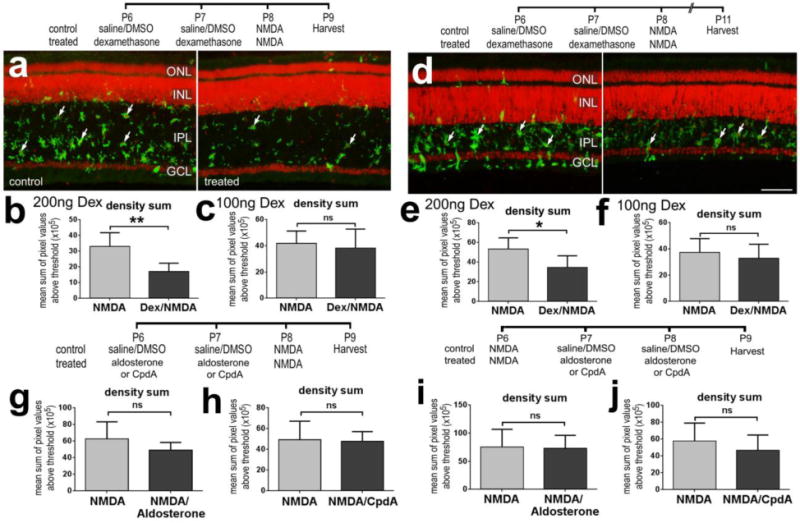
Activation of GCR with Dex before an excitotoxic insult suppresses microglial reactivity in the retina. (a–f) Retinas were obtained from eyes that received 2 consecutive daily injections of 100ng/dose Dex (c,f) or 200ng/dose Dex (a,b,d,e), NMDA at P8, and tissues harvested at P9 or P11. (g and h) Retinas were obtained from eyes that received NMDA at P6 and 2 consecutive daily injections of aldosterone (g), CpdA (h) or vehicle at P7 and P8, and tissues harvested at P9. Retinal sections were labeled with DRAQ5 (red; a,d) and antibodies to CD45 (green; a,d). Arrows indicate reactive microglia in the IPL. As described in the methods, quantitative immunofluorescence was used to measure the mean density sum of CD45-immunofluorescence (b,c,e–j). The histograms illustrate the mean (±SD; n≥5) of the density (intensity) sum. The scale bar (50 μm) in panel d applies to a and d. Significance of difference (*p<0.05, **p<0.01; ns – not significant) was determined by using a paired, two-tailed t test. Abbreviations: ONL – outer nuclear layer, INL – inner nuclear layer, IPL – inner plexiform layer, GCL – ganglion cell layer.
GCR-signaling promotes the survival in retinal interneurons
We next probed for changes in numbers of dying cells which were detected using the TUNEL method to detect dying amacrine and bipolar cells in NMDA-damaged chick retinas (Fischer et al. 1998). We found that 100 and 200ng doses Dex before NMDA-treatment significantly reduced numbers of dying cells at 1 and 3 days after treatment (Fig. 3a–f). Similarly, CpdA significantly reduced number of dying cells when applied before NMDA-induced damage, whereas aldosterone had no effect (Fig. 3c). Interestingly, the 100ng dose of Dex was significantly more effective than the 200ng dose at reducing numbers of dying cells at 1 day after NMDA-treatment (Figs. 3c and 3f). By comparison, there was no significant difference in numbers of dying cells at 3 days after NMDA-treatment using different doses of Dex (Figs. 3d–f). Nevertheless, Dex-treatment had a sustained effect upon suppressing NMDA-mediated cell death, unlike the pro-inflammatory cytokine which transiently reduces numbers of dying cells in NMDA-damaged retinas (Fischer et al. 2015b). These findings suggest that activation of GCR in Müller glia before a severe excitotoxic insult protects inner retinal neurons. Similarly, application of Dex after NMDA-treatment reduces numbers of dying inner retinal neurons (Gallina et al. 2014).
Figure 3.
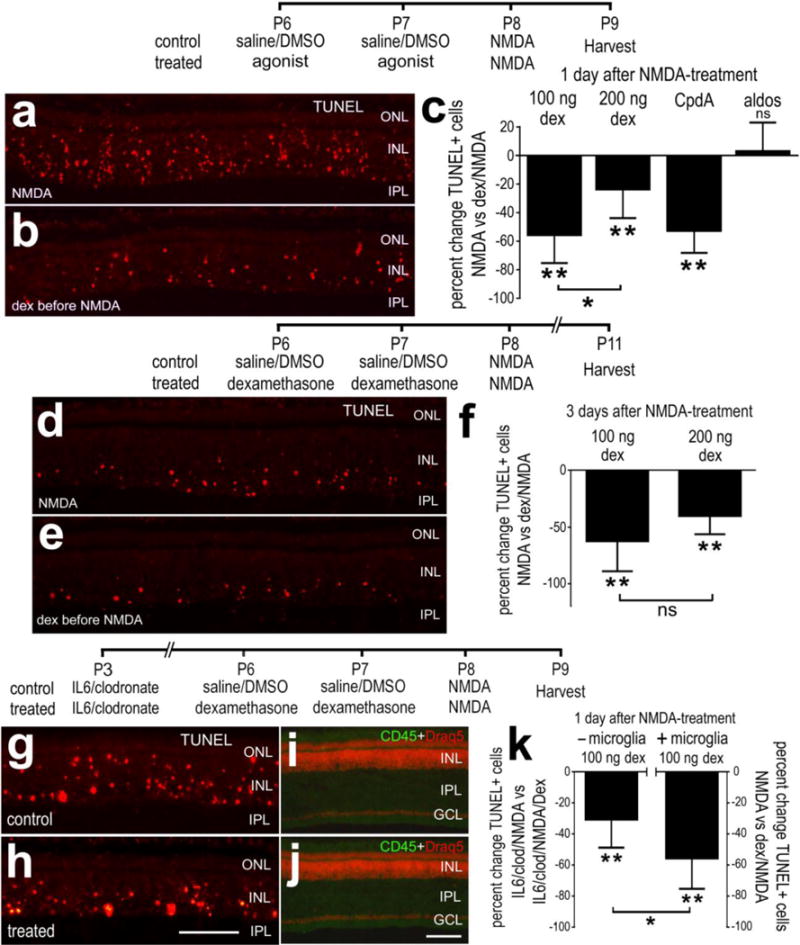
Activation of GCR-signaling before an excitotoxic insult suppresses cell death in NMDA-damaged retinas. Retinas were obtained from eyes that received 2 consecutive daily injections of 100ng/dose Dex (a,b,d,e) or 200ng/dose (c,f), or vehicle at P6 and P7, NMDA at P8, and tissues harvested at P9 or P11. Alternatively, microglia were selectively ablated by using a single intraocular injection of IL6 and clodronate-liposomes at P3 before injections of 100ng Dex/vehicle and NMDA (g–k). Retinal sections were labeled using the TUNEL method (a,b,d,e,g,h), DRAQ5 (red; i,j) and antibodies to CD45 (green; i,j). The scale bar (50 μm) in panel h applies to a,b,d,e,g and h, and the bar in j applies to i and j. c,f,k; The histograms illustrate the mean (±SD; n≥5) percent change in TUNEL-positive cells. Significance of difference (*p<0.05, **p<0.01; ns – not significant) was determined by using Mann-Whitney U test. Abbreviations: ONL – outer nuclear layer, INL – inner nuclear layer, IPL – inner plexiform layer, GCL – ganglion cell layer.
Since reactive microglia/macrophages are known to influence neuronal survival in the retina (Fischer et al. 2015a; Madeira et al. 2015) and Dex inhibits the reactivity of microglia, we examined whether the ablation of microglia/macrophages influences the ability of Dex to influence neuronal survival. We ablated >99% of the retinal microglia/macrophages by applying a single intraocular injection of IL6 and clodronate-liposomes, similar to previous reports (Fischer et al. 2014; Fischer et al. 2015a; Zelinka et al. 2012). We found that 100ng of Dex reduced numbers of dying cells in NMDA-damaged retinas in the absence of microglia/macrophages (Figs. 3g–k). However, the efficacy of Dex-mediated neuroprotection was significantly less in the absence of microglia/macrophages compared to the effects of Dex in the presence of microglia/macrophages (Fig. 3k). These findings suggest that the protective effects of GCR-signaling are mediated, in part, by suppressing the reactivity of microglia.
The survival of ganglion cells is influenced by GCR-signaling
We examined whether the survival of ganglion cells was influenced by the GCR agonist Dex. In the chick retina, colchicine has been shown to destroy ganglion cells when applied within the first week of hatching (Fischer et al. 1999; Morgan 1981). Thus, we damaged ganglion cells with colchicine and examined whether activation of GCR influenced the survival of ganglion cells. Brn3a is expressed by approximately 98% of the ganglion cells (Liu et al. 2000; Xiang et al. 1995; Xiang et al. 1993). We found that there were significant increases in numbers of surviving Brn3a-positive ganglion cells in damaged retinas treated with Dex (Fig. 4). There was nearly a 30% increase in surviving ganglion cells in central regions of the retina, and more than a 2-fold increase in surviving ganglion cells in temporal, nasal and dorsal regions of the retina (Fig. 4).
Figure 4.
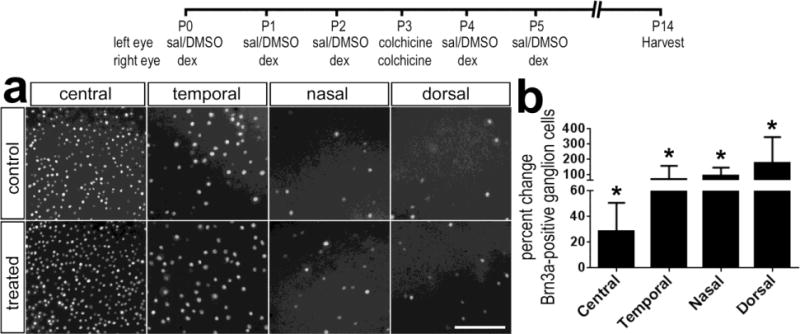
Activation of GCR-signaling protects retinal ganglion against colchicine-mediated damage. Retinas were obtained from eyes that received consecutive daily injections of Dex (100ng/dose) or vehicle at P0, P1 and P2, 50 ng of colchicine at P3, Dex (100ng/dose) or vehicle at P4 and P5, and retinas were harvested at P14. Whole-mounts of the retina were labeled with antibodies to Brn3a (a). The scale bar indicates 50 μm. The histogram in panel b illustrates the mean (±SD; n≥5) percent change in Brn3a-positive ganglion cells per 40,000 μm2 of central, temporal, nasal or dorsal regions of retina. Significance of difference (*p<0.05) was determined by using Mann-Whitney U test.
GCR-signaling and retinal folds and detachments
We have recently reported that NMDA-induced damage coupled to growth factor or cytokine treatment results in retinal folds and focal detachments, which can be prevented by ablating reactive microglia/macrophages (Fischer et al. 2015a). Accordingly, we tested whether Dex influenced the formation of retinal folds and detachments. In retinas treated with FGF2 before NMDA-treatment about one-fifth of the retinal area was folded or detached (Fig. 5). The area of detached retina was reduced by nearly 80% when Dex was co-applied with the FGF2 (Fig. 5).
Figure 5.
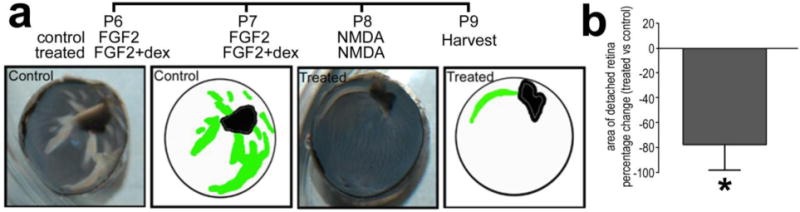
Activation of GCR-signaling prevents the formation of focal detachments or retinal folds that occur in retinas treated with FGF2 and NMDA. Eyes were treated with 2 consecutive daily injections of FGF2 alone (control) or FGF2 + Dex (treated) at P6 and P7, NMDA at P8, and tissues harvested at P9. The detached areas appeared as opacities that were digitally traced and measured by using ImagePro 6.2 (a). The detached retinal area was calculated as a percentage of total retinal area without compensating for concave shape of the eyecup. The histogram illustrates the mean (±SD) percentage change (treat – control) (b). Significance of difference (*p<0.0002; n=5) was determined by using Mann-Whitney U test.
We next examined whether the survival of photoreceptors was influenced by GCR-signaling. We used an experimental model wherein a subretinal injection of saline induced a transient detachment of photoreceptors from the retinal pigmented epithelium (RPE), damage to photoreceptors is incurred, and reactive microglia/macrophages are recruited to the outer retina (Cebulla et al. 2012). We tested whether application of water-soluble Dex (100ng) in the subretinal injection influenced cell survival and glial reactivity in regions of detached retina. We found numerous TUNEL-positive cells in ONL in detached regions of control retinas (Figs. 6a–d). By comparison, there was a significant decrease in the number of TUNEL-positive ONL cells in detached regions of Dex-treated retinas (Figs. 6a–d). In Dex-treated retinas, the numbers of TUNEL-positive cells were greatly reduced in the ONL, but were modestly increased in the inner half of the INL (Figs. 6c,d). These findings suggest that Dex-treatment protects photoreceptors against detachment at the expense of some inner retinal neurons. At the boundary between attached and detached regions of retina there was a sharp transition in numbers of TUNEL-positive cells (Fig. 6a), indicating the highly localized nature of the detachment injury.
Figure 6.
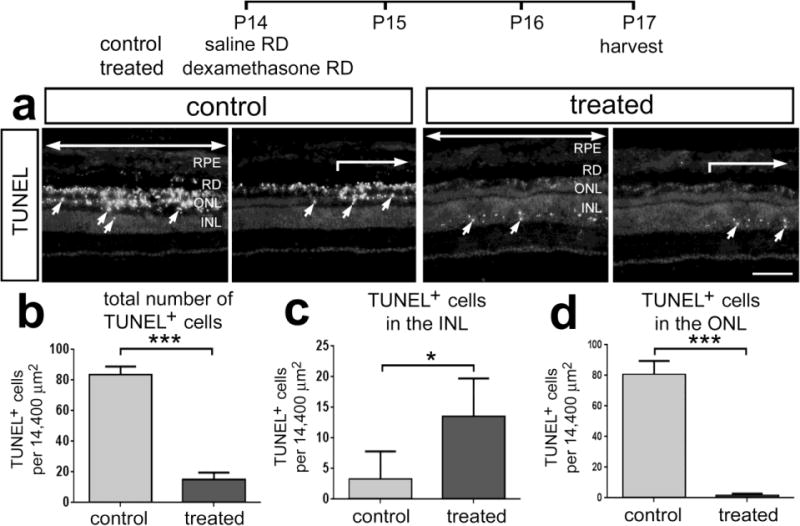
Dex-treatment reduces the loss of photoreceptors following saline-mediated retinal detachment. Retinas were obtained from eyes that received a subretinal injection of saline (control) or saline + soluble Dex (100ng) (treated), and tissues harvested 3 days later. Sections of the retina were labeled using the TUNEL method (a). Large arrows indicate regions of retina where the detachment occurred, and small arrows indicate TUNEL-positive nuclei. Abbreviations: ONL – outer nuclear layer, INL – inner nuclear layer, RPE – retinal pigmented epithelium. Histograms illustrate the mean (±SD) number of TUNEL-positive cells across all layers of the retina (b), INL (c) or ONL (d) in control and Dex-treated retinas. The scale bar denotes 50 μm. Significance of difference (*p<0.05, ***p<0.001, n=4) was determined by using a two-tailed t-test.
Photoreceptors that are stressed or damaged often mistraffick photo-pigments to the inner segments (Zhang et al. 2011), and we have previously shown that mistrafficking of the photo-pigments occurs in detached retina (Cebulla et al. 2012). We tested the hypothesis that GCR signaling would reduce the photo-pigment mistrafficking induced by RD. Mistrafficking of long/medium wavelength (l/m) -opsin was prevalent in cone photoreceptors found in detached regions in both control and treated retinas (Figs. 7a,c). At the boundary between attached and detached regions of retina there was a sharp transition in numbers of photoreceptors with l/m-opsin or rhodopsin mistrafficked to inner segments (Figs. 7a,b). Similar to the mistrafficking of l/m-opsin in cones, rhodopsin-immunoreactivity was found in the inner segments of rod photoreceptors in detached regions in control and Dex-treated retinas (Figs. 7 b,d). Unlike the mistrafficking of l/m-opsin in cone photoreceptors, mistrafficking of rhodopsin in rod photoreceptors was significantly reduced in Dex-treated retinas compared to control retinas; very few rods in detached Dex-treated retinas had rhodopsin mistrafficked to the cell body or axon terminals in the outer plexiform layer (Fig. 7b,c).
Figure 7.
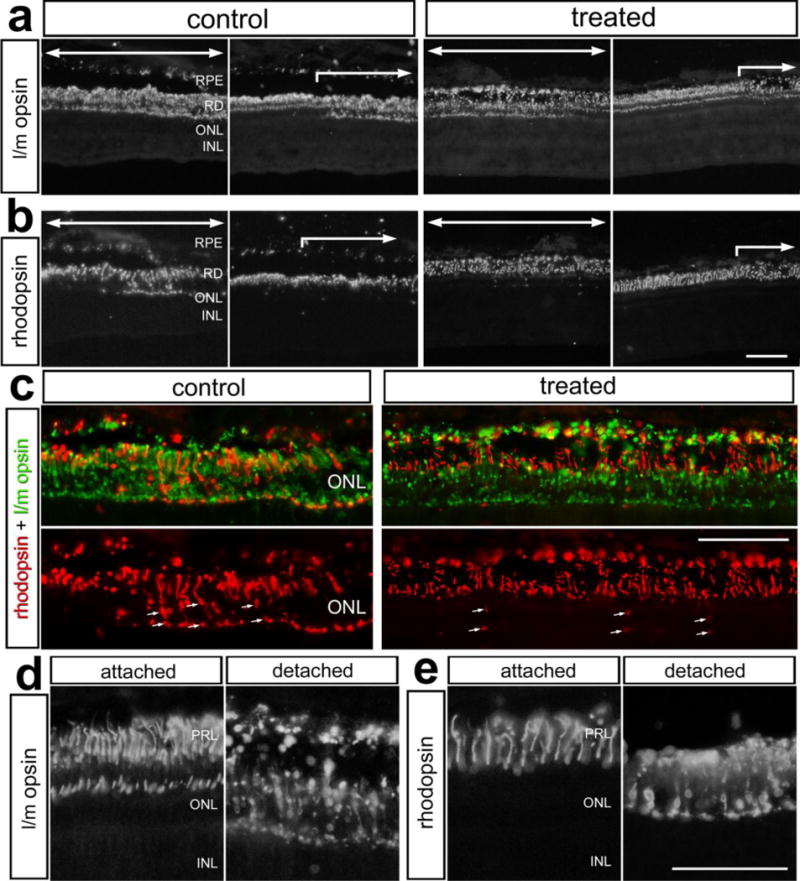
Dex-treatment reduces the mistrafficking of rhodopsin in rod photoreceptors, but had no effect on mistrafficking of l/m-opsin in cone photoreceptors following retinal detachment. Retinas were obtained from eyes that received a subretinal injection of saline (control) or saline + soluble Dex (treated), and tissues harvested 3 days later. Sections of the retina were labeled using antibodies to l/m-opsin (a,c,d) and rhodopsin (b,c,d). The images in c–e illustrate high-magnification fields of view demonstrating mistrafficking of l/m-opsin and rhodopsin to the inner segments of photoreceptors in control retinas, and in treated retinas in c. Large arrows indicate regions of retina where the detachment occurred. The small arrows indicate mistrafficking of rhodopsin (c). The scale bar (50 μm) in panel b applies to a and b, the scale bar (50 μm) in panel c applies to c, and the scale bar (50 μm) in panel e applies to d and e. Abbreviations: RPE – retinal pigmented epithelium, ONL – outer nuclear layer, INL – inner nuclear layer, IPL- inner plexiform layer, PRL – photoreceptor layer.
Glial cells are known to become reactive in response to retinal detachment (Cebulla et al. 2012; Fisher 1994; Fisher and Lewis 2003; Lewis and Fisher 2003; Lewis et al. 1994; Lewis et al. 2005). Thus, we examined whether Dex influenced the microglia/macrophages and Müller glia in detached regions of the retina. Although many reactive microglia/macrophages were present in the INL and IPL, reactive microglia/macrophages were concentrated in the ONL in detached regions of the control retinas (Fig. 8a). Similarly, reactive microglia/macrophages were concentrated in the ONL in detached regions of Dex-treated retinas, but relatively quiescent microglia were present in inner retinal layers (Fig. 8b). Measurement of CD45-immunofluorescence indicated significant decreases in microglial/macrophages reactivity in detached retinas treated with Dex (Figs. 8c,d). At the edge of the detached region of retina that was a sharp transition between relatively quiescent glia and accumulations of reactive microglia/macrophages in the ONL in both control and treated retinas (Figs.8a,b).
Figure 8.
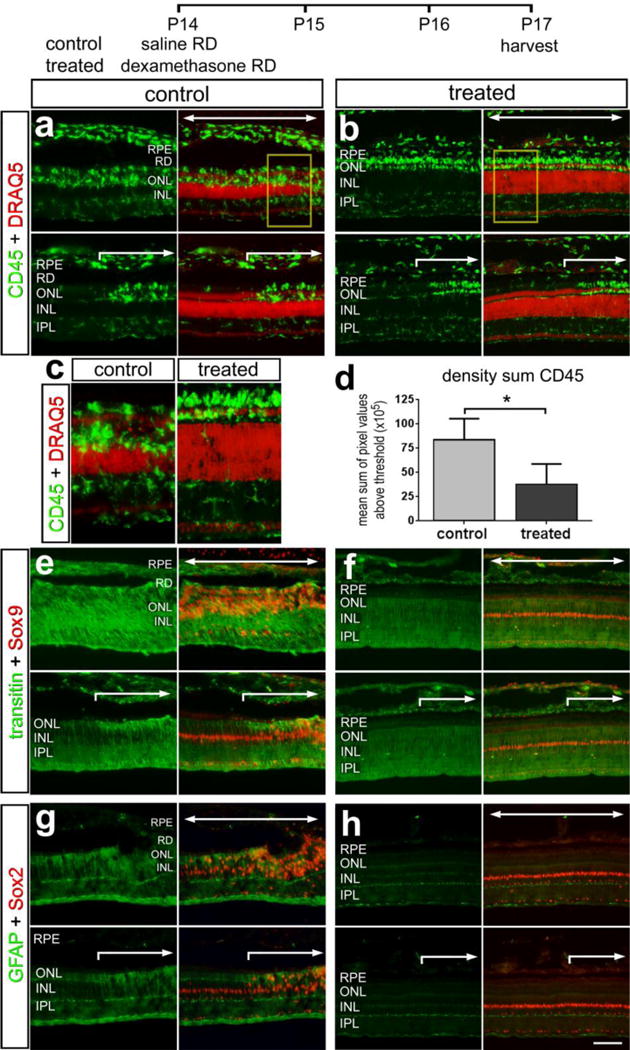
The reactivity of microglia and Müller glia is suppressed by Dex-mediated activation of GCR. Retinas were obtained from eyes that received a subretinal injection of saline (control) or saline + soluble Dex (treated), and tissues harvested 3 days later. Sections of the retina were labeled with DRAQ5 (a and b), and antibodies to CD45 (a and b), Sox9 and transitin (e and f), and Sox2 and GFAP (g and h). c and d; histograms illustrate the mean (±SD) density sum for CD45-immunofluorescence in saline and Dex-treated retinal detachments. Significance of difference (*p<0.05. **p<0.01, n=4) was determined by using a two-tailed t-test. The scale bar in h applies to a, b and e–h. Large arrows indicate regions of retina where the detachment occurred. Abbreviations: RPE – retinal pigmented epithelium, ONL – outer nuclear layer, INL – inner nuclear layer, IPL- inner plexiform layer.
Since GCR is expressed by Müller glia, not microglia/macrophages, we assessed whether the reactivity of Müller glia was influenced by Dex in regions of detached retinas. Müller glia were labeled with antibodies to Sox9 and Sox2 and the intermediate filaments transitin and GFAP respectively. In the chick retina, Müller glia are known to up-regulate transitin and GFAP in response to damage (Fischer and Omar 2005). In detached regions of control retinas levels of transitin and GFAP were elevated, and levels appeared reduced in Müller glia that were adjacent to the region of detachment (Figs. 8e,g). By comparison, levels of transitin and GFAP appeared lower in detached and attached regions of retinas treated with Dex (Figs. 8f,h). There was a dramatic reduction in interkinetic nuclear migration and proliferation of the sox-2 and sox-9 positive Muller glial progenitor cells in the dex-treated retinal detachments.
Changes in gene expression with activation or inhibition of GCR
To better understand how GCR-signaling influences glial reactivity and neuronal survival we probed for changes in expression levels of different cytokines and components of different signaling pathways in undamaged retina. By using qRT-PCR, we found that Dex decreased levels of the pro-inflammatory cytokine il1β and the cytokine-processing enzyme adam17, whereas levels of il6 and tnfα where unaffected (Fig. 9a). By comparison, RU486 had no effect upon retinal levels of il1β, tnfα and adam17, whereas levels of il6 were modestly reduced (Fig. 9a). We have found that inhibition of Notch-signaling in Müller glia can protect retinal neurons against NMDA-induced damage (Ghai et al. 2010). Treatment with Dex significantly reduced levels of notch1 and modestly increased levels of delta1, whereas levels of dll4, jag, hes1 and hes5 were unaffected (Fig. 9b). By comparison, treatment with RU486, increased levels of notch1 and hes1, whereas levels of delta1, dll4, jag and hes5 were unaffected (Fig. 9b). We have recently found that Hedgehog-signaling can have a profound influence upon Müller glia in mature chick retina (Todd & Fischer, unpublished observation). We found that activation of GCR with Dex reduced levels of the Hedgehog-related genes smo, gli1, gli3 and ptch1 (Fig. 9c). By comparison, inhibition of GCR with RU486 increased levels of smo and gli1, whereas levels of gli3 and ptch1 were unaffected (Fig. 9c). The transcription factors ascl1a and pax2 are known to be dynamically regulated in Müller glia that have been stimulated to become progenitor-like (Fischer and Reh 2001; Stanke et al. 2010). We found that Dex-treatment resulted in a small, but significant, decrease in ascl1a and pax2. We found that levels of ascl1a were unaffected by RU486, while levels of pax2 were significantly increased (Fig. 9d).
Figure 9.
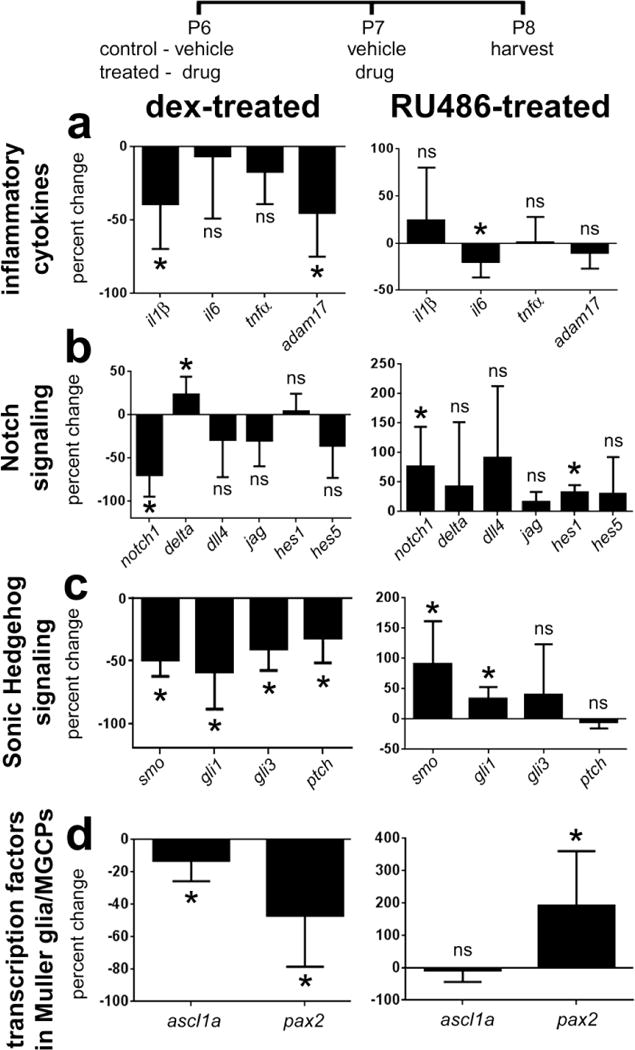
Quantitative RT-PCR was used to measure retinal levels of mRNA in response to GCR-agonist or antagonist. Retinas were obtained from eyes that received two consecutive daily injections of Dex or RU486. Tissues were harvested 24 hrs after the last injection, total RNA extracted from individual retinas, and RNAs processed for qRT-PCR. The histograms illustrate the mean (±SD; n≥5) percent change in mRNA levels for genes related to pro-inflammatory cytokines, Notch-signaling, Sonic Hedgehog-signaling and transcription factors known to be expression by Müller glia/MGPCs. Significance of difference (*p<0.05) was determined by using Mann-Whitney U test.
Discussion
Our findings indicate that GCR-signaling can mediate significant changes in glial reactivity and neuronal survival in the retina following different types of insults. The effects of GCR agonists and antagonists in the retina are predominantly elicited through the Müller glia. We have recently reported that GCR is expressed by Müller glia in the retinas of birds, mice, guinea pigs, canines and humans (Gallina et al. 2014). Further, evidence indicates that GCR agonists directly impact cell-signaling through the Müller glia (Gallina et al. 2014). Thus, neuronal protection and changes microglial reactivity resulting from GCR-signaling are mediated by the Müller glia. In support on the notion that GCR-signaling directly regulates Müller glia, we have reported that MAPK-signaling is antagonized by GCR agonist (Gallina et al. 2014), and that levels of Notch-related genes, Shh-related genes, ascl1a and pax2 are down- or up-regulated by GCR agonist and antagonist, respectively, in normal retinas (current study). These findings suggest that in normal Müller glia, GCR-signaling inhibits the progenitor-like properties of these glia. The progenitor-like properties of Müller glia are promoted by Notch-signaling (Ghai et al. 2010; Hayes et al. 2007), Shh-signaling (Todd and Fischer, unpublished), ascl1a (Fausett and Goldman 2006; Fischer and Reh 2001), and levels of Pax2 are increased by conditions wherein MGPCs are formed (Stanke et al. 2010).
The reactivity of microglia in the retina can be stimulated by many different factors, even by merely puncturing the eye (Fischer et al. 2014). Regardless, of the type of retinal damage, GCR agonist suppressed microglial reactivity and protected retinal neurons against damage. Similarly, the anti-inflammatory effects of glucocorticoids on microglia in the brain are believed to be neuroprotective (Carrillo-de Sauvage et al. 2013; Golde et al. 2003; Nadeau and Rivest 2003). In the CNS, the neuroendocrine system exerts influence on immune responses through activation of steroid hormone receptors (Sternberg 2006). Indeed, microglia are known to express steroid-activated nuclear receptors (Sierra et al. 2008). In damaged tissues, evidence indicates that activation of GCR limits inflammation and stimulates the resolution phase (Goulding 2004). Treatment with Dex can decrease iNOS-mediated toxicity of microglia (Golde et al. 2003), and inhibition of the GCR-signaling can exacerbate cell damage resulting from the pro-inflammatory actions of lipopolysaccharide (Nadeau and Rivest 2003). Further, a recent study has shown that GCR acts on several key processes limiting proinflammatory actions of activated microglia (Carrillo-de Sauvage et al. 2013). Given that activated nuclear GCR in the retina is predominantly found in Müller glia, not microglia (Gallina et al. 2014), we propose that the anti-inflammatory effects of Dex on microglia are indirectly elicited through the Müller glia.
Consistent with the hypothesis that the neuroprotective effects of GCR-signaling are primarily elicited through the Müller glia, we found that Dex continued to reduce numbers of dying cells in NMDA-damaged retinas in the absence of microglia/macrophages. However, the efficacy of Dex-mediated neuroprotection was significantly less when microglia/macrophages where absent compared to the effects of Dex in the presence of microglia/macrophages. These findings suggest that the protective effects of GCR-signaling are mediated, in part, through the Müller glia and by suppressing the reactivity of microglia. However, some studies have shown that corticosterone influences NMDA-mediated currents in culture preparations of different types of neurons (Liu et al. 2007; Sato et al. 2004; Takahashi et al. 2002). These findings indicate that GCR-signaling may directly influence neuronal activity. However, it is unknown whether GCR-expressing glial cells were included in the in vitro preparations.
Different doses of Dex elicited different effects upon the reactivity of microglia. Application of Dex before an excitotoxic insult suppresses microglial reactivity. The higher dose of Dex had long-lasting effects on suppressing microglial reactivity. Our data suggest that microglia/macrophages can be predisposed to be quiescent, since activation of GCR before damage kept the microglia/macrophages quiescent. By comparison, the lower dose of Dex had no significant effect upon microglial reactivity, but was more potent than the higher dose in reducing numbers of dying cells. Elevated or sustained applications of Dex are known to elicit effects opposite to those observed at lower or short-term doses. For example, biphasic effects of Dex have been reported for glycogen metabolism (Zheng et al. 2009), systemic hormone levels (Erman et al. 1987) (Ceda et al. 1987), skeletal development (Cheng et al. 2014), and inflammatory responses of endothelial cells (Prajgrod and Danon 1992). It is possible that the higher dose of Dex has a diminished effect upon neuronal survival because this dose is approaching the level at which the effects of GCR-signaling are reversed.
Protection of neurons against NMDA-induced excitotoxicity was elicited by different doses of GCR-agonist, but the efficacy of neuroprotection at the different doses was not related to the efficacy of suppressed microglial reactivity. The higher dose of Dex was less effective than the lower dose of Dex in suppressing NMDA-induced cell death. Interestingly, the lower dose of Dex that was more neuroprotective than the higher dose, and this lower dose had little effect upon the reactivity of microglia/macrophages in damaged retinas. This suggests that suppression of microglial reactivity may not directly underlie neuroprotection. However, in the absence of microglia/macrophages the neuroprotective effects of the low dose of Dex were diminished compared to the neuroprotective effects in the presence of reactive microglia. These findings suggest that Dex-treated microglia/macrophages, with diminished reactivity, may support neuronal survival following an insult. Interestingly, the ablation of retinal microglia/macrophages does not influence the death of cells in NMDA-damaged retinas (Fischer et al. 2015a). It has been shown that microglia/macrophages acquire two distinct activation patterns, M1 (classical) and M2 (alternative). The activated M1 cells release cytokines and cytotoxic molecules that are important for host defense, destruction of tumor cells and promotion of inflammation (Ding et al. 1988). The M2 cells release anti-inflammatory molecules, and are involved in tissue repair and remodeling while suppressing tissue damage caused by inflammation (Sica et al. 2006). It has been well established that glucocorticoids give rise to M2 microglia/macrophages in vitro (Mantovani et al. 2004). However, it is not known and whether activation of GCR leads to an M2 phenotype for microglia/macrophages in the retina in vivo. Our data suggest that the low dose of Dex might shift the microglia/macrophages towards the M2 phenotype and thereby enhanced neuroprotection.
We have recently reported that the ablation of microglia/macrophages effectively blocks the formation of folds and detachments in damaged retinas, whereas IL6-mediated activation of microglial reactivity exacerbates these folds and detachments (Fischer et al. 2015a). In the present study we show that the folds are also exacerbated by applying FGF2 before NMDA. Consistent with the previous findings, we report here that suppression of microglial reactivity greatly alleviates the folds and detachments in FGF2 and NMDA-damaged retinas, and alleviates the damage that is incurred by saline-induced detachments. Evidence shows that, following retinal detachment, infiltrating macrophages and resident microglia become reactive and accumulate in the retina and in the sub-retinal space, and these reactive monocytes contribute to retinal detachment pathophysiology, including photoreceptor apoptosis (Anderson et al. 1983; Hisatomi et al. 2003; Kaneko et al. 2008; Lewis et al. 2002; Lewis et al. 2005; Nakazawa et al. 2007). We propose that suppressing the reactivity of microglia/macrophages may be an effective treatment to preserve photoreceptors and vision following retinal detachment.
Conclusions
We conclude that GCR-signaling can have profound effects upon neuronal survival in damaged retinas. We propose that the effects of GCR-signaling are manifested predominantly through changes in the Müller glia, which are positive for activated nuclear GCR, and secondarily by changes in microglia, which are negative for nuclear GCR. Collectively, our findings support the hypothesis that suppressed microglial reactivity resulting from Dex-treatment can promote neuronal survival. Activation of GCR-signaling protects inner retinal neurons, including ganglion cells, against cytotoxic damage, and protects photoreceptors from damage during retinal detachment. GCR-signaling through Müller glia may promote neuronal survival by modulating levels of inflammatory cytokines.
Highlights.
- Glucocorticoid signaling in Müller glia suppresses microglial reactivity.
- Glucocorticoid signaling in Müller glia protects amacrine and bipolar cells against excitotoxic damage.
- Glucocorticoid agonists retain the capacity to protect retinal neurons when reactive microglia/macrophages are ablated.
- Glucocorticoid signaling in Müller glia protects ganglion cells against colchicines-induced damage, and photoreceptors against detachment-induced damage.
Acknowledgments
We thank Levi Todd for comments that contributed to the final form of this paper. The transitin (developed by Dr. G. Cole) antibody was obtained from the Developmental Studies Hybridoma Bank developed under auspices of the NICHD and maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA 52242. This work was supported by a grant (EY022030-03) from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson DH, Stern WH, Fisher SK, Erickson PA, Borgula GA. Retinal detachment in the cat: the pigment epithelial-photoreceptor interface. Invest Ophthalmol Vis Sci. 1983;24(7):906–26. [PubMed] [Google Scholar]
- Buschini E, Piras A, Nuzzi R, Vercelli A. Age related macular degeneration and drusen: neuroinflammation in the retina. Prog Neurobiol. 2011;95(1):14–25. doi: 10.1016/j.pneurobio.2011.05.011. [DOI] [PubMed] [Google Scholar]
- Carrillo-de Sauvage MA, Maatouk L, Arnoux I, Pasco M, Sanz Diez A, Delahaye M, Herrero MT, Newman TA, Calvo CF, Audinat E, et al. Potent and multiple regulatory actions of microglial glucocorticoid receptors during CNS inflammation. Cell Death Differ. 2013;20(11):1546–57. doi: 10.1038/cdd.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebulla CM, Zelinka CP, Scott MA, Lubow M, Bingham A, Rasiah S, Mahmoud AM, Fischer AJ. A chick model of retinal detachment: cone rich and novel. PLoS One. 2012;7(9):e44257. doi: 10.1371/journal.pone.0044257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceda GP, Davis RG, Hoffman AR. Glucocorticoid modulation of growth hormone secretion in vitro. Evidence for a biphasic effect on GH-releasing hormone mediated release. Acta Endocrinol (Copenh) 1987;114(4):465–9. doi: 10.1530/acta.0.1140465. [DOI] [PubMed] [Google Scholar]
- Cheng X, Chen JL, Ma ZL, Zhang ZL, Lv S, Mai DM, Liu JJ, Chuai M, Lee KK, Wan C, et al. Biphasic influence of dexamethasone exposure on embryonic vertebrate skeleton development. Toxicol Appl Pharmacol. 2014;281(1):19–29. doi: 10.1016/j.taap.2014.09.014. [DOI] [PubMed] [Google Scholar]
- Chiang CS, Stalder A, Samimi A, Campbell IL. Reactive gliosis as a consequence of interleukin-6 expression in the brain: studies in transgenic mice. Dev Neurosci. 1994;16(3–4):212–21. doi: 10.1159/000112109. [DOI] [PubMed] [Google Scholar]
- De Bosscher K, Beck IM, Haegeman G. Classic glucocorticoids versus non-steroidal glucocorticoid receptor modulators: survival of the fittest regulator of the immune system? Brain Behav Immun. 2010;24(7):1035–42. doi: 10.1016/j.bbi.2010.06.010. [DOI] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Beck IM, Van Molle W, Hennuyer N, Hapgood J, Libert C, Staels B, Louw A, Haegeman G. A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci U S A. 2005;102(44):15827–32. doi: 10.1073/pnas.0505554102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding AH, Nathan CF, Stuehr DJ. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J Immunol. 1988;141(7):2407–12. [PubMed] [Google Scholar]
- Dorrell MI, Friedlander M. Mechanisms of endothelial cell guidance and vascular patterning in the developing mouse retina. Prog Retin Eye Res. 2006;25(3):277–95. doi: 10.1016/j.preteyeres.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Erman A, Pitcock JA, Liston T, Brown P, Baer PG, Nasjletti A. Biphasic effect of dexamethasone on urinary prostaglandins in rats: relation to alterations in renal medulla triglycerides and prostaglandin metabolism. Endocrinology. 1987;121(5):1853–61. doi: 10.1210/endo-121-5-1853. [DOI] [PubMed] [Google Scholar]
- Fausett BV, Goldman D. A role for alpha1 tubulin-expressing Muller glia in regeneration of the injured zebrafish retina. J Neurosci. 2006;26(23):6303–13. doi: 10.1523/JNEUROSCI.0332-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Bongini R. Turning Muller glia into neural progenitors in the retina. Mol Neurobiol. 2010;42(3):199–209. doi: 10.1007/s12035-010-8152-2. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Morgan IG, Stell WK. Colchicine causes excessive ocular growth and myopia in chicks. Vision Res. 1999;39(4):685–97. doi: 10.1016/s0042-6989(98)00178-3. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Omar G. Transitin, a nestin-related intermediate filament, is expressed by neural progenitors and can be induced in Muller glia in the chicken retina. J Comp Neurol. 2005;484(1):1–14. doi: 10.1002/cne.20406. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Omar G, Eubanks J, McGuire CR, Dierks BD, Reh TA. Different aspects of gliosis in retinal Muller glia can be induced by CNTF, insulin and FGF2 in the absence of damage. Molecular Vision. 2004;10:973–986. [PubMed] [Google Scholar]
- Fischer AJ, Reh TA. Muller glia are a potential source of neural regeneration in the postnatal chicken retina. Nat Neurosci. 2001;4(3):247–52. doi: 10.1038/85090. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Reh TA. Exogenous growth factors stimulate the regeneration of ganglion cells in the chicken retina. Dev Biol. 2002;251(2):367–79. doi: 10.1006/dbio.2002.0813. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Ritchey ER, Scott MA, Wynne A. Bullwhip neurons in the retina regulate the size and shape of the eye. Dev Biol. 2008;317(1):196–212. doi: 10.1016/j.ydbio.2008.02.023. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Scott MA, Ritchey ER, Sherwood P. Mitogen-activated protein kinase-signaling regulates the ability of Müller glia to proliferate and protect retinal neurons against excitotoxicity. Glia. 2009a;57(14):1538–1552. doi: 10.1002/glia.20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Scott MA, Tuten W. Mitogen-activated protein kinase-signaling stimulates Muller glia to proliferate in acutely damaged chicken retina. Glia. 2009b;57(2):166–81. doi: 10.1002/glia.20743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Scott MA, Zelinka C, Sherwood P. A novel type of glial cell in the retina is stimulated by insulin-like growth factor 1 and may exacerbate damage to neurons and Muller glia. Glia. 2010a;58(6):633–49. doi: 10.1002/glia.20950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Seltner RL, Poon J, Stell WK. Immunocytochemical characterization of quisqualic acid- and N-methyl-D-aspartate-induced excitotoxicity in the retina of chicks. J Comp Neurol. 1998;393(1):1–15. [PubMed] [Google Scholar]
- Fischer AJ, Skorupa D, Schonberg DL, Walton NA. Characterization of glucagon-expressing neurons in the chicken retina. J Comp Neurol. 2006;496(4):479–94. doi: 10.1002/cne.20937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Zelinka C, Gallina D, Scott MA, Todd L. Reactive microglia and macrophage facilitate the formation of Muller glia-derived retinal progenitors. Glia. 2014;62(10):1608–1628. doi: 10.1002/glia.22703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Zelinka C, MIlani-Nejad N. Reactive retinal microglia, neuronal survival and the formation of retinal folds and detachments Glia in press. 2015a doi: 10.1002/glia.22752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Zelinka C, Milani-Nejad N. Reactive retinal microglia, neuronal survival, and the formation of retinal folds and detachments. Glia. 2015b;63(2):313–27. doi: 10.1002/glia.22752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Zelinka C, Scott MA. Heterogeneity of glia in the retina and optic nerve of birds and mammals. PLoS One. 2010b;5(6):e10774. doi: 10.1371/journal.pone.0010774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SK, Anderson DH. In: Cellular effects of detachment on the neural retina and the retinal pigmented epithelium. Ryan SJ, editor. Retina St Louis: C.V Mosby; 1994. pp. 2035–2061. [Google Scholar]
- Fisher SK, Lewis GP. Muller cell and neuronal remodeling in retinal detachment and reattachment and their potential consequences for visual recovery: a review and reconsideration of recent data. Vision Res. 2003;43(8):887–97. doi: 10.1016/s0042-6989(02)00680-6. [DOI] [PubMed] [Google Scholar]
- Friedlander M, Dorrell MI, Ritter MR, Marchetti V, Moreno SK, El-Kalay M, Bird AC, Banin E, Aguilar E. Progenitor cells and retinal angiogenesis. Angiogenesis. 2007;10(2):89–101. doi: 10.1007/s10456-007-9070-4. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Imagawa T, Uehara M. Fine structure of the retino-optic nerve junction in the chicken. Tissue Cell. 2001;33(2):129–34. doi: 10.1054/tice.2000.0152. [DOI] [PubMed] [Google Scholar]
- Gallina D, Zelinka C, Fischer AJ. Glucocorticoid receptors in the retina, Müller glia and the formation of Müller glia-derived progenitors. Development. 2014;141:3340–3351. doi: 10.1242/dev.109835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghai K, Zelinka C, Fischer AJ. Serotonin released from amacrine neurons is scavenged and degraded in bipolar neurons in the retina. J Neurochem. 2009;111(1):1–14. doi: 10.1111/j.1471-4159.2009.06270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghai K, Zelinka C, Fischer AJ. Notch signaling influences neuroprotective and proliferative properties of mature Muller glia. J Neurosci. 2010;30(8):3101–12. doi: 10.1523/JNEUROSCI.4919-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde S, Coles A, Lindquist JA, Compston A. Decreased iNOS synthesis mediates dexamethasone-induced protection of neurons from inflammatory injury in vitro. Eur J Neurosci. 2003;18(9):2527–37. doi: 10.1046/j.1460-9568.2003.02917.x. [DOI] [PubMed] [Google Scholar]
- Goulding NJ. The molecular complexity of glucocorticoid actions in inflammation - a four-ring circus. Curr Opin Pharmacol. 2004;4(6):629–36. doi: 10.1016/j.coph.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Hayes S, Nelson BR, Buckingham B, Reh TA. Notch signaling regulates regeneration in the avian retina. Dev Biol. 2007;312(1):300–11. doi: 10.1016/j.ydbio.2007.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisatomi T, Sakamoto T, Sonoda KH, Tsutsumi C, Qiao H, Enaida H, Yamanaka I, Kubota T, Ishibashi T, Kura S, et al. Clearance of apoptotic photoreceptors: elimination of apoptotic debris into the subretinal space and macrophage-mediated phagocytosis via phosphatidylserine receptor and integrin alphavbeta3. Am J Pathol. 2003;162(6):1869–79. doi: 10.1016/s0002-9440(10)64321-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko H, Nishiguchi KM, Nakamura M, Kachi S, Terasaki H. Characteristics of bone marrow-derived microglia in the normal and injured retina. Invest Ophthalmol Vis Sci. 2008;49(9):4162–8. doi: 10.1167/iovs.08-1738. [DOI] [PubMed] [Google Scholar]
- Karlstetter M, Ebert S, Langmann T. Microglia in the healthy and degenerating retina: insights from novel mouse models. Immunobiology. 2010;215(9–10):685–91. doi: 10.1016/j.imbio.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Klein MA, Moller JC, Jones LL, Bluethmann H, Kreutzberg GW, Raivich G. Impaired neuroglial activation in interleukin-6 deficient mice. Glia. 1997;19(3):227–33. doi: 10.1002/(sici)1098-1136(199703)19:3<227::aid-glia5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Langmann T. Microglia activation in retinal degeneration. J Leukoc Biol. 2007;81(6):1345–51. doi: 10.1189/jlb.0207114. [DOI] [PubMed] [Google Scholar]
- Lebrun-Julien F, Duplan L, Pernet V, Osswald I, Sapieha P, Bourgeois P, Dickson K, Bowie D, Barker PA, Di Polo A. Excitotoxic death of retinal neurons in vivo occurs via a non-cell-autonomous mechanism. J Neurosci. 2009;29(17):5536–45. doi: 10.1523/JNEUROSCI.0831-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis GP, Charteris DG, Sethi CS, Fisher SK. Animal models of retinal detachment and reattachment: identifying cellular events that may affect visual recovery. Eye (Lond) 2002;16(4):375–87. doi: 10.1038/sj.eye.6700202. [DOI] [PubMed] [Google Scholar]
- Lewis GP, Fisher SK. Up-regulation of glial fibrillary acidic protein in response to retinal injury: its potential role in glial remodeling and a comparison to vimentin expression. Int Rev Cytol. 2003;230:263–90. doi: 10.1016/s0074-7696(03)30005-1. [DOI] [PubMed] [Google Scholar]
- Lewis GP, Guerin CJ, Anderson DH, Matsumoto B, Fisher SK. Rapid changes in the expression of glial cell proteins caused by experimental retinal detachment. Am J Ophthalmol. 1994;118(3):368–76. doi: 10.1016/s0002-9394(14)72962-9. [DOI] [PubMed] [Google Scholar]
- Lewis GP, Sethi CS, Carter KM, Charteris DG, Fisher SK. Microglial cell activation following retinal detachment: a comparison between species. Mol Vis. 2005;11:491–500. [PubMed] [Google Scholar]
- Liu L, Wang C, Ni X, Sun J. A rapid inhibition of NMDA receptor current by corticosterone in cultured hippocampal neurons. Neurosci Lett. 2007;420(3):245–50. doi: 10.1016/j.neulet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Liu W, Khare SL, Liang X, Peters MA, Liu X, Cepko CL, Xiang M. All Brn3 genes can promote retinal ganglion cell differentiation in the chick. Development. 2000;127(15):3237–47. doi: 10.1242/dev.127.15.3237. [DOI] [PubMed] [Google Scholar]
- Madeira MH, Boia R, Santos PF, Ambrosio AF, Santiago AR. Contribution of Microglia-Mediated Neuroinflammation to Retinal Degenerative Diseases. Mediators Inflamm. 2015;2015:673090. doi: 10.1155/2015/673090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–86. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Morgan IG. Intraocular colchicine selectively destroys immature ganglion cells in chicken retina. Neurosci Lett. 1981;24(3):255–60. doi: 10.1016/0304-3940(81)90167-1. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Glucocorticoids play a fundamental role in protecting the brain during innate immune response. J Neurosci. 2003;23(13):5536–44. doi: 10.1523/JNEUROSCI.23-13-05536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T, Hisatomi T, Nakazawa C, Noda K, Maruyama K, She H, Matsubara A, Miyahara S, Nakao S, Yin Y, et al. Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc Natl Acad Sci U S A. 2007;104(7):2425–30. doi: 10.1073/pnas.0608167104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkowa M, Giralt M, Lago N, Camats J, Carrasco J, Hernandez J, Molinero A, Campbell IL, Hidalgo J. Astrocyte-targeted expression of IL-6 protects the CNS against a focal brain injury. Exp Neurol. 2003;181(2):130–48. doi: 10.1016/s0014-4886(02)00051-1. [DOI] [PubMed] [Google Scholar]
- Prajgrod G, Danon A. Biphasic regulation by dexamethasone of IL-1- and LPS-stimulated endothelial prostacyclin production. Agents Actions. 1992;36(1–2):70–6. doi: 10.1007/BF01991231. [DOI] [PubMed] [Google Scholar]
- Sato S, Osanai H, Monma T, Harada T, Hirano A, Saito M, Kawato S. Acute effect of corticosterone on N-methyl-D-aspartate receptor-mediated Ca2+ elevation in mouse hippocampal slices. Biochem Biophys Res Commun. 2004;321(2):510–3. doi: 10.1016/j.bbrc.2004.06.168. [DOI] [PubMed] [Google Scholar]
- Shen W, Fruttiger M, Zhu L, Chung SH, Barnett NL, Kirk JK, Lee S, Coorey NJ, Killingsworth M, Sherman LS, et al. Conditional Mullercell ablation causes independent neuronal and vascular pathologies in a novel transgenic model. J Neurosci. 2012;32(45):15715–27. doi: 10.1523/JNEUROSCI.2841-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42(6):717–27. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Sierra A, Gottfried-Blackmore A, Milner TA, McEwen BS, Bulloch K. Steroid hormone receptor expression and function in microglia. Glia. 2008;56(6):659–74. doi: 10.1002/glia.20644. [DOI] [PubMed] [Google Scholar]
- Stanke J, Moose HE, El-Hodiri HM, Fischer AJ. Comparative study of Pax2 expression in glial cells in the retina and optic nerve of birds and mammals. J Comp Neurol. 2010;518(12):2316–33. doi: 10.1002/cne.22335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol. 2006;6(4):318–28. doi: 10.1038/nri1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Kimoto T, Tanabe N, Hattori TA, Yasumatsu N, Kawato S. Corticosterone acutely prolonged N-methyl-d-aspartate receptor-mediated Ca2+ elevation in cultured rat hippocampal neurons. J Neurochem. 2002;83(6):1441–51. doi: 10.1046/j.1471-4159.2002.01251.x. [DOI] [PubMed] [Google Scholar]
- Van Rooijen N. The liposome-mediated macrophage ‘suicide’ technique. J Immunol Methods. 1989;124(1):1–6. doi: 10.1016/0022-1759(89)90178-6. [DOI] [PubMed] [Google Scholar]
- van Rooijen N. Liposome-mediated elimination of macrophages. Res Immunol. 1992;143(2):215–9. doi: 10.1016/s0923-2494(92)80169-l. [DOI] [PubMed] [Google Scholar]
- Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174(1–2):83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Wang M, Ma W, Zhao L, Fariss RN, Wong WT. Adaptive Muller cell responses to microglial activation mediate neuroprotection and coordinate inflammation in the retina. J Neuroinflammation. 2011;8:173. doi: 10.1186/1742-2094-8-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won MH, Kang TC, Cho SS. Glial cells in the bird retina: immunochemical detection. Microsc Res Tech. 2000;50(2):151–60. doi: 10.1002/1097-0029(20000715)50:2<151::AID-JEMT7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Xiang M, Zhou L, Macke JP, Yoshioka T, Hendry SH, Eddy RL, Shows TB, Nathans J. The Brn-3 family of POU-domain factors: primary structure, binding specificity, and expression in subsets of retinal ganglion cells and somatosensory neurons. J Neurosci. 1995;15(7 Pt 1):4762–85. doi: 10.1523/JNEUROSCI.15-07-04762.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Zhou L, Peng YW, Eddy RL, Shows TB, Nathans J. Brn-3b: a POU domain gene expressed in a subset of retinal ganglion cells. Neuron. 1993;11(4):689–701. doi: 10.1016/0896-6273(93)90079-7. [DOI] [PubMed] [Google Scholar]
- Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retin Eye Res. 2009;28(5):348–68. doi: 10.1016/j.preteyeres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Zelinka CP, Scott MA, Volkov L, Fischer AJ. The Reactivity, Distribution and Abundance of Non-Astrocytic Inner Retinal Glial (NIRG) Cells Are Regulated by Microglia, Acute Damage, and IGF1. PLoS One. 2012;7(9):e44477. doi: 10.1371/journal.pone.0044477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Zhang N, Baehr W, Fu Y. Cone opsin determines the time course of cone photoreceptor degeneration in Leber congenital amaurosis. Proc Natl Acad Sci U S A. 2011;108(21):8879–84. doi: 10.1073/pnas.1017127108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XF, Liu L, Zhou J, Miao MY, Zhou JR, Zhu D, Xia ZF, Jiang CL. Biphasic effects of dexamethasone on glycogen metabolism in primary cultured rat hepatocytes. J Endocrinol Invest. 2009;32(9):756–8. doi: 10.1007/BF03346532. [DOI] [PubMed] [Google Scholar]