

Abstract
Polyglutamine (polyQ) diseases are a family of dominantly transmitted neurodegenerative disorders caused by an abnormal expansion of CAG trinucleotide repeats in the protein-coding regions of the respective disease-causing genes. Despite their simple genetic basis, the etiology of these diseases is far from clear. Over the past two decades, Drosophila has proven to be successful in modeling this family of neurodegenerative disorders, including the faithful recapitulation of pathological features such as polyQ length-dependent formation of protein aggregates and progressive neuronal degeneration. Additionally, it has been valuable in probing the pathogenic mechanisms, in identifying and evaluating disease modifiers, and in helping elucidate the normal functions of disease-causing genes. Knowledge learned from this simple invertebrate organism has had a large impact on our understanding of these devastating brain diseases.
Common neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) have complicated etiologies. Although environmental factors have been increasingly suspected to play a role in these diseases, the causes for the majority of cases are unclear, and only a small proportion is linked to specific genetic factors. In this regard, the family of polyglutamine (polyQ) diseases, also called glutamine repeat diseases, stands out for their relatively simple genetic basis, thus providing a model for more complex neurodegenerative diseases.
I. Polyglutamine diseases
I-1. The family of polyglutamine diseases
Currently, there are nine known polyQ diseases (Table 1), including Huntington’s disease (HD), Dentatorubral-pallidoluysian atrophy (DRPLA), spinobulbar muscular atrophy (SBMA), spinocerebellar ataxia type 1 (SCA1), type 2 (SCA2), type 3 (SCA3 or Machado-Joseph disease or MJD1), type 6 (SCA6), type 7 (SCA7) and type 17 (SCA17)1-14. All these diseases are caused by an abnormal expansion of a CAG repeat encoding a glutamine (Q) track in the protein-coding region of the mutated alleles of the respective disease genes (Table-1). For example, HD, the best-known polyQ disease, is caused by an abnormal expansion of CAG repeats in the exon 1 (HTTex1) of Huntingtin (HTT, Figure 1). In healthy individuals, the number of CAG repeats in HTT varies from 6 to 34. In contrast, in HD patients, the mutated allele is always expanded to more than 35 repeats1, 15, 16. DRPLA is caused by an unstable expansion of CAG repeats to a range of 49 to 88 in the middle of the Atrophin-1 gene, which normally has 6 to 35 CAG repeats17-21.
Table I.
Polyglutamine Diseases
| Disease | Gene |
Drosophila homolog (Annotation symbol) |
Transmission |
Glutamine
size |
repeat |
|---|---|---|---|---|---|
|
| |||||
| Normal | Patients | ||||
| Dentatorubral-pallidoluysian atrophy (DRPLA) |
Atrophin-1 (ATN1) | Atro (also called Grunge)(CG17108) |
Autosomal dominant |
6-35 | 49-88 |
|
| |||||
| Huntington’s disease (HD) | Huntingtin (HTT) | dHtt (CG9995) | Autosomal dominant |
6-35 | 36-121 |
|
| |||||
| Spinobulbar muscular atrophy (SBMA, Kennedy disease) |
Androgen receptor (AR) | Estrogen-related receptor (CG7404) |
X-linked, Dominant | 9-36 | 38-62 |
|
| |||||
| Spinocerebellar ataxia type 1 (SCA1) |
Ataxin-1 (ATXN1) | Ataxin 1 (CG4547) | Autosomal dominant |
6-44 | 39-82 |
|
| |||||
| Spinocerebellar ataxia type 2 (SCA2) |
Ataxin-2 (ATXN2) | Ataxin-2 (CG5166) | Autosomal dominant |
15-31 | 36-63 |
|
| |||||
| Spinocerebellar ataxia type 3 (SCA3), Machado-Joseph disease, (MJD1) |
Ataxin-3 (ATXN3) | - | Autosomal dominant |
12-40 | 55-84 |
|
| |||||
| Spinocerebellar ataxia type 6 (SCA6) |
a1A-voltage-dependent calcium channel subunit (CACNA1A ) |
Cacophony (CG43368) |
Autosomal dominant |
4-18 | 21-33 |
|
| |||||
| Spinocerebellar ataxia type 7 (SCA7) |
Ataxin-7 (ATXN7) | - | Autosomal dominant |
4-35 | 37-306 |
|
| |||||
| Spinocerebellar ataxia type 17 (SCA17) |
TATA-binding protein (TBP) |
Tbp (CG9874) | Autosomal dominant |
29-42 | 45-63 |
Figure 1. HTT proteins and HD mutation.

(A) Schematic illustration of the structure of amino acid glutamine (Q), which is encoded by the tri-nucleotide CAG.
(B) HD is caused by an abnormal expansion of the glutamine tract (polyQ) located near the N-terminus of HTT protein.
(C) Schematics of predicted secondary structures of human and Drosophila HTT proteins. Both are composed mainly of HEAT repeat (represented as cylinder boxes in the diagram, also see D).
(D) Illustration of the proposed structure of the HEAT repeat, a ~40 amino acid long hairpin-like protein motif.
Notably, all these disorders are dominantly transmitted and share several characteristic genetic as well as clinical features that are linked to the variation in the number of CAG repeats, such as phenotypic heterogeneity, an inverse relationship between the repeat length and the age of disease onset, and the phenomenon of genetic anticipation. The variable length of CAG repeats can only be partially responsible for the phenotypic heterogeneity, as patients with the same length of CAG repeats often show different phenotypic manifestations14, 19, 22-24. In the case of HD, although people with CAG repeats of 40 or longer invariably develop the disease, those carrying the CAG repeats in the range of 36-39 have reduced penetrance14, 22-28. Together, these observations suggest that in addition to CAG repeats, additional environmental factors and/or genetic modifiers exist that also affect disease pathogenesis.
I-2. Expanded polyQ tract causes dominant neuronal toxicity
It is generally believed that the expanded polyQ tract confers on their host proteins a dominant gain-of-function effect that is toxic to neurons14, 22-24, 26-28. First, all of these diseases are dominantly inherited, which is usually associated with a gain of function. Second, patients carrying loss of function mutations in polyQ disease genes show phenotypes that are different from neurodegeneration. For example, although patients with SBMA caused by polyQ expansion in the androgen receptor (AR) show some signs of loss of receptor activity29, SBMA cannot be solely due to loss of AR function, as patients with inactivation mutations in AR have a different phenotype (testicular feminization or androgen insensitivity syndrome) that does not include neuronal degeneration30, 31. Third, CAG expansion usually does not interfere with the normal expression of disease-causing genes. Finally, numerous studies in different animal models all support the gain-of-toxicity hypothesis in polyQ diseases32, 33 (see below).
I-3. Protein aggregates, a unifying pathological feature with an unclear pathogenic role
Abnormal protein aggregates (i.e., compact protein deposits) are a shared pathological hallmark of almost all neurodegenerative disorders, including extracellular plaques and intracellular tangles observed in AD and Lewy bodies in PD27, 34. Similarly, nuclear and cytoplasmic aggregates have been found in brain tissues of human patients in the majority of polyQ diseases and in corresponding established animal models1, 25, 27, 28, 34-39. This unifying pathological feature implicates a potential common pathogenic mechanism involving aggregates.
It is believed that once polyQ length exceeds the pathogenic threshold, mutated disease proteins become prone to misfold and adopt abnormal conformations that resist degradation by cellular clearance machineries such as the ubiquitin-proteasome system (UPS) and autophagy22, 27, 28, 34, 40. This idea is also supported by the observation that these protein aggregates contain 20S proteasome and molecular chaperones and are typically ubiquitinated41-43. It is proposed that the expanded polyQ tracts can be organized into polar zipper-like β-sheet structures held together by hydrogen bonding between the main chain and the side chain amides, with longer glutamine repeats leading to increasing stability of this association44.
Consistently, the propensity of the mutant proteins to form aggregates is tightly linked to the length of the polyQ tract27, 28, 45. For example, in cultured striatal cells, expressed HTTex1 proteins with normal polyQ lengths (<34) remained soluble while those with pathogenic polyQ lengths (>36) formed aggregates in a time- and polyQ length-dependent manner, with longer polyQ lengths enabling faster aggregation45.
Given the tight link between polyQ length and both the pathogenesis of these diseases and with aggregate formation, it is natural to hypothesize that the aggregates themselves are the toxic agents that kill neurons. However, despite being a common pathogenic feature, the role of aggregates in neuronal degeneration remains highly controversial. On the basis of different studies, aggregates have been assigned diverse roles such as neurotoxic agents, beneficial factors, or simply by-products of the diseases22, 27, 34, 40, 45. For example, the regional distribution of aggregates in tissues from polyQ disease patients does not always correspond to the sites of degeneration46-52. In addition, in HD mice, aggregates either appear in large quantities in cells that are spared in HD, or are detected with very low frequency (<1%) in the striatum where neuronal loss is prominent42, 53-55. Moreover, SCA1 transgenic mice that express mutant Ataxin 1 (ATXN1), the disease-causing gene, with 77 glutamines, but lacking the self-association region, develop ataxia and Purkinje cell pathology but without apparent nuclear aggregates56.
To date, it is generally believed that formation of aggregates is a dynamic process involving many smaller oligomeric species that are likely to be more toxic, whereas the large aggregates might be inert or even protective22, 27, 34, 40, 45. However, the nature of the toxic aggregate species and the exact role of aggregates in the pathogenesis of polyQ diseases remain to be determined.
I-4. Selective neurodegeneration in polyQ diseases
Another intriguing feature of polyQ diseases is the selective neuronal degeneration in the brain. All the identified disease genes are expressed ubiquitously. However, each disease more or less affects a specific subset of neurons13, 14, 23, 24, 34. For example, Purkinje cells are the primary target of degeneration in SCA157, whereas dentate neurons are the primary site of cerebellar pathology in SCA34. DRPLA mainly causes a combined degeneration of the dentatofugal and pallidofugal regions in the central nervous system20, 24, 58, 59, whereas in HD, the medium spiny projection neurons in the caudate and putamen are most notably affected60. This observation suggests that besides the expanded polyQ tract, other regional-, cell type- and protein-specific factors also account for pathogenesis.
I-5. Unclear pathogenic mechanisms underlying polyQ diseases
Over the past two decades, studies in different model systems have led to a growing number of hypotheses on the pathogenic mechanisms underlying polyQ diseases, from aggregates and apoptosis to transcriptional dysregulation and mitochondrial dysfunction, to malfunctioned cellular clearance machineries, among many others61-73. In addition, increasing evidence, supported by the observation of selective neuronal degeneration in polyQ diseases, have led to the hypothesis that the alteration of normal cellular functions of disease genes also plays a role in disease pathogeneses24, 38, 61, 62, 70. These diverse mechanisms might not be mutually exclusive. Rather, given the complexity of the diseases, it is likely that in each disease, multiple mechanisms contribute to different stages of disease initiation and progression. Nevertheless, to date, despite extensive studies, except for the consensus that the expanded polyQ tract is the culprit behind all the polyQ diseases, it is still not clear which of these molecular mechanisms plays an initiating role, which are secondary, and how they collectively contribute to the selective neuronal degeneration in each of the diseases.
II. Drosophila melanogaster, an excellent model organism
II-1. The fruit fly: small insect, big promise
Drosophila has been an excellent animal model to uncover the function of many evolutionarily conserved proteins74-80. Many genes essential for development are well conserved between the fly and human. As an experimental organism, Drosophila has been subjected to thorough genetic analysis for over a century, and its developmental biology is very well understood75. Many powerful experimental tools and in vivo assays have been perfected in the fly, such as easy and convenient methods to generate transgenic flies and manipulate its genome and the UAS/GAL4 binary expression system for targeted overexpression or knockdown of any gene in selected tissues75, 80-85, all of which allow convenient genetic manipulation in whole animals or in specific tissues. Furthermore, the ease of raising flies in large quantities and their short life cycle make Drosophila amenable to large-scale genetic screens, allowing the identification of essential genes and the isolation of novel components in signaling pathways. In fact, the functions of many important genes, including entire signaling pathways, such as the Wingless/Wnt and Notch signaling pathways, were first elucidated in the fly74-80. As such, this small insect has evolved into a favorite model organism for the functional analyses of many basic biological questions.
Drosophila has proven to be a valuable system to uncover the function of human neurodegenerative disease genes80. For example, fly homologs for many human neurodegenerative disease genes exist, including the AD genes Presenilin and amyloid precursor protein (APP)86-89. Loss of fly APP causes behavioral defects that can be compensated for by a functional human APP transgene, demonstrating the evolutionarily conserved function of fly APP 86. Analyses of the fly presenilin homolog have provided the first evidence that Presenilin is involved in the Notch signaling pathway and required for its proper processing87, 88, 90.
Conversely, earlier mutagenesis screens in the fly have led to the isolation of many novel mutants 91, such as swiss cheese92, bubblegum93, spongecake and eggroll94, which show late-onset progressive degeneration of the adult nervous system resembling various human diseases. For example, mutants for bubblegum, which encodes a fly homolog of human very long chain fatty acids (VLCFAs) acyl CoA synthetase93, show elevated levels of VLCFAs, as seen in the human disease adrenoleukodystrophy (ALD) 93. Unsaturated fatty acids have been shown to lower the excessive VLCFAs in ALD. Feeding the bubblegum mutant flies with glyceryl trioleate oil, an unsaturated fatty acid, can block the accumulation of excess VLCFAs 93. Recent screens in Drosophila have led to the identification of many additional neurodegenerative disease genes such as nmnat80, 95. As summarized in many reviews, Drosophila has becoming an important model organism for studying neurodegenerative diseases22, 96-105.
II-2. Drosophila eye, an excellent model tissue
Over the years, the Drosophila eye has emerged as a favorite experimental system to elucidate biological questions and model human diseases. First, the adult eye is not essential for the viability or fertility of the animal, allowing manipulations that severely disrupt eye development. Second, the adult eye phenotype can be easily examined under a dissecting microscope. Third, the structure of the Drosophila eye has been well characterized, and its developmental process extensively analyzed, thus a particular eye phenotype can be linked to a specific developmental process106-110. Fourth, eye-specific tools, such as the eyeless and glass promoters, enable eye-specific genetic manipulations, including targeted knockdown and ectopic expression (See Table II). Furthermore, a large percentage of essential genes and almost all the important signaling pathways in the fly are required for proper eye patterning. Finally, as a neuronal tissue, the Drosophila eye is well suited to model neurodegenerative diseases.
Table II.
Tissue-specific Gal4 drivers
| Promoter (Gal4 driver) |
Promoter specificity |
|---|---|
|
GMR-Gal4 (glass) |
All cells in and posterior to the MF 116 |
|
sevenless- Gal4 |
R1, R3, R4, R6, R7 and cone cells 280, 281 |
|
eyeless- Gal4 |
All cells in the eye imagincal disc, beginning early in development, prominently anterior to the MF 114 |
| elav-Gal4 | All central and peripheral nervous system 282-284 |
|
scabrous- Gal4 |
All central and peripheral nervous system 285 |
| 24B (Gal4) | Presumptive mesoderm and muscle cells 81 |
| dpp-Gal4 | Epithelial cells, along the anterior/posterior border of imaginal discs 286, 287 |
MF, morphogenetic furrow.
The wild-type adult fly eye is a beautifully organized lattice structure consisting of about 800 ommatidia (Figure 2)106-110. Within each ommatidium, there are eight neuronal photoreceptor cells surrounded by other non-neuronal accessory cells, including pigment cells and cone cells (Figure 2B and 2C). These cells can be easily recognized, which allows easy detection of even minor developmental defects (Figure 2B and 2C and Figure 3). The integrity of the internal photoreceptor cells can be easily visualized and quantified using the corneal pseudopupil technique without further dissection111, greatly facilitating phenotype evaluation in large-scale screens (Figure 3D-G).
Figure 2. The wildtype Drosophila eye structure.

(A) Scanning electron micrograph of a wild-type adult eye.
(B) Tangential section of one ommatidium unit. High-magnification view. Neuronal photoreceptor cells (black) are surrounded by pigment cells (red).
(C) Illustration of an ommatidium structure. The identity of each photoreceptor cell (black) is labeled. Pigment cells are painted in red.
Figure 3. Progressive neurodegeneration in a Drosophila HD model and its suppression by a modifier gene.
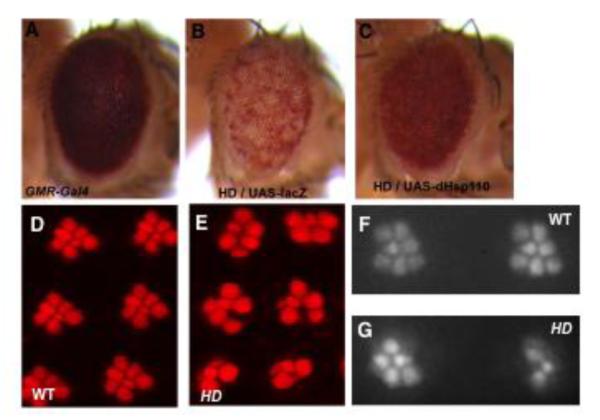
(A-C) 30-day-old adult fly eyes. (A) GMR-Gal4 control. (B and C) HD model that expresses HTT exon 1 (HTTex1)-Q93 together with (B) a LacZ control or (C) wildtype dHsp110 protein. Note the dramatic de-pigmentation of the eye in (B), indicating the significant loss of underlying eye tissues, which is clearly suppressed by the co-expression of dHsp110 (C) but not LacZ (B).
(D-G) Examination of photoreceptor cells in 7-day-old adult eyes, (D and E) visualized after dissection and immunofluorescent staining for F-Actin, or (F and G) visualized directly using pseudopupil technique without dissection. The seven well-organized photoreceptors in (D, F) wildtype (WT) were partially lost in (E, G) HTTex1-Q93 (HD) flies. Note that the pseudopupil method (F and G) produces comparable resolution for photoreceptor cells in the eye as that obtained by the more tedious dissection and staining approach (D and E).
The adult eye is derived from a sac of single layered epithelial cells called the eye imaginal disc. The eye differentiation process, visible as an indented morphogenetic furrow (MF), is initiated during 3rd instar larval stage from the posterior end of the eye disc and gradually moves within several days to the anterior end to specify the neuronal photoreceptor cells and other accessory cells, a process that lasts several days106-110, 112, 113.
III. Drosophila models of polyQ diseases
III-1. Experimental designs for modeling polyQ diseases in Drosophila
In Drosophila, one highly successful approach to model human diseases is to generate transgenic flies for a wildtype or mutant human disease gene and characterize the resulting phenotypes in the eye and other tissues. This is usually followed by phenotype-based modifier screens to uncover perturbed signaling pathways and novel pathogenic factors. The targeted overexpression or knockdown of selected genes is mostly achieved using the UAS/Gal4 binary expression system81, directed by the established tissue-specific Gal4 lines (Table II). For example, the promoter region from eyeless directs gene expression in all cells anterior to the MF, where cells are proliferating and no differentiation occurs114, 115. Promoters from elav and appl target gene expression to all specified neurons, including photoreceptor cells. GMR- (Glass-Multiple-Repeats) promoter is expressed in all cells within and posterior to the MF and continues to be expressed at high levels in these cells into adulthood116-118. Because of this, GMR-Gal4 is one of the most commonly used drivers to direct the continuous expression of a transgene in the eye. Importantly, the temporal expression pattern of GMR-Gal4 mirrors the highly ordered differentiation process of the eye during development, and cells at the posterior end of the eye disc mature roughly 2 days earlier than those in the anterior end.
III-2. Drosophila models recapitulate the pathological features of polyQ diseases
Two landmark studies, one on an SCA3 model by Warrick et al. from Nancy Bonini’s group and the other on HD by Jackson et al., convincingly show that the main pathogenic features of polyQ diseases can be faithfully recapitulated in Drosophila, thus establishing this tiny insect as an excellent organism to model these debilitating diseases119, 120.
Using the UAS/Gal4 system, Warrick et al. generated transgenic fly lines expressing a truncated human SCA3-causative gene Ataxin 3 (ATXN3) with a normal repeat of 27 glutamines (SCA3tr-Q27) or a pathogenic repeat of 78 glutamines (SCA3tr-Q78)120. Almost simultaneously, Jackson et al. reported the first fly HD model based on an N-terminal human HTT fragment containing the first 142 amino acids (a.a.) with a polyQ tract of normal (Q2) or pathogenic lengths directly under the control of the eye-specific GMR promoter (GMR-HTT1-142-Q75 (HD-Q75) or GMR-HTT1-142-Q120 (HDQ120))119. In both models, only the mutant proteins, but not controls of normal polyQ lengths, induce toxicity that is clearly illustrated in the eye. For SCA3 flies, when driven by GMR-Gal4, progeny from strong SCA3tr-Q78 lines show abnormally thin and severely de-pigmented eyes that are especially fragile and easily collapse, primarily due to the severe loss of underlying eye cells. However, during earlier development, the stereotypic differentiation and patterning of the pigment and neuronal photoreceptor cells all proceed normally, and the abnormality becomes detectable only after the completion of eye development, suggesting a late-onset loss of eye cells in these flies. Consistent with this conclusion, progeny from weak SCA3tr-Q78 lines show morphologically normal eyes at eclosion, but over time gradually lose eye pigmentation. More tellingly, the depigmentation starts from the posterior end of the eye and progressively spreads to the anterior end, nicely mirroring the temporal expression pattern of the GMR-Gal4 driver and providing a vivid illustration of progressive cellular degeneration, a feature that has been reproduced in other fly models of polyQ diseases (Figure 3B shows a HD model as an example).
Similar, although milder, eye degeneration also develops in the HD model: both HD-Q75 and HD-Q120 flies show normal eye morphology and intact ommatidial structure at eclosion (day 0), but by day 10, a significant subset of rhabdomeres are disrupted, with more severe disruption in HD-Q120 than in HD-Q75 flies.
Taking advantage of the flexibility of the UAS/Gal4 binary system, Warrick et al. further targeted the expression of truncated ATXN3 proteins to several other tissues and cell types using a battery of tissue-specific Gal4 lines. In all the tested Gal4 lines, SCA3tr-Q27 does not induce any obvious effect, whereas SCA3tr-Q78 show robust cell type-specific and dosage-sensitive toxicity. For example, when directed to differentiated neurons (by the pan-neuronal driver elav-Gal4), strong SCAtr-Q78 lines do not produce viable adult offspring. Progeny from the weak lines, corresponding to lower level of transgene expression, do survive to adulthood with normal external morphology, but they have significantly shortened lifespan, and over time their brains shrink in size, and the photoreceptors in the eye progressively degenerate.
The toxicity of SCA3tr-Q78 is not just restricted to neurons, as its expression in muscle (targeted by 24B Gal4), even from weak transgenic lines, causes animal lethality. In contrast, no toxicity is observed when SCA3tr-Q78 is expressed in epithelial cells (by dpp-Gal4), supporting cell type-specific toxicity of the SCA3tr-Q78 protein.
Importantly, both SCA3tr and HD flies also develop aggregates in an age-dependent manner, another pathologic feature of polyQ diseases. For example, at the subcellular level, while SCA3tr-Q27 maintains exclusive cytoplasmic distribution, SCA3tr-Q78 gradually translocates from the cytoplasm to the nucleus and forms prominent aggregates that grow larger over time. At the biochemical level, in addition to mutant SCA3tr-Q78, these aggregates also contain chaperones and other ubiquitinated proteins and can form highly compact structures that resist harsh treatments such as boiling in strong detergents solutions121, 122. Further, aggregates can develop in almost all cells that express SCA3tr-Q78, not only in the neurons and pigment cells that are vulnerable to SCA3tr-Q78 protein, but also in cells, such as epithelial cells, in which it is not toxic, suggesting that the aggregates alone do not necessarily cause degeneration120.
Following these studies, additional models of polyQ diseases, including SCA1, SCA7, SCA17, DRPLA and SBMA, have been subsequently established in Drosophila102, 123-130. These fly models largely recapitulate the main pathological features of the polyQ diseases, including progressive neuronal degeneration, cell type-specific toxicity of the mutant proteins, and aggregate formation. Detailed characterizations of these fly models have provided new insights into the complicated mechanisms of these diseases.
III-3. Lessons from Drosophila models of polyQ diseases
The polyQ track alone is toxic
To examine whether the polyQ tract alone, in the absence of any disease protein context, is sufficient to induce neurodegeneration, Marsh and Thompson’s group generated transgenic flies expressing a peptide with either 22 (Q22) or 108 (Q108) glutamines flanked by only a few amino acids at both ends131. Tested with a similar set of Gal4 lines used in the SCA3tr study120, only the Q108 peptide can elicit strong deleterious effects. A similar study by Kazemi-Esfarjani and Benzer, who generated flies expressing peptides with either 20 (Q20-HA) or 127 (Q127-HA) glutamines tagged with a short HA epitope, produced similar results132.
PolyQ expansion might affect but does not abolish the normal function of the disease proteins
A question in polyQ diseases is whether polyQ expansion affects the normal function of its host protein. To explore this question, Marsh et al. used disheveled (dsh), a Drosophila gene of the Wingless/Wnt signaling pathway with well-characterized mutant phenotypes whose gene product is a ubiquitously expressed protein with a native 28-glutamine tract133-135. For the engineered dsh transgenes that express Dsh with no (Dsh-Q0), 27 (Dsh-Q27), or 108 (Dsh-Q108) glutamines under its own native promoter, Dsh-Q27 can fully rescue the phenotypes of flies with a null dsh mutation, while both Dsh-Q0 and Dsh-108Q have only partial rescue efficiency, suggesting that the polyQ tract is not critical for the normal function of Dsh but is necessary for its full biological activity in vivo131. Similarly, in both Drosophila and mammalian systems, pathogenic ATXN1 (SCA1-Q82) shows conserved functional interactions with the same group of binding partners (e.g., scaffold protein 14-3-3, transcription regulators Capicua and Senseless/Gfi-1) as wildtype ATXN1 (SCA1-Q2), although with altered affinity, to regulate transcription and neurogenesis136-138. In addition, human Atrophin-1 with 118Q (Atrophin-1-Q118) still functions as a transcriptional co-repressor in vivo, similar to the fly Atrophin-1 homolog (atro) and wildtype Atrophin-1, but with reduced activity139. Moreover, pathogenic ATXN3 in SCA3 flies (SCA3-Q78 or Q84) retains a neuronal protective function through a proteasome-mediated mechanism, resembling that observed in normal ATXN3130. Lastly, in full-length AR-based fly SBMA model, AR with expanded polyQ can translocate into the nucleus and activate transcription in an androgen-dependent manner125, 127. Similar observations have also been documented for other polyQ disease genes. For example, in mouse and human samples, the polyQ tract in HTT is not essential for HTT’s function, but both the complete removal of the polyQ or its expansion partially affects HTT’s full activity139-146, post-translational modifications (PTMs) and/or stability, such as the inflammatory kinase IKK-mediated phosphorylation of HTT, which in turn regulates additional PTMs and fate of HTT protein147. Together, they suggest that in most cases, the polyQ does not abolish the normal function of host protein but might play a modulatory role for its full functionality.
Protein context determines the aggregation dynamics and toxicity of polyQ proteins
Examination of Drosophila polyQ models reveals that although the length of the polyQ tract is the main determining pathogenic factor, with the longer polyQ being associated with faster aggregation and stronger neurodegeneration, protein context plays a prominent role in the aggregation property and toxicity of the disease proteins. This is clearly exemplified in the SBMA and the extensively studied HD models. In full-length AR-based SBMA model, the toxicity of pathogenic AR requires its binding with its ligand androgen, which induces AR translocation into the nucleus to activate transcription125-127, whereas the toxicity elicited by a truncated AR (ARtr-Q112) is not androgen-dependent123. In HD, the polyQ tract, encoded in HTT’s exon 1, is near the very N-terminus of the encoded large HTT protein (~3,144 a.a.). Multiple naturally occurring N-terminal HTT fragments, potentially arising from proteolytic processing or aberrant splicing, have been documented in patient samples and in animal models148-160. Because of this, in addition to full-length HTT (HttFL), large numbers of HD models based on truncated HTT with various lengths of polyQ have been established161-166. In all these HD models, HTT variants with normal lengths of polyQ mainly localize to the cytoplasm, do not form aggregates and do not induce toxicity. Although majority of HTT variants with pathogenic polyQ lengths cause toxicity, their effects vary significantly161-166. For example, HttFL-Q128, which induces mild late-onset neurodegeneration, remains in the cytoplasm and does not form aggregates even in neurons of older flies164. However HTTex1-Q93 forms prominent cytoplasmic aggregates and causes severe degeneration (Figure 5 as an example), while Htt1-336-Q128 forms large aggregates primarily in the nucleus162, 164. In a carefully controlled study to compare the pathogenic potential and biophysical properties of the 7 naturally occurring N-terminal HTT fragments all carrying a Q120 tract, it has been shown that the shortest N-terminal HTTex1 fragment (90 a.a. plus 120 glutamines, HTT1-90-Q120) is the most toxic, most aggregation-prone, exhibiting unique biochemical properties and having the most potent amyloid seeding ability. On the other hand, the longer HTTex1 fragments either developed only lower levels of aggregates at a slower pace (e.g., HTT1-108-Q120) or remained diffused in the cytoplasm, hardly forming any visible aggregates at all (e.g., HTT1-469-Q120)161.
Figure 5. Aggregate formation by mutant HTT in Drosophila.

Formation of aggregates by mutant HTT protein can be modeled and studied in (A) cultured Drosophila cells and (B-D) adult fly eyes. In these studies, mutant HTT exon 1 fragment is revealed by eGFP tag fused in frame at its C-terminus.
(A) A double-labeling image of cultured Drosophila cells that express HTTex1-Q46. Aggregates (bright dots in top picture) are evident in some of the cells. The overall morphology of these cells are marked by staining for cytoskeletal protein F-actin (bottom picture in red), which reveals the sequestration of F-actin in these aggregates (bright dots in bottom picture).
(B-D) Images of same adult fly eyes illuminated by (top panels) bright light to show the overall eye morphology and by (bottom panels) fluorescent light to reveal the presence of eGFP-label HTTex1 aggregates, respectively.
(B) No fluorescent signal in the eye of a wildtype control fly (normal) that did not express human HTT protein.
(C) No clear aggregates in the eye of a transgenic fly that expressed HTTex1 with 23 glutamine (HTTex1-Q23).
(D) Numerous aggregates (bright dots) in the eye of a transgenic fly that expressed mutant HTTex1 with 103 glutamine (HTTex1-Q103).
Native functions of the disease genes in pathogenesis
Increasing evidence from fly- and mammalian-based studies suggest that the native functions of the disease genes directly affect pathogenesis. For example, in both SCA1 and SBMA models that express full-length human ATXN1 or AR, although the expanded proteins (SCA1-82Q or AR-Q52) cause stronger toxicity, overexpression of wildtype ATXN1or AR (SCA1-30Q or AR-Q12) also leads to neurodegeneration124, 125. In addition, the toxicity of ATXN1 in both flies and mice relies on its conserved AXH domain and requires its interaction with its endogenous binding partners such as Capicua, Senseless and 14-3-3, which control neurogenesis and ATXN1 stability136-138. Studies of SCA1 mouse models led to similar conclusions136, 137, 167. In the fly SBMA model, the AR-induced toxicity is ligand-dependent, requiring the presence of androgen or other known agonists125, 127. Furthermore, the native functions of AR, including its ability to bind target DNA sequences and recruit transcriptional coregulators, are essential for its toxicity125. In contrast, in SCA3 flies, overexpression of full-length wildtype ATXN3 (SCA3-Q27), encoding a protein with both ubiquitin binding motifs and ubiquitin protease activity, shows no deleterious effects and instead can potently suppress polyQ-induced neurodegeneration130. Because the pathogenic ATXN3 (SCA3-Q84) still retains this intrinsic neuroprotective function130, it raises the question as to how two opposite activities in the same protein counteract each other during disease pathogenesis. Nevertheless, these findings reveal the importance of the disease genes’ normal function in pathogenesis.
Axonal trafficking defect in polyQ diseases
When examining different fly models of polyQ diseases, both the Goldstein and Littleton groups have observed strong axonal trafficking defects in flies expressing polyQ-expanded proteins, but not in controls163, 168. For example, in larval motor neurons of wild type and control flies, cargoes such as synaptic proteins, vesicles, and mitochondria are effectively delivered to the neuromuscular junctions (NMJs) through the axon. In contrast, in polyQ flies (e.g., Htt1-548-Q128) that develop cytoplasmic aggregates, but not in lines (e.g., SCA3tr-Q78 and Q127-HA) that form exclusively nuclear aggregates, diminished delivery efficiency of the cargoes are observed, accompanied by prominent accumulation of aggregates with trapped synaptic organelles and mutant proteins along the swollen axon track. These animals also display sluggish movement, indicating the substantially compromised function of motor neurons. Further, reducing the dosage of key components of dynein- and kinesin-based motor complexes can strongly enhance this “axonal jamming” phenotype168. Given that neuronal cells have a particularly high reliance on axonal transport for long-distance delivery of essential constituents that maintain the survival and functionality of axonal projections, these findings implicate compromised axonal trafficking as one underlying contributing factor for polyQ diseases.
Transcriptional dysregulation in polyQ diseases
Notably, several polyQ disease proteins function either as transcription factors (e.g., TBP and AR) or transcription coregulators (e.g., Atxain-7 and Atrophin-1), or have been implicated extensively in transcriptional regulation (e.g., ATXN1 and HTT)136-138, 169-182. Such a convergence on transcription implicates a role of transcriptional dysregulation in disease pathogenesis, a hypothesis that has been supported by many studies from both fly and mouse models, as exemplified by the Capicua- and Senseless-mediated ATXN1 toxicity discussed earlier138-140. In addition, in the fly SBMA model, misappropriation of target gene expression by pathogenic AR (AR-Q52) is suspected to play a major role in its toxicity125. As another example, in mammalian cells, HTTex1 can inhibit histone acetyltransferases (HAT) activities of the transcriptional co-activator CREB-binding protein (CBP) and p300/CBP-associated factor (P/CAF), which modulate the accessibility of chromatin to sequence-specific transcription factors, resulting in reduced levels of acetylated histone H3 and H4 in mammalian cells176, 183, 184. In Drosophila, the robust neurodegeneration of HTTex1-Q48 and HTTex1-Q93 flies can be arrested by manipulating the cellular levels of histone acetylation, either pharmacologically (i.e., feeding with histone deacetyltransferase (HDAC) inhibitors SAHA and butyrate) or genetically. This result not only underscores the in vivo importance of transcriptional dysregulation in HD pathogenesis, but also nicely demonstrates the feasibility of using fly models to screen for potential bioactive compounds against polyQ diseases.
Aberrant neurotransmission and calcium homeostasis in HD pathogenesis
Both the Littleton and Botas groups have observed abnormal neurotransmission defects in HD flies. For example, in the giant fiber neuronal circuit, which controls escape response and flight initiation of the adult animals, Htt1-548-Q128 but not control Htt1-548-Q0 flies show increased neuronal activity163. However, given the severe degeneration and axonal blockade phenotypes in Htt1-548-Q128 flies, it is not clear whether the observed electrophysiological defects are a source of toxicity or just a secondary effect downstream of other cellular abnormalities. HTTFL-128Q flies, which develop mild late-onset neurodegeneration, also show abnormal electrophysiologic responses as early as in the third-instar larval NMJ before any apparent neuronal degeneration, showing an aberrantly higher level of resting presynaptic Ca2+ levels accompanied with increased neurotransmission release. As these animals do not develop detectable aggregates, evidence of axonal blockade, or translocation of HTTFL-128Q protein to the nucleus even in older animals, these data imply a cytoplasm-derived toxicity distinct from that induced by aggregate formation or impairments in axonal trafficking or transcription164. Further, genetic manipulations that blunt synaptic transmission or lower presynaptic Ca2+ levels can suppress the electrophysiological defects and neurodegeneration in HTTFL-128Q flies164. As aberrant calcium signaling has been observed in HD patients and mouse models53, 149, 153, 185-187, these findings suggest that abnormal Ca2+ homeostasis and Ca2+-dependent neurotransmission release may be early pathogenic events in HD, preceding aggregate formation, axonal blockade, and transcriptional dysregulation.
IV. Identification of genetic modifiers of polyQ diseases using Drosophila
With the successful generation of polyQ disease models in Drosophila, a wave of studies has followed to identify potential genetic modifiers of polyQ-associated toxicity and aggregate formation. The robust phenotypes manifested in fly polyQ models, especially the prominent eye degeneration and animal lethality, offer a convenient functional readout to test whether the toxicity can be influenced by specific genetic manipulations. In this regard, the easily accessible and assessable adult eye is frequently the tissue-of-choice in modifier screens. For a given fly polyQ model, eye color and external eye morphology can be directly evaluated under a dissection microscope, while the integrity of internal photoreceptor cells can be easily quantified using the corneal pseudopupil technique111, thus allowing for quick assessment of a large number of candidate modifier genes (examples in Figure 3 and Figure 5).
IV-1. Diverse molecular pathways modulate polyQ toxicity and aggregate formation
Through candidate-based approaches or unbiased forward genetic screens, a large number of genes involved in diverse molecular pathways have been isolated as genetic modifiers of polyQ diseases122-124, 132, 188-192. For example, Steffan et al. showed that different posttranslational modifications on HTTex1, such as ubiquitination or small ubiquitin-like modifier (SUMO)ylation, resulted in opposite effects on the pathogenic and biochemical properties of the HTTex1-Q97 protein193. These effects could be reversed genetically by manipulating the genes involved in SUMOylation or ubiquitination193. Separately, through large-scale mutagenesis screens, Kazemi-Esfarjani and Benzer isolated the chaperone DNAJ1(dHDj1/Hsp40) and other genes as modifiers of Q127 toxicity132, 189. Similarly Fernandez-Funez et al. identified SCA1 modifiers with roles in protein folding (DNAJ1), UPS, transcriptional regulation, and RNA processing, among others124, while Bilen and Bonini isolated 18 modifier genes for SCA3tr-Q78 with functions that converge on protein misfolding188. Separately, using an image-based genome-wide RNA interference (RNAi) screen in Drosophila S2 cells that stably express eGFP-tagged HTTex1-Q46 (Figure 5A), Zhang et al. isolated over 100 genes whose depletion modulates aggregate formation by HTTex1-Q46. Functionally, these aggregation modulators are associated with diverse cellular processes such as protein folding (e.g., Hsp110), transcriptional regulation (e.g., Rpd3), signal transduction (e.g., Tor), and others. Among them, several (e.g., DNAJ1, Sin3A, Sec61a) have been isolated previously as toxicity modifiers, suggesting that although aggregates are not directly responsible for toxicity, the misfolding process is intimately associated with toxicity, probably due to the production of intermediate oligomer species. Thus, aggregation of polyQ proteins such as HTTex1 is not only determined by polyQ length and protein context, but also by other cellular and genetic factors. Identification of these factors could allow systematic dissection of the molecular networks governing the formation and toxicity of aggregates.
IV-2. Common and disease-specific disease modifiers
Among the large number of genetic modifiers identified from different fly screens, except for a few common hits (e.g., chaperones, see below), most modifiers of different polyQ diseases do not overlap. For example, one study noted that none of the tested genetic suppressors isolated from other fly polyQ models rescue the lethality of HTT1-548-Q128 flies163. To examine whether these modifiers exert similar effects across different polyQ disease models, Branco et al. performed a comparative analysis on SCA1-Q82 and HTT1-336-Q128 models162. While many genetic modifiers for SCA1-82Q similarly affect HTT1-336-128Q flies, others show no effect and a few of them even behave in a contradictory manner. As an example, the serine/threonine kinase Akt1 has been shown to act as an enhancer of SCA1-Q82 toxicity, but with HTT1-336-128Q, it behaves as a suppressor. These findings highlight the importance of protein context in polyQ diseases, and also indicate that both common and distinct mechanisms affect their origin and progression.
IV-3. Protein folding machinery in polyQ toxicity
An emerging theme from multiple modifier screens is the convergence on molecular chaperones, which are an important cellular protection mechanism against cellular stress and protein misfolding194, 195. Powered by its ATPase activity, Hsp70 chaperones operate through ATP-dependent iterative cycles of substrate binding and release, thereby preventing aggregation of misfolded proteins and promoting their folding to the stable, functional state. Hsf1, a conserved stress-responsive master transcriptional regulator195, controls the expression of these chaperones. Optimal functionality of Hsp70 depends on its co-factors, Hsp40 and Hsp110, which stimulate Hsp70’s ATPase activity and accelerate the exchange of ADP for ATP in Hsp70, respectively, thereby facilitating the chaperone cycle of binding and refolding of sequestered clients196-198. Recently, the Hsp40/Hsp70/Hsp110 chaperone triad has been shown to also act as the long-speculated metazoan disaggregase, with the capacity to extract and refold substrates from protein aggregates199, 200.
In support of the importance of proper protein folding in polyQ toxicity, these chaperones have been independently identified as strong suppressors of polyQ diseases. DNAJ1, the fly homolog of human Hsp40, has been isolated multiple times as a strong suppressor of toxicity caused by different polyQ proteins, and DNAJ1 together with Hsp110 and Hsf1 are also among the top suppressors of aggregate formation by HTTex1-Q46122, 124, 132, 166. Moreover, manipulation of endogenous Hsp110 level, either alone or together with DNAJ1, can significantly affect the neurodegenerative phenotypes of HD flies (Figure 3A-C)166, 201. Additionally, over-expression of HspA1L, a human Hsp70 protein, potently rescues the eye degeneration and lethality of SCA3 flies, demonstrating the conserved role of Hsp70 in preventing protein misfolding diseases121, 130. Moreover, both HspA1L and endogenous Hsp70 and DNAJ1 proteins are highly enriched in SCA3tr-Q78-positive nuclear aggregates. However, overexpression of these chaperones, either alone or together, cannot alter the onset, size or number of nuclear aggregates, but instead significantly increase the soluble monomeric portion of SCA3tr-Q78, suggesting that these chaperones modulate toxicity by altering the biochemical properties of SCA3tr-Q78122. Consistently, a comparative analysis of the reported genetic modifiers of SCA1 and HD models shows that whereas some genetic modifiers can alter the formation of nuclear aggregates, their effect on aggregation does not correlate with their effect on the toxicity of the proteins162. Collectively, these results support protein misfolding as the molecular basis of polyQ diseases, although aggregates per se might not be the specific agent responsible for the toxicity. Importantly, mammalian-based studies have confirmed the protective effects of Hsp70 and Hsp40 chaperones against polyQ-induced toxicity202-204. Exploiting chaperone machineries might be a potentially effective therapeutic strategy against these protein-misfolding diseases.
V. Drosophila facilitates drug discovery and mammalian-based studies
Currently, there are no effective preventive therapies or drugs against polyQ diseases. Increasingly, the established Drosophila polyQ models are being employed as convenient in vivo tools to facilitate drug design and to prioritize candidate modifiers from mammalian-based screens.
V-1. Drosophila in drug discovery
By testing in the established Drosophila polyQ models, a growing list of bioactive compounds, from HDAC inhibitors to Lithium, have been shown to be effective in alleviating the toxicity of polyQ-expanded proteins205-215. For example, directly feeding Congo red and cystamine, two compounds that are effective in reducing aggregation of mutant HTT in neuronal PC12 cells, to HTTex1-Q48 flies can significantly suppress the eye degeneration and animal lethality206. Separately, overexpression of designed suppressor peptides, which can inhibit aggregation of mutant HTT in mammalian COS-1 cells, also inhibits the aggregation and rescues the neurodegenerative phenotypes of Q48 and Q108 flies 216. Further, in both the fly and mammalian cells, pharmacological stimulation of Hsp70 chaperone promotes the clearance of pathogenic AR and mitigates its toxicity204.
One promising family of drug candidates are inhibitors of the mechanistic target of rapamycin (mTOR), a master regulator of cellular metabolic pathways and a strong inhibitor of autophagy. Autophagy is a key cellular clearance mechanism against protein aggregates217-220. In Drosophila, activation of autophagy by inhibiting mTOR genetically or pharmacologically (e.g., mTOR inhibitors rapamycin) markedly suppresses HTTex1-Q120 neurodegeneration221, a protective effect that has been subsequently confirmed in mouse models of HD, SCA3 and other protein misfolding diseases221-227. These results demonstrate the potential of exploiting autophagy in treating polyQ diseases and validate the fly polyQ models as a convenient in vivo tool for drug selection.
V-2. Using Drosophila to facilitate mammalian-based studies
A growing number of candidate disease modifiers are being identified from mammalian-based genetic and proteomic studies. For example, HTT alone already has more than 1,000 reported HTT interacting proteins (HIPs)62, 228-233. Thus, one pressing challenge is to evaluate the in vivo relevance of these candidate modifiers to disease pathogenesis. Drosophila polyQ models have proven to be effective for such studies, especially in helping to assess the large number of binding partners and downstream targets of the disease proteins isolated from large-scale screens. For example, fly SCA1 models have provided important in vivo evidence in determining the functional importance of several ATXN1 interactors (e.g., Capicua, Senseless and 14-3-3) and AKT signaling in mediating SCA1 neurodegeneration136-138. In two recent proteomic studies, fly HD models have been applied to test a selected group of high-confidence mammalian HIPs for their effectiveness in modulating the neuronal dysfunction induced by mutant HTT230, 232.
VI. Using Drosophila to dissect the normal functions of polyQ disease genes
As more evidence link the normal functions of disease genes directly to pathogenesis, a better understanding of these genes’ endogenous functions become highly relevant for disease studies61, 62, 124, 125, 136, 167, 181, 182, 229, 234-238. Most of the polyQ disease genes are conserved in Drosophila (Table 1). Among them, SBMA (AR), SCA6 (CACNA1A) and SCA17 (TBP) genes have well-defined functions239-242. Characterization of the fly homologs of other less-understood genes, such as ATXN1, Atrophin-1 and HTT, as briefly summarized below, have helped our understanding of these human diseases.
VI-1. ATXN1 regulates transcription and controls neuronal development and survival
ATXN1 represents an excellent example of using the fly to uncover functional roles for human disease genes and underlying pathogenic mechanisms136-138. As descried earlier, both the physical and functional interactions between ATXN1 and its binding partners (e.g., Senseless, Capicua and 14-3-3) and the signaling pathways involved (e.g., 14-3-3 and AKT) are conserved in Drosophila. For example, in flies, overexpression of ATXN1 leads to very similar neuronal defects as that induced by its Drosophila homolog (dAtx-1), including eye abnormality and bristle loss. Further, dAtx-1 physically interacts with and down-regulates Senseless, a transcription factor required for the development of sensory organs (e.g., bristles) in flies136. Importantly, a similar functional interaction is conserved between ATXN1 and vertebrate Senseless homolog Gfi-1, a gene important for the survival of Purkinje cells. In these cases, the functional importance of these interactions in SCA1 pathogenesis have almost always been revealed first using fly model and subsequently validated in mouse SCA1 models or human patient samples, for example decreased expression of Gfi-1 is shown to exacerbate the pathogenesis of SCA1 mice136. Thus, fly-based studies, coupled with validation in mammalian systems, have demonstrated that ATXN1, through its interaction with multiple partners, regulate transcription and neurogenesis that are critical for SCA1 pathogenesis136-138.
VI-2. Atrophin-1 is a versatile transcriptional co-repressor
Flies carrying mutations for atro, the Atrophin-1 homolog, show diverse developmental abnormalities, including excessive neurogenesis, polarity defects, and split thorax. These phenotypes are characteristic of defects in multiple signaling pathways such as Notch, Frizzled/PCP and JNK139, 243, 244. During embryogenesis, atro mutants show a plethora of patterning phenotypes that reveal Atro’s critical role in restricting the boundary of embryo segmentation, a regulation mainly achieved through transcriptional regulation. Most tellingly, atro mutants show strong genetic interaction with a bona fide transcription repressor, even-skipped, and Atro is essential for its repressive activity. Moreover, when directly tethered to DNA using a reporter assay in Drosophila embryos, both Atro as well as wildtype and polyQ-expanded human Atrophin-1 repress transcription in vivo139. Together, they demonstrate that Atrophin-1 functions as a versatile transcriptional co-repressor in multiple signaling pathways and diverse cellular processes. Subsequent studies in mammalian systems have since validated this finding, showing that Atrophin-1 is a corepressor for nuclear receptors (NRs)245-247.
VI-3. Characterization of Drosophila HTT homolog
HTT, an enigmatic protein
Both human HTT and its Drosophila homologue (dhtt) encode large proteins (HTT: 3,144a.a.; dHtt: 3,583 a.a.) that contain no obvious functional domains to offer clues of their normal cellular functions1,248, 249. Structurally, both are composed of a string of HEAT (Huntingtin, Elongation factor 3, A subunit of protein phosphatase 2A and TOR1) repeats, a 40-a. a. long, anti-parallel helical structural motif of unknown function (Figure 1)249-252. Extensive studies since its identification in 1993 have led to many proposed roles for HTT, including endocytosis, transcriptional regulation, trafficking, and cell death, among others. It is now generally believed that HTT acts as a scaffold to integrate inputs from many cellular signals and coordinate cellular responses, although its exact physiological functions remain controversial61-63, 67, 228, 229, 252.
Mice lacking HTT die in early embryonic stages (day E7.5)253-255. Surprisingly, flies carrying the null dhtt allele (dhtt-ko) are homozygous viable and develop normally into adults with no apparent developmental defects. Because flies develop ex utero, this phenotypic discrepancy could be attributed partially to the difference in embryogenic processes between flies and mice, in particular their divergent reliance on extraembryonic tissues (e.g., placenta). An elegant study by the Zeitlin group has demonstrated that the early lethality of HTT knockout (KO) mice is largely due to HTT’s essential role in extraembryonic tissues, not in the embryo per se256.
Expression of dHtt can rescue a spindle orientation defect observed in HTT-depleted mammalian cells. A similar although milder spindle phenotype has also been observed in dhtt-ko flies257. Furthermore, resonating with observations from early mammalian studies258, analysis of dhtt null flies has revealed a potential involvement of dhtt in epigenetic control, as dhtt shows genetic interactions with heterochromatin genes and components of chromatin remodeling complex and can facilitate the global demethylation of histone H3K4259. These findings suggest that HTT has conserved functions in regulating mitotic spindle orientation and epigenetic regulation.
In mice, targeted KO of HTT in postnatal brain or reduced levels of HTT expression both lead to prominent apoptosis, severe brain degeneration and rapid loss of brain volume142, 260. Although dhtt-ko flies are viable as adults, they have shortened lifespan and an accelerated decline of mobility as they age, accompanied by mild axonal defects in the brain, indicating a role for dhtt in neuronal function249. Furthermore, dhttko flies are vulnerable to additional stresses, showing greatly exacerbated mobility decline, neuronal loss and early death when challenged by the ectopically expressed HTTex1-Q93249. Together, these findings demonstrate that HTT has a conserved neuronal protective role and its function is critical for maintaining neuronal viability in higher species. However, little is known exactly how HTT carries out its essential neuroprotective role, and how polyQ expansion affects HTT’s normal functions and contributes to disease development.
HTT is a scaffold protein for selective macroautophagy
Using both Drosophila and mammalian systems, our group in collaboration with the Cuervo group and independently, the Steffan group recently showed that HTT plays an important role in selective autophagy, an important cellular protective mechanism146, 261. Macroautophagy is a cellular catabolic process that involves the formation of double-membrane structures called autophagosomes to enclose cytosolic constituents and deliver them into the lysosome for their eventual degradation262, 263. Initiation of the autophagy cascade is controlled by a serine/threonine kinase ULK1, and the formation of the autophagosome requires a critical structural component LC3. During starvation stress, a strong autophagic response leads to nonselective bulk engulfment of nonessential cellular materials for recycling, although autophagy during starvation may also be selective. Under nutrient-rich and starvation conditions, selective autophagy mainly targets specific substrates such as protein aggregates (also called aggrephagy), lipid droplets (lipophagy) and damaged organelles such as mitochondria (mitophagy) and peroxisomes (pexophagy)262, 263. Moreover, selective autophagy often involves receptors such as p62/SQSTM1, which bind to both LC3 and ubiquitinated substrates, thereby facilitating their sequestration into the autophagosome for eventual degradation264-267.
Starting with genetic analyses in Drosophila and further characterized biochemically in mammalian systems, we showed that HTT is required for stress-induced selective autophagy. Furthermore, HTT regulates the functions of the kinase ULK1 to control the activation of autophagy in response to stresses, and also modulates the interaction between the autophagy receptor p62 and K63-ubiquitinated substrates. Thus, by acting as a scaffold, HTT orchestrates both autophagic activation and effective sequestration of specific cargos into autophagosomes, thereby achieving efficient autophagic response against stresses146. The Steffan group also reported autophagic defects in dhtt-ko flies with accumulation of Ref(2)p, the Drosophila p62 homolog, in addition to a build-up of p62 in the striatum of HTT-KO mouse brains with aging, extending HTT’s selective autophagic relevance to mammals in vivo. They also demonstrated physical interaction of HTT with multiple autophagy proteins including ULK1 complex proteins ULK1, FIP200, and mATG13, mammalian Atg8s GABARAPL1 and LC3B, and mitophagy receptors p62, BNIP3 and NIX, suggesting a role for HTT in mitophagy261. Together, these findings demonstrate a conserved role of HTT in an important cellular protective mechanism as a selective autophagic scaffold. Interestingly, more recent studies support the role of HTT in regulating additional aspects of the autophagy pathway, and reciprocally, implicate autophagy in HD pathogenesis268. In particular, samples from HD patients and mouse HD models show an “empty autophagosome” phenotype arising from defective cargo recognition145. Furthermore, in mice, complete removal of the polyQ stretch from endogenous HTT enhances neuronal autophagy and animal longevity140. Combined with our finding that HTT regulates selective autophagy, these observations raise an intriguing possibility that the polyQ expansion in HTT compromises its own cellular protective role, which in turn contributes to HD pathogenesis. A detailed understanding of HTT’s roles in coordinating autophagy and other cellular processes will not only provide a comprehensive functional atlas of this large, enigmatic protein, but also help dissect the pathogenic mechanisms underlying HD.
VII. Challenges and Promises
Over the past two decades, Drosophila has proven to be a valuable system to model various human neurodegenerative diseases22, 96-105, as illustrated by the successful creation of these polyQ disease models and by the significant role of the fruit fly in the functional dissection of other brain disease genes such as PD genes Parkin and Pink1269-272.
Despite its many successes, there are limitations in utilizing Drosophila for studying polyQ and other brain diseases, especially considering the vast differences between flies and humans, including differences in the complexity of the regulatory elements and proteins encoded in their genomes, their developmental process and physiology, brain structures and neurotransmitters utilized, among others. In addition, homologs for some disease genes (e.g., Ataxin-3 and PD gene αSynuclein) are missing in Drosophila. Also, because most fly models rely on overexpression of human disease genes, the physiological relevance of the findings from such overexpression studies need to be validated. Thus, it is important to consider these limiting factors when integrating the many lessons learned from the fly for human diseases.
By evaluating the large numbers of fly polyQ models, it also becomes clear that the assay conditions, the tissues targeted, and the protein context of the studied mutations (i.e., polyQ tract) all have major influences on the phenotypic outcomes and the conclusions deduced. This is clearly exemplified in the very different toxicity and aggregating behaviors observed in HTTex1- and HTTFL-based fly HD models161-166, and between ARtr- and ARFL-based SBMA models123, 125. Importantly, studies from other model organisms reach very similar conclusions. For example, the fast-progressing mouse R6/2 model for HD, which is based on overexpression of expanded HTTex1, exhibits rapid neuronal loss but no apparent degeneration of the striatum, the region most affected in HD273, whereas several genomic HTTFL-based HD models (e.g., YAC72, BACHD and Q175 knockin) show slow but selective degeneration of the striatum accompanied by progressive motor and physiological phenotypes that recapitulate the human disease more closely53, 274-278. Considering this, and given the increasingly appreciated role of these disease genes’ native functions in the pathogenic process, it might be preferable to focus the studies on disease models derived from full-length proteins, as such models likely better recapitulate the whole series of pathogenic events that lead to disease. Moreover, in studying animal models of human diseases, it is important to consider the potential influence of different genetic backgrounds, such as the presence of the disease gene homologs (e.g., dHtt and dAtx-1 for HD and SCA1 studies in the fly) and their associated signaling pathways (e.g., the conserved nuclear receptor pathway for SBMA studies using Drosophila125, 279). For example, HTTex1-Q93 induces stronger toxicity in dhtt-ko flies than in wildtype control background249.
Among future challenges in fly-based studies, one is to evaluate existing disease models and identify the ones most closely resembling the pathogenic events in humans, perhaps by analyzing and comparing the alterations of gene expression profiles, so as to optimize the models to more closely parallel the human disease conditions. It is equally important to take advantage of the power of Drosophila genetics and carry out detailed characterization of the endogenous functions of the disease genes. Another challenge is to integrate the findings from fly models with the large number of data sets from mammalian-based studies, so as to pin down the early molecular events most relevant to disease pathogenesis, and to identify the most promising pathways for therapeutic intervention. Given that Drosophila is highly amenable to genetic manipulation, aided with a great number of sophisticated experimental tools available in this model organism, these challenges might also become opportunities to make more effective Drosophila models for human disease studies in the future.
In perspective, although the genetic cause for polyQ diseases is simple, our understanding of these disorders is still far from complete and their pathogenesis has proven to be far more complicated, as revealed by findings from different model systems including Drosophila. Yet, questions compounding the studies on polyQ disorders, including the role of aggregates and the involvement of disease genes’ normal cellular functions, are important concerns similarly confronting other genetically complicated diseases such as AD and PD. Currently, there are no disease-modifying therapies available for these brain degenerative disorders. The remarkably faithful recapitulation of pathological features of human brain diseases in Drosophila as well as the valuable knowledge learned about the normal functions of disease genes have and will continue to help our pursuit of a clear understanding of the molecular mechanisms underlying these devastating brain diseases, and will ultimately aid our search for targeted and effective therapeutic approaches.
Highlights.
. Drosophila models can recapitulate main pathological features of polyQ diseases
. Study on fly disease models reveals important principles regarding pathogenesis
. In addition to polyQ tract, protein context also determines the disease outcome
. Altered native functions of polyQ disease genes can be important pathogenic factor
. Drosophila is valuable for dissecting disease genes’ native functions
Figure 4. Mutant HTT protein forms age-dependent aggregates in the fly brain.
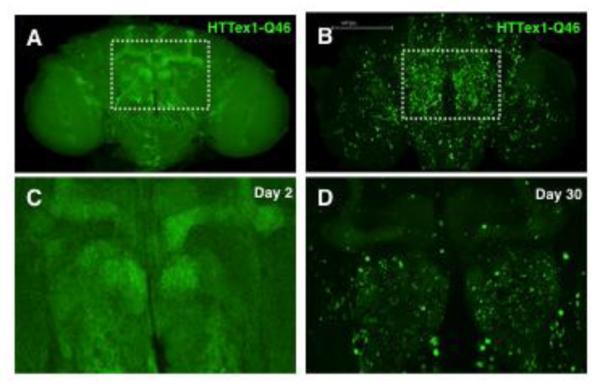
Confocal images of adult brains expressing HTTex1 with a 46 glutamine tract (HTTex1-Q46) at (A) day 2 and (B) day 30. (C and D) High-magnification views of the regions highlighted above. HTTex1-Q46 protein is evenly dispersed in mushroom bodies and other structures in young brains but forms prominent aggregates by day 30.
Acknowledgements
We apologize for the many works not being cited in this review due to space limitation. We thank Pedro Fernandez-Funez, Tom Lloyd, Gabriela Riva David-Morrison, J. Lawrence Marsh, Joan S. Steffan and anonymous reviewers for critical reading and insightful input. This work was supported by NIH grant R01-NS069880 (S.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 2.David G, et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet. 1997;17:65–70. doi: 10.1038/ng0997-65. [DOI] [PubMed] [Google Scholar]
- 3.Imbert G, et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet. 1996;14:285–291. doi: 10.1038/ng1196-285. [DOI] [PubMed] [Google Scholar]
- 4.Kawaguchi Y, et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet. 1994;8:221–228. doi: 10.1038/ng1194-221. [DOI] [PubMed] [Google Scholar]
- 5.Koob MD, et al. Rapid cloning of expanded trinucleotide repeat sequences from genomic DNA. Nat Genet. 1998;18:72–75. doi: 10.1038/ng0198-72. [DOI] [PubMed] [Google Scholar]
- 6.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 7.Orr HT, et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4:221–226. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- 8.Pulst SM, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet. 1996;14:269–276. doi: 10.1038/ng1196-269. [DOI] [PubMed] [Google Scholar]
- 9.Sanpei K, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet. 1996;14:277–284. doi: 10.1038/ng1196-277. [DOI] [PubMed] [Google Scholar]
- 10.Zhuchenko O, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura K, et al. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet. 2001;10:1441–1448. doi: 10.1093/hmg/10.14.1441. [DOI] [PubMed] [Google Scholar]
- 12.Bauer PO, Nukina N. The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J Neurochem. 2009;110:1737–1765. doi: 10.1111/j.1471-4159.2009.06302.x. [DOI] [PubMed] [Google Scholar]
- 13.Cummings CJ, Zoghbi HY. Fourteen and counting: unraveling trinucleotide repeat diseases. Hum Mol Genet. 2000;9:909–916. doi: 10.1093/hmg/9.6.909. [DOI] [PubMed] [Google Scholar]
- 14.Ross CA, et al. Polyglutamine pathogenesis. Philos Trans R Soc Lond B Biol Sci. 1999;354:1005–1011. doi: 10.1098/rstb.1999.0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andrew SE, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat Genet. 1993;4:398–403. doi: 10.1038/ng0893-398. [DOI] [PubMed] [Google Scholar]
- 16.Snell RG, et al. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat Genet. 1993;4:393–397. doi: 10.1038/ng0893-393. [DOI] [PubMed] [Google Scholar]
- 17.Deka R, et al. Normal CAG repeat variation at the DRPLA locus in world populations. Am J Hum Genet. 1995;57:508–511. [PMC free article] [PubMed] [Google Scholar]
- 18.Ikeuchi T, et al. Dentatorubral-pallidoluysian atrophy: clinical features are closely related to unstable expansions of trinucleotide (CAG) repeat. Ann Neurol. 1995;37:769–775. doi: 10.1002/ana.410370610. [DOI] [PubMed] [Google Scholar]
- 19.Ikeuchi T, et al. Dentatorubral-pallidoluysian atrophy (DRPLA): close correlation of CAG repeat expansions with the wide spectrum of clinical presentations and prominent anticipation. Semin Cell Biol. 1995;6:37–44. doi: 10.1016/1043-4682(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 20.Koide R, et al. Unstable expansion of CAG repeat in hereditary dentatorubral- pallidoluysian atrophy (DRPLA) Nat Genet. 1994;6:9–13. doi: 10.1038/ng0194-9. [DOI] [PubMed] [Google Scholar]
- 21.Komure O, et al. DNA analysis in hereditary dentatorubral-pallidoluysian atrophy: correlation between CAG repeat length and phenotypic variation and the molecular basis of anticipation. Neurology. 1995;45:143–149. doi: 10.1212/wnl.45.1.143. [DOI] [PubMed] [Google Scholar]
- 22.Gusella J, MacDonald M. No post-genetics era in human disease research. Nat Rev Genet. 2002;3:72–79. doi: 10.1038/nrg706. [DOI] [PubMed] [Google Scholar]
- 23.La Spada AR, Paulson HL, Fischbeck KH. Trinucleotide repeat expansion in neurological disease. Ann Neurol. 1994;36:814–822. doi: 10.1002/ana.410360604. [DOI] [PubMed] [Google Scholar]
- 24.Ross CA. When more is less: pathogenesis of glutamine repeat neurodegenerative diseases. Neuron. 1995;15:493–496. doi: 10.1016/0896-6273(95)90138-8. [DOI] [PubMed] [Google Scholar]
- 25.Bates GP, Harper P, Jones L. Huntington’s Disease. 3rd Edition Oxford Medical Publications; 2002. [Google Scholar]
- 26.Gusella JF, MacDonald ME. Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nat Rev Neurosci. 2000;1:109–115. doi: 10.1038/35039051. [DOI] [PubMed] [Google Scholar]
- 27.Ross CA. Intranuclear neuronal inclusions: a common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron. 1997;19:1147–1150. doi: 10.1016/s0896-6273(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 28.Sisodia SS. Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell. 1998;95:1–4. doi: 10.1016/s0092-8674(00)81743-2. [DOI] [PubMed] [Google Scholar]
- 29.Warner CL, et al. X-linked spinomuscular atrophy: a kindred with associated abnormal androgen receptor binding. Neurology. 1992;42:2181–2184. doi: 10.1212/wnl.42.11.2181. [DOI] [PubMed] [Google Scholar]
- 30.Brown TR, et al. Deletion of the steroid-binding domain of the human androgen receptor gene in one family with complete androgen insensitivity syndrome: evidence for further genetic heterogeneity in this syndrome. Proc Natl Acad Sci U S A. 1988;85:8151–8155. doi: 10.1073/pnas.85.21.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quigley CA, et al. Complete deletion of the androgen receptor gene: definition of the null phenotype of the androgen insensitivity syndrome and determination of carrier status. J Clin Endocrinol Metab. 1992;74:927–933. doi: 10.1210/jcem.74.4.1347772. [DOI] [PubMed] [Google Scholar]
- 32.Lin X, Cummings CJ, Zoghbi HY. Expanding our understanding of polyglutamine diseases through mouse models. Neuron. 1999;24:499–502. doi: 10.1016/s0896-6273(00)81104-6. [DOI] [PubMed] [Google Scholar]
- 33.Sipione S, Cattaneo E. Modeling huntington’s disease in cells, flies, and mice. Molecular neurobiology. 2001;23:21–51. doi: 10.1385/MN:23:1:21. [DOI] [PubMed] [Google Scholar]
- 34.Ross CA, Poirier MA. Opinion: What is the role of protein aggregation in neurodegeneration? Nat Rev Mol Cell Biol. 2005;6:891–898. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- 35.DiFiglia M, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 36.Schilling G, et al. Nuclear accumulation of truncated atrophin-1 fragments in a transgenic mouse model of DRPLA. Neuron. 1999;24:275–286. doi: 10.1016/s0896-6273(00)80839-9. [DOI] [PubMed] [Google Scholar]
- 37.Skinner PJ, et al. Ataxin-1 with an expanded glutamine tract alters nuclear matrix- associated structures. Nature. 1997;389:971–974. doi: 10.1038/40153. [DOI] [PubMed] [Google Scholar]
- 38.Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 39.Paulson HL. Protein fate in neurodegenerative proteinopathies: polyglutamine diseases join the (mis)fold. Am J Hum Genet. 1999;64:339–345. doi: 10.1086/302269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 41.Cummings CJ, et al. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- 42.Davies SW, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 43.Paulson HL, et al. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333–344. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 44.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci U S A. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 46.Gutekunst CA, et al. Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holmberg M, et al. Spinocerebellar ataxia type 7 (SCA7): a neurodegenerative disorder with neuronal intranuclear inclusions. Hum Mol Genet. 1998;7:913–918. doi: 10.1093/hmg/7.5.913. [DOI] [PubMed] [Google Scholar]
- 48.Huynh DP, Del Bigio MR, Ho DH, Pulst SM. Expression of ataxin-2 in brains from normal individuals and patients with Alzheimer’s disease and spinocerebellar ataxia 2. Ann Neurol. 1999;45:232–241. doi: 10.1002/1531-8249(199902)45:2<232::aid-ana14>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 49.Koyano S, et al. Neuronal intranuclear inclusions in spinocerebellar ataxia type 2: triple-labeling immunofluorescent study. Neuroscience letters. 1999;273:117–120. doi: 10.1016/s0304-3940(99)00656-4. [DOI] [PubMed] [Google Scholar]
- 50.Kuemmerle S, et al. Huntington aggregates may not predict neuronal death in Huntington’s disease. Ann Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- 51.Li M, et al. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol. 1998;44:249–254. doi: 10.1002/ana.410440216. [DOI] [PubMed] [Google Scholar]
- 52.Li M, et al. Nonneural nuclear inclusions of androgen receptor protein in spinal and bulbar muscular atrophy. Am J Pathol. 1998;153:695–701. doi: 10.1016/S0002-9440(10)65612-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hodgson JG, et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 54.Reddy PH, et al. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet. 1998;20:198–202. doi: 10.1038/2510. [DOI] [PubMed] [Google Scholar]
- 55.Sathasivam K, et al. Formation of polyglutamine inclusions in non-CNS tissue. Hum Mol Genet. 1999;8:813–822. doi: 10.1093/hmg/8.5.813. [DOI] [PubMed] [Google Scholar]
- 56.Klement IA, et al. Ataxin-1 nuclear localization and aggregation: role in polyglutamine- induced disease in SCA1 transgenic mice [see comments] Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 57.Zoghbi HY. Spinocerebellar ataxia type 1. Clin Neurosci. 1995;3:5–11. [PubMed] [Google Scholar]
- 58.Ross CA, et al. Huntington’s disease and dentatorubral-pallidoluysian atrophy: proteins, pathogenesis and pathology. Brain Pathol. 1997;7:1003–1016. doi: 10.1111/j.1750-3639.1997.tb00898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ross CA, et al. Huntington disease and the related disorder, dentatorubral pallidoluysian atrophy (DRPLA) Medicine (Baltimore) 1997;76:305–338. doi: 10.1097/00005792-199709000-00001. [DOI] [PubMed] [Google Scholar]
- 60.Vonsattel JP, et al. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 61.Cattaneo E, et al. Loss of normal huntingtin function: new developments in Huntington’s disease research. Trends Neurosci. 2001;24:182–188. doi: 10.1016/s0166-2236(00)01721-5. [DOI] [PubMed] [Google Scholar]
- 62.Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease. Nat Rev Neurosci. 2005;6:919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- 63.Caviston JP, Holzbaur EL. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009;19:147–155. doi: 10.1016/j.tcb.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davies JE, Sarkar S, Rubinsztein DC. The ubiquitin proteasome system in Huntington’s disease and the spinocerebellar ataxias. BMC Biochem. 2007;8(Suppl 1):S2. doi: 10.1186/1471-2091-8-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Feany MB, La Spada AR. Polyglutamines stop traffic: axonal transport as a common target in neurodegenerative diseases. Neuron. 2003;40:1–2. doi: 10.1016/s0896-6273(03)00600-7. [DOI] [PubMed] [Google Scholar]
- 66.Gil JM, Rego AC. Mechanisms of neurodegeneration in Huntington’s disease. Eur J Neurosci. 2008;27:2803–2820. doi: 10.1111/j.1460-9568.2008.06310.x. [DOI] [PubMed] [Google Scholar]
- 67.Imarisio S, et al. Huntington’s disease: from pathology and genetics to potential therapies. Biochem J. 2008;412:191–209. doi: 10.1042/BJ20071619. [DOI] [PubMed] [Google Scholar]
- 68.Rubinsztein DC, Carmichael J. Huntington’s disease: molecular basis of neurodegeneration. Expert Rev Mol Med. 2003;5:1–21. doi: 10.1017/S1462399403006549. [DOI] [PubMed] [Google Scholar]
- 69.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 70.Zoghbi HY, Orr HT. Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1. J Biol Chem. 2009;284:7425–7429. doi: 10.1074/jbc.R800041200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zoghbi HY, Orr HT. Polyglutamine diseases: protein cleavage and aggregation. Curr Opin Neurobiol. 1999;9:566–570. doi: 10.1016/S0959-4388(99)00013-6. [DOI] [PubMed] [Google Scholar]
- 72.Bilen J, Liu N, Burnett BG, Pittman RN, Bonini NM. MicroRNA pathways modulate polyglutamine-induced neurodegeneration. Mol Cell. 2006;24:157–163. doi: 10.1016/j.molcel.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 73.Rubinsztein DC. How does the Huntington’s disease mutation damage cells? Sci Aging Knowledge Environ. 2003;2003:PE26. doi: 10.1126/sageke.2003.37.pe26. [DOI] [PubMed] [Google Scholar]
- 74.Arias AM. Drosophila melanogaster and the development of biology in the 20th century. Methods Mol Biol. 2008;420:1–25. doi: 10.1007/978-1-59745-583-1_1. [DOI] [PubMed] [Google Scholar]
- 75.Bellen HJ, Tong C, Tsuda H. 100 years of Drosophila research and its impact on vertebrate neuroscience: a history lesson for the future. Nat Rev Neurosci. 2010;11:514–522. doi: 10.1038/nrn2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bier E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat Rev Genet. 2005;6:9–23. doi: 10.1038/nrg1503. [DOI] [PubMed] [Google Scholar]
- 77.Rubin GM, Lewis EB. A brief history of Drosophila’s contributions to genome research. Science. 2000;287:2216–2218. doi: 10.1126/science.287.5461.2216. [DOI] [PubMed] [Google Scholar]
- 78.Rubin GM, et al. Comparative genomics of the eukaryotes. Science. 2000;287:2204–2215. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spradling A, et al. New roles for model genetic organisms in understanding and treating human disease: report from the 2006 Genetics Society of America meeting. Genetics. 2006;172:2025–2032. doi: 10.1093/genetics/172.4.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wangler MF, Yamamoto S, Bellen HJ. Fruit Flies in Biomedical Research. Genetics. 2015 doi: 10.1534/genetics.114.171785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 82.Rubin GM, Spradling AC. Genetic transformation of Drosophila with transposable element vectors. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- 83.Venken KJ, et al. Versatile P[acman] BAC libraries for transgenesis studies in Drosophila melanogaster. Nat Methods. 2009;6:431–434. doi: 10.1038/nmeth.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Venken KJ, He Y, Hoskins RA, Bellen HJ. P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science. 2006;314:1747–1751. doi: 10.1126/science.1134426. [DOI] [PubMed] [Google Scholar]
- 85.Venken KJ, et al. MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat Methods. 2011;8:737–743. doi: 10.1038/nmeth.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Luo L, Tully T, White K. Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for Appl gene. Neuron. 1992;9:595–605. doi: 10.1016/0896-6273(92)90024-8. [DOI] [PubMed] [Google Scholar]
- 87.Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila [see comments] Nature. 1999;398:522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- 88.Ye Y, Fortini ME. Characterization of Drosophila Presenilin and its colocalization with Notch during development. Mech Dev. 1998;79:199–211. doi: 10.1016/s0925-4773(98)00169-5. [DOI] [PubMed] [Google Scholar]
- 89.Fortini ME, Skupski MP, Boguski MS, Hariharan IK. A survey of human disease gene counterparts in the Drosophila genome. J Cell Biol. 2000;150:F23–30. doi: 10.1083/jcb.150.2.f23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Song W, et al. Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc Natl Acad Sci U S A. 1999;96:6959–6963. doi: 10.1073/pnas.96.12.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Benzer S. From the gene to behavior. Jama. 1971;218:1015–1022. [PubMed] [Google Scholar]
- 92.Kretzschmar D, Hasan G, Sharma S, Heisenberg M, Benzer S. The swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. J Neurosci. 1997;17:7425–7432. doi: 10.1523/JNEUROSCI.17-19-07425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Min KT, Benzer S. Preventing neurodegeneration in the Drosophila mutant bubblegum. Science. 1999;284:1985–1988. doi: 10.1126/science.284.5422.1985. [DOI] [PubMed] [Google Scholar]
- 94.Min KT, Benzer S. Spongecake and eggroll: two hereditary diseases in Drosophila resemble patterns of human brain degeneration. Curr Biol. 1997;7:885–888. doi: 10.1016/s0960-9822(06)00378-2. [DOI] [PubMed] [Google Scholar]
- 95.Zhai RG, et al. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 2006;4:e416. doi: 10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bilen J, Bonini NM. Drosophila as a model for human neurodegenerative disease. Annu Rev Genet. 2005;39:153–171. doi: 10.1146/annurev.genet.39.110304.095804. [DOI] [PubMed] [Google Scholar]
- 97.Bonini NM. Drosophila as a genetic approach to human neurodegenerative disease. Parkinsonism Relat Disord. 2001;7:171–175. doi: 10.1016/s1353-8020(00)00054-7. [DOI] [PubMed] [Google Scholar]
- 98.Bonini NM, Fortini ME. Human neurodegenerative disease modeling using Drosophila. Annu Rev Neurosci. 2003;26:627–656. doi: 10.1146/annurev.neuro.26.041002.131425. [DOI] [PubMed] [Google Scholar]
- 99.Driscoll M, Gerstbrein B. Dying for a cause: invertebrate genetics takes on human neurodegeneration. Nat Rev Genet. 2003;4:181–194. doi: 10.1038/nrg1018. [DOI] [PubMed] [Google Scholar]
- 100.Feany MB. Studying human neurodegenerative diseases in flies and worms. J Neuropathol Exp Neurol. 2000;59:847–856. doi: 10.1093/jnen/59.10.847. [DOI] [PubMed] [Google Scholar]
- 101.Marsh JL, Thompson LM. Drosophila in the study of neurodegenerative disease. Neuron. 2006;52:169–178. doi: 10.1016/j.neuron.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 102.Chan HY, Bonini NM. Drosophila models of polyglutamine diseases. Methods Mol Biol. 2003;217:241–251. doi: 10.1385/1-59259-330-5:241. [DOI] [PubMed] [Google Scholar]
- 103.Krench M, Littleton JT. Modeling Huntington disease in Drosophila: Insights into axonal transport defects and modifiers of toxicity. Fly. 2013;7:229–236. doi: 10.4161/fly.26279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lu B. Recent advances in using Drosophila to model neurodegenerative diseases. Apoptosis. 2009 doi: 10.1007/s10495-009-0347-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lu B, Vogel H. Drosophila models of neurodegenerative diseases. Annu Rev Pathol. 2009;4:315–342. doi: 10.1146/annurev.pathol.3.121806.151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Treisman JE, Heberlein U. Eye development in Drosophila: formation of the eye field and control of differentiation. Curr Top Dev Biol. 1998;39:119–158. doi: 10.1016/s0070-2153(08)60454-8. [DOI] [PubMed] [Google Scholar]
- 107.Wolff T, Ready DF. The beginning of pattern formation in the Drosophila compound eye: the morphogenetic furrow and the second mitotic wave. Development. 1991;113:841–850. doi: 10.1242/dev.113.3.841. [DOI] [PubMed] [Google Scholar]
- 108.Wolff T, Ready DF. Pattern formation in the Drosophila retina. In: Martinez-Arias MBA, editor. The development of Drosophila melanogaster. Cold Spring Harbor Press; Cold Spring Harbor: 1993. pp. 1277–1326. [Google Scholar]
- 109.Tomlinson A, Ready DF. Neuronal differentiation in Drosophila ommatidium. Dev Biol. 1987;120:366–376. doi: 10.1016/0012-1606(87)90239-9. [DOI] [PubMed] [Google Scholar]
- 110.Tomlinson A, Ready DF. Cell fate in the Drosophila ommatidium. Dev Biol. 1987;123:264–275. doi: 10.1016/0012-1606(87)90448-9. [DOI] [PubMed] [Google Scholar]
- 111.Franceschini N. In: Information Processing in the Visual Sysytem of Arthropods. Wehner R, editor. Vol. 1972. Springer; Berlin: 1972. pp. 75–82. [Google Scholar]
- 112.Cagan RL, Ready DF. The emergence of order in the Drosophila pupal retina. Dev Biol. 1989;136:346–362. doi: 10.1016/0012-1606(89)90261-3. [DOI] [PubMed] [Google Scholar]
- 113.Steller H, Grether ME. Programmed cell death in Drosophila. Neuron. 1994;13:1269–1274. doi: 10.1016/0896-6273(94)90413-8. [DOI] [PubMed] [Google Scholar]
- 114.Karim FD, Rubin GM. Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development. 1998;125:1–9. doi: 10.1242/dev.125.1.1. [DOI] [PubMed] [Google Scholar]
- 115.Quiring R, Walldorf U, Kloter U, Gehring WJ. Homology of the eyeless gene of Drosophila to the Small eye gene in mice and Aniridia in humans. Science. 1994;265:785–789. doi: 10.1126/science.7914031. [DOI] [PubMed] [Google Scholar]
- 116.Ellis MC, O’Neill EM, Rubin GM. Expression of Drosophila glass protein and evidence for negative regulation of its activity in non-neuronal cells by another DNA-binding protein. Development. 1993;119:855–865. doi: 10.1242/dev.119.3.855. [DOI] [PubMed] [Google Scholar]
- 117.Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development. 1994;120:2121–2129. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- 118.Moses K, Rubin GM. Glass encodes a site-specific DNA-binding protein that is regulated in response to positional signals in the developing Drosophila eye. Genes Dev. 1991;5:583–593. doi: 10.1101/gad.5.4.583. [DOI] [PubMed] [Google Scholar]
- 119.Jackson GR, et al. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron. 1998;21:633–642. doi: 10.1016/s0896-6273(00)80573-5. [DOI] [PubMed] [Google Scholar]
- 120.Warrick JM, et al. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93:939–949. doi: 10.1016/s0092-8674(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 121.Warrick JM, et al. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 122.Chan HY, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM. Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet. 2000;9:2811–2820. doi: 10.1093/hmg/9.19.2811. [DOI] [PubMed] [Google Scholar]
- 123.Chan HY, Warrick JM, Andriola I, Merry D, Bonini NM. Genetic modulation of polyglutamine toxicity by protein conjugation pathways in Drosophila. Hum Mol Genet. 2002;11:2895–2904. doi: 10.1093/hmg/11.23.2895. [DOI] [PubMed] [Google Scholar]
- 124.Fernandez-Funez P, et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- 125.Nedelsky NB, et al. Native functions of the androgen receptor are essential to pathogenesis in a Drosophila model of spinobulbar muscular atrophy. Neuron. 2010;67:936–952. doi: 10.1016/j.neuron.2010.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pandey UB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 127.Takeyama K, et al. Androgen-dependent neurodegeneration by polyglutamine- expanded human androgen receptor in Drosophila. Neuron. 2002;35:855–864. doi: 10.1016/s0896-6273(02)00875-9. [DOI] [PubMed] [Google Scholar]
- 128.Napoletano F, et al. Polyglutamine Atrophin provokes neurodegeneration in Drosophila by repressing fat. EMBO J. 2011;30:945–958. doi: 10.1038/emboj.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nisoli I, et al. Neurodegeneration by polyglutamine Atrophin is not rescued by induction of autophagy. Cell Death Differ. 2010;17:1577–1587. doi: 10.1038/cdd.2010.31. [DOI] [PubMed] [Google Scholar]
- 130.Warrick JM, et al. Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol Cell. 2005;18:37–48. doi: 10.1016/j.molcel.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 131.Marsh JL, et al. Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila. Hum Mol Genet. 2000;9:13–25. doi: 10.1093/hmg/9.1.13. [DOI] [PubMed] [Google Scholar]
- 132.Kazemi-Esfarjani P, Benzer S. Genetic suppression of polyglutamine toxicity in Drosophila. Science. 2000;287:1837–1840. doi: 10.1126/science.287.5459.1837. [DOI] [PubMed] [Google Scholar]
- 133.Seto ES, Bellen HJ. The ins and outs of Wingless signaling. Trends Cell Biol. 2004;14:45–53. doi: 10.1016/j.tcb.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 134.Siegfried E, Perrimon N. Drosophila wingless: a paradigm for the function and mechanism of Wnt signaling. Bioessays. 1994;16:395–404. doi: 10.1002/bies.950160607. [DOI] [PubMed] [Google Scholar]
- 135.Siegfried E, Wilder EL, Perrimon N. Components of wingless signalling in Drosophila. Nature. 1994;367:76–80. doi: 10.1038/367076a0. [DOI] [PubMed] [Google Scholar]
- 136.Tsuda H, et al. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell. 2005;122:633–644. doi: 10.1016/j.cell.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 137.Chen HK, et al. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell. 2003;113:457–468. doi: 10.1016/s0092-8674(03)00349-0. [DOI] [PubMed] [Google Scholar]
- 138.Lam YC, et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127:1335–1347. doi: 10.1016/j.cell.2006.11.038. [DOI] [PubMed] [Google Scholar]
- 139.Zhang S, Xu L, Lee J, Xu T. Drosophila atrophin homolog functions as a transcriptional corepressor in multiple developmental processes. Cell. 2002;108:45–56. doi: 10.1016/s0092-8674(01)00630-4. [DOI] [PubMed] [Google Scholar]
- 140.Zheng S, et al. Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genet. 2010;6:e1000838. doi: 10.1371/journal.pgen.1000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zheng S, Ghitani N, Blackburn JS, Liu JP, Zeitlin SO. A series of N- terminal epitope tagged Hdh knock-in alleles expressing normal and mutant huntingtin: their application to understanding the effect of increasing the length of normal Huntingtin’s polyglutamine stretch on CAG140 mouse model pathogenesis. Molecular brain. 2012;5:28. doi: 10.1186/1756-6606-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.White JK, et al. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat Genet. 1997;17:404–410. doi: 10.1038/ng1297-404. [DOI] [PubMed] [Google Scholar]
- 143.Van Raamsdonk JM, Murphy Z, Slow EJ, Leavitt BR, Hayden MR. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005;14:3823–3835. doi: 10.1093/hmg/ddi407. [DOI] [PubMed] [Google Scholar]
- 144.Van Raamsdonk JM, et al. Loss of wild-type huntingtin influences motor dysfunction and survival in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005;14:1379–1392. doi: 10.1093/hmg/ddi147. [DOI] [PubMed] [Google Scholar]
- 145.Martinez-Vicente M, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nature neuroscience. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rui YN, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nature cell biology. 2015;17:262–275. doi: 10.1038/ncb3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Thompson LM, et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. 2009;187:1083–1099. doi: 10.1083/jcb.200909067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gafni J, et al. Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J Biol Chem. 2004;279:20211–20220. doi: 10.1074/jbc.M401267200. [DOI] [PubMed] [Google Scholar]
- 149.Goffredo D, et al. Calcium-dependent cleavage of endogenous wild-type huntingtin in primary cortical neurons. J Biol Chem. 2002;277:39594–39598. doi: 10.1074/jbc.C200353200. [DOI] [PubMed] [Google Scholar]
- 150.Goldberg YP, et al. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet. 1996;13:442–449. doi: 10.1038/ng0896-442. [DOI] [PubMed] [Google Scholar]
- 151.Graham RK, et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 152.Hackam AS, et al. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol. 1998;141:1097–1105. doi: 10.1083/jcb.141.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kim YJ, et al. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc Natl Acad Sci U S A. 2001;98:12784–12789. doi: 10.1073/pnas.221451398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lunkes A, et al. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell. 2002;10:259–269. doi: 10.1016/s1097-2765(02)00602-0. [DOI] [PubMed] [Google Scholar]
- 155.Ratovitski T, et al. N-terminal proteolysis of full-length mutant huntingtin in an inducible PC12 cell model of Huntington’s disease. Cell Cycle. 2007;6:2970–2981. doi: 10.4161/cc.6.23.4992. [DOI] [PubMed] [Google Scholar]
- 156.Sathasivam K, et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A. 2013;110:2366–2370. doi: 10.1073/pnas.1221891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Toneff T, et al. Comparison of huntingtin proteolytic fragments in human lymphoblast cell lines and human brain. J Neurochem. 2002;82:84–92. doi: 10.1046/j.1471-4159.2002.00940.x. [DOI] [PubMed] [Google Scholar]
- 158.Wellington CL, et al. Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington’s disease. J Neurosci. 2002;22:7862–7872. doi: 10.1523/JNEUROSCI.22-18-07862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wellington CL, Hayden MR. Caspases and neurodegeneration: on the cutting edge of new therapeutic approaches. Clin Genet. 2000;57:1–10. doi: 10.1034/j.1399-0004.2000.570101.x. [DOI] [PubMed] [Google Scholar]
- 160.Wellington CL, et al. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem. 2000;275:19831–19838. doi: 10.1074/jbc.M001475200. [DOI] [PubMed] [Google Scholar]
- 161.Barbaro BA, et al. Comparative study of naturally occurring huntingtin fragments in Drosophila points to exon 1 as the most pathogenic species in Huntington’s disease. Hum Mol Genet. 2015;24:913–925. doi: 10.1093/hmg/ddu504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Branco J, et al. Comparative analysis of genetic modifiers in Drosophila points to common and distinct mechanisms of pathogenesis among polyglutamine diseases. Hum Mol Genet. 2008;17:376–390. doi: 10.1093/hmg/ddm315. [DOI] [PubMed] [Google Scholar]
- 163.Lee WC, Yoshihara M, Littleton JT. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc Natl Acad Sci U S A. 2004;101:3224–3229. doi: 10.1073/pnas.0400243101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Romero E, et al. Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron. 2008;57:27–40. doi: 10.1016/j.neuron.2007.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Weiss KR, Kimura Y, Lee WC, Littleton JT. Huntingtin aggregation kinetics and their pathological role in a Drosophila Huntington’s disease model. Genetics. 2012;190:581–600. doi: 10.1534/genetics.111.133710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhang S, Binari R, Zhou R, Perrimon N. A genomewide RNA interference screen for modifiers of aggregates formation by mutant Huntingtin in Drosophila. Genetics. 2010;184:1165–1179. doi: 10.1534/genetics.109.112516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Duvick L, et al. SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron. 2010;67:929–935. doi: 10.1016/j.neuron.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gunawardena S, et al. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- 169.Chen-Plotkin AS, et al. Decreased association of the transcription factor Sp1 with genes downregulated in Huntington’s disease. Neurobiol Dis. 2006;22:233–241. doi: 10.1016/j.nbd.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 170.Dunah AW, et al. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 171.Kegel KB, et al. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J Biol Chem. 2002;277:7466–7476. doi: 10.1074/jbc.M103946200. [DOI] [PubMed] [Google Scholar]
- 172.Li SH, et al. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol. 2002;22:1277–1287. doi: 10.1128/mcb.22.5.1277-1287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhai W, Jeong H, Cui L, Krainc D, Tjian R. In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell. 2005;123:1241–1253. doi: 10.1016/j.cell.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 174.Zuccato C, et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J Neurosci. 2007;27:6972–6983. doi: 10.1523/JNEUROSCI.4278-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog Neurobiol. 2007;81:294–330. doi: 10.1016/j.pneurobio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 176.Zuccato C, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 177.Zuccato C, et al. Progressive loss of BDNF in a mouse model of Huntington’s disease and rescue by BDNF delivery. Pharmacol Res. 2005;52:133–139. doi: 10.1016/j.phrs.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 178.Zuccato C, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 179.Helmlinger D, Hardy S, Eberlin A, Devys D, Tora L. Both normal and polyglutamine- expanded ataxin-7 are components of TFTC-type GCN5 histone acetyltransferase- containing complexes. Biochem Soc Symp. 2006:155–163. doi: 10.1042/bss0730155. [DOI] [PubMed] [Google Scholar]
- 180.Helmlinger D, et al. Ataxin-7 is a subunit of GCN5 histone acetyltransferase- containing complexes. Hum Mol Genet. 2004;13:1257–1265. doi: 10.1093/hmg/ddh139. [DOI] [PubMed] [Google Scholar]
- 181.McMahon SJ, Pray-Grant MG, Schieltz D, Yates JR, 3rd, Grant PA. Polyglutamine-expanded spinocerebellar ataxia-7 protein disrupts normal SAGA and SLIK histone acetyltransferase activity. Proc Natl Acad Sci U S A. 2005;102:8478–8482. doi: 10.1073/pnas.0503493102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Palhan VB, et al. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci U S A. 2005;102:8472–8477. doi: 10.1073/pnas.0503505102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Steffan JS, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 184.Steffan JS, et al. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Panov AV, et al. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 186.Tang TS, et al. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003;39:227–239. doi: 10.1016/s0896-6273(03)00366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zeron MM, et al. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 188.Bilen J, Bonini NM. Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet. 2007;3:1950–1964. doi: 10.1371/journal.pgen.0030177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kazemi-Esfarjani P, Benzer S. Suppression of polyglutamine toxicity by a Drosophila homolog of myeloid leukemia factor 1. Hum Mol Genet. 2002;11:2657–2672. doi: 10.1093/hmg/11.21.2657. [DOI] [PubMed] [Google Scholar]
- 190.Kanuka H, et al. Cytosol-endoplasmic reticulum interplay by Sec61alpha translocon in polyglutamine-mediated neurotoxicity in Drosophila. Proc Natl Acad Sci U S A. 2003;100:11723–11728. doi: 10.1073/pnas.1934748100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lu B, et al. Identification of NUB1 as a suppressor of mutant Huntington toxicity via enhanced protein clearance. Nat Neurosci. 2013;16:562–570. doi: 10.1038/nn.3367. [DOI] [PubMed] [Google Scholar]
- 192.Latouche M, et al. A conditional pan-neuronal Drosophila model of spinocerebellar ataxia 7 with a reversible adult phenotype suitable for identifying modifier genes. J Neurosci. 2007;27:2483–2492. doi: 10.1523/JNEUROSCI.5453-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Steffan JS, et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 194.Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 195.Craig EA, Weissman JS, Horwich AL. Heat shock proteins and molecular chaperones: mediators of protein conformation and turnover in the cell. Cell. 1994;78:365–372. doi: 10.1016/0092-8674(94)90416-2. [DOI] [PubMed] [Google Scholar]
- 196.Dragovic Z, Broadley SA, Shomura Y, Bracher A, Hartl FU. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. The EMBO journal. 2006;25:2519–2528. doi: 10.1038/sj.emboj.7601138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Raviol H, Sadlish H, Rodriguez F, Mayer MP, Bukau B. Chaperone network in the yeast cytosol: Hsp110 is revealed as an Hsp70 nucleotide exchange factor. The EMBO journal. 2006;25:2510–2518. doi: 10.1038/sj.emboj.7601139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Shaner L, Sousa R, Morano KA. Characterization of Hsp70 binding and nucleotide exchange by the yeast Hsp110 chaperone Sse1. Biochemistry. 2006;45:15075–15084. doi: 10.1021/bi061279k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Rampelt H, et al. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. The EMBO journal. 2012;31:4221–4235. doi: 10.1038/emboj.2012.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Shorter J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PloS one. 2011;6:e26319. doi: 10.1371/journal.pone.0026319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kuo Y, Ren S, Lao U, Edgar BA, Wang T. Suppression of polyglutamine protein toxicity by co-expression of a heat-shock protein 40 and a heat-shock protein 110. Cell Death Dis. 2013;4:e833. doi: 10.1038/cddis.2013.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bailey CK, Andriola IF, Kampinga HH, Merry DE. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Human molecular genetics. 2002;11:515–523. doi: 10.1093/hmg/11.5.515. [DOI] [PubMed] [Google Scholar]
- 203.Kobayashi Y, et al. Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem. 2000;275:8772–8778. doi: 10.1074/jbc.275.12.8772. [DOI] [PubMed] [Google Scholar]
- 204.Wang AM, et al. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol. 2013;9:112–118. doi: 10.1038/nchembio.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Agrawal N, et al. Identification of combinatorial drug regimens for treatment of Huntington’s disease using Drosophila. Proc Natl Acad Sci U S A. 2005;102:3777–3781. doi: 10.1073/pnas.0500055102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Apostol BL, et al. A cell-based assay for aggregation inhibitors as therapeutics of polyglutamine-repeat disease and validation in Drosophila. Proc Natl Acad Sci U S A. 2003;100:5950–5955. doi: 10.1073/pnas.2628045100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Arribat Y, et al. A huntingtin peptide inhibits polyQ-huntingtin associated defects. PLoS One. 2013;8:e68775. doi: 10.1371/journal.pone.0068775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Bortvedt SF, McLear JA, Messer A, Ahern-Rindell AJ, Wolfgang WJ. Cystamine and intrabody co-treatment confers additional benefits in a fly model of Huntington’s disease. Neurobiol Dis. 2010;40:130–134. doi: 10.1016/j.nbd.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gohil VM, et al. Meclizine is neuroprotective in models of Huntington’s disease. Hum Mol Genet. 2011;20:294–300. doi: 10.1093/hmg/ddq464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Jia DD, et al. Lithium chloride alleviates neurodegeneration partly by inhibiting activity of GSK3beta in a SCA3 Drosophila model. Cerebellum. 2013;12:892–901. doi: 10.1007/s12311-013-0498-3. [DOI] [PubMed] [Google Scholar]
- 211.Jung J, Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science. 2007;315:1857–1859. doi: 10.1126/science.1139517. [DOI] [PubMed] [Google Scholar]
- 212.Nagai Y, et al. Prevention of polyglutamine oligomerization and neurodegeneration by the peptide inhibitor QBP1 in Drosophila. Hum Mol Genet. 2003;12:1253–1259. doi: 10.1093/hmg/ddg144. [DOI] [PubMed] [Google Scholar]
- 213.Schulte J, Sepp KJ, Wu C, Hong P, Littleton JT. High-content chemical and RNAi screens for suppressors of neurotoxicity in a Huntington’s disease model. PloS one. 2011;6:e23841. doi: 10.1371/journal.pone.0023841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Sontag EM, et al. Methylene blue modulates huntingtin aggregation intermediates and is protective in Huntington’s disease models. J Neurosci. 2012;32:11109–11119. doi: 10.1523/JNEUROSCI.0895-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Yi J, et al. Sodium valproate alleviates neurodegeneration in SCA3/MJD via suppressing apoptosis and rescuing the hypoacetylation levels of histone H3 and H4. PLoS One. 2013;8:e54792. doi: 10.1371/journal.pone.0054792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kazantsev A, et al. A bivalent Huntingtin binding peptide suppresses polyglutamine aggregation and pathogenesis in Drosophila. Nat Genet. 2002;30:367–376. doi: 10.1038/ng864. [DOI] [PubMed] [Google Scholar]
- 217.Huang K, Fingar DC. Growing knowledge of the mTOR signaling network. Seminars in cell & developmental biology. 2014;36:79–90. doi: 10.1016/j.semcdb.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Ravikumar B, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 222.Cortes CJ, Qin K, Cook J, Solanki A, Mastrianni JA. Rapamycin delays disease onset and prevents PrP plaque deposition in a mouse model of Gerstmann-Straussler-Scheinker disease. J Neurosci. 2012;32:12396–12405. doi: 10.1523/JNEUROSCI.6189-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Frederick C, et al. Rapamycin ester analog CCI-779/Temsirolimus alleviates tau pathology and improves motor deficit in mutant tau transgenic mice. J Alzheimers Dis. 2015;44:1145–1156. doi: 10.3233/JAD-142097. [DOI] [PubMed] [Google Scholar]
- 224.Jiang T, et al. Temsirolimus promotes autophagic clearance of amyloid-beta and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol Res. 2014;81:54–63. doi: 10.1016/j.phrs.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 225.Jiang T, et al. Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology. 2014;85:121–130. doi: 10.1016/j.neuropharm.2014.05.032. [DOI] [PubMed] [Google Scholar]
- 226.Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011;6:e25416. doi: 10.1371/journal.pone.0025416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Menzies FM, et al. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain. 2010;133:93–104. doi: 10.1093/brain/awp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Borrell-Pages M, Zala D, Humbert S, Saudou F. Huntington’s disease: from huntingtin function and dysfunction to therapeutic strategies. Cell Mol Life Sci. 2006;63:2642–2660. doi: 10.1007/s00018-006-6242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci. 2003;28:425–433. doi: 10.1016/S0968-0004(03)00168-3. [DOI] [PubMed] [Google Scholar]
- 230.Kaltenbach LS, et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007;3:e82. doi: 10.1371/journal.pgen.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146–154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 232.Shirasaki DI, et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron. 2012;75:41–57. doi: 10.1016/j.neuron.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Goehler H, et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington’s disease. Mol Cell. 2004;15:853–865. doi: 10.1016/j.molcel.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 234.Emamian ES, et al. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron. 2003;38:375–387. doi: 10.1016/s0896-6273(03)00258-7. [DOI] [PubMed] [Google Scholar]
- 235.Friedman MJ, et al. Polyglutamine domain modulates the TBP-TFIIB interaction: implications for its normal function and neurodegeneration. Nat Neurosci. 2007;10:1519–1528. doi: 10.1038/nn2011. [DOI] [PubMed] [Google Scholar]
- 236.Helmlinger D, et al. Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol. 2006;4:e67. doi: 10.1371/journal.pbio.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Lim J, et al. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature. 2008;452:713–718. doi: 10.1038/nature06731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Monks DA, et al. Overexpression of wild-type androgen receptor in muscle recapitulates polyglutamine disease. Proc Natl Acad Sci U S A. 2007;104:18259–18264. doi: 10.1073/pnas.0705501104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Cummings CJ, Zoghbi HY. Trinucleotide repeats: mechanisms and pathophysiology. Annu Rev Genomics Hum Genet. 2000;1:281–328. doi: 10.1146/annurev.genom.1.1.281. [DOI] [PubMed] [Google Scholar]
- 240.van Roon-Mom WM, Reid SJ, Faull RL, Snell RG. TATA-binding protein in neurodegenerative disease. Neuroscience. 2005;133:863–872. doi: 10.1016/j.neuroscience.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 241.Matsumoto T, et al. The androgen receptor in health and disease. Annu Rev Physiol. 2013;75:201–224. doi: 10.1146/annurev-physiol-030212-183656. [DOI] [PubMed] [Google Scholar]
- 242.Rajakulendran S, Kaski D, Hanna MG. Neuronal P/Q-type calcium channel dysfunction in inherited disorders of the CNS. Nat Rev Neurol. 2012;8:86–96. doi: 10.1038/nrneurol.2011.228. [DOI] [PubMed] [Google Scholar]
- 243.Erkner A, et al. Grunge, related to human Atrophin-like proteins, has multiple functions in Drosophila development. Development. 2002;129:1119–1129. doi: 10.1242/dev.129.5.1119. [DOI] [PubMed] [Google Scholar]
- 244.Fanto M, et al. The tumor-suppressor and cell adhesion molecule Fat controls planar polarity via physical interactions with Atrophin, a transcriptional co repressor. Development. 2003;130:763–774. doi: 10.1242/dev.00304. [DOI] [PubMed] [Google Scholar]
- 245.Wang L, Charroux B, Kerridge S, Tsai CC. Atrophin recruits HDAC1/2 and G9a to modify histone H3K9 and to determine cell fates. EMBO Rep. 2008;9:555–562. doi: 10.1038/embor.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Wang L, Rajan H, Pitman JL, McKeown M, Tsai CC. Histone deacetylase-associating Atrophin proteins are nuclear receptor corepressors. Genes Dev. 2006;20:525–530. doi: 10.1101/gad.1393506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Wang L, Tsai CC. Atrophin proteins: an overview of a new class of nuclear receptor corepressors. Nucl Recept Signal. 2008;6:e009. doi: 10.1621/nrs.06009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Li Z, Karlovich CA, Fish MP, Scott MP, Myers RM. A putative Drosophila homolog of the Huntington’s disease gene. Hum Mol Genet. 1999;8:1807–1815. doi: 10.1093/hmg/8.9.1807. [DOI] [PubMed] [Google Scholar]
- 249.Zhang S, Feany MB, Saraswati S, Littleton JT, Perrimon N. Inactivation of Drosophila Huntingtin affects long-term adult functioning and the pathogenesis of a Huntington’s disease model. Disease models & mechanisms. 2009;2:247–266. doi: 10.1242/dmm.000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Andrade MA, Bork P. HEAT repeats in the Huntington’s disease protein. Nat Genet. 1995;11:115–116. doi: 10.1038/ng1095-115. [DOI] [PubMed] [Google Scholar]
- 251.Andrade MA, Petosa C, O’Donoghue SI, Muller CW, Bork P. Comparison of ARM and HEAT protein repeats. J Mol Biol. 2001;309:1–18. doi: 10.1006/jmbi.2001.4624. [DOI] [PubMed] [Google Scholar]
- 252.MacDonald ME. Huntingtin: alive and well and working in middle management. Sci STKE. 2003;2003:pe48. doi: 10.1126/stke.2003.207.pe48. [DOI] [PubMed] [Google Scholar]
- 253.Duyao MP, et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science. 1995;269:407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- 254.Nasir J, et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- 255.Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- 256.Dragatsis I, Efstratiadis A, Zeitlin S. Mouse mutant embryos lacking huntingtin are rescued from lethality by wild-type extraembryonic tissues. Development. 1998;125:1529–1539. doi: 10.1242/dev.125.8.1529. [DOI] [PubMed] [Google Scholar]
- 257.Godin JD, et al. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron. 2010;67:392–406. doi: 10.1016/j.neuron.2010.06.027. [DOI] [PubMed] [Google Scholar]
- 258.Seong IS, et al. Huntingtin facilitates polycomb repressive complex 2. Hum Mol Genet. 2010;19:573–583. doi: 10.1093/hmg/ddp524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Dietz KN, et al. The Drosophila Huntington’s disease gene ortholog dhtt influences chromatin regulation during development. Hum Mol Genet. 2015;24:330–345. doi: 10.1093/hmg/ddu446. [DOI] [PubMed] [Google Scholar]
- 260.Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- 261.Ochaba J, et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci U S A. 2014;111:16889–16894. doi: 10.1073/pnas.1420103111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol. 2012;2012:736905. doi: 10.1155/2012/736905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12:836–841. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 265.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Molecular cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 267.Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell death and differentiation. 2013;20:21–30. doi: 10.1038/cdd.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Martin DD, Ladha S, Ehrnhoefer DE, Hayden MR. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2014 doi: 10.1016/j.tins.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 269.Clark IE, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 270.Greene JC, et al. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Park J, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 272.Yang Y, et al. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Mangiarini L, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 274.Gray M, et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Heikkinen T, et al. Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington’s disease. PloS one. 2012;7:e50717. doi: 10.1371/journal.pone.0050717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Menalled LB, et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PloS one. 2012;7:e49838. doi: 10.1371/journal.pone.0049838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Farrar AM, et al. Cognitive deficits in transgenic and knock-in HTT mice parallel those in Huntington’s disease. Journal of Huntington’s disease. 2014;3:145–158. doi: 10.3233/JHD-130061. [DOI] [PubMed] [Google Scholar]
- 278.Menalled LB. Knock-in mouse models of Huntington’s disease. NeuroRx. 2005;2:465–470. doi: 10.1602/neurorx.2.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.King-Jones K, Thummel CS. Nuclear receptors--a perspective from Drosophila. Nat Rev Genet. 2005;6:311–323. doi: 10.1038/nrg1581. [DOI] [PubMed] [Google Scholar]
- 280.Basler K, Hafen E. Dynamics of Drosophila eye development and temporal requirements of sevenless expression. Development. 1989;107:723–731. doi: 10.1242/dev.107.4.723. [DOI] [PubMed] [Google Scholar]
- 281.Bowtell DD, Lila T, Michael WM, Hackett D, Rubin GM. Analysis of the enhancer element that controls expression of sevenless in the developing Drosophila eye. Proc Natl Acad Sci U S A. 1991;88:6853–6857. doi: 10.1073/pnas.88.15.6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Bier E, Ackerman L, Barbel S, Jan L, Jan YN. Identification and characterization of a neuron-specific nuclear antigen in Drosophila. Science. 1988;240:913–916. doi: 10.1126/science.3129785. [DOI] [PubMed] [Google Scholar]
- 283.Robinow S, Campos AR, Yao KM, White K. The elav gene product of Drosophila, required in neurons, has three RNP consensus motifs. Science. 1988;242:1570–1572. doi: 10.1126/science.3144044. [DOI] [PubMed] [Google Scholar]
- 284.Robinow S, White K. The locus elav of Drosophila melanogaster is expressed in neurons at all developmental stages. Dev Biol. 1988;126:294–303. doi: 10.1016/0012-1606(88)90139-x. [DOI] [PubMed] [Google Scholar]
- 285.Mlodzik M, Hiromi Y, Weber U, Goodman CS, Rubin GM. The Drosophila seven-up gene, a member of the steroid receptor gene superfamily, controls photoreceptor cell fates. Cell. 1990;60:211–224. doi: 10.1016/0092-8674(90)90737-y. [DOI] [PubMed] [Google Scholar]
- 286.Blackman RK, Sanicola M, Raftery LA, Gillevet T, Gelbart WM. An extensive 3′ cis-regulatory region directs the imaginal disk expression of decapentaplegic, a member of the TGF-beta family in Drosophila. Development. 1991;111:657–666. doi: 10.1242/dev.111.3.657. [DOI] [PubMed] [Google Scholar]
- 287.Raftery LA, Sanicola M, Blackman RK, Gelbart WM. The relationship of decapentaplegic and engrailed expression in Drosophila imaginal disks: do these genes mark the anterior-posterior compartment boundary? Development. 1991;113:27–33. doi: 10.1242/dev.113.1.27. [DOI] [PubMed] [Google Scholar]