

SUMMARY
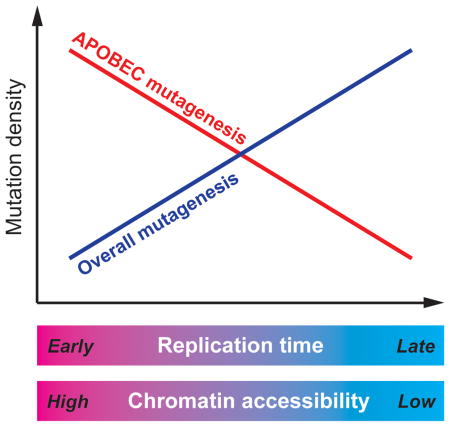
An antiviral component of the human innate immune system - the APOBEC cytidine deaminases – was recently identified as a prominent source of mutations in cancers. Here, we investigated the distribution of APOBEC-induced mutations across the genomes of 119 breast and 24 lung cancer samples. While the rate of most mutations is known to be elevated in late replicating regions that are characterized by reduced chromatin accessibility and low gene density, we observed a marked enrichment of APOBEC mutations in early-replicating regions. This unusual mutagenesis profile may be associated with a higher propensity to form single-strand DNA substrates for APOBEC enzymes in early-replicating regions and should be accounted for in statistical analyses of cancer genome mutation catalogues aimed at understanding the mechanisms of carcinogenesis as well as highlighting genes that are significantly mutated in cancer.
Graphical abstract
INTRODUCTION
Recent advances in sequencing of human cancer genomes have implicated a subfamily of the human APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) cytidine deaminases in cancer mutagenesis (Nik-Zainal et al., 2012; Roberts et al., 2012). Normally, APOBECs function to restrict retroviruses and retrotransposons via deamination of cytidines resulting in hypermutation or/and degradation of the retroelement’s single-stranded DNA replication intermediate (Refsland and Harris, 2013). However, APOBECs can also mutate a host’s DNA when it persists in single-stranded form, likely resulting in the significant number of apparent APOBEC-induced mutations observed in many types of human cancer (Alexandrov et al., 2013; Burns et al., 2013; Roberts et al., 2013). The conclusion that one or multiple APOBECs induced these mutations was supported by the mutations’ observed tendency to occur in clusters in a strand-coordinated fashion (Alexandrov et al., 2013; Nik-Zainal et al., 2012; Roberts et al., 2013; Roberts et al., 2012) and by a high enrichment of the APOBEC mutagenesis signature, TCW→TTW or TCW→TGW (mutated nucleotide underlined, W=A or T), among clustered as well as scattered mutations. The strand-coordinated clusters observed in these studies agreed with the expected pattern of mutations caused by an APOBEC acting processively on a long ssDNA substrate, where cytidine deaminations can only occur on the same DNA strand. Unlike in mutation clusters, scattered mutations could be caused by APOBEC-induced cytidine deamination in shorter ssDNA stretches. The origin of the ssDNA substrates for APOBEC mutagenesis in cancer genomes is still to be determined (reviewed in (Roberts and Gordenin, 2014)). The main difficulty in this task is the complexity and variability of environmental and genetic factors that influence the accumulation of mutations over the lifetime of cancer. One useful approach toward understanding mutagenesis in cancer emerged recently due to the progress of the Epigenome Roadmap and ENCODE projects (Kellis et al., 2014; Raney et al., 2011). The genome-wide distributions of multiple epigenomic features, such as replication timing, chromatin accessibility and transcription were determined and cataloged for cell lines originating from different human tissue types. The distributions of these features subsequently proved to be good predictors of regional differences in mutation density in cancers originated from the same tissues (Lawrence et al., 2013; Polak et al., 2015). The profiling of mutations in cancer genomes against these epigenomic features can guide future research of mutagenic mechanisms in model systems and also help in dissecting the relative roles of mutagenesis and selection in the accumulation of cancer driver and passenger mutations (Lawrence et al., 2013; Polak et al., 2014). Here, we analyzed the genomic localization of both clustered and scattered APOBEC mutations across lung and breast cancer genomes and their correlation with the location of epigenomic features including replication timing, chromatin accessibility, and transcription. We found that the relationship between the location of APOBEC-induced mutations and these epigenomic features is reversed compared to other mutation types.
RESULTS
Mutation clusters enriched with APOBEC-signature mutations are more frequent in early-replicating regions
We assessed the trinucleotide sequence context and base substitution of each mutation in 24 lung (Imielinski et al., 2012) and 119 breast cancer (Alexandrov et al., 2013) genomes to annotate mutations consistent with the APOBEC signature (i.e. TCW→TGW or TCW→TTW). We also identified mutation clusters based on inter-mutation distance, excluding complex mutations as described earlier (Roberts et al., 2013; Roberts et al., 2012). Similar to prior observations (Alexandrov et al., 2013; Nik-Zainal et al., 2012; Roberts et al., 2013; Roberts et al., 2012) strand-coordinated clusters in which all mutations occurred in either cytosines (C-coordinated) or in guanines (G-coordinated) of the same strand were highly enriched with APOBEC signature mutations. Clusters of three or more mutations all displayed equally high APOBEC enrichment regardless of the number of mutations indicating that they contained at most a small fraction of incidental non-APOBEC mutations (Figure S1). We next examined the genomic positions of these APOBEC-enriched C- or G-coordinated clusters relative to replication timing and chromatin accessibility (Figure 1) and found a high abundance of such clusters in the early replicating regions of the genomes, which preferentially contain accessible chromatin and active transcription. This observed distribution of APOBEC-induced mutations in relationship to replication timing is reversed compared to the known distribution of most other somatic mutations in cancer, which have been shown to be prevalent in late-replicating heterochromatinized regions of the genome (Donley and Thayer, 2013; Koren et al., 2012; Lawrence et al., 2013; Liu et al., 2013; Polak et al., 2014; Schuster-Bockler and Lehner, 2012; Sima and Gilbert, 2014).
Figure 1. The distribution of APOBEC-signature mutation clusters relative to epigenomic features in cancer genomes.

(see Figure S1 and text for defining the subgroup of clusters used in this analysis.)
(A–D) The distribution of C- or G-strand coordinated clusters with at least 3 mutations relative to DNA replication timing in lung (A) and breast (B) cancer genomes, and relative to chromatin accessibility in lung (C) and breast (D) cancer genomes. Bins on the horizontal axis were obtained by sorting all genome positions by the values of the genomic feature (DNA replication time or chromatin accessibility) and dividing into four non-overlapping equal-sized windows. The deviation from the uniform distribution of clusters in genomic space was confirmed by Cochran-Armitage test (the P-values were calculated under the null hypothesis that all bins would contain an equal fraction of clusters).
Similar to clustered mutations, scattered APOBEC-signature mutations show elevated density in early-replicating regions
Next, we inquired whether the genome-wide distribution of all APOBEC-induced mutations relative to replication timing and chromatin accessibility would be similar to that observed for clustered mutations. While not all mutations consistent with the APOBEC signature are actually induced by APOBEC, samples with higher enrichments of the APOBEC signature will contain greater fractions of mutations that in fact have been induced by APOBEC. Thus, the APOBEC-signature mutations in samples with high enrichment would more accurately depict the genome-wide distribution of APOBEC mutagenesis. We therefore calculated the enrichment of individual samples with the APOBEC signature mutations as described before (Roberts et al., 2013), to determine the extent to which APOBEC enzymes were operating in a given sample. We analyzed the distribution of APOBEC-signature mutations with respect to replication timing and chromatin accessibility using linear regression. We found that regression coefficients (i.e. slopes of the regression lines) are inversely proportional to the APOBEC sample enrichment (Figure 2, Figure S2), indicating that the density of mutations actually induced by APOBEC increases in early-replicating, chromatin accessible regions. This inverse proportionality was invariably observed, even when the two subcategories of APOBEC-signature mutations (TCW→TTW and TCW→TGW) were analyzed separately (Figure S3). All sample-specific enrichment values used in the analyses are listed in Table S1.
Figure 2. Dependence of the normalized density of APOBEC-signature mutations from replication timing of a cancer genome region.

(A,B) Samples with low or no enrichment with APOBEC mutation signature (fold enrichment <2) display a positive correlation between the normalized density of APOBEC-signature mutations and replication timing (an example for lung cancer is shown in panel A, an example for breast cancer is shown in panel B).
(C,D) Samples with high enrichment with APOBEC mutation (fold enrichment ≥2) display a negative correlation between the normalized density of APOBEC-signature mutations and replication timing (an example for lung cancer is shown in panel C, an example for breast cancer is shown in panel D). Bins on the horizontal axes in A–D panels were obtained by sorting all genome positions by the values of a genomic feature (DNA replication time or chromatin accessibility) and then dividing into ten non-overlapping equal-sized windows.
(E,F) In general, the slopes of this regression are anti-correlated with APOBEC-signature enrichment (lung cancer data is shown in panel E and breast cancer data is shown in panel F). Similar analyses with respect to chromatin accessibility are shown on Figure S2 and analyses performed separately for the two subcategories of APOBEC-signature mutations (TCW→TTW and TCW→TGW) are shown on Figure S3. The sample specific enrichment values used for the analyses in Figure 2 and Figure S3 are shown in Table S1.
To extend the model into all samples and to take into account C→T and C→G mutations occurring in the TCW motif, but induced by other mutagens, we introduced a linear model that explicitly allows for two classes of APOBEC-signature mutations: mutations induced by APOBEC and mutations due to other mechanisms. The regression coefficient for mutations actually induced by APOBEC is a free parameter of this model. This model allows us to infer the dependency of mutations in fact induced by APOBEC on epigenomic variables. This analysis is conservative because it assumes that APOBEC never induces mutations outside of the motif. As seen from Figure 3a (see also Table S1) these mutations show a strong preference towards early-replicating regions (all P-values for replication timing and chromatin accessibility of lung and breast cancer are below 0.001, see exact values in Table S1). Collectively, our observations establish that the distribution of APOBEC-induced mutations in cancer genomes is reversed in comparison to the bulk of mutations produced by other mechanisms.
Figure 3. Increased density of the APOBEC-signature mutations in early-replicating regions of genome is confirmed by the linear model considering heterogeneity of mutational mechanisms and by the analysis of mutations in multiple exomes of several cancer types.

(A) The dependency of APOBEC-induced mutation density (inferred from the linear model that allows for heterogeneity of mutational mechanisms - see Experimental Procedures) on replication timing and on APOBEC-signature mutation enrichment in lung cancer samples. DNA replication time is presented in ENCODE units of measure, linearly scaled in the range [0 to 90] and binned as in Figure 2. The vertical axis shows mutation density per 10M window, calculated as a number of mutations in the window normalized by a number of TCW motifs in the window and by a total number of mutations in the sample (see formula in Experimental Procedures). See Table S1 for all calculated model parameters.
(B) The median of differences in APOBEC-signature mutation enrichments between early- and late-replicating regions of exomes from samples of six types of cancer in which an enrichment with the APOBEC mutation signature was statistically significant in both early- and late-replicating regions. Cancer types are abbreviated as in TCGA: bladder urothelial carcinoma (BLCA), breast invasive carcinoma (BRCA), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), head and neck squamous cell carcinoma (HNSC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC). * P<0.05, ** P<0.01, *** P<0.001. Exact values: PBLCA=0.0011, PBRCA=0.038, PCESC=0.023, PHNSC=0.49, PLUAD=0.0028, PLUSC=0.0007.
APOBEC-signature mutations in the exomes from The Cancer Genome Atlas (TCGA) are more prevalent in early-replicating regions
In addition to the exploration of WGS cancer samples, we applied our analysis to a large dataset of somatic mutations that occurred in the exomes of six cancer types known to be highly mutated by APOBEC enzymes (Roberts et al 2013 NatGenet) (obtained from the TCGA;Broad GDAC Firehose standard data run of Feb. 15, 2014 http://gdac.broadinstitute.org/runs/stddata__2014_02_15/ ). Although, exomes constitute only around 1% of the genome, the number of sequenced exomes in TCGA is large in comparison with the number of available complete cancer genomes. For this simplified analysis we used two genome tracks identified as universally early- or universally late-replicating regions, based on available replication timing data for multiple cell types (Pedersen et al., 2013). We then created two lists of mutation calls, falling into each of these tracks and calculated the enrichments with APOBEC-signature for the two groups of mutations of each samples. Samples with statistically significant APOBEC enrichment for both early- and late-replicating regions were used to evaluate the impact of replication timing. In agreement with the results for WGS analysis, the APOBEC enrichments for mutation calls falling into early-replicating regions exceeded enrichments for calls from late replicating regions for breast and lung cancers as well as for two other cancer types, cervical and bladder carcinomas (Figure 3b). We note that the lack of a statistically significant difference for head and neck squamous cell carcinomas could be due to its small sample size within the exome mutation catalogue or could reflect differences in the mechanisms underlying APOBEC mutagenesis in this cancer type.
The observed genome distribution of APOBEC-signature mutations between early- and late-replicating regions is not affected by transcription
Because ssDNA associated with transcription is an established target of several AID/APOBEC family members and early-replication regions are also gene dense, we examined whether the enrichment of APOBEC-induced mutations in these regions could be dependent on transcription. To this end, we compared the distribution of APOBEC-signature mutations to replication timing separately for transcribed and non-transcribed regions of the genome. APOBEC-induced mutations predominated in early replicating regions of the genome for non-transcribed as well as in transcribed regions with no detectable difference between the two trends (Figure 4a,b). Within transcribed regions, the distribution of APOBEC-signature mutations between early- and late-replicating regions was also similar between transcribed and non-transcribed strands of DNA (Figure 4c,d). These results held when the analysis was repeated with all samples, including those without a statistically significant enrichment of the APOBEC mutation signature, with one exception – a minor difference between transcribed and non-transcribed strands for breast cancer (Figure S4)
Figure 4. The anti-correlation between the density of APOBEC-signature mutation and replication timing in cancer genomes is independent on transcription.

(A–D) Anti-correlation of APOBEC-enrichment versus replication timing regression slopes in samples with different APOBEC-signature enrichment determined separately for transcribed and non-transcribed regions of the lung (A) and breast (B) cancer genomes and for transcribed and non-transcribed strands of DNA in the lung (C) and breast (D) cancer genomes, including only samples with statistically significant enrichment with APOBEC mutation signature. Exact P-values: (a) 0.055, (b) 0.34, (c) 0.14, (d) 0.17. See Figure S3 for a similar comparison including all samples.
DISCUSSION
Altogether, our results indicate that APOBEC-induced mutations occur preferentially in early replicating regions, which themselves are enriched with active chromatin. We suggest that the main cause of the observed effect is the necessity for DNA to be in a single-stranded state in order to be mutated by APOBEC enzymes. One source of ssDNA in early replicating regions could be simply the higher levels of transcription in these areas. The APOBEC relative, Activation-induced Cytidine Deaminase (AID) is known to require transcription to mediate immunoglobulin hypermutation in B-cells (reviewed in (Liu and Schatz, 2009)) and both AID and APOBEC3G in yeast appear to target transcription when expressed in yeast (Taylor et al., 2014). However, our analysis shows that the density of APOBEC-signature mutations were equal between transcribed and non-transcribed regions (Figures 4 and S3), which suggests that replication timing could be the primary factor affecting the chance of cytidine deamination by one of APOBEC enzymes. The dependence on replication timing appears robust and universal. It was detectable even in the exome datasets of 5 out of 6 cancer types known to have a high presence of APOBEC mutagenesis and even when only universally late- or universally early replicating regions were used in the analysis (Figure 3b). We speculate that increased DNA fragility in early replicating regions may produce more ssDNA substrate for APOBEC enzymes. Early replicating, highly transcribed regions of cancer genomes are known to be associated with changes stemming from chromosome breakage, such as copy number variation, chromosome rearrangements, fragility and loss of heterozygosity (Barlow et al., 2013; Koren et al., 2012; Pedersen and De, 2013; Sima and Gilbert, 2014). An increased frequency of DNA breakage would in turn be expected to produce more hypermutable ssDNA as the repair of these breaks often involve formation of ssDNA through either 5′→3′ resection (Mimitou and Symington, 2011; Roberts et al., 2012) or uncoupled conservative replication (Malkova and Ira, 2013; Saini et al., 2013; Sakofsky et al., 2014). Consistent with this idea, ssDNA formed during DNA double strand break repair is prone to DNA damage induced mutation in yeast model systems. Additionally, APOBEC mutagenesis in cancer is increased in the vicinity of chromosome rearrangement breakpoints (Drier et al., 2013; Nik-Zainal et al., 2012; Roberts et al., 2012). While a mechanistic explanation for the correlation between the location of APOBEC mutations and structural alternations in cancer genomes remains to be established, we propose that these events may originate from a common source relating to replication timing. Importantly, the targeting of APOBEC-induced mutations to early replicating regions appears to be a distinct mechanism from the specific localization of the AID to active promoters and super-enhancers during B-cell transcription (Meng et al., 2014; Qian et al., 2014). In activated B-cells, AID-induced double strand breaks and kataegic sites associate with a relatively small number of transcribed promotors and enhancers (Meng et al., 2014; Qian et al., 2014), resulting in a limited over-representation of the AID signature motif (WRC, where W=A or T and R=G or A) among all mutations in B-cell derived tumors and no observed AID activity in other tumor types (Alexandrov et al., 2013). Contrastingly, APOBEC-induced mutations occur in greater abundance and widely spread across cancer genomes, without an apparent preference to any regulatory sequences. Reportedly <6% of APOBEC-induced kataegic events occur near transcriptional start sites compared to 82% for AID-induced events (Qian et al., 2014), while APOBECs favor early replicating regions of the genome, which in B-cells, are devoid of AID-induced DSBs (Barlow et al., 2013). It remains to establish the specific cellular processes accounting for the unique correlation of APOBEC mutagenesis with gene dense early replicating regions of active chromatin, however, this association could already be used in the search for genes significantly mutated in cancers to accurately define background mutation rates for APOBEC signature mutations specific to genomic regions of interest (Lawrence et al., 2013).
EXPERIMENTAL PROCEDURES
The mutation data for 24 lung adenocarcinomas are from (Imielinski et al., 2012) and mutation data for 119 breast cancers are from (Alexandrov et al., 2013). We annotated mutation clusters as well as APOBEC-signature mutations as described previously (Roberts et al., 2013). Briefly, mutation clusters were identified and mutation signatures assigned after filtering out mutations (usually <10% of total) falling within regions identified as simple repeats in simpleRepeat.txt.gz from http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/. Additionally, groups of very closely (10 nt or less) spaced mutation events, which are often caused by single act of synthesis by error prone polymerase, were counted as a single mutation event. The TCW APOBEC mutation signature was defined as TCW→TTW or TCW→TGW mutation events. Enrichment with APOBEC mutation signature was calculated as an overrepresentation of the signature compared to random mutagenesis as in (Roberts et al., 2013).
We used epigenomic data generated by the ENCODE project (Raney et al., 2011). For the analysis of lung cancer and breast cancer genomes we used DNA replication timing for IMR90 and MCF-7 cell lines, and DNase I hypersensitivity data obtained for NHEK and MCF-7 cell lines, respectively. A single-sample distribution of the APOBEC induced mutation density relative to genomic features were estimated by a simple linear regression model where a particular genomic feature was considered as the only independent variable. The independent variable was sampled as follows: all genome positions where sorted by the values of the genomic feature, then divided into ten adjacent non-overlapped equal-sized windows (bins). The number of APOBEC-signature mutations in a window was normalized by the number of TCW motifs in a bin and by the total number of APOBEC-signature mutations in a sample (referred to as the normalized density throughout this paper).
In a multi-sample linear regression model of genome-wide mutation density, we considered separately APOBEC- and background mutagenesis. This model was defined as follows:
where x - genomic position; f(x) – the value of epigenomic feature at x; α(s) - the fraction of APOBEC-induced mutations in a sample as estimated from the APOBEC-signature mutation enrichment of the sample s: α(s)=1−1/e(s); βA0,βA1, are model coefficients corresponding to mutations that were in fact induced by APOBEC, βN0, βN1 are coefficients corresponding to mutations not caused by APOBEC independently of the presence of the APOBEC signature; M – the mutation density calculated as the number of mutations in the window normalized by the number of TCW motifs in a window and by the total number of mutations in a sample. The number of mutations and values of genomic features were respectively summed or averaged over 10M non-overlapping windows along the genome. The results for smaller window sizes are qualitatively similar.
Somatic mutations in exomes were obtained from TCGA (Broad GDAC Firehose standard data run of Feb. 15, 2014 http://gdac.broadinstitute.org/runs/stddata__2014_02_15/ ). Each nucleotide position with the exomes was classified into early- or late-replication region according to (Pedersen et al., 2013). Enrichment of APOBEC signature mutations in early- and late-replicating regions was calculated separately and as described before (Roberts et al., 2013). Samples with statistically insignificant APOBEC enrichment in any replicating regions were excluded from this analysis.
A statistical significance of the difference between transcribed and non-transcribed regions and strands was calculated as follows: data from different regions or strand was merged with introduction of the indicator variable describing a source of the data. A linear regression was calculated and the value of statistical significance was extracted from the regression results as the significance of the coefficient relating to the indicator variable.
Supplementary Material
Acknowledgments
This work was supported by NIH grant R01 MH101244 (S.R.S), Intramural Research Program of the National Institutes of Health (NIH), National Institute of Environmental Health Sciences, project Z1AES103266 (D.A.G.), the Pathway to Independence Award 4R00ES022633-02 from the National Institute of Environmental Health Sciences (NIH/NIEHS) (S.A.R.), and the Russian Science Foundation grant 14-24-00155 (M.D.K.).
Footnotes
AUTHOR CONTRIBUTIONS
M.D.K., P.P., S.A.R., and L.J.K. analyzed the data. J.S. provided data elements key for analysis; M.D.K. and S.R.S. designed the analysis. M.D.K. wrote the first draft of the paper. S.R.S., S.A.R, and D.A.G. contributed to writing. D.A.G. and S.R.S. jointly supervised the work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow JH, Faryabi RB, Callen E, Wong N, Malhowski A, Chen HT, Gutierrez-Cruz G, Sun HW, McKinnon P, Wright G, et al. Identification of early replicating fragile sites that contribute to genome instability. Cell. 2013;152:620–632. doi: 10.1016/j.cell.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013;45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donley N, Thayer MJ. DNA replication timing, genome stability and cancer: late and/or delayed DNA replication timing is associated with increased genomic instability. Paper presented at: Seminars in cancer biology; Elsevier; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drier Y, Lawrence MS, Carter SL, Stewart C, Gabriel SB, Lander ES, Meyerson M, Beroukhim R, Getz G. Somatic rearrangements across cancer reveal classes of samples with distinct patterns of DNA breakage and rearrangement-induced hypermutability. Genome Res. 2013;23:228–235. doi: 10.1101/gr.141382.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, Ward LD, Birney E, Crawford GE, Dekker J, et al. Defining functional DNA elements in the human genome. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:6131–6138. doi: 10.1073/pnas.1318948111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Polak P, Nemesh J, Michaelson JJ, Sebat J, Sunyaev SR, McCarroll SA. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet. 2012;91:1033–1040. doi: 10.1016/j.ajhg.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, De S, Michor F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nature communications. 2013;4:1502. doi: 10.1038/ncomms2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Schatz DG. Balancing AID and DNA repair during somatic hypermutation. Trends in immunology. 2009;30:173–181. doi: 10.1016/j.it.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Malkova A, Ira G. Break-induced replication: functions and molecular mechanism. Curr Opin Genet Dev. 2013;23:271–279. doi: 10.1016/j.gde.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng FL, Du Z, Federation A, Hu J, Wang Q, Kieffer-Kwon KR, Meyers Robin M, Amor C, Wasserman Caitlyn R, Neuberg D, et al. Convergent Transcription at Intragenic Super-Enhancers Targets AID-Initiated Genomic Instability. Cell. 2014;159:1538–1548. doi: 10.1016/j.cell.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. DNA end resection--unraveling the tail. DNA Repair (Amst) 2011;10:344–348. doi: 10.1016/j.dnarep.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BS, De S. Loss of heterozygosity preferentially occurs in early replicating regions in cancer genomes. Nucleic Acids Res. 2013;41:7615–7624. doi: 10.1093/nar/gkt552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BS, Yang IV, De S. CruzDB: software for annotation of genomic intervals with UCSC genome-browser database. Bioinformatics. 2013;29:3003–3006. doi: 10.1093/bioinformatics/btt534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P, Karlic R, Koren A, Thurman R, Sandstrom R, Lawrence MS, Reynolds A, Rynes E, Vlahovicek K, Stamatoyannopoulos JA, et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015;518:360–364. doi: 10.1038/nature14221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P, Lawrence MS, Haugen E, Stoletzki N, Stojanov P, Thurman RE, Garraway LA, Mirkin S, Getz G, Stamatoyannopoulos JA, et al. Reduced local mutation density in regulatory DNA of cancer genomes is linked to DNA repair. Nature biotechnology. 2014;32:71–75. doi: 10.1038/nbt.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon KR, Resch W, Liang G, Tang Z, Mathé E, Benner C, et al. B Cell Super-Enhancers and Regulatory Clusters Recruit AID Tumorigenic Activity. Cell. 2014;159:1524–1537. doi: 10.1016/j.cell.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raney BJ, Cline MS, Rosenbloom KR, Dreszer TR, Learned K, Barber GP, Meyer LR, Sloan CA, Malladi VS, Roskin KM, et al. ENCODE whole-genome data in the UCSC genome browser (2011 update) Nucleic Acids Res. 2011;39:D871–875. doi: 10.1093/nar/gkq1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refsland EW, Harris RS. The APOBEC3 family of retroelement restriction factors. Curr Top Microbiol Immunol. 2013;371:1–27. doi: 10.1007/978-3-642-37765-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Gordenin DA. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer. 2014;14:786–800. doi: 10.1038/nrc3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Malc E, Mieczkowski PA, et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell. 2012;46:424–435. doi: 10.1016/j.molcel.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, Ira G, Haber JE, Lobachev KS, Malkova A. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature. 2013;502:389–392. doi: 10.1038/nature12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, Roberts SA, Malc E, Mieczkowski PA, Resnick MA, Gordenin DA, Malkova A. Break-induced replication is a source of mutation clusters underlying kataegis. Cell reports. 2014;7:1640–1648. doi: 10.1016/j.celrep.2014.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster-Bockler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488:504–507. doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- Sima J, Gilbert DM. Complex correlations: replication timing and mutational landscapes during cancer and genome evolution. Curr Opin Genet Dev. 2014;25:93–100. doi: 10.1016/j.gde.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BJ, Wu YL, Rada C. Active RNAP pre-initiation sites are highly mutated by cytidine deaminases in yeast, with AID targeting small RNA genes. eLife. 2014;3:e03553. doi: 10.7554/eLife.03553. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.