

Abstract
Background
Micro ribonucleic acid (miR) dysregulation in the myocardium has been implicated in cardiac remodeling after injury or stress.
Objectives
This study sought to explore the role of miR in human CD34+ cell (hCD34+) dysfunction in vivo after transplantation into the myocardium under ischemia-reperfusion (I-R) conditions.
Methods
In response to inflammatory stimuli, the miR array profile of endothelial progenitor cells (EPC) was analyzed using a polymerase chain reaction-based miR microarray. MiR-377 expression was assessed in myocardial tissue from human patients with heart failure (HF). We investigated the effect of miR-377 inhibition on hCD34+ cell angiogenic proteome profile, in vitro and on cardiac repair and function after I-R injury in immunodeficient mice.
Results
The miR array data from EPCs in response to inflammatory stimuli indicate changes in numerous miR with a robust decrease in miR-377. Human cardiac biopsies from HF patients showed significant increase in miR-377 expression compared to nonfailing control hearts. Proteome profile of hCD34+ cells transfected with miR-377 mimics showed significant decrease in proangiogenic proteins versus nonspecific control transfected cells. We also validated that serine/threonine kinase 35 is a target of miR-377 using a dual-luciferase reporter assay. In a mouse model of myocardial I-R, intramyocardial transplantation of miR-377-silenced hCD34+ cells in immunodeficient mice, promoting neovascularization (at 28 days, post-I-R) and lower interstitial fibrosis, leading to improved left ventricular (LV) function.
Conclusions
These findings indicate that HF increases miR-377 in the myocardium, which is detrimental to stem cell function, and transplantation of miR-377 knockdown hCD34+ cells into ischemic myocardium promoted their angiogenic ability, attenuating LV remodeling and cardiac fibrosis.
Keywords: endothelial progenitor cells, heart failure, neovascularization
Introduction
Heart failure (HF) has been classified as an epidemic of the 21st century and is now the major cause of morbidity in the United States (1). A number of cardiac pathophysiological conditions, including myocardial infarction (MI) and ischemia reperfusion (I-R) injury that lead to HF, are associated with activation of inflammatory mediators in the heart (2).
Despite our belief that the heart has limited regenerative capacity, murine endothelial progenitor cells (EPC) and human CD34+ (hCD34+) cell-based therapy provide substantial clinical benefits for ischemic diseases, such as chronic angina, MI, and HF (3,4). These cells, through paracrine-mediated growth factor secretion, induce neo-angiogenesis/vasculogenesis and therefore augment cardiac cell survival and function (5). The major tissue factors for the poor clinical outcome include hostile ischemia and chronic inflammatory tissue microenvironments into which the cells are introduced.
Recently, micro ribonucleic acids (miR) (∼20-22 nucleotide small RNAs) have elicited substantial interest as regulators of and therapeutic targets for HF (6). MiR dysregulation after cardiac injury has been implicated in several biological processes involved in cardiovascular diseases (7,8). However, studies to understand the deleterious effect of miR dysregulation on hCD34+ cells or murine EPCs in response to an inflammatory stimulus and its implications on cardiac regeneration and repair after myocardial transplantation are limited.
So far, the role of miR-377 in hCD34+ cell or EPC biology and function has never been explored. Moreover, it is unknown whether altering miR-377 expression affects hCD34+ cell-induced angiogenesis in ischemic myocardial tissue. Therefore, determining the role of miR-377 in hCD34+ cell or EPC biology and function and its molecular mechanisms could be of major significance for stem cell-based therapy aimed at regeneration of heart tissue.
In this study, we first sought to determine the alterations of miR in mouse EPCs under inflammatory conditions and cardiac biopsies from human HF patients. We further tested whether knockdown of miR-377 in hCD34+ cells could affect their angiogenic response in ischemic myocardium.
Methods
EPCs isolated from bone marrow of C57BL/6J male mice (Jackson Laboratories, Bar Harbor, Maine) were cultured as described previously (9). Detailed methodology is provided in the Online Appendix. EPCs were subjected to lipopolysaccharide (LPS) treatment (25 ng/ml) for 12 h (to stimulate inflammatory response). Untreated cells served as controls. MiR expression was analyzed using a polymerase chain reaction (PCR)-based miR microarray platform covering a total of 352 mouse miR. Data analysis was performed using web-based software for miR PCR array system.
Heart tissue samples were obtained from failing human hearts at the time of transplantation at the Houston Methodist DeBakey Heart and Vascular Center, Houston Methodist Hospital, Houston, Texas, and immediately frozen in liquid nitrogen and stored at -80°C until use. Normal tissue samples were obtained from donor hearts not used for transplantation and were collected and stored in the same manner. All tissues were collected under an approved protocol by the Houston Methodist Research Institutional Review Board.
Isolation of Cells and Study Design
Cardiomyocytes were isolated from 12-week-old C57BL/6J male mice after 3 days of myocardial I-R by Langendorff perfusion and Thompson's procedure (10). For isolation of adult cardiac endothelial cells, CD31+ cells were isolated via magnetic-activated cell sorting separation using anti-biotin CD31 microbeads (Miltenyi Biotec, San Diego, California) and CD31- cells were depleted. Purity of CD31+ cells was confirmed by immunohistochemistry for CD31 on cytospins of freshly isolated CD31+/CD31- cardiac cells.
To determine the role of miR-377 in hCD34+ cells angiogenic property, the condition medium collected from culturing hCD34+ cells transfected for 48 h with either miR-377 mimic (60 nm/l), inhibitor (60 nm/l: anti-miR-377), miR mimic, or miR inhibitor negative control was analyzed using Proteome Profiler™ (human angiogenesis antibody array, R&D Systems, Minneapolis, Minnesota) per the manufacturer's instructions. Data were analyzed via ImageQuant LAS 4000 software (GE Healthcare, Waukesha, Wisconsin). The detailed transfection method is provided in the Online Appendix.
Human umbilical vein endothelial cells (HUVECs) were co-transfected with either miR-377 mimic (60 nm/l) or miR-377 inhibitor (60 nm/l) or miR mimic or inhibitor negative control (60 nm/l), and a reporter plasmid containing the three prime untranslated region (3′UTR) of serine/threonine kinase 35 (STK35) (HmiT003085-MT06, 100 ng) or mutated 3′UTR of STK35 (CS-HmiT003085b-MT06-01, 100 ng), or the corresponding control empty luciferase reporter vector (CmiT000001-MT06 100 ng, GeneCopoeia, Rockville, Maryland) was mixed with Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific Inc., Tewksbury, Massachusetts) and added to 48-well plate containing HUVECs (1.3 × 104). After 24-h transfection, luciferase activity was assessed using the Dual-Luciferase reporter assay kit (Promega Corp., Madison, Wisconsin) per manufacturer's protocol.
Twelve-week-old, severe combined immunodeficiency (SCID) mice were subjected to myocardial ischemia for 30 min by temporary ligation of the left anterior descending coronary artery followed by reperfusion for 28 days as described previously (11,12). Immediately after ischemia was completed and reperfusion begun, mice received intramyocardial injection of hCD34+ cells (5×104) transfected with either miR-377 inhibitor or miR inhibitor negative control at 3 different sites in the ischemia area. The hCD34+ cells were labeled using PKH-26 red fluorescent cell linker kit per manufacturer's protocol. Transplanted hCD34+ cells were detected at 24 h after injection by immunofluorescence and left ventricular (LV) functional changes and structural remodeling were evaluated on 28 days after I-R.
Statistical Analysis
Data are presented as mean ± SE. An unpaired Student t test was performed between 2 groups of mice to determine statistical significance. When involving more than 2 groups, analysis of variance with Tukey post-hoc test was used to analyze the data. Probability (p) values of < 0.05 were considered a significant difference.
Results
Our previous study showed that prolonged inflammatory response in the myocardium is detrimental for EPC function (9). To determine the miR profile of EPCs under myocardial inflammatory conditions, we treated bone marrow-derived EPCs (mouse) with LPS (25 ng/ml) for 12 h and performed quantitative reverse transcription PCR (qRT-PCR)-based miR array analysis. The miR array data analysis showed that several miRs were differentially expressed with robust decreases in miR-377 in EPCs treated with LPS (Figures 1A and 1B [well 7C]). We further validated miR-377 expression in LPS-treated hCD34+ cells (Figure 1C) using qRT-PCR. The results consistently showed significant decreases in miR-377 expression upon LPS treatment (p < 0.05 vs. control-untreated cells). Further, we confirmed similar results in the mouse EPC (Online Figure 1A) and HUVECs (Online Figure 1B) upon LPS treatment versus control (p < 0.05). The miR array data (heatmap and expression) of all the miR analyzed is depicted in Online Figure 2.
Figure 1. MiR Array Analysis of EPCs in Response to Inflammatory Stimuli.

(A) Heat map shows miR expression in control and LPS-treated EPCs. (B) Location of the miRs in the heat map and their relative expression. (C) Validation of miR-377 expression in control and LPS-treated hCD34+ cells by qRT-PCR (normalized to control U6; n = 3; *p < 0.05). EPC = endothelial progenitor cells; LPS = lipopolysaccharide; miR = micro ribonucleic acid; qRT-PCR = reverse transcription polymerase chain reaction.
MiR-377 Expression in Human Failing Hearts
To determine HF's effect on miR-377 expression, cardiac biopsies were collected from the LV free wall of ischemia patients at the Houston Methodist DeBakey Heart and Vascular Center. The qRT-PCR results showed that the miR-377 expression is significantly upregulated in human heart tissues samples from HF patients compared to patients with noncardiac-related ailments (p < 0.05) (Figure 2).
Figure 2. miR-377 Expression in Failing Human Heart.

As measured by qRT-PCR, miR-377 expression is significantly upregulated in failing versus nonfailing human heart samples (normalized to control U6; n = 5; *p < 0.05). Abbreviations as in Figure 1.
To determine ischemia-induced miR-377 expression in different cardiac cell types, we isolated cardiomyocytes, endothelial cells (CD31+ve), and CD31-ve cells from mouse hearts with sham or I-R procedure, 3 days post-surgery, and assessed miR-377 expression by qRT-PCR analysis. I-R injury increased miR-377 expression in all the cardiac cell types versus levels in sham-operated mice (p < 0.05) (Figures 3A through 3C). These data, together with the miR-377 expression in the human failing heart, prompted us to evaluate the effect of increased miR-377 in the myocardium on hCD34+ cells that will be introduced into the myocardium for cell-based therapy.
Figure 3. Upregulated miR-377 Expression in Ischemic Mouse Hearts.

In (A) cardiomyocytes, (B) CD31+ve cells, and (C) CD31-ve cells isolated from mouse heart subjected to I-R or sham operation, I-R significantly upregulated MiR-377 expression (normalized to control U6; n = 6; *p < 0.05). I-R = ischemia-reperfusion; other abbreviations as in Figure 1.
Previous reports have suggested that the beneficial effect of hCD34+ cell therapy is mediated through paracrine secretion of growth factors that aid in angiogenic response (13). To determine the effect of miR-377 on paracrine secretion of angiogenic factors in hCD34+ cells, proteome profile of condition medium of hCD34+ cells transfected with miR-377 mimic or miR-377 inhibitor or respective negative controls was performed using a human angiogenesis array. Figure 4 shows that miR-377 mimic treatment significantly decreased secretion of various proangiogenic factors such as vascular endothelial growth factor (VEGF), matrix metalloproteinase-9, platelet-derived growth factor-AA, hepatocyte growth factor, insulin-like growth factor-binding protein-1, endocrine gland-derived -VEGF, endothelin-1, angiopoietin-1, and angiopoietin-2 when compared to the miR mimic negative control transfected hCD34+ cells conditioned media (p < 0.05) (Figures 4A through 4C). Some of the antiangiogenic proteins like serpin E1, serpin F1, thrombospondin-1, uPA, platelet factor 4, angiogenin, angiostatin, and artemin were significantly higher in the conditioned media collected from miR-377 mimic-treated hCD34+ cells as compared to miR mimic negative control-transfected hCD34+ cell-conditioned media. On the contrary, miR-377 inhibitor treatment showed higher proangiogenic factor and lower antiangiogenic factor secretion compared to miR-377 mimic-treated cell-conditioned media (p <0.05) (Figures 4A through 4C).
Figure 4. Proteome Profile of Conditioned Media from hCD34+ Cells.
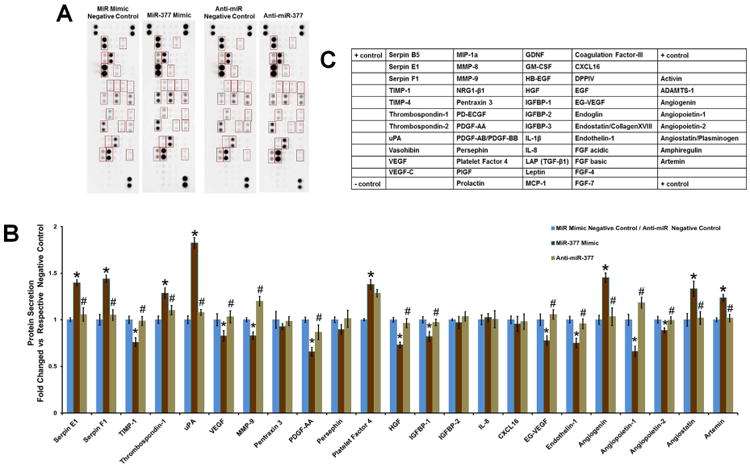
(A) Human angiogenesis antibody array analysis on condition media collected after 48 h from miR-377 mimic or inhibitor or respective negative control transfected hCD34+ cells. (B) Values were normalized to controls in the blot and expressed as fold change versus respective control (n = 3; *p < 0.05, miR-377 mimic vs. miR mimic negative control; #p < 0.05, miR-377 inhibitor vs. miR-377 mimic). (C) Layout of the antibodies on the human angiogenesis array blots. Abbreviations as in Figure 1.
Vasculogenesis is a dynamic process involving migration, differentiation, endothelial cell engraftment, and vascular tube formation (14). To first determine the effect of miR-377 on migratory response of endothelial cells, we transfected HUVECs with miR-377 mimic or miR-377 inhibitor or respective negative controls and assessed their migration towards a well-known stimulant VEGF using modified Boyden chamber migration assay as described earlier (11). HUVECs transfected with miR-377 mimic demonstrated significantly lower migration in response to VEGF stimulation compared to control nonspecific transfection (p < 0.05) whereas HUVEC transfected with miR-377 inhibitor showed significantly higher migration in response to VEGF stimulation as compared to miR inhibitor negative control.(p < 0.05) (Figure 5A).
Figure 5. miR-377 mimic Inhibit the HUVEC Migration and Vascular Tube Formation.

(A) Migratory response of HUVECs transfected with miR-377 mimic or inhibitor or respective negative control toward VEGF gradient was measured by modified Boyden chamber migration assay. (B) Matrigel angiogenesis assay, tube formation by HUVEC transfected with miR-377 mimic or inhibitor or respective negative control, and relative quantification of tube length. *p < 0.05, miR-377 mimic versus miR mimic negative control; #p < 0.05, miR-377 inhibitor versus miR inhibitor negative control. HUVEC = human umbilical vein endothelial cell; VEGF = vascular endothelial growth factor; other abbreviations as in Figure 1.
Furthermore, we analyzed the effect of miR-377 on morphogenesis of HUVECs into vascular tubes. HUVECs transfected with miR-377 mimic showed significantly reduced ability and those transfected with miR-377 inhibitor displayed significantly increased ability to form vascular tubes compared to their respective negative controls (each p < 0.05) (Figure 5B).
STK35 3′UTR: Direct Target of miR-377
MiRs bind to 3′UTR of target messenger RNAs (mRNA) and regulate gene expression via post-transcriptional, translational repression or mRNA destabilization. To identify potential target genes involved in miR-377-mediated regulation, specifically those involved in angiogenic processes, we performed computational miR target prediction analysis using the miR databases and target prediction tools, TargetScan, miRBase, and PicTar. STK35, also known as CLIK1, STK35L1 and listed as one of the top potential targets for miR-377, contains complementary seed sequence in its 3′UTR that is highly conserved among human, chimpanzee, mouse, rat, guinea pig, rabbit, dog, cow, elephant, and horse (Figure 6A). Unlike other cytoplasmic kinases, STK35 is a novel kinase, mainly localized in the nucleus and nucleolus, that binds to nuclear actin (15); thus, it might play a critical role in directly modulating gene transcription machinery (16). A previous study showed that VEGF-stimulation in endothelial cells up-regulates STK35 expression and that STK35 knockdown leads to diminished angiogenic ability of endothelial cells (15).
Figure 6. Validation of STK35 as miR-377 Target.

(A) Computational miR target prediction analysis shows that serine/threonine kinase 35 (STK35) has complementary sequences for miR-377 that is highly conserved (white fonts) in most mammals. STK35 messenger RNA (B) and protein expression (C) were measured in the miR-377 mimic or inhibitor or respective controls transfected HUVEC by qRT-PCR and western blot. (D) Quantitative data for dual-luciferase reporter assay. n = 3; *p < 0.05, miR-377 mimic versus miR mimic negative control; #p < 0.05, miR-377 inhibitor versus miR inhibitor negative control. 3′UTR = three prime untranslated region; other abbreviations as in Figures 1 and 5.
We first examined the impact of miR-377 mimic and miR-377 inhibitor on STK35 mRNA and protein expression in HUVECs. The effect of miR-377 mimic or inhibitor transfection on miR-377 expression was confirmed by qRT-PCR in HUVECs and hCD34+ cells (Online Figures 3A and 3B). The effect of miR-377 on STK35 expression was validated by both qRT-PCR and western blot methods. Levels of STK35 mRNA and protein expression were decreased by miR-377 mimic and increased by miR-377 inhibitor compared to their respective negative controls (p < 0.05) (Figures 6B and 6C).
Next, to validate whether STK35 3′UTR is a direct target of miR-377, HUVECs were transfected with a dual-luciferase reporter vector containing the 3′-UTR of STK35 or mutated 3′-UTR of STK35 along with miR-377 mimic or miR-377 inhibitor or its respective negative controls. MiR-377 inhibitor significantly increased luciferase activity versus miR inhibitor negative control treated cells, while cells transfected with miR-377 mimic showed ∼40% decrease in luciferase activity compared to miR mimic negative control treated cells (both p < 0.05) (Figure 6D). No change was observed in mutated STK35-treated cells with both miR-377 mimic/inhibitor compared to their respective negative control transfections. These data indicate that STK35 is a direct target of miR-377.
Our in vitro data revealed that miR-377 has a negative influence on hCD34+ cell secretion of proangiogenic paracrine factors coupled with reduced endothelial migration and morphogenesis. To assess the effect of miR-377 inhibition on hCD34+ cells-mediated cardiac repair, we evaluated capillary density and fibrosis in the myocardium and LV function on day 28 after myocardial I-R injury followed by PKH26-labeled hCD34+ cells transplantation (Online Figure 4 showing transplanted cells in the myocardium). The mice receiving hCD34+ cells treated with anti-miR-377 negative control showed a trend towards increased capillary density versus sham-operated mice (p < 0.05) (Figures 7A, 7B, and 7D); mice receiving anti-miR-377 treated hCD34+ cells had a further increase in the number of capillaries compared to mice receiving hCD34+ cells treated with anti-miR negative control (p < 0.05) (Figures 7B through 7D).
Figure 7. Myocardium Capillary Density.
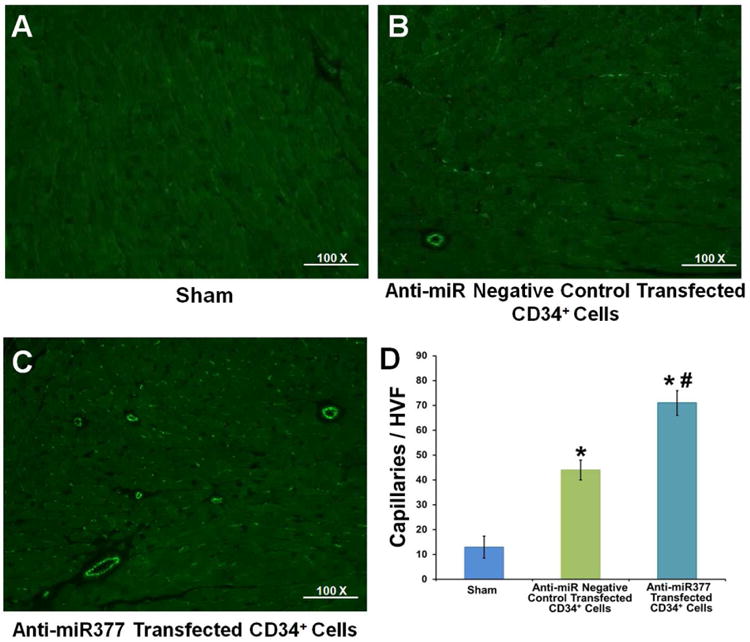
Representative immunofluorescence images taken within the infarct border zone of mice receiving (A) sham treatment or hCD34+ cells treated with (B) anti-miR negative control or (C) anti-miR-377. (D) Graph depicting capillary density across treatments presented as the number of CD31+ cells (n = 4; *p < 0.05, hCD34+ cells treated with anti-miR negative control or anti-miR377 vs. sham-operated group; #p < 0.05, hCD34+ cells treated with anti-miR377 vs. hCD34+ cells treated with anti-miR negative control group). Other abbreviations as in Figure 1.
Furthermore, we assessed the relevance of anti-miR-377 transfected hCD34+ cells on LV remodeling and functional recovery. Although not statistically significant, infarct size after 28 days post-I-R was reduced in mice receiving anti-miR-377 hCD34+ cells (Figures 8A, 8B, 8C, and 8D). Mice receiving anti-miR-377 treated hCD34+ cells showed significantly lower interstitial fibrosis in the myocardium compared to controls (p < 0.05) (Figures 8E, 8F, 8G, and 8H).
Figure 8. Cardiac Fibrosis and LV Functional Recovery after I-R.

Trichrome-stained heart sections from mice with sham operations (A, E) or transplanted with hCD34+ cells treated with anti-miR negative control (B,F) or hCD34+ cells treated with anti-miR-377 (C,G) (28 days post-I-R). Quantitative analysis of infarct size (D), fibrosis area (H), percent ejection fraction (%EF) (I), and percent fractional shortening (%FS) (J) was conducted 28 days post-I-R. n = 4; *p < 0.05, hCD34+ cells treated with anti-miR negative control versus sham-operated group; #p < 0.05, hCD34+ cells treated with anti-miR377 versus hCD34+ cells treated with anti-miR negative control. Abbreviations as in Figures 1 and 3.
Also, LV function analysis using echocardiography at 28 days post-I-R showed decreased LV function versus sham-operated mice (p < 0.05) (Figures 8I and 8J). Mice receiving anti-miR-377 treated hCD34+ cells demonstrated significant improvement in LV function with increased percent ejection fraction and fractional shortening compared to mice receiving negative control treated cells (p < 0.05) (Figures 8I and 8J). These data suggest that miR-377 inhibition in hCD34+ cells promotes their angiogenic ability in the myocardium and attenuates myocardial I-R-induced fibrosis and LV dysfunction.
Discussion
Current medical therapies have improved prognosis of patients with MI. Despite these advancements, significant mortality and increased risk for reoccurrence of HF remain in survivors (17). The underlying causes include microvasculature abnormalities due to endothelial dysfunction, loss of cardiomyocytes, and adverse remodeling due to fibrosis. Hence, developing therapeutics to preserve or regenerate damaged myocardial tissue might open novel avenues for treating patients with heart diseases. Accumulating evidence suggests that miRs are involved in several pathophysiological processes and their dysregulation has been implicated in cardiac diseases (18-20). Additionally, they are used as diagnostic or prognostic markers in various diseases (21). However, studies to determine the effect of cardiac miR (induced upon injury) on the stem/progenitor cells that are transplanted for tissue regeneration are limited. Therefore, therapeutic strategies to augment progenitor cell-based myocardial repair, especially in the context of tissue ischemia, might be clinically relevant and crucial to the success of cell-based therapy.
In the present study, we observed that patients with ischemic heart disease (IHD) have substantial upregulation of miR-377 in biopsy samples from myocardial tissue. In contrast, hCD34+ cells exposed to inflammatory stimuli for 12 h show significant decreases in miR-377 expression. Interestingly, miR-377 mimics' treatment diminished proangiogenic paracrine secretion in hCD34+ cells and reduced endothelial cells' tube formation ability. These data suggest that the decrease in miR-377 expression in response to inflammatory stimuli might be a compensatory mechanism to protect endothelial cell function.
Furthermore, reports have indicated that miR-377 has been negatively implicated in various pathological conditions, such as diabetic nephropathy (22), oxidative stress (23), lung fibrosis, hydrogen peroxide-induced premature senescence (24), and cancer (25-28). Our findings, for the first time, demonstrated that miR-377 critically regulates endothelial cell function in an inflammatory setting. Therefore, we speculated that strategies to modulate miR-377 to enhance hCD34+ cell-mediated myocardial repair might be suitable for therapeutic intervention in the setting of IHD. We demonstrated this by showing that transplantation of anti-miR-377-treated hCD34+ cells enhanced neovascularization, reduced interstitial fibrosis, and attenuated LV dysfunction. These in vivo changes corroborated well with the in vitro observations that miR-377 inhibitor treatment enhanced hCD34+ cells function. A recent study showed that miR-377 knockdown in mouse mesenchymal stem cells promoted their angiogenic ability in mouse MI models (29). Our data demonstrated that anti-miR-377 treatment enhances the angiogenic potential of hCD34+ cells, supporting Wen et al. (29); together demonstrating that miR-377 plays a negative role in IHD, in line with various other pathophysiological conditions.
We know miRs bind to target mRNAs through imperfect base pairing and are involved in post-transcriptional regulation of gene expression by affecting mRNA stability and translation. Based on computational analysis, we identified STK35 as a potential target gene. We observed that miR-377 mimic treatment in hCD34+ cells decreases proangiogenic paracrine factors in association with reduced STK35 expression. On the contrary, miR-377 inhibitor treatment had the opposite effect. More importantly, we validated that STK35 mRNA is a direct target of miR-377. These data, together with previous reports, suggest that the negative effects of miR-377 on hCD34+ cells might be potentially mediated through STK35 signaling.
Enhanced levels of STK35 have been reported in tissues obtained from colorectal cancer patients (30) and shown to be altered in a rodent model of Parkinson disease (31). Additionally, VEGF stimulates STK35 expression in endothelial cells (15). A well-known proangiogenic cytokine, VEGF promotes EPC migration, differentiation, and angiogenic function. Our data showed that VEGF treatment in HUVECs significantly decreased miR-377 expression; furthermore, VEGF mRNA expression decreased with miR-377 mimic treatment in HUVECs (Online Figure 5). Computational analysis revealed that VEGF-A is yet another potential target of miR-377, as validated in a previous study on mesenchymal stem cells (29). Taken together, these data suggest that a negative feedback loop might exist in regulating miR-377 and VEGF-A expression. Kinases are of great interest because they serve as targets in cancer therapeutics. Therefore, strategies involving pharmacological, genetic, or miR-based manipulation to modulate STK35-mediated angiogenesis in cardiomyopathies or cancer might be clinically relevant.
Beyond STK35, miR-377 has other experimentally verified targets relevant to cardiovascular diseases, such as VEGF-A (29) and hemeoxygenase-1 (HO-1) (23), all shown to be expressed in endothelial (progenitor) cells. Wang and associates (22) also demonstrated that miR-377 plays a role in the pathogenesis of diabetic nephropathy through translational repression of target genes-superoxide dismutase (SOD) 1 and 2 and p21/Cdc42/Rac1-activated kinase 1 (22). Our study showed that miR-377 negatively regulated hCD34+ cell function in association with reduced STK35. Taken together, by regulating the expression of STK35, VEGF-A, HO-1, and SOD1/2, miR-377 has deleterious effects on endothelial cell biology and function, possibly through inflammation and oxidative stress.
Considering miRs' potential role in a multitude of human diseases (18-20,33-39), targeting miR-377 represents an exciting prospect for therapeutic applications to limit cardiac remodeling and enhance hCD34+ cell-based tissue repair post-MI, after stroke, or in other ischemic events, or to inhibit aberrant angiogenesis in cancer.
Limitations
More studies are required to determine the role of miR-377 in inflammation/oxidative stress under myocardial ischemic conditions. We showed that miR-377 expression was increased in various cardiac cell types isolated from mouse hearts after I-R injury. However, we did not explore its role in different cell types. Previous studies have shown the possibility of a pleotropic role of some miR in different cell types (32). Therefore, further work is warranted to clarify the role of miR-377 in progenitor cell mobilization, cardiomyocyte cell death, and function during HF, therefore uncovering potential therapeutic modalities to modulate fibrogenesis process, LV remodeling, and repair post-ischemic injury.
Conclusions
We demonstrated for the first time that: 1) miR-377, which increases in the myocardium of patients with IHD, negatively influences hCD34+ angiogenic paracrine factors in vitro; and 2) anti-miR-377 treatment in hCD34+ cells enhanced their function and attenuated LV dysfunction after intramyocardial transplantation in SCID mice with myocardial I-R injury. Thus, knockdown of miR-377 appears to be a feasible approach to protect against LV remodeling and might prove to be an attractive therapeutic strategy for cell-based therapy in patients with HF.
Supplementary Material
Online Figure 1. MiR-377 Expression in LPS Treated Mouse EPC and HUVECs.
MiR-377 expression in the control and LPS treated (A) mouse EPC and (B) HUVEC were validated by qRT-PCR (normalized to control U6, n=6, *P < 0.05).
Online Figure 2. Heat Map Showing Differentially Expressed MiRNAs in LPS Treated Mouse EPC.
EPCs were treated with LPS (25 ng/ml) for 12 hrs and miRNA expression was analysed using PCR-based miRNA microarray platform covering a total of 352 mouse miRNAs.
Online Figure 3. MiR-377 Expression in MiR-377 Mimic, Inhibitor or miR Mimic or Inhibitor Negative Controls Treated hCD34+ Cells and HUVECs.
(A) hCD34+ cells and (B) HUVECs were transfected with miR-377 mimic, inhibitor or miR mimic or inhibitor negative controls and were subject to qRT-PCR. Data was normalized to internal U6 control and expressed as fold change vs respective negative controls. n=6, (*P<0.05, miR-377 mimic vs miR mimic negative control; #P<0.05, miR-377 inhibitor vs miR inhibitor negative control).
Online Figure 4. Presence of Transplanted miR Mimic Negative Control and MiR-377 Knockdown hCD34+ Cells Labelled with PKH26 in the Mice Myocardium at 3 Day After IR.
(A, B) PKH26+ labelled hCD34+ cells after transplantation into the mice myocardium HCD34+ cells (PKH26 positive, red florescence) and DAPI (blue) for nuclear staining.
Online Figure 5. Effect of VEGF on MiR-377 Expression.
(A) HUVECs were treated with VEGF and miR-377 expression was determined by qRT-PCR (normalized to control U6, n=6). (B) HUVECs were transfected with miR-377 mimic or Inhibitor or miR mimic or inhibitor negative controls and VEGF mRNA level was determined by qRT-PCR. (n=6, ¶P<0.05, VFGF treated vs control untreated: *P<0.05, miR-377 mimic vs miR mimic negative control; #P<0.05, miR-377 inhibitor vs miR inhibitor negative control).
Central Illustration. MicroRNA-377 Knockdown Human CD34+ Cells Enhances Cardiac Function.

Perspectives.
Competency in Medical Knowledge
The adverse microenvironment of ischemic tissue detrimentally affects the survival of mobilized/transplanted hCD34+ cells, which could compromise their full benefits in cell-based therapy.
Translational Outlook
Therefore developing therapeutic strategies to preserve stem cell function or regenerate damaged myocardial tissue might open novel scientific avenues for treating patients with heart diseases. This study demonstrated that transplantation of miR-377 knockdown hCD34+ cells into the ischemia myocardium of mice promoted cardiac repair and attenuated LV dysfunction.
Acknowledgments
This work was supported, in part, by the National Institutes of Health (NIH) grants 1R01HL116729 (to P.K.); HL091983, HL053354, HL108795 and HL108806 (to R.K.), American Heart Association Grant-in-aid GRNT 25860041 (to P.K.), American Heart Association Postdoctoral Fellowship 15POST25710392 (to R.A.T).
Abbreviations and Acronyms
- EPC
endothelial progenitor cell
- HF
heart failure
- HUVEC
human umbilical vein endothelial cell
- I-R
ischemia-reperfusion
- MI
myocardial infarction
- miR
micro ribonucleic acid
Footnotes
All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–e322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 2.Huebener P, Abou-Khamis T, Zymek P, et al. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J Immunol. 2008;180:2625–33. doi: 10.4049/jimmunol.180.4.2625. [DOI] [PubMed] [Google Scholar]
- 3.Schachinger V, Erbs S, Elsasser A, et al. Improved clinical outcome after intracoronary administration of bone-marrow-derived progenitor cells in acute myocardial infarction: final 1-year results of the REPAIR-AMI trial. Eur Heart J. 2006;27:2775–83. doi: 10.1093/eurheartj/ehl388. [DOI] [PubMed] [Google Scholar]
- 4.Losordo DW, Henry TD, Davidson C, et al. Intramyocardial, autologous CD34+ cell therapy for refractory angina. Circ Res. 2011;109:428–36. doi: 10.1161/CIRCRESAHA.111.245993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinnaird T, Stabile E, Burnett MS, et al. Local delivery of marrow-derived stromal cells augments collateral perfusion through paracrine mechanisms. Circulation. 2004;109:1543–9. doi: 10.1161/01.CIR.0000124062.31102.57. [DOI] [PubMed] [Google Scholar]
- 6.Joladarashi D, Thandavarayan RA, Babu SS, Krishnamurthy P. Small engine, big power: microRNAs as regulators of cardiac diseases and regeneration. Int J Mol Sci. 2014;15:15891–911. doi: 10.3390/ijms150915891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Rooij E, Sutherland LB, Thatcher JE, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105:13027–32. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bostjancic E, Zidar N, Stajer D, Glavac D. MicroRNAs miR-1, miR-133a, miR-133b and miR-208 are dysregulated in human myocardial infarction. Cardiology. 2010;115:163–9. doi: 10.1159/000268088. [DOI] [PubMed] [Google Scholar]
- 9.Krishnamurthy P, Thal M, Verma S, et al. Interleukin-10 deficiency impairs bone marrow-derived endothelial progenitor cell survival and function in ischemic myocardium. Circ Res. 2011;109:1280–9. doi: 10.1161/CIRCRESAHA.111.248369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song J, Zhang XQ, Wang J, et al. Regulation of cardiac myocyte contractility by phospholemman: Na+/Ca2+ exchange versus Na+ -K+ -ATPase. Am J Physiol Heart Circ Physiol. 2008;295:H1615–25. doi: 10.1152/ajpheart.00287.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ Res. 2009;104:e9–18. doi: 10.1161/CIRCRESAHA.108.188243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krishnamurthy P, Lambers E, Verma S, et al. Myocardial knockdown of mRNA-stabilizing protein HuR attenuates post-MI inflammatory response and left ventricular dysfunction in IL-10-null mice. FASEB J. 2010;24:2484–94. doi: 10.1096/fj.09-149815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mackie AR, Losordo DW. CD34-positive stem cells: in the treatment of heart and vascular disease in human beings. Tex Heart Inst J. 2011;38:474–85. [PMC free article] [PubMed] [Google Scholar]
- 14.Caiado F, Dias S. Endothelial progenitor cells and integrins: adhesive needs. Fibrogenesis Tissue Repair. 2012;5:4. doi: 10.1186/1755-1536-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goyal P, Behring A, Kumar A, Siess W. STK35L1 associates with nuclear actin and regulates cell cycle and migration of endothelial cells. PloS One. 2011;6:e16249. doi: 10.1371/journal.pone.0016249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grummt I. Actin and myosin as transcription factors. Curr Opin Genet Dev. 2006;16:191–6. doi: 10.1016/j.gde.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 17.Braunwald E. Heart failure. JACC Heart Fail. 2013;1:1–20. doi: 10.1016/j.jchf.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Dorn GW., 2nd MicroRNAs in cardiac disease. Transl Res. 2011;157:226–35. doi: 10.1016/j.trsl.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sessa WC. MicroRNA regulation of cardiovascular functions. Arterioscler Thromb Vasc Biol. 2011;31:2369. doi: 10.1161/ATVBAHA.111.238311. [DOI] [PubMed] [Google Scholar]
- 20.van Rooij E, Sutherland LB, Liu N, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006;103:18255–60. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roncarati R, Viviani Anselmi C, Losi MA, et al. Circulating miR-29a, among other up-regulated microRNAs, is the only biomarker for both hypertrophy and fibrosis in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2014;63:920–7. doi: 10.1016/j.jacc.2013.09.041. [DOI] [PubMed] [Google Scholar]
- 22.Wang Q, Wang Y, Minto AW, et al. MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB J. 2008;22:4126–35. doi: 10.1096/fj.08-112326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beckman JD, Chen C, Nguyen J, et al. Regulation of heme oxygenase-1 protein expression by miR-377 in combination with miR-217. J Biol Chem. 2011;286:3194–202. doi: 10.1074/jbc.M110.148726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maes OC, Sarojini H, Wang E. Stepwise up-regulation of microRNA expression levels from replicating to reversible and irreversible growth arrest states in WI-38 human fibroblasts. J Cell Physiol. 2009;221:109–19. doi: 10.1002/jcp.21834. [DOI] [PubMed] [Google Scholar]
- 25.Zhang R, Luo H, Wang S, et al. MicroRNA-377 inhibited proliferation and invasion of human glioblastoma cells by directly targeting specificity protein 1. Neuro Oncol. 2014;16:1510–22. doi: 10.1093/neuonc/nou111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lowery AJ, Miller N, Devaney A, et al. MicroRNA signatures predict oestrogen receptor, progesterone receptor and HER2/neu receptor status in breast cancer. Breast Cancer Res. 2009;11:R27. doi: 10.1186/bcr2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melkamu T, Zhang X, Tan J, Zeng Y, Kassie F. Alteration of microRNA expression in vinyl carbamate-induced mouse lung tumors and modulation by the chemopreventive agent indole-3-carbinol. Carcinogenesis. 2010;31:252–8. doi: 10.1093/carcin/bgp208. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Volinia S, Bonome T, et al. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer. Proc Natl Acad Sci U S A. 2008;105:7004–9. doi: 10.1073/pnas.0801615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wen Z, Huang W, Feng Y, et al. MicroRNA-377 regulates mesenchymal stem cell-induced angiogenesis in ischemic hearts by targeting VEGF. PloS One. 2014;9:e104666. doi: 10.1371/journal.pone.0104666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Capra M, Nuciforo PG, Confalonieri S, et al. Frequent alterations in the expression of serine/threonine kinases in human cancers. Cancer Res. 2006;66:8147–54. doi: 10.1158/0008-5472.CAN-05-3489. [DOI] [PubMed] [Google Scholar]
- 31.Hourani M, Berretta R, Mendes A, Moscato P. Genetic signatures for a rodent model of Parkinson's disease using combinatorial optimization methods. Methods Mol Biol. 2008;453:379–92. doi: 10.1007/978-1-60327-429-6_20. [DOI] [PubMed] [Google Scholar]
- 32.Kumarswamy R, Thum T. Non-coding RNAs in cardiac remodeling and heart failure. Circ Res. 2013;113:676–89. doi: 10.1161/CIRCRESAHA.113.300226. [DOI] [PubMed] [Google Scholar]
- 33.Han M, Yang Z, Sayed D, et al. GATA4 expression is primarily regulated via a miR-26b-dependent post-transcriptional mechanism during cardiac hypertrophy. Cardiovasc Res. 2012;93:645–54. doi: 10.1093/cvr/cvs001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hullinger TG, Montgomery RL, Seto AG, et al. Inhibition of miR-15 Protects Against Cardiac Ischemic Injury. Circ Res. 2012;110:71–81. doi: 10.1161/CIRCRESAHA.111.244442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porrello ER, Johnson BA, Aurora AB, et al. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ Res. 2011;109:670–9. doi: 10.1161/CIRCRESAHA.111.248880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med. 2011;208:549–60. doi: 10.1084/jem.20101547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suarez Y, Sessa WC. MicroRNAs as novel regulators of angiogenesis. Circ Res. 2009;104:442–54. doi: 10.1161/CIRCRESAHA.108.191270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Y, Ago T, Zhai P, et al. Thioredoxin 1 negatively regulates angiotensin II-induced cardiac hypertrophy through upregulation of miR-98/let-7. Circ Res. 2011;108:305–13. doi: 10.1161/CIRCRESAHA.110.228437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao Y, Srivastava D. A developmental view of microRNA function. Trends Biochem Sci. 2007;32:189–97. doi: 10.1016/j.tibs.2007.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Figure 1. MiR-377 Expression in LPS Treated Mouse EPC and HUVECs.
MiR-377 expression in the control and LPS treated (A) mouse EPC and (B) HUVEC were validated by qRT-PCR (normalized to control U6, n=6, *P < 0.05).
Online Figure 2. Heat Map Showing Differentially Expressed MiRNAs in LPS Treated Mouse EPC.
EPCs were treated with LPS (25 ng/ml) for 12 hrs and miRNA expression was analysed using PCR-based miRNA microarray platform covering a total of 352 mouse miRNAs.
Online Figure 3. MiR-377 Expression in MiR-377 Mimic, Inhibitor or miR Mimic or Inhibitor Negative Controls Treated hCD34+ Cells and HUVECs.
(A) hCD34+ cells and (B) HUVECs were transfected with miR-377 mimic, inhibitor or miR mimic or inhibitor negative controls and were subject to qRT-PCR. Data was normalized to internal U6 control and expressed as fold change vs respective negative controls. n=6, (*P<0.05, miR-377 mimic vs miR mimic negative control; #P<0.05, miR-377 inhibitor vs miR inhibitor negative control).
Online Figure 4. Presence of Transplanted miR Mimic Negative Control and MiR-377 Knockdown hCD34+ Cells Labelled with PKH26 in the Mice Myocardium at 3 Day After IR.
(A, B) PKH26+ labelled hCD34+ cells after transplantation into the mice myocardium HCD34+ cells (PKH26 positive, red florescence) and DAPI (blue) for nuclear staining.
Online Figure 5. Effect of VEGF on MiR-377 Expression.
(A) HUVECs were treated with VEGF and miR-377 expression was determined by qRT-PCR (normalized to control U6, n=6). (B) HUVECs were transfected with miR-377 mimic or Inhibitor or miR mimic or inhibitor negative controls and VEGF mRNA level was determined by qRT-PCR. (n=6, ¶P<0.05, VFGF treated vs control untreated: *P<0.05, miR-377 mimic vs miR mimic negative control; #P<0.05, miR-377 inhibitor vs miR inhibitor negative control).