

Summary
Pulmonary hypertension (PH) is a deadly vascular disease with enigmatic molecular origins. We found that vascular extracellular matrix (ECM) remodeling and stiffening are early and pervasive processes that promote PH. In multiple pulmonary vascular cell types, such ECM stiffening induced the microRNA-130/301 family via activation of co-transcription factors YAP/TAZ. MicroRNA-130/301 controlled a PPARγ-APOE-LRP8 axis, promoting collagen deposition and LOX-dependent remodeling and further up-regulating YAP/TAZ via a mechanoactive feedback loop. In turn, ECM remodeling controlled pulmonary vascular cell crosstalk via such mechanotransduction, modulation of secreted vasoactive effectors, and regulation of associated microRNA pathways. In vivo, pharmacologic inhibition of microRNA-130/301, APOE, or LOX activity ameliorated ECM remodeling and PH. Thus, ECM remodeling, as controlled by the YAP/TAZ-miR-130/301 feedback circuit, is an early PH trigger and offers combinatorial therapeutic targets for this devastating disease.
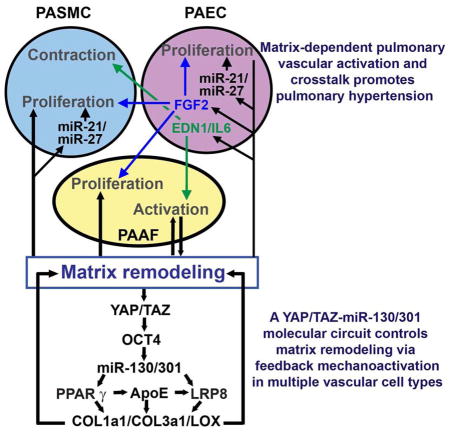
Graphical Abstract
Introduction
Pulmonary hypertension (PH) is a deadly vascular disease of increasing prevalence worldwide (Chan and Loscalzo, 2008). PH can be induced by myriad triggers, and a growing number of molecular pathways as well as pathogenic crosstalk between pulmonary vascular cell types have been described to influence PH. A majority of the molecular targets chosen for clinical testing primarily affect end-stage disease phenotypes (Boutet et al., 2008; Stenmark and Rabinovitch, 2010). However, they fail to target the enigmatic origins active at early disease time points and thus neither reverse nor prevent PH.
Aberrant collagen and elastin expression (Mecham et al., 1987; Poiani et al., 1990) in the vascular extracellular matrix (ECM) at end-stage PH has long been recognized. Vascular stiffness in the proximal and distal pulmonary arterial tree occurs in various forms of PH (Lammers et al., 2012; Wang and Chesler, 2011), and stiffness is an index of disease progression (Gan et al., 2007). Discrete pharmacologic manipulation of vascular ECM can ameliorate disease (Cowan et al., 2000; Kerr et al., 1984; Kerr et al., 1987; Nave et al., 2014). Yet, deeper molecular insights into the causative relationship between vascular ECM remodeling and PH are only just emerging, both at earlier time points of disease and end-stage. In general, ECM remodeling is a complex process, occurring through changes in the balance between collagen and elastin deposition, matrix degradation, and matrix remodeling via collagen crosslinking enzymes such as lysyl oxidase (LOX) (Schafer and Werner, 2008). In that context of matrix remodeling, two related transcriptional coactivators, YAP (Yes Associated Protein 1) and TAZ (Transcriptional Coactivator with PDZ-Binding Motif), are crucial for mechanotransduction, a process that converts extracellular mechanical cues into intracellular signaling (Dupont et al., 2011; Piccolo et al., 2014) and is known to regulate cellular proliferation, survival, polarity, organ size, and the ECM, particularly in development and cancer progression. More recently, YAP/TAZ have been implicated in vascular development (Zhou, 2014) and pulmonary parenchymal fibrosis (Liu et al., 2015). However, key concepts remain undefined: 1) the mechanosensitive pathways that regulate ECM remodeling particularly in the pulmonary vasculature and 2) their exact relation to PH.
MicroRNAs (miRNAs) are essential mediators of multiple cellular processes involving cell-cell and cell-matrix interactions (Valastyan and Weinberg, 2011). Yet, we have little prior knowledge regarding the biomechanical effects of ECM on any non-coding RNAs, beyond special contexts of cancer (Mouw et al., 2014). Furthermore, crosstalk among miRNAs, the ECM, and downstream pulmonary vascular phenotypes has been largely unexplored. Previously, we described the proliferative and vasoconstrictive actions of miR-130/301 in PH (Bertero et al., 2014a; Bertero et al., 2014b). Yet, beyond these functions, we have now found a prominent component of their related gene targets associated with ECM biology. Therefore, in this study, we interrogated whether arteriolar ECM modification is an early driver of PH progression, regulated by a complex network of mechanosensitive factors involving miR-130/301.
Results
PH is characterized by a programmatic shift in fibrotic gene expression with early and sustained arteriolar collagen remodeling
To characterize initially the relevance of ECM biology in PH, we performed a transcriptomic analysis of PH lung tissue from mice (chronic hypoxia with administration of the VEGF receptor antagonist SU5416) (Ciuclan et al., 2011). RNA-sequencing coupled with pathway enrichment revealed dysregulated genes involved in ECM plasticity specifically (index pathway #4 ECM organization, #7 ECM receptor interaction; Table S1) as well as pathways indirectly associated with ECM stiffening and the collagen crosslinking enzyme LOX (index #1 and #18; Table S1). As a result, such mice displayed increased pulmonary collagen content and fibrillar collagen, as reflected by in situ Picrosirius Red stain (Fig. 1A), collagen isoform and LOX transcript analysis (Fig. S1A), Lox-dependent collagen crosslinking activity (Fig. S1B), and biochemical analysis of pulmonary collagen content (Fig. 1B). In correlation, atomic force microscopy revealed an increase in pulmonary arteriolar stiffness in PH mice (Fig. 1C). In a separate animal model, PH induced by monocrotaline exposure in rats led to similar increases in pulmonary vascular collagen content (total and fibrillar) (Fig. 1D–E; Fig. S1C–D). Of note, Lox expression was increased throughout the PH arteriolar wall, evident in the intimal, medial, and adventitial layers (Fig. 1F) and generally consistent with the location of fibrillar collagen (Fig. 1D, particularly D21 of disease). Moreover, in monocrotaline-induced PH in rats, the early phases of PH development (3 days post-monocrotaline exposure) were characterized by increased arteriolar fibrillar collagen (Fig. 1D–E) and Lox activity (Fig. S1D) prior to hemodynamic PH manifestation (as assessed by right ventricular systolic pressure, RVSP) (Fig. S1E) or increase of medial thickening (Fig. S1F). To prove the relevance of these findings across various forms of PH, six additional animal models of hemodynamically confirmed PH (RVSP measurements, data not shown) also displayed evidence of alterations in pulmonary vascular ECM remodeling -- including hypoxia-driven models [mice exposed to chronic hypoxia alone (Fig. S1A–B), rats exposed to chronic hypoxia + SU5416 (Fig. S1G), and VHL-null mice (Fig. S1H–I)]; inflammatory-driven models (mice expressing transgenic IL-6 (Fig. S1J–K) and Schistosoma mansoni–infected mice [Fig. S1L–M]); and a surgical lamb model of congenital heart disease (Fig. S1N). Finally, similar vascular ECM remodeling and Lox up-regulation were observed in human pulmonary arterial hypertension (PAH) tissue [cohorts described in (Bertero et al., 2014b), Fig. 1G–H]. Together, these results demonstrate that arteriolar ECM stiffening is an early and pervasive process in PH and is associated with a programmatic shift in a network of ECM-related genes.
Figure 1. Pulmonary arteriolar ECM remodeling and stiffening is an early hallmark of PH.

A–C) Mice were exposed to hypoxia +/− SU5416 (10% O2 for 3 weeks) in order to induce PH. A) Picrosirius Red stain of mouse lung tissues was imaged in parallel light to display total collagen content (top) or orthogonal light to display fibrillar collagen (bottom) (<100μm vessel diameter; 10 vessels/animal). B) Increased total collagen and fibrillar collagen (decreased Soluble/Insoluble ratio) in PH lung were demonstrated by Sircol assay. C) Atomic force microscopy revealed increased pulmonary arteriolar (<50 μm) stiffness in PH lung (n=3 mice) versus untreated (n=4 mice); horizontal lines denotes median; symbols denote individual PA measurements. P-value calculated by Mann Whitney U testing. D–F) Monocrotaline was administered to rats (3 weeks, n=6–8/time point) to induce PH. Picrosirius Red stain (D) and Sircol assay (E) revealed increased arteriolar fibrillar collagen (starting at D3) evident prior to hemodynamic disease (see Fig. S1K). Increased Lox in intimal and media of rat PH pulmonary arterioles was demonstrated by immunofluorescence (F). G) By Picrosirius Red stain of human lung, quantification of < 200μm vessels (10 vessels/patient) revealed increased collagen deposition and fibrillar collagen expression in PAH. H) Lox was increased in intima and media of human PAH lung arterioles, shown by immunofluorescence. Data are expressed as mean ± SEM (*P<0.05; ** P<0.01, *** P<0.001, **** P<0.0001). Scale bars 50 μm. See also Fig. S1.
ECM stiffness is a mechanical stimulus for miR-130/301 family expression via YAP/TAZ signaling
Among the predicted pool of target genes for the PH-associated miR-130/301 family (miR-130a/b; miR-301a/b, and miR-454), we observed a broad component of factors related to ECM remodeling (Fig. 2A, encircled genes). To delineate further the connections among this miRNA family, the ECM, and PH, we constructed in silico a “fibrosis network” based on curated seed genes known to be causatively involved in ECM remodeling (Table S2) and their first degree interactors (Table S3). The final “fibrosis network” (Fig. 2A, Table S3) displayed substantial identity (Fig. 2A, encircled and enlarged genes) with the similarly curated and previously described PH gene network (Bertero et al., 2014b). Furthermore, miR-130/301 members were highly ranked (#5 among all conserved miRNAs in TargetScan 6.2) by “miRNA spanning score” (based on number and architectural distribution of predicted miRNA targets, see Experimental Procedures), thus indicating their control over multiple pathways in the fibrosis network (Fig. 2A, Table S4). Thus, as predicted by network analysis, the miR-130/301 family carries both overlapping and systems-wide effects in both the PH and fibrosis networks.
Figure 2. ECM stiffening induces miR-130/301 for downstream modulation of collagen deposition and remodeling through a PPARγ-ApoE-LRP8 axis.

A) A fibrosis network, composed of known fibrotic genes and their closest first degree interactors (left, Tables S2–S3) shares a large portion of its members with a PH disease network [right, as previously described [(Bertero et al., 2014b)]. Color-coding denotes architectural network clusters. Enlarged nodes are shared by both networks (70 nodes), and encircled genes are miR-130/301 direct targets (per Targetscan 6.2), thus highlighting a prominent fibrotic component among the miR-130/301 targets. miR-130/301 was ranked among the top five miRNA by “spanning score” (Bertero et al., 2014b) in both network contexts, reflecting the functional overlap and this miRNA family’s shared, systems-level control over both networks. B–C) miR-130/301 expression was quantified in human PAAFs cultured in hydrogel of varying stiffness (B) and transfected with siRNAs (YAP/TAZ versus si-NC control) (C). D) miR-130/301 was increased in human PAAFs overexpressing YAP (pYAP) versus a control vector (pGFP). E) ChIP-qPCR confirmed the presence of OCT4 binding sites (P1-4) in distinct miR-130/301 family member promoter regions near the transcription start site (proximal promoter 1Kb). Results are expressed as percent of total input DNA prior to IP. F–G) PAAFs were transfected with miR-NC, miR-130a, tiny-LNA-NC or tiny-LNA-130 and cultivated in soft or stiff matrix. Forced miR-130a expression or stiff matrix increased collagen transcripts, LOX, and CTGF, a marker of ECM stiffening and fibrosis (F). In high ECM stiffness, tiny-LNA-130 decreased this fibrotic gene cohort (G). H) By RT-qPCR in PAAFs, constitutive PPARγ (pPPARγ) reversed the miR-130a-induced up-regulation of collagen and LOX. I) By immunoblot, miR-130a, matrix stiffening, and PPARγ knockdown decreased ApoE while inhibition of miR-130/301 (tiny-LNA-130) increased ApoE in stiff conditions. J) PAAFs exposed to exogenous ApoE were resistant to miR-130a-induced up-regulation of collagen, CTGF, and LOX. K) Lox activity was measured in PAAFs cultured in soft or stiff matrix and treated with miR-130a +/− exogenous ApoE or miR-130/301 inhibitor (tiny-LNA-130). L) By immunoblot, LRP8 was reduced by miR-130a or matrix stiffening and preserved by miR-130/301 inhibition. M) siRNA knockdown of both LRP8 and PPARγ, increased collagen and CTGF. In all panels, mean expression in control groups (0.2 KPa, miR-NC, Tiny-LNA-NC or si-NC in soft matrix) was assigned a fold change of 1, to which relevant samples were compared. Data are expressed as mean ± SD (*P<0.05; ** P<0.01). See also Fig. S2–S3.
To begin to evaluate the connection of miR-130/301 to the ECM, we found that miR-130/301 members were up-regulated in pulmonary arterial adventitial fibroblasts (PAAFs) after culture in hydrogels of increasing stiffness (Fig. 2B). Stiff matrix also up-regulated YAP in PAAFs (Fig. S2A) without concomitant alterations of total or phosphorylated forms of the LATS or MST1/2 kinases, factors previously linked to YAP activation in some mechanosensitive contexts (Piccolo et al., 2014). Nonetheless, given the known link between mechanotransduction and YAP/TAZ (Dupont et al., 2011), we postulated that YAP and TAZ may control the downstream miR-130/301 mechanosensitive response. Only siRNA knockdown of YAP and TAZ together (Fig. 2C), but not separately (Fig. S2B–D), reversed miR-130/301 up-regulation, indicating the dependence upon coordinated activation of YAP/TAZ. Conversely, forced expression of YAP in PAAFs was sufficient to induce miR-130/301 (Fig. 2D). No YAP/TAZ-specific TEAD binding site was predicted in the promoter regions of these miRNAs. Instead, we postulated that YAP/TAZ may induce miR-130/301 via increase of the transcription factor POU5F1/OCT4, a target of YAP/TAZ (Lian et al., 2010) and a factor that up-regulates miR-130/301 in hypoxia (Bertero et al., 2014b). To address definitively the mechanism of regulation of miR-130/301 by POU5F1/OCT4, ChIP-qPCR demonstrated active OCT4 binding sites in distinct promoter regions of the miR-130/301 genomic loci (Fig. 2E). In correlation, POU5F1/OCT4 was induced by matrix stiffness in PAAFs and reversed by YAP/TAZ knockdown (Fig. S2E). Additionally, knockdown of POU5F1/OCT4 (Fig. S2F) reversed miR-130/301 mechanoinduction (Fig. S2G) as well as reversed induction of miR-130/301 by YAP overexpression (data not shown). Similar findings were observed in other pulmonary vascular cell types (Fig. S2H–M). Together, these results indicate that, across cell types, coordinated up-regulation of the miR-130/301 family by mechanical ECM remodeling is mediated by a unique YAP/TAZ- and POU5F1/OCT4-dependent pathway.
The miR-130/301 family controls a cohort of factors relevant to fibrosis in the lung
Downstream of YAP/TAZ, we examined the consequent effects of miR-130/301 on known fibrotic factors within the lung as well as biochemical properties of the ECM. In cultured PAAFs, either forced miR-130a expression or high ECM stiffness increased transcripts encoding fibrillar collagen isoforms, LOX, and CTGF, a direct target of YAP/TAZ (Dupont et al., 2011) and marker of ECM stiffening and fibrosis (Fig. 2F), as well as increased collagen production (Fig. S3A). Similar findings were observed in other pulmonary vascular cell types (Fig. S3B–D). Conversely, in high ECM stiffness, short locked nucleic acid inhibitory oligonucleotides (“tiny-LNAs”) with antisense complementarity to the miR-130/301 seed sequence (tiny-LNA-130) decreased transcript expression of this fibrotic gene cohort (Fig. 2G) and decreased collagen production (Fig. S3A). Thus, these results define miR-130/301 as a broad molecular regulator of ECM modification.
miR-130/301 controls collagen deposition and remodeling via the PPARγ-APOE-LRP8 axis
To elucidate molecular factors facilitating collagen deposition and remodeling downstream of miR-130/301, the peroxisome proliferator-activated receptor gamma (PPARγ) was studied – a known target of miR-130/301 in control of cellular proliferation (Bertero et al., 2014b), with a known role in ECM modification in other contexts (Wei et al., 2012). Constitutive PPARγ disrupted the miR-130/301-mediated up-regulation of collagen and LOX in PAAFs (Fig. 2H). Notably, miR-130a overexpression or PPARγ knockdown (Fig. 2I) decreased apolipoprotein E (ApoE), a direct target of PPARγ (Hansmann et al., 2008) involved in ECM remodeling (Kothapalli et al., 2012). In contrast, miR-130/301 inhibition in stiff ECM increased ApoE (Fig. 2I), and PAAFs exposed to media enriched with ApoE were similarly resistant to miR-130/301 activity (Fig. 2J–K). Downstream of PPARγ and ApoE, PPARγ knockdown alone in soft conditions increased the aforementioned cohort of fibrotic genes (Fig. S3E). Conversely, constitutive PPARγ expression (pPPARγ) or ApoE treatment disrupted the miR-130/301-mediated up-regulation of collagen production (Fig. S3F) and the miR-130/301-mediated up-regulation of Lox activity (Fig. 2K). Of note, the regulation of Lox activity by miR-130/301 corresponded with consistent alterations of expression of cleaved, activated Lox in these cultured PAAFs. (data not shown). Thus, the miR-130/301 family critically relies on PPARγ and ApoE to control collagen deposition and remodeling.
Given the role of ApoE and a predicted miR-130/301 binding site in the ApoE receptor LRP8 transcript [TargetScan 6.2 (Friedman et al., 2009)], we considered that LRP8 may also function prominently here. Via luciferase reporter assay, we confirmed LRP8 as a direct miR-130/301 target (Fig. S3G). In cultured PAAFs, miR-130a decreased LRP8 expression, while inhibition of miR-130/301 increased LRP8 (Fig. 2L). As with PPARγ knockdown, LRP8 knockdown increased collagen expression and Lox activity (Fig. S3H–J). Importantly, knockdown of PPARγ and LRP8 together induced a more robust response than either knockdown alone, indicating their coordinated roles in controlling collagen expression (Fig. 2M and Fig. S3I–J).
Finally, we postulated that miR-130/301 may be central to a feedback loop amplifying fibroblast activation and ECM remodeling in PH. PAAFs were transfected with miR-130a mimic oligonucleotides or siRNAs for PPARγ and LRP8 or infected with a lentivirus carrying a YAP transgene (pYAP) and cultured in soft matrix to remodel the ECM. Cells were then removed from this ECM and replaced with non-transfected, naive PAAFs (Fig. 3A; analyses of remodeled ECM in Fig. 3B–C and Fig. S4A). After 16 hours, ECM remodeled by miR-130a or YAP promoted YAP nuclear localization (Fig. 3D–E and Fig. S4B) and increased POU5F1/OCT4 (Fig. S4C), miR-130/301 (Fig. 3F–H), and downstream fibrosis-relevant genes (Fig. S4C–D) in naive PAAFs. Conversely, when ECM remodeling was inhibited by miR-130/301 or YAP/TAZ knockdown, a decrease was observed in YAP nuclear localization (Fig. 3D–E), POU5F1/OCT4 (Fig. S4C, E), miR-130/301 (Fig. 3F–G), and the fibrosis gene cohort (Fig. S4E). Importantly, the ECM modifications activating downstream fibrosis-relevant genes and miR-130/301 were reversed by ApoE (Fig. 3B, D, I and Fig. S4F). Taken together, these results reveal that miR-130/301 and YAP/TAZ act in a feedback-driven, self-amplifying regulatory loop that integrates multiple direct target genes including PPARγ and LRP8 in order to regulate coordinately ECM remodeling and stiffening (see Graphical Abstract).
Figure 3. A YAP/TAZ-miR-130/301 feedback loop promotes ECM modification to induce dysregulated pulmonary vascular crosstalk.

A) Schema of the experimental procedure. B) ECM staining by Picrosirius Red revealed that miR-130a increased fibrillar collagen, while miR-130/301 inhibition (tiny-LNA-130) or ApoE reversed such effects. C) Similarly, YAP (pYAP) increased fibrillar collagen as compared with control (pGFP), while knockdown of YAP/TAZ (siYAP/TAZ) decreased fibrillar collagen. D–E) In that context, immunofluorescence for YAP (red) and nuclei (DAPI, blue) was performed on naïve PAAFs plated on ECM remodeled by the indicated conditions. Nuclear stain relative to cytosolic stain of YAP was quantified (n=3 experiments with ten 20x fields analyzed per experiment). F–H) RT-qPCR revealed that ECM remodeled by miR-130a (F) or by siPPARγ+LRP8 (G) or by YAP (H) up-regulated miR-130/301 in naïve cells plated on remodeled ECM. Conversely, ECM remodeled by tiny-LNA-130 (F) or by si-YAP/TAZ (G) down-regulated miR-130/301 in naïve cells. I) miR-130/301 induction in naïve cells cultured on matrix remodeled by miR-130a was reversed by ApoE. J–M) Direct cellular effects of miR-130a/YAP-dependent stiffness were demonstrated by increased proliferation (BrdU-staining) of PAECs (J), PASMCs (K), and PAAFs (L) when culturing in ECM remodeled by PAAFs overexpressing miR-130a or YAP. Such ECM also increased LOX activity in PAAFs (M). N–S) Pulmonary vascular cell crosstalk dependent on ECM remodeling was revealed by modulation of a panel of vasoactive genes in PAECs via stiff ECM (N). Conditioned media from PAECs cultured in such stiff matrix increased proliferation in naïve PAECs (O), PASMCs (P), and PAAFs (Q), and was reversed by a FGF2 blocking antibody (FGF2 Ab). Conditioned media from PAECs cultured in stiff ECM increased contraction of PASMCs (R) and PAAFs (S). Contraction was partially reversed by ambrisentan or an IL-6 antibody (IL6 Ab) alone, and more robustly reversed by their combination. In all panels, mean expression in control groups (miR-NC, Tiny-LNA-NC, or si-NC in soft matrix) was assigned a fold change of 1, to which relevant samples were compared. Data are expressed as mean ± SD (*P<0.05; ** P<0.01). See also Fig. S4.
miR-130/301-dependent ECM remodeling controls vascular cell proliferation and pulmonary vascular crosstalk
Next, we postulated that miR-130/301-dependent ECM remodeling controls specific downstream molecular functions and PH-relevant cellular phenotypes. ECM remodeled by forced expression of miR-130a or YAP in PAAFs increased proliferation of naive PAECs (Fig. 3J) and PASMCs (Fig. 3K). In naive PAAFs plated in these matrices, ECM modification also up-regulated proliferation (Fig. 3L) and Lox activity (Fig. 3M). Correlating with these findings, in naïve PAECs, PASMCs, and PAAFs (Fig. S4G–J), ECM remodeling induced the expression of other miRNAs, miR-21 and miR-27a – miRNAs which are active in controlling pulmonary vascular cell proliferation (Fig. S4K–M) and previously implicated in PH pathogenesis in vivo (Bi et al., 2015; White et al., 2012). Beyond direct control of proliferation, in PAECs, ECM stiffening induced a panel of secreted vasoactive effectors implicated in PH, including the pro-proliferative cytokine FGF2, the vasoconstrictor EDN1, and the inflammatory cytokine IL-6. ECM remodeling also down-regulated endothelial nitric oxide synthase NOS3 (Fig. 3N). Conditioned media from PAECs cultivated in stiff matrix activated the proliferation of naïve PAECs (Fig. 3O), PASMCs (Fig. 3P) and PAAFs (Fig. 3Q); such proliferation was partially reversed by a FGF2 blocking antibody. PAEC conditioned media also activated PASMC (Fig. 3R) and PAAF (Fig. 3S) contraction. Implicating END1 and IL-6 as causative factors, contraction was partially reversed by the endothelin receptor antagonist ambrisentan or an IL-6 blocking antibody alone and, to a better extent, by these agents together (Fig. 3R, S). Thus, the YAP/TAZ miR-130/301 circuit remodels ECM for control of associated miRNA-dependent cellular proliferation as well as pulmonary vascular cell crosstalk related to contractile response.
The YAP/TAZ-miR-130/301 circuit promotes matrix remodeling and PH in vivo
We wanted to determine whether the YAP/TAZ-miR-130/301 circuit is active in PH in vivo and correlates with markers of arteriolar ECM remodeling. As PH progressed in monocrotaline-exposed rats (Fig. 4A–F and Fig. S5), in situ arteriolar staining displayed a strong positive correlation among YAP, POU5F1/OCT4, and miR-130a expression. By immunofluorescence, miR-130a expression was also associated with increased proliferation (PCNA nuclear stain in multiple vessel layers but particularly in the outer layers, Fig. 4E) and increased Lox expression (staining throughout the vessel wall, Fig. 4F). Importantly, correlating with early and sustained ECM remodeling (Fig. 1D–E), staining of all markers was increased at every disease time point, including early D3. Similar increases in YAP and miR-130a were observed in human PH patients [as described in (Bertero et al., 2014b)] (Fig. 4G–H).
Figure 4. The YAP/TAZ-miR-130/301 molecular circuit is active in rodent and human examples of PH.

A–F) Monocrotaline was administered to rats (3 weeks, n=6–8/time point) to induce PH (see Fig. 1). In serial sections of pulmonary arterioles (A), quantification of stain intensity (B) and percentage of positively stained cells (C) revealed a correlation between miR-130a, Yap, and Oct4 expression (D). E–F) Immunofluorescence stain confirmed an increase of PCNA (E) and Lox (F) expression in miR-130a positive cells G–H) In serial sections of human pulmonary arterioles, a similar relationship was observed between increased miR-130a and YAP in PAH. Data are expressed as mean ± SD (*P<0.05; ** P<0.01). Scale bars 50 μm. See also Fig. S5.
To determine whether miR-130a alone promotes vascular matrix remodeling in vivo, miR-130a mimic oligonucleotides were delivered in mice via serial intrapharyngeal delivery. miR-130a reduced target gene expression (i.e., LRP8), increased collagen deposition and remodeling (Fig. 5A, B, D), and promoted Lox expression and activity (Fig. 5B–C). Notably, miR-130a exposure was also accompanied by vascular YAP nuclear localization and YAP-dependent gene expression (i.e., CTGF) (Fig. 5A–B), consistent with our in vitro evidence of a feedback loop between ECM remodeling and endogenous YAP/TAZ-miR-130/301 induction. These events ultimately increased proliferation in both endothelial and smooth muscle compartments (PCNA staining, Fig. 5E) and promoted PH (Fig. 5F–H), as reflected by RVSP, right ventricular remodeling (RV/LV+S ratio), and pulmonary arteriolar muscularization. Conversely, experiments were designed to prevent collagen remodeling via serial treatment with a pharmacologic inhibitor of LOX (Levental et al., 2009), β-aminopropionitrile (BAPN). In miR-130-exposed mice, inhibition of LOX by BAPN reversed indices of ECM remodeling (Fig. 5A–D). Consistent with an ECM-YAP feedback loop, BAPN also reversed YAP activation, blunted the ongoing effects of miR-130a on LRP8 repression and collagen deposition and remodeling (Fig. 5A–B), and ultimately reversed miR-130a-dependent proliferation (Fig. 5E) and downstream PH manifestations (Fig. 5F–H). Thus, miR-130a is sufficient to promote pulmonary vascular ECM remodeling in PH and is dependent upon a feedback loop via the ECM and YAP/TAZ.
Figure 5. miR-130a induces YAP/TAZ-miR-130/301 to promote pulmonary vascular ECM remodeling in a LOX-dependent manner.

Along with SU5416, mice received 4 weekly administrations of miR-NC or miR-130a and were treated either with daily BAPN or vehicle. A) In situ staining of mouse lung demonstrated that BAPN blunted miR-130a-specific induction of LRP8, ECM remodeling (Picrosirius Red), and medial thickening (α– SMA), thus leading to decreased YAP nuclear localization. B) By RT-qPCR, fibrillar collagen, Lox, and YAP-dependent gene expression (i.e., CTGF), were increased in miR-130a-diseased lung, but were blunted by BAPN. C–E) Lox activity (C), biochemical indices of collagen remodeling (D), and proliferation (PCNA staining in arteriolar CD31+ and α-SMA+ cells) (E) were increased in diseased lung, but blunted by BAPN. F–H) BAPN decreased the miR-130-mediated increase in PH severity, as quantified by RVSP (F), Fulton index (RV/LV+S) (G), and pulmonary arteriolar muscularization (H). Data are expressed as mean ± SEM (*P<0.05; ** P<0.01). Normalized values are expressed as arbitrary units (A.U.) in (A, C). Scale bars 50 μm.
To determine whether inhibition of miR-130/301 ameliorates vascular ECM remodeling in PH, hypoxia+SU5416-exposed mice and monocrotaline-exposed rats were examined (Fig. 6). Rodents were serially administered a “shortmer” oligonucleotide recognizing the miR-130/301 seed sequence (Short-130), thus inhibiting all miR-130/301 members in vivo, as demonstrated in mouse lung (Bertero et al., 2014b) and rat lung (Fig. S5). In monocrotaline-exposed rats, Short-130 reversed the decrease of the miR-130/301 targets Lrp8 and Pparγ in diseased control pulmonary arterioles (Short-NC) (Fig. 6A and Fig. S5). This was accompanied by consequent decreases in Lox expression (Fig. 6B, Fig. S5), Lox activity (Fig. 6C), and collagen deposition and fibrillar collagen expression (Fig. 6A, B, D). Again reflecting the ECM-YAP feedback loop, miR-130/301 inhibition and its effects on ECM remodeling also decreased YAP nuclear localization (Fig. 6A) and YAP activation (reflected by decreased Ctgf, Fig. 6B). Together, these effects culminated in reduced cellular proliferation (decreased PCNA in intimal and medial vessel layers, Fig. 6E) and decreased hemodynamic and histologic severity of PH (Fig. 6F–G). Similarly, in hypoxia+SU5416-exposed mice where PH is known to improve with Short-130 treatment (Bertero et al., 2014b), miR-130/301 inhibition reversed collagen deposition and fibrillar collagen remodeling (Fig. 6H) and blunted hypoxia-induced vascular stiffening, as demonstrated by atomic force microscopy (Fig. 6I). Furthermore, although whole lung transcriptomics likely captured only a subset of the miR-130/301 targets affecting the diseased pulmonary vasculature, such analyses of mouse lung with hypoxia+SU5416-induced PH revealed a generalized de-repression of miR-130/301 targets by Short-130 (data not shown) and a generalized de-repression of hypoxia induced genes (Table S5), many of which overlap with known genes in the fibrosis network and ECM plasticity, in general. Pathway enrichment of genes revealed pronounced representation of several pathways known to be involved in fibrosis (Fig. 6J, Table S6). Thus, rather than relying on one single gene target or pathway, the miR-130/301 family induces a programmatic molecular shift toward the fibrotic pathophenotype in vivo. In sum, miR-130/301 is both necessary and sufficient to promote pulmonary vascular stiffening and consequent PH manifestations.
Figure 6. miR-130/301 inhibition disrupts YAP/TAZ-miR-130/301 signaling and reverses a program of ECM remodeling and PH.

A–G) Following monocrotaline exposure, rats were treated with Short-NC or Short-130. A) By in situ stain, Short-130 reversed monocrotaline-mediated changes in target genes Pparγ and Lrp8, decreased collagen deposition and remodeling (Picrosirius Red), and medial thickening (α-SMA). In turn, Yap nuclear localization decreased. B) RT-qPCR demonstrated a decrease in fibrillar collagen isoforms, Lox, and Ctgf in Short-130-treated lung. C) Short-130 decreased Lox activity in PH lung. D) Short-130 decreased monocrotaline-induced collagen deposition (left) and fibrillar collagen content (right) in PH lung. E) Co-immunofluorescence microscopy revealed YAP-positive proliferating cells (PCNA/YAP double-positive stain) in diseased pulmonary arterioles (Short-NC). Short-130 reduced the number of PCNA/YAP double-positive cells in CD31+ and CD31− compartments. F–G) Short-130 decreased the miR-130/301-mediated increase in PH severity, as quantified by RVSP (F) and right ventricular hypertrophy (G). H–I) After two weeks of PH induction with hy-poxia+SU5416, mice were serially injected with Short-NC or Short-130 along with hypox-ia+SU5416 for two more weeks. Short-130 decreased collagen deposition and fibrillar collagen (Picrosirius Red) (H). Atomic force microscopy revealed increased pulmonary arteriolar (<50 μm) stiffness in PH, but Short-130 blunted this alteration (I). Black lines denote median; symbols denote individual PA measurements (n=4 normoxic mice vs n=3/hypoxic group). P-values were calculated by Kruskal-Wallis testing followed by Mann Whitney U post-hoc analysis; a significance cutoff of <0.008 was based on Bonferroni correction. (J) Transcriptomic analyses of whole mouse lung were performed after exposure to hypoxia+SU5416 treated with Short-NC (n=3) or Short-130 (n=3) versus mice in normoxia+SU5416 (Control; n=3). Genes from this transcriptomic analysis were identified according to modulation by both hypoxia and miR-130/301 inhibition. Pathway enrichment identified ECM modification as the pathway with the lowest hypergeometric p-value. Genes are color-coded according to membership in the five pathways with lowest p-values. Enlarged genes are shared by the fibrosis network (see Fig. 2A). Data are expressed as mean ± SEM (*P<0.05; ** P<0.01). Scale bars 50 μm. See also Fig. S5.
Therapeutic targeting of downstream ApoE or Lox reduces ECM remodeling and blunts the YAP/TAZ-miR-130/301 feedback loop in order to ameliorate PH in vivo
Finally, we wanted to define more definitively the in vivo ECM-YAP/TAZ-miR-130/301 feedback mechanism by pharmacologic manipulation of downstream ECM regulators, ApoE and Lox, in PH. To assess the importance of ApoE activity in PH, mice were treated with daily ingestion of the liver-X nuclear hormone receptor (LXR) agonist GW3965, a pharmacologic inducer of ApoE (Pencheva et al., 2014), simultaneously with hypoxia. Alternatively, similar to experiments with miR-130a-induced PH (Fig. 5), BAPN was administered in PH mice either simultaneously with hypoxia (“prevention”) or after disease development (“reversal”). In both cases of GW3965 or BAPN treatment, downstream collagen deposition and remodeling (Fig. 7A, E; Fig. S6) as well as Lox expression (Fig. 7A, E; Fig. S6) and activity (Fig. 7B, F) were inhibited. In the context of these changes in ECM, GW3965 and BAPN treatment also reduced YAP nuclear localization (Fig. 7A, E) and activation (reflected by decreased Ctgf, Fig. S6), decreased miR-130/301 expression (Fig. 7C, G), and reversed miR-130/301-dependent Pparγ and Lrp8 down-regulation (Fig. S6). Consistent with miR-130/301 manipulation in vivo, these feedback events culminated in decreased cellular proliferation in CD31+ and α-SMA+ arteriolar cells (PCNA staining, Fig. 7D, H) and improved downstream hemodynamic and histologic indices of PH (Fig. S6; Fig. 7I–K). Thus, we conclude that the YAP-TAZ-miR-130/301 circuit and its downstream network of targets are programmed both to respond to and to promote ECM remodeling in PH and represent an integrally linked set of targets for potential tailored therapy in this disease.
Figure 7. Pharmacologic inhibition of LOX or activation of APOE disrupts YAP/TAZ-miR-130/301 signaling to reduce vascular ECM remodeling and PH.

A–D) Mice were treated with BAPN either simultaneously with hypoxia (prevention) or after hypoxic disease induction (reversal). In prevention and rescue experiments, BAPN blunted hypoxia-mediated increases of vascular Lox, collagen remodeling (Picrosirius Red), medial thickening (α-SMA) (A), and Lox activity (B). In correlation with these ECM modifications, BAPN decreased Yap nuclear localization (A), miR-130/301 (C), and proliferation (PCNA-positive) of arteriolar CD31+ and α-SMA+ cells as compared with diseased controls (D). E–K) Hypoxic mice were treated with the LXR agonist GW3965 by prevention protocol. Similar to BAPN, the LXR agonist GW3965 blunted hypoxia-mediated increases of Lox, collagen remodeling (Picrosirius Red), medial thickening (α-SMA) (E), and Lox activity (F). Consistent with such ECM alterations, GW3965 decreased Yap nuclear localization (E), miR-130/301 (G), and downstream pulmonary vascular proliferation (PCNA stain in arteriolar CD31+ and α-SMA+ cells) compared with diseased controls (H). Consequently, GW3965 ameliorated PH severity, as quantified by RVSP (I), right ventricular hypertrophy (Fulton index, RV/LV+S) (J), and arteriolar muscularization (K). Data are expressed as mean ± SEM (*P<0.05; ** P<0.01). Normalized values are expressed as arbitrary units (A.U.) in (A, B, E, F). Scale bars 50 μm. See also Fig. S6.
Discussion
Mechanical forces act through a YAP/TAZ-miR-130/301 feedback loop to promote PH via ECM remodeling and vascular stiffening at both early and late time points of disease. In turn, vascular stiffening controls a number of vascular cell phenotypes and crosstalk mechanisms and thus plays a crucial role in PH pathogenesis. This work highlights both the fundamental significance yet complex control of ECM plasticity in PH and the attractive potential of tailoring therapy to both this molecular circuit and the related downstream PPARγ-APOE-LRP8-LOX axis.
Previous studies have implicated specific molecules related to the ECM in PH (Merklinger et al., 2005; Nave et al., 2014; Nickel et al., 2015; Wang et al., 2014). Yet, questions persist of whether ECM remodeling is merely an end-stage feature of PH and whether massive fibrosis is necessary to induce pathogenic outcomes. Our work clarify these points by reporting the early development of ECM remodeling in PH and supporting the notion that dynamic changes of ECM mediated via the YAP/TAZ-miR-130/301 circuit can have relevant pathogenic consequences both early and late in disease. Specifically, in vitro, miR-130/301 members are responsive to modest alterations (1 kPa) of matrix stiffness (Fig. 2B, Fig. S2H, Fig. S2K). Moreover, based on our timed confocal microscopy data, subtle changes in arteriolar stiffening in vivo without massive fibrosis correlate with activation of this pathogenic circuit, particularly at early disease time points (Fig. 4, Fig. S5). Notably, under conditions where stiffness abates (i.e., transferring cultured cells from stiff to soft matrix), we found down-regulation of miR-130/301, suggesting their role as more dynamic mechanical stress response factors in the pulmonary vessel (data not shown). Unlike early-stage disease, control of the YAP/TAZ-miR-130/301 circuit at later disease time points may be much less linear and aligns well with the “multi-hit” hypothesis of the origins of PH (Chan and Loscalzo, 2008). That is, considering that miR-130/301 members are up-regulated by stiffness as well as by hypoxia, inflammatory cytokines, and deficiencies of certain factors genetically associated with PH (Bertero et al., 2014a; Bertero et al., 2014b), these effects of disparate disease exposures on YAP/TAZ-miR-130/301, coupled with a potent ECM-YAP/TAZ positive feedback loop, support a model of self-sustaining and anatomic “spreading” of ECM remodeling as PH progresses. Therefore, it remains an intriguing question as to how and at what time points do other factors and/or diseases clinically associated with PH interface with the YAP/TAZ-miR-130/301 circuitry and ECM biology.
Controlling how cells adapt to their external space (Piccolo et al., 2014), the multi-faceted functionality of YAP/TAZ also sheds light on the interactions of disparate vascular cell types with environmental cues and with each other in PH. Our data indicate that the miRNA-dependent actions of YAP/TAZ are pervasive throughout multiple cell types of the pulmonary vasculature and multiple time points of PH. Such a ubiquitous presence suggests an expansive repertoire of environmental cues that may depend upon these molecules for vascular function, both in health and disease. For example, since shear stress can induce YAP/TAZ (Kim et al., 2014), miR-130/301 may also be responsive to increased pulmonary vascular flow such as in cases of congenital heart disease where PH secondary to shunting predominates. Yet, despite the shared dependence on YAP/TAZ–miR-130/301, downstream events can still be cell-type specific, resulting from the modulation of a cohort of downstream pathways and cellular crosstalk patterns -- some of which overlap and some of which differ among PAAFs, PAECs, and PASMCs (Fig. 3). Considering this degree of specificity and its adjustable, feedback-driven properties, the YAP/TAZ circuit may be partly responsible for individualized “tuning” of ECM remodeling, depending upon each PH subtype or temporal stage. Given such inherent control over the molecular tuning of the vascular ECM, it will be useful in the future to define the pathways beyond LATS and MST1/2 kinase signaling (Fig. S2A) that are involved in pulmonary vascular YAP/TAZ mechanoactivation. More generally, it will be intriguing to define the role(s) of the YAP/TAZ-miR-130/301 circuit in other diseased sites throughout the pulmonary vascular tree (Lammers et al., 2012), the peripheral vasculature, and other organ systems plagued by fibrotic states.
Downstream of the YAP/TAZ-miR-130/301 axis, the involvement of the PPARγ-APOE-LRP8-LOX pathway, downstream secreted factors, and additional miRNAs further define the molecular hierarchy and pathway overlap under the control of the ECM. In addition to the relation between the adventitial fibroblast and the vascular ECM, our results describe key molecular signaling pathways that facilitate the direct interplay between the ECM with endothelial and smooth muscle cells as well as the ECM-dependent crosstalk among these cell types. These actions are consistent with previously reported pro-proliferative phenotypes in PH, such as for PPARγ and APOE (Bertero et al., 2014b; Hansmann et al., 2008) and consistent with fibrotic phenotypes in other contexts such as for miR-21 (Huang et al., 2015). On the other hand, the regulation of the associated miRNAs miR-21 and miR-27a as well as the vasoactive factors FGF2, IL-6, and EDN1 now better defines the complete cellular pathobiology in PH relevant to ECM remodeling. Much future work remains, as it is likely that an even more complex and wide-reaching interactome exists among YAP/TAZ, miR-130/301, and their targets, all coincident with large portions of the same fibrotic gene network (Fig. 2A). As a result, it is possible that other aspects of ECM remodeling such as collagen degradation and turnover may feed into this mechanosensitive microRNA circuit. With such studies, it may be possible to address whether certain of these overlapping pathophenotypes linked to YAP/TAZ-miR-130/301 (i.e., fibrosis and proliferation) can be separated from one another during PH and if so, whether those contexts would result in distinct differences in disease manifestation.
Finally, the paradigm of the YAP/TAZ-miR-130/301 circuit in ECM remodeling may be applied to current suboptimal clinical management in PH. From a diagnostic perspective, it may be feasible to identify new populations at risk for PH via targeted molecular screening of YAP/TAZ or miR-130/301 activity. From a therapeutic perspective, targeting the YAP/TAZ-miR-130/301 circuit and downstream effectors could influence ECM remodeling at both early and late stages of PH, thus presenting an opportunity for disease prevention or reversal. Our findings may also allow for re-purposing of a related set of pharmacologic agents in PH, applied previously in unrelated contexts and/or used in isolation rather than coordinated combination. For instance, our data reveal that the LXR agonist GW3965 ameliorates ECM remodeling and PH in vivo (Fig. 7, Fig. S6). While clinical use of LXR agonists has focused on their roles in lipid and cholesterol metabolism (Im and Osborne, 2011), application of their actions in ECM remodeling beyond cancer metastasis (Pencheva et al., 2014) has yet to be explored. Furthermore, consistent with prior work (Kerr et al., 1984; Nave et al., 2014), our findings demonstrate the utility of LOX inhibition by BAPN for partially ameliorating PH (Fig. 7, Fig. S6). Importantly, because it inhibits future collagen cross-links rather than lysing existing ones, BAPN may be inadequate to induce a complete reversal of ECM remodeling; yet, in combination with GW3965, a more robust synergistic effect may be possible. Cyclic YAP-like peptides that interrupt YAP-TEAD interactions in oncogenesis (Zhou et al., 2014) could also carry therapeutic potential in PH. Therefore, when considered with our data demonstrating the efficacy of repressing miR-130/301 via shortmers (Fig. 6) and even PPARγ [via rosiglitazone as described in (Bertero et al., 2014b)], these findings suggest that tailored, ECM-based drug combinations may be effective for additive or synergistic control of overall PH manifestation.
In sum, our findings define the central pathogenic importance in PH of the YAP/TAZ-miR-130/301 circuit, at early and late stages of disease, in both responding to and promoting ECM remodeling via the broad control of a fibrotic gene/miRNA network and downstream cellular crosstalk. Coupled with the feasibility of therapeutically manipulating multiple targets in this pathway, these results support the application of cooperative therapeutic maneuvers that ameliorate vascular stiffness and thus prevent or reverse multiple forms of human PH.
Experimental Procedures
Study Approval
All animal experiments were approved by the Harvard Center for Comparative Medicine, the University of Colorado, Denver, and the Committees on Animal Research of the University of California, San Francisco. All experimental procedures involving the use of human tissue were approved by Institutional Review Boards at Partners Healthcare, Boston Children’s Hospital, and University of California, Los Angeles, as well as the New England Organ Bank. Ethical approval for this study and informed consent conformed to the standards of the Declaration of Hel-sinki. For formalin-fixed paraffin-embedded lung samples, human PH specimens from unused or discarded surgical samples and non-diseased human lung specimens from the New England Organ Bank have been described (Bertero et al., 2014).
Oligonucleotides and Transfection
Pre-miRNA oligonucleotides (pre-miR-130a, negative control pre-miR-NC1, and premiR-NC2) and custom-designed tiny LNA oligonucleotides (tiny-130: 5′-ATTGCACT-3′ and tiny-NC: 5′-TCATACTA-3′) were purchased from Thermo Scientific/Ambion and Exiqon, respectively. siR-NAs for PPARγ (sc-29455), OCT4/POU5F1 (sc-5279), and scrambled control (sc-44236) were purchased from Santa Cruz Biotechnology. On Target Plus siRNAs for YAP (J-012200-07), TAZ (WWTR1) (J-016083-05), LRP8 (J-011802-05), and scrambled control (D-001810-01) were purchased from Dharmacon (GE Healthcare). PAAFs, PAECs, and PASMCs were plated in collagen-coated plastic (50ug/mL) and transfected 24h later at 70–80% confluence using pre-miRNA (5nM), tiny-LNA (20nM), or siRNA (25nM) and Lipofectamine 2000 reagent (Thermo Scientific), according to the manufacturers’ instructions. Eight hours after transfection, cells were trypsinized and re-plated in hydrogel. Key siRNA experiments were replicated with two independent siRNA sequences targeting YAP (J-012200-05, Dharmacon), TAZ (J-016083-06, Dharmacon), PPARγ (sc-44220, Santa Cruz Biotechnology), and LRP8 (J-011802-06, Dharmacon) as well as scrambled controls (sc-37007 Santa Cruz Biotechnology and D-001810-02 Dharmacon).
Forced pulmonary expression of miR-130a in lungs of mice in vivo
Eight-week-old mice (C57Bl6) were injected with SU5416 (20 mg/kg; Sigma-Aldrich), followed by 4 intrapharyngeal injections (once by week) of 1nmol of miR-control (pre-miR-NC) or miR-130a (pre-miR-130a) mixed in 100 uL PBS solution containing 5% Lipofectamine 2000 (Thermo Scientific). Such intrapharyngeal injections led to effective delivery to the pulmonary arterioles, as we previously described in detail (Bertero et al., 2014b). BAPN (Sigma-Aldrich) was dissolved in water and administered daily in indicated mouse cohorts (30mg/kg/day). Three days after the last oligonucle-otide injection, right heart catheterization was performed as previously described (Parikh et al., 2012), followed by harvest of lung tissue for RNA extraction or paraffin embedding, as described above.
Inhibition of miR-130/301 in a rat model of monocrotaline-induced PH
Male Sprague-Dawley rats (10–14 week old) were injected with 60 mg/kg monocrotaline at time 0 followed by 5 intraperitoneal injections (every 3 days) of control or miR-130/301 shortmer oligonucleotides (20mg/kg/dose; Regulus). Three days after the last injection, right heart catheterization was performed followed by harvest of lung tissue for RNA extraction or paraffin embedding.
Inhibition of miR-130/301 in a mouse model of PH
Eight-week-old mice (C57Bl6) were injected with SU5416 (20 mg/kg/dose/week; Sigma-Aldrich), followed by exposure to normobaric hypoxia (10% O2; OxyCycler chamber, Biospherix Ltd, Redfield, NY) for 2 weeks. After two weeks and confirmation of PH development in five mice (right heart catheterization), mice were further treated with hypoxia + SU5416, along with 3 in-trapharyngeal injections (every 4 days) of control or miR-130/301 shortmer oligonucleotides, designed as fully modified antisense oligonucleotides complementary to the seed sequence of the miR-130/301 miRNA family (10mg/kg; Regulus). Specifically, the control and miR-130/301 shortmer oligonucleotides were non-toxic, lipid-permeable, high-affinity oligonucleotides. The miR-130/301 shortmer carried a sequence complementary to the active site of the miR-130/301 miRNA family, containing a phosphorothioate backbone and modifications (fluoro, methoxyethyl, and bicyclic sugar) at the sugar 2′ position. Such intrapharyngeal injections led to effective delivery to the pulmonary arterioles, as we previously described in detail (Bertero et al., 2014). Three days after the last injection, right heart catheterization was performed followed by harvest of lung tissue for RNA extraction or paraffin embedding, as described above.
Treatment of hypoxic mice with BAPN
Mice were exposed to normoxia or chronic hypoxia, and treated with BAPN (Sigma-Aldrich, 30mg/kg/day as described above) or vehicle control, either simultaneously with hypoxia (“prevention”) or after disease development (“reversal”) (Fig. S6A). Specifically, normoxic mice and hypoxic mice were treated either with BAPN or vehicle for 4 weeks (“prevention”). Alternatively, mice were exposed to hypoxia for 2 weeks to induce PH and then treated with BAPN along with 2 more weeks of hypoxic exposure (“reversal”). After these time periods, right heart catheterization was performed followed by harvest of lung tissue for RNA extraction or paraffin embedding.
Treatment of hypoxic mice with oral ingestion of the liver-X nuclear hormone receptor (LXR) agonist GW3965
To determine the effects of the LXR agonist GW3965 (Sigma-Aldrich) and consequent APOE induction on PH development, as previously described (Pencheva et al., 2014), mice were exposed to normobaric hypoxia and simultaneously assigned to control chow or chow supplemented with GW3965 (Research Diets, Inc.) at doses of 100 mg of drug per kilogram of mouse per day (based on average daily intake of 3.5 gm of chow). After three weeks, right heart catheterization was performed followed by harvest of lung tissue for RNA extraction or paraffin embedding.
Statistics
Cell culture experiments were performed at least three times and at least in triplicate for each replicate. The number of animals in each group was calculated to measure at least a 20% difference between the means of experimental and control groups with a power of 80% and standard deviation of 10%. The number of unique patient samples for this study was determined primarily by clinical availability. In situ expression/histologic analyses of both rodent and human tissue, and pulmonary vascular hemodynamics in mice and rats were performed in a blinded fashion. Numerical quantifications for in vitro experiments using cultured cells or in situ quantifications of transcript/miRNA expression represent mean ± standard deviation (SD). Numerical quantifications for physiologic experiments using rodents or human reagents represent mean ± standard error of the mean (SEM). Immunoblot images are representative of experiments that have been repeated at least three times. Micrographs are representative of experiments in each relevant cohort. Unless otherwise stated, paired samples were compared by a 2-tailed Student’s t test, and multiple group comparisons were performed by one-way ANOVA and post-hoc Bon-ferroni testing. For comparison among non-normally distributed data (i.e., AFM data), a Mann-Whitney U test was used for paired samples. For multiple comparisons in Fig. 6I, Kruskal-Wallis testing was followed by post-hoc between group Mann Whitney U tests. Of note, for such multiple comparisons, we utilized a P-value significance cutoff of <0.008, based on a Bonferroni correction of p (0.05) for the number of comparisons (6) among these 4 cohorts of mice. Otherwise, a P-value less than 0.05 was considered significant. Correlation analyses were performed by Pearson correlation coefficient calculation.
Please see Supplemental for further details of standardized and/or previously described meth-ods and reagents.
Supplementary Material
Acknowledgments
We thank J. Androsavich for technical assistance. This work was supported by the NIH [grants HL096834, HL124021 (S.Y.C.); HL67841 (S.M.B.); HL61284 (J.R.F.)] McArthur-Radovsky, Lerner, Harris, and Watkins Funds (S.Y.C.); Pulmonary Hypertension Association (S.Y.C.); American Heart Association (Post-doctoral Award, T.B.); and Foundation Bettencourt-Schueller (T.B.).
Footnotes
Accession Codes
RNA-Seq experimental data and associated designs were deposited in the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under series GSE61828.
Author Contributions
T.B., K.A.C., and S.Y.C. conceived and designed the experiments. K.A.C. and V.K. performed the in silico analyses. T.B., Y.L., C.M.H., P.D., S.A., A.H., Y.Y.Z., and L.E.F. performed the experiments. B.B.G., R.K., R.S., R.S., W.D.W., D.J.R., S.M.B., S.F., J.R.F., S.O.V., K.J.H., A.B.W. performed the work to obtain human and animal PH reagents. B.B. and B.N.C. constructed the anti-miR reagents. T.B., K.A.C., C.M.H., P.D., L.E.F., and S.Y.C. wrote the manuscript. All authors participated in interpreting the results and revising the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bertero T, Cottrill K, Krauszman A, Lu Y, Annis S, Hale A, Bhat B, Waxman AB, Chau BN, Kuebler WM, et al. The microRNA-130/301 family controls vasoconstriction in pulmonary hypertension. J Biol Chem. 2014a;290:2069–2085. doi: 10.1074/jbc.M114.617845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertero T, Lu Y, Annis S, Hale A, Bhat B, Saggar R, Saggar R, Wallace WD, Ross DJ, Vargas SO, et al. Systems-level regulation of microRNA networks by miR-130/301 promotes pulmonary hypertension. J Clin Invest. 2014b;124:3514–3528. doi: 10.1172/JCI74773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi R, Bao C, Jiang L, Liu H, Yang Y, Mei J, Ding F. MicroRNA-27b plays a role in pulmonary arterial hypertension by modulating peroxisome proliferator-activated receptor gamma dependent Hsp90-eNOS signaling and nitric oxide production. Biochem Biophys Res Commun. 2015;460:469–475. doi: 10.1016/j.bbrc.2015.03.057. [DOI] [PubMed] [Google Scholar]
- Boutet K, Montani D, Jais X, Yaici A, Sitbon O, Simonneau G, Humbert M. Therapeutic advances in pulmonary arterial hypertension. Ther Adv Respir Dis. 2008;2:249–265. doi: 10.1177/1753465808094762. [DOI] [PubMed] [Google Scholar]
- Chan SY, Loscalzo J. Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol. 2008;44:14–30. doi: 10.1016/j.yjmcc.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, Good R, Stringer R, Jones P, Morrell NW, Jarai G, et al. A Novel Murine Model of Severe Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2011;184:1171–1182. doi: 10.1164/rccm.201103-0412OC. [DOI] [PubMed] [Google Scholar]
- Cowan K, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med. 2000;6:698–702. doi: 10.1038/76282. [DOI] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan CT, Lankhaar JW, Westerhof N, Marcus JT, Becker A, Twisk JW, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Noninvasively assessed pulmonary artery stiffness predicts mortality in pulmonary arterial hypertension. Chest. 2007;132:1906–1912. doi: 10.1378/chest.07-1246. [DOI] [PubMed] [Google Scholar]
- Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T, Wang L, Morrell NW, et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118:1846–1857. doi: 10.1172/JCI32503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, He Y, Li J. Microrna-21: a central regulator of fibrotic diseases via various targets. Current pharmaceutical design. 2015;21:2236–2242. doi: 10.2174/1381612820666141226095701. [DOI] [PubMed] [Google Scholar]
- Im SS, Osborne TF. Liver x receptors in atherosclerosis and inflammation. Circ Res. 2011;108:996–1001. doi: 10.1161/CIRCRESAHA.110.226878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JS, Riley DJ, Frank MM, Trelstad RL, Frankel HM. Reduction of chronic hypoxic pulmonary hypertension in the rat by beta-aminopropionitrile. J Appl Physiol. 1984;57:1760–1766. doi: 10.1152/jappl.1984.57.6.1760. [DOI] [PubMed] [Google Scholar]
- Kerr JS, Ruppert CL, Tozzi CA, Neubauer JA, Frankel HM, Yu SY, Riley DJ. Reduction of chronic hypoxic pulmonary hypertension in the rat by an inhibitor of collagen production. Am Rev Resp Dis. 1987;135:300–306. doi: 10.1164/arrd.1987.135.2.300. [DOI] [PubMed] [Google Scholar]
- Kim KM, Choi YJ, Hwang JH, Kim AR, Cho HJ, Hwang ES, Park JY, Lee SH, Hong JH. Shear stress induced by an interstitial level of slow flow increases the osteogenic differentiation of mesenchymal stem cells through TAZ activation. PLoS One. 2014;9:e92427. doi: 10.1371/journal.pone.0092427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothapalli D, Liu SL, Bae YH, Monslow J, Xu T, Hawthorne EA, Byfield FJ, Castagnino P, Rao S, Rader DJ, et al. Cardiovascular protection by ApoE and ApoE-HDL linked to suppression of ECM gene expression and arterial stiffening. Cell Rep. 2012;2:1259–1271. doi: 10.1016/j.celrep.2012.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers S, Scott D, Hunter K, Tan W, Shandas R, Stenmark KR. Mechanics and Function of the Pulmonary Vasculature: Implications for Pulmonary Vascular Disease and Right Ventricular Function. Compr Physiol. 2012;2:295–319. doi: 10.1002/cphy.c100070. [DOI] [PubMed] [Google Scholar]
- Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, Chinnaiyan A, Israel MA, Goldstein LS, Abujarour R, et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010;24:1106–1118. doi: 10.1101/gad.1903310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Lagares D, Choi KM, Stopfer L, Marinkovic A, Vrbanac V, Probst CK, Hiemer SE, Sisson TH, Horowitz JC, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2015;308:L344–357. doi: 10.1152/ajplung.00300.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecham RP, Whitehouse LA, Wrenn DS, Parks WC, Griffin GL, Senior RM, Crouch EC, Stenmark KR, Voelkel NF. Smooth muscle-mediated connective tissue remodeling in pulmonary hypertension. Science. 1987;237:423–426. doi: 10.1126/science.3603030. [DOI] [PubMed] [Google Scholar]
- Merklinger S, Wagner R, Spiekerkoetter E, Hinek A, Knutsen R, Kabir M, Desai K, Hacker S, Wang L, Cann G, et al. Increased fibulin-5 and elastin in S100A4/Mts1 mice with pulmonary hypertension. Circulation Res. 2005;97:596–604. doi: 10.1161/01.RES.0000182425.49768.8a. [DOI] [PubMed] [Google Scholar]
- Mouw JK, Yui Y, Damiano L, Bainer RO, Lakins JN, Acerbi I, Ou G, Wijekoon AC, Levental KR, Gilbert PM, et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat Med. 2014;20:360–367. doi: 10.1038/nm.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave AH, Mizikova I, Niess G, Steenbock H, Reichenberger F, Talavera ML, Veit F, Herold S, Mayer K, Vadasz I, et al. Lysyl oxidases play a causal role in vascular remodeling in clinical and experimental pulmonary arterial hypertension. Arterio Thromb Vasc Biol. 2014;34:1446–1458. doi: 10.1161/ATVBAHA.114.303534. [DOI] [PubMed] [Google Scholar]
- Nickel NP, Spiekerkoetter E, Gu M, Li CG, Li H, Kaschwich M, Diebold I, Hennigs JK, Kim KY, Miyagawa K, et al. Elafin Reverses Pulmonary Hypertension via Caveolin-1-Dependent Bone Morphogenetic Protein Signaling. Am J Respir Crit Care Med. 2015;191:1273–1286. doi: 10.1164/rccm.201412-2291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh VN, Jin RC, Rabello S, Gulbahce N, White K, Hale A, Cottrill KA, Shaik RS, Waxman AB, Zhang YY, et al. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation. 2012;125:1520–1532. doi: 10.1161/CIRCULATIONAHA.111.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pencheva N, Buss CG, Posada J, Merghoub T, Tavazoie SF. Broad-spectrum therapeutic suppression of metastatic melanoma through nuclear hormone receptor activation. Cell. 2014;156:986–1001. doi: 10.1016/j.cell.2014.01.038. [DOI] [PubMed] [Google Scholar]
- Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94:1287–1312. doi: 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- Poiani GJ, Tozzi CA, Yohn SE, Pierce RA, Belsky SA, Berg RA, Yu SY, Deak SB, Riley DJ. Collagen and elastin metabolism in hypertensive pulmonary arteries of rats. Circ Res. 1990;66:968–978. doi: 10.1161/01.res.66.4.968. [DOI] [PubMed] [Google Scholar]
- Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9:628–638. doi: 10.1038/nrm2455. [DOI] [PubMed] [Google Scholar]
- Stenmark KR, Rabinovitch M. Emerging therapies for the treatment of pulmonary hypertension. Pediatr Crit Care Med. 2010;11:S85–90. doi: 10.1097/PCC.0b013e3181c76db3. [DOI] [PubMed] [Google Scholar]
- Valastyan S, Weinberg RA. Roles for microRNAs in the regulation of cell adhesion molecules. J Cell Sci. 2011;124:999–1006. doi: 10.1242/jcs.081513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Zhang H, Li M, Frid MG, Flockton AR, McKeon BA, Yeager ME, Fini MA, Morrell NW, Pullamsetti SS, et al. MicroRNA-124 controls the proliferative, migratory, and inflammatory phenotype of pulmonary vascular fibroblasts. Circ Res. 2014;114:67–78. doi: 10.1161/CIRCRESAHA.114.301633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Chesler NC. Pulmonary vascular wall stiffness: An important contributor to the increased right ventricular afterload with pulmonary hypertension. Pulm Circ. 2011;1:212–223. doi: 10.4103/2045-8932.83453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Bhattacharyya S, Jain M, Varga J. Regulation of Matrix Remodeling by Peroxisome Proliferator-Activated Receptor-gamma: A Novel Link Between Metabolism and Fibrogenesis. Open Rheumatol J. 2012;6:103–115. doi: 10.2174/1874312901206010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K, Loscalzo J, Chan SY. Holding our breath: The emerging and anticipated roles of microRNA in pulmonary hypertension. Pulm Circ. 2012;2:278–290. doi: 10.4103/2045-8932.101395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J. An emerging role for Hippo-YAP signaling in cardiovascular development. J Biomed Res. 2014;28:251–254. doi: 10.7555/JBR.28.20140020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hu T, Xu Z, Lin Z, Zhang Z, Feng T, Zhu L, Rong Y, Shen H, Luk JM, et al. Targeting Hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides. FASEB J. 2014;29:724–732. doi: 10.1096/fj.14-262980. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.