

Abstract
Patients with chemo-refractory acute myeloid leukemia (AML) have a dismal prognosis. Chimeric antigen receptor T (CART) cell therapy has produced exciting results in CD19+ malignancies and may overcome many of the limitations of conventional leukemia therapies. We developed CART cells to target CD33 (CART33) using the anti-CD33 single chain variable fragment used in gemtuzumab ozogamicin (clone My96) and tested the activity and toxicity of these cells. CART33 exhibited significant effector functions in vitro and resulted in eradication of leukemia and prolonged survival in AML xenografts. CART33 also resulted in human lineage cytopenias and reduction of myeloid progenitors in xenograft models of hematopoietic toxicity, suggesting that permanently expressed CD33-specific CART cells would have unacceptable toxicity. To enhance the viability of CART33 as an option for AML, we designed a transiently expressed mRNA anti-CD33 CAR. Gene transfer was carried out by electroporation into T cells and resulted in high-level expression with potent but self-limited activity against AML. Thus our preclinical studies show potent activity of CART33 and indicate that transient expression of anti-CD33 CAR by RNA modification could be used in patients to avoid long-term myelosuppression. CART33 therapy could be used alone or as part of a preparative regimen prior to allogeneic transplantation in refractory AML.
Introduction
Acute myeloid leukemia (AML) is the most common form of acute leukemia in adults.1 There have been only modest changes in the treatment of AML over the past 40 years and the prognosis remains poor with average 5-year survival rates of 20%.2 Allogeneic stem cell transplantation remains the only potentially curative treatment in relapsed or high-risk disease but is associated with significant morbidity and mortality3; in addition, outcomes for patients transplanted with refractory disease are poor4 and almost half of patients with relapsed disease are chemo-refractory and thus not suitable for transplantation.3,5 Novel strategies to induce remission are therefore crucial to improving the outcome of AML.
Chimeric antigen receptors (CARs) are synthetic polypeptides that are composed of an extracellular domain derived from a single chain variable fragment (scFv) isolated from a monoclonal antibody, a hinge region, a transmembrane domain, and an intracellular signaling moiety with a costimulatory domain.6,7 T cells bearing CAR (CART cells) combine the antigen specificity of a monoclonal antibody with the potent effector functions of T cells and may overcome the limitations of conventional AML cytotoxic therapy.
CART cell therapy has recently emerged as a potentially curative therapy in B-lineage malignancies. In early phase clinical trials, anti-CD19 CART cells resulted in a very impressive antitumor effect and long-term remissions in chronic lymphocytic leukemia, acute lymphocytic leukemia and diffuse large B-cell lymphoma.8–15
CD19 represents an ideal target for B-cell malignancies, because it is homogeneously expressed on malignant cells, its off-target expression is limited to normal B cells and because patients can tolerate prolonged B-cell aplasia. To our knowledge, there are no cell surface antigens that are unique to malignant myeloid blasts and that spare normal hematopoietic cells, implying that potent CAR-directed therapy for AML is likely to be myelotoxic. Indeed, we have shown that the preclinical antileukemic activity of anti-CD123 CART cells comes at the cost of significant myeloablation.
CD33 is a transmembrane receptor of the SIGLEC family that is expressed on myeloid cells.16,17 CD33 is known to be expressed on AML blasts and on normal myeloid progenitors.18–20 Gemtuzumab ozogamicin (GO), an anti-CD33 monoclonal antibody conjugated to the immunotoxin calicheamicin, was the only new drug approved in AML in the past decade but was voluntarily withdrawn in 2010 due to unclear benefits in postmarketing clinical trials. However, a meta-analysis of subsequent clinical trials showed that the combination of chemotherapy with GO is associated with improvement in relapse and survival rates at 6 years,21 indicating that CD33 likely is a viable target in AML.
We developed a CAR based on the GO scFv (referred to as CART33). Here we report the preclinical activity of CART33 and compare it with our previously published interleukin-3Rα-specific CART123.22 We show that CART33 cells are able to eradicate human AML and myelodysplastic syndrome blasts, while resulting in significant myelotoxicity in mouse xenografts. To minimize the risk of long-term hematopoietic toxicity, we developed transiently expressed mRNA-modified CART33 with an increased therapeutic index that could be used in future clinical trials.
Materials and Methods
Generation of CART cells
The pTRPE anti-CD33-41BB-CD3 zeta (CAR33) plasmid DNA was generated by cloning the light and heavy chain of the humanized and murine anti-human CD33 scFv derived from GO (clone my96) into the previously described murine CART19 plasmid vector.23 Normal donor T cells were positively selected using anti-CD4 and anti-CD8 microbeads (Miltenyi, San Diego, CA, USA), mixed at 1:1 ratio and expanded in vitro using anti-CD3/CD28 Dynabeads (Invitrogen, Life Technologies, Grand Island, NY, USA, added on the first day of culture) with interleukin-2 at 50 IU/ml. T cells were transduced with lentiviral supernatant 1 day following stimulation at a multiplicity of infection of 3. The anti-CD3/CD28 Dynabeads were removed on day 6 and T cells were grown in T-cell media (X-vivo 15 media, human serum 5%, penicillin, streptomycin and glutamax) for up to 15 days and then cryopreserved for future experiments. Prior to all experiments, T cells were thawed and rested overnight at 37 °C. Production of CART123 cells was previously described.22
Generation of mRNA-modified CART33
The CAR construct from the pTRPE anti-CD33-41BB-CD3z plasmid was sub-cloned into the pDA vector24 as previously published. In vitro transcription was performed using mMESSAGE mMachineT7 ULTRA Transcription Kit (Ambion, Life Technologies). The RNA was purified using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). RNA-CAR33 was electroporated into T cells as previously described.24–26 Electroporation was done using an ECM830 Electro Square Wave Porator (Harvard Apparatus BTX, Holliston, MA, USA).
Cells
The MOLM14 cell line was obtained from the ATCC (Manassas, VA, USA) and maintained in R10 media (RPMI media,10% fetal calf serum, penicillin, and streptomycin). MOLM14-cells transduced with luciferase-GFP under the control of the EF1α promoter (MOLM14-Luc) were used in some experiments as indicated. De-identified primary human AML and myelodysplastic syndrome (MDS) bone marrow specimens were obtained from the University of Pennsylvania Stem Cell and Xenograft Core facility. All samples were obtained after informed, written consent. For all functional studies, AML cells were thawed at least 12 h before analysis and rested overnight at 37 °C. MDS bone marrow samples were positively selected for CD34+ cells using MACSQuant columns (Miltenyi).
Flow cytometry analysis
Anti-human antibodies were purchased from Biolegend (San Diego, CA, USA), eBioscience (San Diego, CA, USA) or BD Biosciences (San Jose, CA, USA). Cells were isolated from in vitro culture or from animals, washed once in phosphate-buffered saline supplemented with 2% fetal calf serum and stained at 4 °C after blockade of Fc receptors. For cell number quantitation, Countbright beads (Invitrogen) were used according to the manufacturer's instructions (Invitrogen). In all analyses, the population of interest was gated based on time gating, followed by forward vs side scatter characteristics, followed by singlet gating, and live cells were gated using Live Dead Aqua (Invitrogen). Surface expression of anti-CD33 CAR was detected by staining with an Alexa Fluor 647-conjugated goat anti-mouse F(ab')2 antibody from Jackson Immunoresearch (West Grove, PA, USA). Flow cytometry was performed on a four-laser Fortessa analyzer (BD Biosciences). All analyses were performed using FlowJo X10.0.7r2.
T-cell function assays
T-cell degranulation and intracellular cytokine assays
Degranulation assays were performed as previously described.27 Briefly, T cells were incubated with target cells at a 1:5 ratio. After staining for CAR expression, antibodies against CD107a, CD28, CD49d and monensin were added at the time of incubation. After 4 h, cells were harvested and stained for CD3 and Live Dead staining (Invitrogen). Cells were fixed and permeabilized (FIX & PERM Cell Fixation & Cell Permeabilization Kit, Life Technologies) and intracellular cytokine staining was then performed.
Proliferation assays
T cells were washed and re-suspended at 1 × 107/ml in 100 ul of phosphate-buffered saline and labeled with 100 ul of carboxyfluorescein succinimidyl ester (CFSE) 2.5 uM (Life Technologies) for 5 min at 37 °C. The reaction was then quenched with cold R10, and the cells were washed three times. Targets were irradiated at a dose of 100 Gy. T cells were incubated at a 1:1 ratio with irradiated target cells for 120 h. Cells were then harvested, stained for CD3, CAR and Live Dead aqua (Invitrogen), and Countbright beads (Invitrogen) were added prior to flow cytometric analysis.
Cytotoxicity assays
MOLM14-Luc cells or CFSE (Invitrogen) labeled primary AML samples were used for cytotoxicity assay as previously described.28 In brief, targets were incubated at the indicated ratios with effector T cells for 4 or 16 h. Killing was calculated either by bioluminescence imaging on a Xenogen IVIS-200 Spectrum camera (PerkinElmer, Hopkinton, MA, USA) or by flow cytometry. For the latter, cells were harvested; Countbright beads and 7-AAD (Invitrogen) were added prior to analysis. Residual live target cells were CFSE+ 7-AAD–. For MDS, T cells were incubated with CD34-selected bone marrow at 1:1 ratio for 4 or 24 h as indicated, and cytotoxicity was then measured by flow cytometry or by fluorescence in situ hybridization using a probe to detect 5q deletion or monosomy 5 (EGR1 (5q31)/ D5S23, D5S721 (5p15.2)).
Secreted cytokine measurement
Effector and target cells were incubated at a 1:1 ratio in T-cell media for 24 or 72 h as indicated. Supernatant was harvested and analyzed by 30-plex Luminex array according to the manufacturer's protocol (Invitrogen).
In vivo experiments
NOD-SCID-γ chain−/− (NSG) and NSG mice transgenic for human interleukin-3, stem cell factor and granulocyte macrophage colony-stimulating factor (NSG-S) originally obtained from Jackson Laboratories were purchased from the Stem Cell and Xenograft Core (University of Pennsylvania). All experiments were performed on protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Schemas of the utilized xenograft models are discussed in detail in the relevant figures and the Results section. Cells (MOLM 14-Luc, primary AML cells or T cells) were injected in 200 ul of phosphate-buffered saline at the indicated concentration into the tail veins of mice. Bioluminescent imaging was performed using a Xenogen IVIS-200 Spectrum camera. Images were acquired and analyzed using Living Image version 4.4 (Caliper LifeSciences, Inc., PerkinElmer). Humanized immune system mice were created at the Stem Cell and Xenograft Core by injection of fetal liver CD34+ cells into newborn NSG mice and were used at approximately 8 weeks of age.
Histology and immunohistochemistry
Formalin-fixed, paraffin-embedded sections from mouse femurs were stained with hematoxylin and eosin and counterstained with antibodies to human CD45 (M0701, Dako) and human CD34 (PA0212, QBEND10). Immunohistochemistry staining of 28 formalin-fixed paraffin-embedded normal human tissues (adipose, adrenal, appendix, cerebellum, cervix, colon, endometrium, esophagus, fat, heart, kidney, liver, lymph node, lung, muscle, ovary, pancreas, parathyroid, placenta, prostate, salivary, spinal cord, spleen, stomach, testis, thymus, thyroid, tonsil) was done in order to evaluate off-tumor expression of CD33 (triplicates). This was performed on a Leica Bond-III instrument (Leica Biosystems, Buffalo Grove, IL, USA) using the Bond Polymer Refine Detection System (Leica Biosystems). Antibodies against CD33 (Novocastra/Leica, NCL-L-CD33, Leica Biosystems) were used undiluted. Heat-induced epitope retrieval was done for 20 min with ER2 solution (Leica Microsystems AR9640, Leica Biosystems). Images were digitally acquired using the Aperio ScanScope (Leica Biosystems).
Statistical considerations
All statistics were performed using GraphPad Prism 6 (La Jolla, CA, USA) for windows, version 6.04.
Results
CD33 as a target in AML and CART33 constructs
To verify the clinical relevance of CD33 as a target for immunotherapy in AML, we first assessed the level of expression of CD33 in AML. Consistent with the literature,16,20,29–31 CD33 was expressed on the majority of AML blasts in primary AML samples as well as in bone marrows from MDS patients (Figures 1a and b). CD33 was also expressed on myeloid lineages in normal bone marrow and on resident macrophages in the liver, lung and kidneys (Figure 1c). To test the efficacy of CART33, we designed four constructs derived from the original murine clone my96 and from a humanized version of the same scFv. Two constructs utilized an immunoglobulin G4 (IgG4) hinge and two constructs used CD8 hinge (Figure 1d, Supplementary Figure S1).
Figure 1.
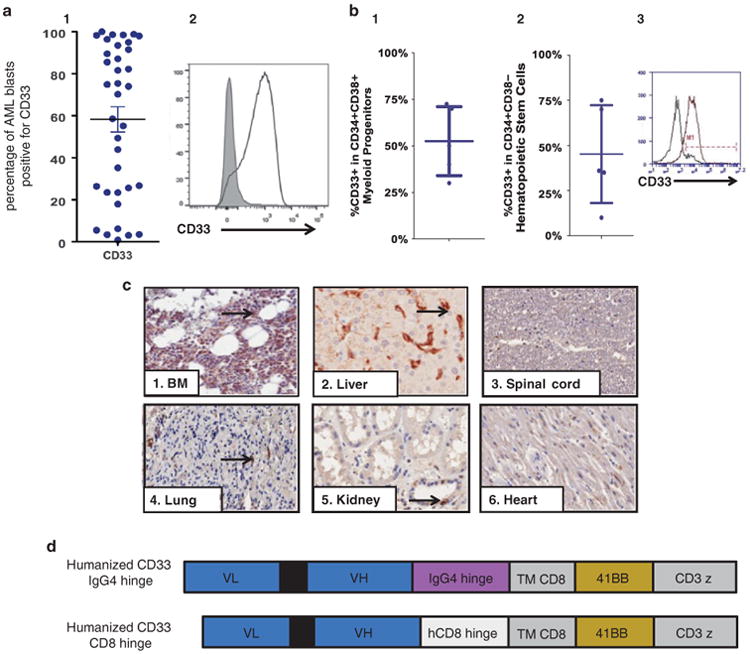
Expression of CD33 on malignant and normal myeloid cells. (a) CD33 is expressed in AML. (1) CD33 is expressed on blasts in most patient samples with AML (AML blasts were gated using standard side scatterlow CD45dim characteristics, n = 36) (2) Histogram mean fluorescence intensity (MFI) from an AML patient (gated on AML blasts). (b) CD33 is expressed in bone marrow from MDS patients. (1) CD33 is expressed on the CD34+CD38+ compartment in MDS patients. (2) CD33 is expressed on the CD34+CD38– hematopoietic stem cell compartment in MDS patients. (3) Histogram MFI from a MDS patient (gated on CD34+CD38- cells). (c) Tissue expression of CD33. CD33 is expressed on normal bone marrow myeloid progenitors (1), liver Kupffer cells (2), no expression on neurological tissue (3), occasional lung macrophage expression (4), occasional myeloid cells in the kidneys (5) and non-specific cardiac staining (6). (d) CAR constructs used in this study. All are second-generation CARs composed of: an extracellular domain (the light-to-heavy orientation of the scFV clone MY96 of GO), a hinge derived from either human CD8 or IgG4 as indicated, a transmembrane domain (TM) derived from CD8, 41BB (CD137) costimulatory domain and CD3zeta intracellular signaling domain.
CART33 therapy shows potent in vitro activity against AML cell lines, primary AML samples and MDS
We tested the activity of the four different CART33 constructs in vitro and compared each to the CART123 that we previously described.22 Humanized CART33 was consistently superior to murine CART33 (data not shown), and therefore all subsequent studies were performed on the two humanized CART33 constructs. As a model tumor, we employed the MOLM14 cell line (CD33 and CD123 expression on MOLM14 is shown in Supplementary Figure S2a). Incubation of CART33 with MOLM14 resulted in significant degranulation (Figure 2a), potent cytotoxicity at low effector:target ratios (Figure 2b), extensive proliferation (Figure 2c) and robust cytokine production (Figure 2d, Supplementary Figures S3 and S4) that was significantly higher than incubation with the control T-cell leukemia cell line Jurkat. The majority of CART33 cells produced two or more cytokines per cell after incubation with MOLM14, in a similar pattern to potent non-specific stimulation with PMA/ionomycin (Figure 2e). This function has been associated with superior in vivo activity.32 Furthermore, CART33 cells resulted in significant in vitro activity against primary AML samples (data not shown). CART33 with IgG4 hinge resulted in superior cytotoxicity compared with CART33 with CD8 hinge (Supplementary Figure S5). All of these in vitro experiments are representative of four independent experiments. CART33 and CART123 cells used in these experiments were produced from the same donor and expanded together under the same controlled conditions. They had similar CD4/CD8 ratio (Supplementary Figure S6).
Figure 2.
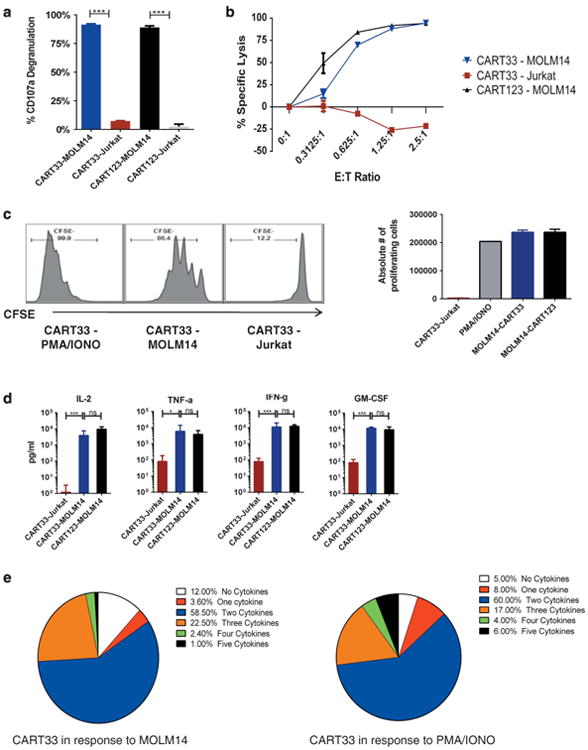
CART33 cells exhibit robust in vitro effector functions in response to the CD33+ cell line MOLM14 (plots are representative of four independent experiments). (a) CART33 and CART123 cells undergo specific degranulation to the CD33+/CD123+ MOLM14. CART33, CART123 and untransduced T cells (UTD) were incubated with the CD33+/CD123+ cell line MOLM14, PMA/ionomycin as a positive non-specific T-cell stimulant (not shown) and the control T-cell ALL cell line Jurkat, in the presence of CD49d, CD28 costimulatory molecules and monensin. CD107a degranulation was measured by flow cytometry after 4 h of incubation. (b) CART33 cells result in specific killing of MOLM14 cells that is comparable to CART123. CART33, CART123 and UTD were incubated with MOLM14-luc or Jurkat-luc for 24 h at different E:T ratios as indicated, and bioluminescence imaging was then performed as a measure of residual living cells. (c) CART33 cells undergo specific proliferation to MOLM14 and not to Jurkat. T cells were labeled with CFSE and incubated with MOLM14, PMA/ionomycin as a positive nonspecific T-cell stimulant or with Jurkat as a negative control, for 120 h. The number of proliferating T cells was significantly higher in response to MOLM14 as compared with Jurkat and was comparable to CART123. (d) CART33 cells produce robust levels of cytokines in response to MOLM14, comparable to CART123 cells. CART33, CART123 and UTD cells were incubated with MOLM14, Jurkat and PMA/ionomycin for 24 h. Supernatant was then harvested, and a 30-plex Luminex assay was performed. Levels of the rest of cytokines are presented in Supplementary Figure S4. There was some variability in cytokine production between the donors, but when averaged out, CART123 and CART33 produced similar levels of cytokines. (e) CART33 cells produce more than one cytokine per cell in response to MOLM14. CART33, CART123 and UTD cells were incubated with MOLM14, or PMA/ionomycin. The cells were then fixed and permeabilized, stained for five different cytokines (tumor necrosis factor alpha, interferon gamma, granulocyte macrophage colony-stimulating factor, macrophage inflammatory protein 1b and interleukin-2), and flow cytometric analyses were performed. The majority of CART33 cells produce more than one cytokine in response to MOLM14, similar to their response to PMA/Ionomycin.
CART33 also exhibited significant in vitro activity in MDS. Selected MDS samples were positive for CD33 and CD123 and harbored a specific cytogenetic abnormality. CART33 activity was evident by specific degranulation of CART33 in response to CD34-enriched bone marrow samples from MDS patients (Figure 3a), specific killing after a 24-h incubation of CART33 with CD34-enriched MDS samples (1:1 effector: target ratio, Figure 3b) and the specific reduction of the malignant clone (measured by fluorescence in situ hybridization) after 4 h of incubation (Figure 3c).
Figure 3.
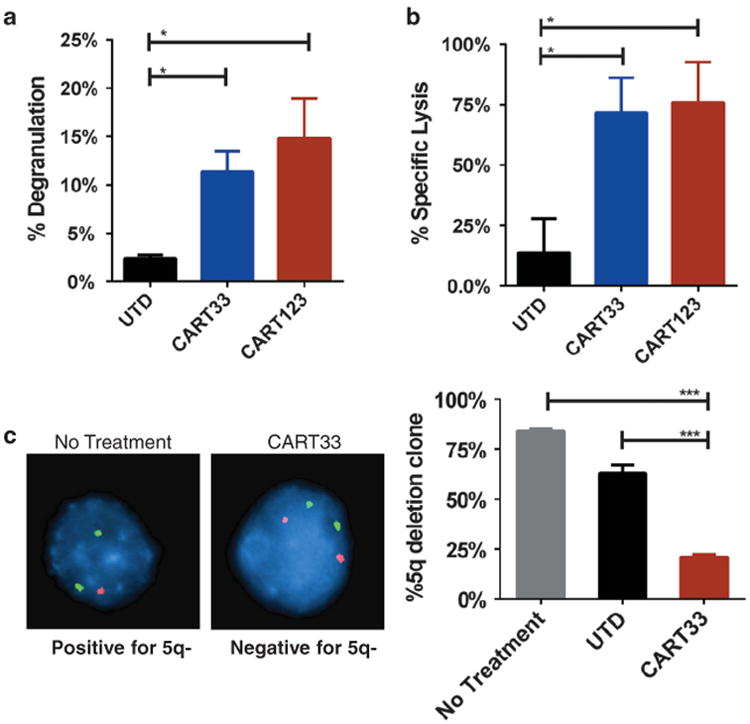
CART33 and CART123 cells exhibit antitumor activity in MDS. (a) CART33 and CART123 cells undergo specific degranulation in response to bone marrow cells from MDS patients. Bone marrow samples from MDS patients were CD34 enriched (≥85% purity) and then incubated with CART33, CART123 or UTD cells at E:T ratio of 1:5 for 4 h, in the presence of CD49d, CD28 co-stimulation and monensin. CD107a degranulation was then measured by flow cytometry. (b) CART33 and CART123 cells kill CD34-enriched bone marrow cells from MDS patients. CD34-enriched bone marrow from patients with MDS were incubated with either UTD, CART33 or CART123 for 24 h, and then live leukemic cells were measured by flow cytometry. There was a significant reduction in live CD45dimCD34+ cells in samples treated with CART33 or CART123. (c) Treatment with CART33 results in specific killing of the MDS clone. CD34-enriched bone marrow sample from a patient with MDS and 5q deletion was incubated with CART33, UTD cells or with no treatment at 1:1 E:T ratio for 4 h. Sample was then harvested, and fluorescence in situ hybridization for 5q- was performed. There was significant reduction in the 5q- clone percentage in the group treated with CART33 when compared with UTD and No treatment groups (as T cells comprised 50% of the sample, interphase nuclei were corrected by a factor of two). Results are representative of three experiments.
CART33 therapy results in reduction of leukemia burden and survival advantages in MOLM14 Engrafted xenografts
To test the in vivo activity of CART33, NSG mice were injected with MOLM14-Luc (Figure 4a). After confirmation of engraftment by bioluminescence imaging, mice received a single injection of CART33 or control untransduced T cells (UTD) at different dose levels. Mice were then followed with serial imaging, and disease burden was quantified using bioluminescence. Mice treated with control T cells succumbed quickly to disease, whereas mice treated with CART33 cells showed significant reduction of the disease and a survival advantage (Figures 4b–d). Furthermore, we observed a dose-dependent response (Supplementary Figure S7) in CART33-treated mice and superior antileukemic activity of CART33 with IgG4 hinge compared with CART33 with CD8 hinge (Supplementary Figure S8). For all subsequent experiments, only CART33 with an IgG4 hinge were used.
Figure 4.
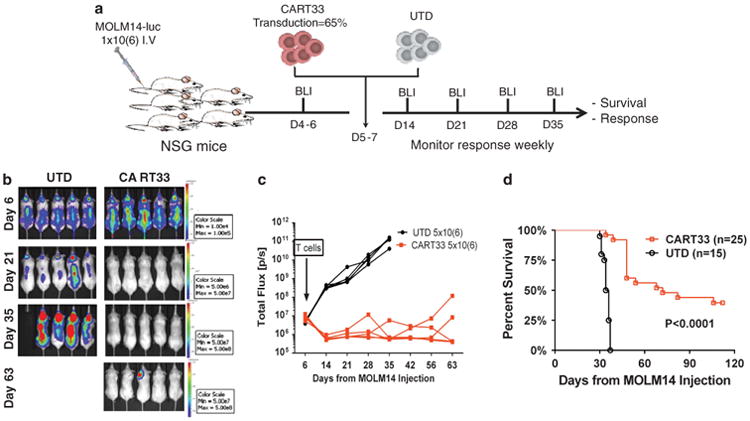
CART33 treatment results in reduction in disease burden and prolonged survival in MOLM14-engrafted xenografts (three independent experiments). (a) Experiment schema: NOD-SCID-gamma chain knockout (NSG) mice were injected with the AML cell line MOLM14 1×106 intravenously and imaged for engraftment after 4–6 days. Between day 5 and 7, mice were treated with CART33 (5×106), or control vehicle (UTD cells 5 × 106). The mice were followed with serial weekly imaging to assess the burden of AML. (b) Representative Images of tumor burden by bioluminescent imaging (BLI) from one experiment. (c) CART33 treatment results in reduction in leukemia in MOLM14-engrafted xenografts. Tumor burden over time by BLI; data from one experiment (n= 5 per group), each mouse is represented by a line. (d) Composite survival of three independent experiments. Treatment with CART33 resulted in significant survival advantages when compared with treatment with UTD.
CART33 therapy results in eradication of leukemia in primary AML xenografts and in long-term survival
Primary leukemia cells are likely more clonally heterogeneous than immortalized cell lines and hence are more representative of the human disease. We evaluated the efficacy of CART33 therapy in primary AML xenografts. NSG-S mice were injected with primary AML samples expressing CD33 and CD123 (Supplementary Figure S2b). We used NSG-S mice in this setting as engraftment efficiency of human AML has been shown to be improved using this model.33 Disease burden was quantified in the peripheral blood by retro-orbital bleedings (Figure 5a). Engraftment was defined as ≥ 0.5–1% circulating huCD45+ cells and was typically achieved 2–4 weeks after injection of the leukemic cells. These mice were then treated with a single injection of CART33, CART123 or UTD cells (1 × 105 via tail vein injection). Leukemia was eradicated within 4 weeks of CART33 or CART123 injection (Figures 5b and c) and long-term survival was demonstrated (Figure 5d).
Figure 5.
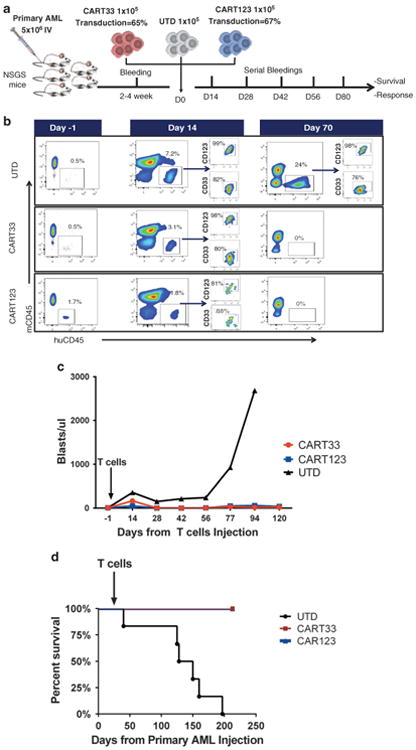
CART33 treatment results in leukemia eradication and long-term disease-free survival in primary AML engrafted xenografts. (a) Experiment schema: NSG mice transgenic for the human cytokines stem cell factor, interleukin-3, granulocyte macrophages colony-stimulating factor (NSG-S mice) were injected with a primary AML sample (5 × 106 intravenously (IV)). Engraftment was confirmed by retro-orbital bleeding after 2–4 weeks, and then mice were treated with CART33, CART123 or control vehicle (UTD cells). Total number of T cells injected was 1 ×105 IV. CART33 and CART123 cells had comparable transduction efficacy. The mice were followed with serial retro-orbital bleedings to assess the burden of leukemia. (b) Analysis of peripheral blood from mice treated with UTD, CART33 or CART123 at baseline, day 14 and day 70 after T cells injection. AML was not detected in mice treated with CART33 or CART123 starting 4 weeks after T-cell treatment. (c) Summary of disease burden measured by blasts/ul from retro-orbital bleedings at different time points as indicated. (d) Survival of mice treated with CART33, CART123 or UTD (n=8 per group) (P < 0.001 when treatment with CART33 or CART123 is compared with UTD).
CART33 treatment results in hematopoietic toxicity
As CD33 is known to be expressed on myeloid progenitors, albeit to lower levels compared with leukemic cells,16,20,31 we investigated the impact of CART33 on normal hematopoiesis. We used two different models to assess hematopoietic toxicity of CART33. Humanized immune system mice postnatally engrafted with human fetal CD34+ cells were bled to confirm engraftment and then were treated with CART33, CART123 or UTD cells (Figure 6a). Mice were bled weekly for 4 weeks. Mice were then euthanized, and bone marrows from these mice were collected for analysis. As expected, based on CD33 expression on myeloid lineage, these mice developed reduction in peripheral blood myeloid cells, including monocytes, compared with mice treated with UTD cells. Analysis of the bone marrow 4 weeks after treatment showed reduction of the CD34+CD38 – hematopoietic stem cell and the CD34+CD38+ myeloid progenitors by flow cytometry (Figure 6b) or immunohistochemistry (Figure 6c). The humanized immune system model is biased toward B-cell lineage, and so we employed a second model that is more myeloid biased. Here bone marrow from normal adult donors was T cell depleted and injected into busulfan-conditioned NSG-S mice. Autologous CART33, CART123 or UTD cells were generated by transducing peripheral blood T cells from the marrow donor with the relevant lentivirus (Figure 6d). After confirming engraftment of these mice, they were treated with autologous CART33, CART123 or UTD cells and followed with weekly bleedings for a total of 3 weeks. Mice were then euthanized, and tissues were harvested and analyzed. Similar to the humanized immune system xenografts, we observed reduction in peripheral blood myeloid cells and monocytes and in the CD34+ marrow compartments in mice treated with CART33 as well as CART123 cells (Figures 6e and f). We observed earlier reduction in monocytes in mice treated with CART33 compared with CART123, likely due to the higher levels of expression of CD33 on monocytes, compared with CD123 (Supplementary Figure S9).
Figure 6.
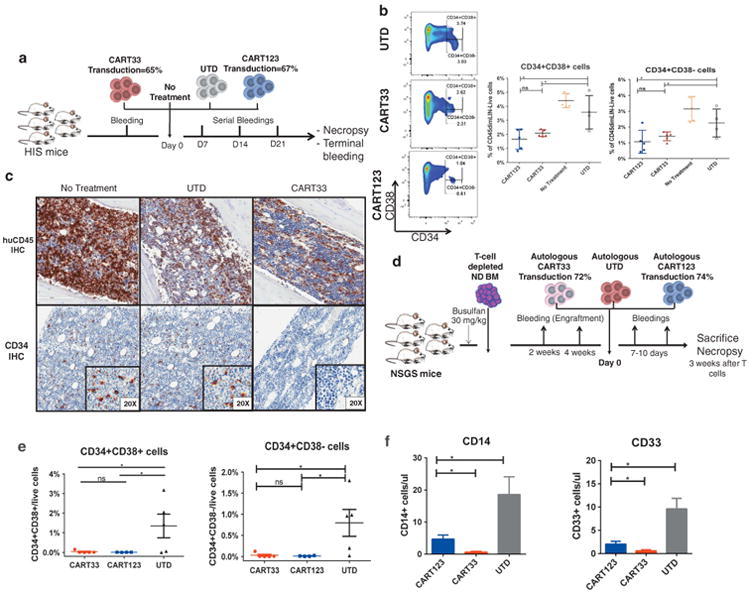
CART33 and CART123 treatment results in similar hematopoietic toxicity in two different humanized xenografts models (representative of six independent experiments). (a) Schema of Model no. 1: humanized immune system (HIS) mice were bled retro-orbitally 6–8 weeks after injection of human CD34+ cells derived from the fetal liver to confirm engraftment of human cells. Mice were then treated with either CART33, CART123, UTD cells (1 ×106 cells each) or with no treatment and followed by serial weekly retro-orbital bleedings. Mice were then euthanized on day 28, and organs were harvested and analyzed. (b) CART33 or CART123 treatment results in significant reduction in myeloid progenitors (CD34+CD38+) and in hematopoietic stem cells (CD34+CD38 –). Representative FACS plots and summary statistics from bone marrow analysis by flow cytometry on day 28 at the conclusion of the experiment, gated on singlets, huCD45dim, Lineage negative. (c) CART33 treatment results in reduction of the CD34+ compartment in the bone marrow by immunohistochemistry. Sections of the femur were taken from the mice on day 28 after treatment with UTD cells or CART33 cells. huCD45 and CD34 staining by immunohistochemistry was performed. No difference in huCD45 between control T cells and CART33, although both these groups show less huCD45 staining likely consistent with an allogeneic human-anti-human effect. There was specific reduction of CD34+ cells in mice treated with CART33. (d) Schema of Model no. 2: NSGS mice received busulfan intraperitoneally followed by 2 × 106 T-cell depleted bone marrow cells from a normal donor the following day. Engraftment was confirmed by flow cytometric analysis of peripheral blood after 4 weeks, and mice were then treated with 1 × 106 autologous CART33, autologous CART123 or UTD cells. Mice were then followed with retro-orbital bleeding on days 7 and 14 and were euthanized for necropsy on day 21. (e) CART33 or CART123 treatment results in significant reduction in myeloid progenitors (CD34+CD38+) and in hematopoietic stem cells (CD34+CD38 –). Representative plot of bone marrow analysis by flow cytometry on day 21. CART33 and CART123 treatment resulted in significant reduction in myeloid progenitors (CD34+CD38+) and in hematopoietic stem cells (CD34+CD38 –), Gated on huCD45dim, Lin – . (f) Summary statistics from peripheral blood analysis by flow cytometry on day 14.
Transient RNA-modified ‘biodegradable’ CART33 therapy results in potent but transient antileukemia activity
As CD33 is expressed on normal hematopoietic cells and tissue resident macrophages (Figures 1a and c), it is important to validate the potential therapeutic index prior to clinical application. We therefore developed RNA-modified CART33. Electroporation of T cells with RNA-modified CAR33 resulted in high level expression of CAR that gradually diminished over 7 days (Figures 7a and b). When compared with lentivirally transduced CART33 (LV-CART33), RNA-modified CART33 cells have similar but transient in vitro activity. Incubation of MOLM14 with RNA-CART33 cells resulted in specific cytotoxicity comparable to LV-CART33 at E:T ratios of 1:1 and 2:1. Importantly, cytotoxicity decreased with time postelectroporation (Figure 7c). We then tested the combination of RNA-CART33 therapy with chemotherapy in vivo. NSG mice engrafted with MOLM14 were treated with either the combination of cyclophosphamide (60 mg/kg intraperitoneally) for two doses plus RNA-CART33 or cyclophosphamide plus UTD. (10×106 T cells were given intravenously in three doses, Figure 7d). Cyclophosphamide was used as a lympho-depleting regimen and has been shown to enhance RNA-modified CART cell persistence.24,25 RNA-modified CART33 therapy resulted in improved leukemic control and survival advantages (Figure 7e).
Figure 7.
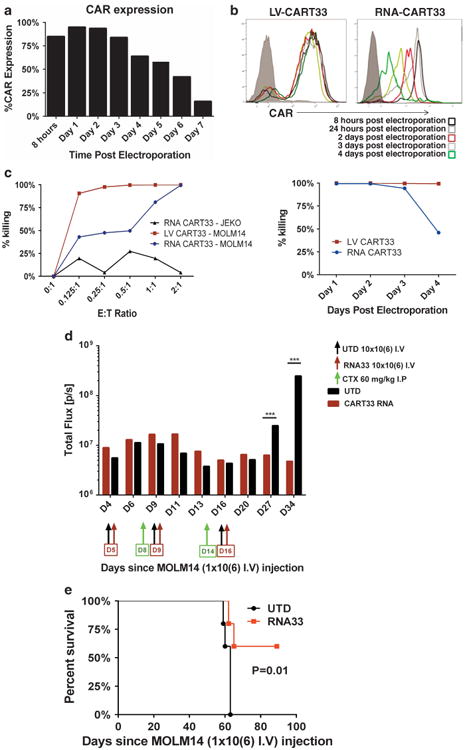
RNA-modified CART33 cells have transient CAR expression and result in significant in vitro and in vivo activity. (a) Transient expression of CAR following RNA modification. RNA-modified CAR33 expression as correlated with time postelectroporation. CAR expression reaches the peak at 24 h postelectroporation and gradually decreases after that. (b) The mean fluorescence intensity (MFI) of CAR expression in mRNA CART33 compared with lentivirally transduced CART33. The MFI of CAR expression decreases with time postelectroporation, while there is stable expression of CAR in lentivirally transduced T cells. (c) RNA-CART33 treatment results in specific killing against MOLM14 that is comparable to LV-transduced CART33. RNA-modified CART33 and LV-transduced CART33 cells were incubated with the CD33-positive cell line MOLM14-luc and a control mantle cell lymphoma cell line JEKO-luc at different E:T ratios as indicated. Bioluminescence imaging was performed after 24 h as a measure of living cells. This experiment was repeated at different time points postelectroporation of T cells. Twenty-four hours postelectroporation, RNA CART33 resulted in the most specific killing of MOLM14 cell line (comparable to LV-transduced CART33) that decreased with time post electroporation. (d) RNA-CART33 therapy combined with lympho-depleting chemotherapy results in further reduction of leukemic burden in MOLM14-engrafted xenografts. NSG mice were injected with MOLM14-luc (1 × 106 intravenously (IV)) and imaged to confirm engraftment 4 days later. Mice were then randomized to receive either RNA-CART33 with cyclophosphamide or UTD cells with cyclophosphamide (60 mg/kg intraperitoneally). T cells were given at a dose of 10 ×106 IV on days 5, 9 and 16. Lympho-depleting doses of cyclophosphamide were given on days 8 and 14, prior to T-cell infusion. (e) RNA-CART33 treatment combined with lympho-depleting chemotherapy result in prolonged survival compared with control T cells with chemotherapy.
Discussion
In this study, we report the preclinical activity and safety of CART33 therapy in AML and propose a safety mechanism of ‘biodegradable’, transiently expressed CART33 as an approach to avoid prolonged toxicity when used in patients with refractory AML. This is the first preclinical report of the activity of CART33 treatment that includes comprehensive survival and toxicity data incorporating multiple mouse models as well as the antileukemic activity of RNA-modified CART33 cells. In this report, we use a second-generation 41BB-costimulated CART33, with an scFv derived from GO. CART33 cells exhibited potent effector functions against AML cell line and primary AML samples, including specific killing at low E:T ratios, degranulation, profound proliferation and robust cytokine production, and were also active against a MDS clone after only 4 h of incubation at 1:1 ratio. Treatment with CART33 also resulted in eradication of AML and increased survival in both MOLM14 and primary AML xenografts after a single infusion. We observed the expected myeloid hematopoietic toxicity with CART33 in two different humanized mouse models. Because of the potential myelotoxicity and concerns for CD33 expression on resident tissue macrophages, we developed RNA-modified CART33 cells and show potent, but transient, in vitro activity. We also show an antileukemic effect by combining RNA-CART33 and lympho-depleting chemotherapy.
Other preclinical reports of CART cell therapy in AML have been published.22,34–36 In addition to CD123-directed CART cells, CD44v6-directed CARs were also shown to have preclinical activity, but target expression is heterogeneous on leukemic blasts and is also expressed on monocytes and keratinocytes.34 The preclinical activity of anti-CD33 chimeric receptor expressing EBV-CTL37 and anti-CD33 CAR cytokine-induced killer (CIK) cells36 have also been reported.
Our results differ from those of other groups that have published on anti-CD33-modified T cells in AML. Pizzitola et al.36 reported the preclinical activity of CIK cells genetically modified to express CD33 CAR. A direct comparison of results is problematic, but we note that, in vitro, cytotoxicity of CIK CAR33 was inferior to our CART33 despite incubation at much higher E:T ratios. Pizzitola et al.36 used KG-1-engrafted NSG mice and treated them with three doses of 10 × 106 redirected CIK cells with their best response being slowing of disease progression, while in our study a single dose of CART33 (dose of ≤ 5 × 106) led to remissions and survival benefits. Although CIK CAR33 cells were reported to induce modest hematological toxicity, it is conceivable that this was simply a reflection of their diminished activity. These striking differences could be related to a myriad of variables that are known to affect CAR function and antitumor activity. Pizzitola et al.36 used a third-generation construct that is derived from a different scFV, CD28/OX40 costimulatory domains, CIK as effectors, retroviral transduction, high-dose cytokines and OKT3 for stimulation and expansion. Our construct, on the other hand, is a second-generation CAR that uses 41BB costimulatory domain; our vector is a lentivirus vector and T cells are expanded using anti CD3/CD28 beads. In the present study, we used a similar backbone to that of our clinical CART19 studies. Although the optimal CAR design remains unknown, our clinical results from the CART19 trials have shown T-cell persistence and long-term remissions in patients with relapsed refractory acute lymphoblastic leukemia.8–10
Our experiments also show that the IgG4 hinge is superior to the CD8 hinge in this particular model and with this particular scFv/target combination. The IgG4 molecule is significantly different from the CD8 molecule. It contains three times more amino acids, which could result in a more flexible hinge. This is an important observation that is different from our previous experience with CART1923 (where the use of CD8 hinge was superior) and suggests that empirical CAR design is warranted for each new target.23,38,39
The hematopoietic toxicity and reduction in myeloid progenitor as well as peripheral blood cytopenia we observed with CART33 in these preclinical studies is expected based on CD33 expression on leukemic as well as myeloid progenitors and was similar to that observed after CART123 treatment.
The CD33 expression on resident macrophages and especially on Kupffer cells raises concerns for hepatotoxicity when CART33 is applied in clinical trials. However, it is unknown whether Kupffer cell depletion leads to hepatotoxicity. Although GO was associated with hepatotoxicity and veno-occlusive disease,40 this could have been related to calicheamicin, as other CD33-targeted therapies were not associated with significant hepatotoxicity; treatment of acute lymphoblastic leukemia with inotozumab ozogamicin (an anti-CD22 antibody conjugated to calicheamicin) resulted in similar rates of hepatotoxicity to that seen with GO.41,42
Two studies reported on the feasibility of CART cell therapy in AML.43,44 Ritchie et al.43 published a study of anti-Le-Y CART cell therapy in AML, indicating that this approach is feasible in highly proliferative malignancy such as AML. Importantly, an AML patient was recently treated with CIK-CART33 in the Chinese PLA General Hospital. Although the efficacy of their CART33 was transient, only mild fluctuations in bilirubin were noted, indicating that this approach may be safe in patients with refractory AML.44
Nonetheless, potential for off-target toxicity with the use of CART33 mandates the incorporation of transiently expressed rather than permanent CAR in clinical trials, particularly with potent constructs. Our group has had extensive experience with RNA-modified CARs in preclinical models,24,25 and a phase I trial of RNA-modified antimesothelin CART cells in patients with solid tumor at our institution showed that this approach is safe and feasible.45 Hence, we developed RNA-modified CART33 as a way to transiently express ‘biodegradable’ CAR in order to mitigate potential off-target toxicity.
We then tested the efficacy of combining multiple infusions of RNA-modified CART33 cells with cyclophosphamide as lympho-depleting chemotherapy in AML mouse xenografts (Figure 7d). This combination resulted in deeper and longer responses and in survival advantages for these mice.
Collectively, our observations highlight several potential translational applications for RNA-modified CART33 therapy. This can be used alone or in combination with chemotherapy to render patients with relapsed refractory AML transplant eligible. In addition, RNA-modified CART33 can potentially be incorporated in conditioning regimens prior to allogeneic stem cell transplantation. The use of RNA-CART33 would allow subsequent stem cell transplantation. In contrast, due to the persistence of LV-CART33 cells, a donor graft would likely be rejected.
Once the safety and feasibility of a biodegradable CART33 has been demonstrated in patients, future strategies could include lentiviral CART33 with an ‘off’ switch, such as a suicide gene.46 Furthermore, our findings suggest that both CART33 and CART123 are effective against AML and open up new therapeutic horizons in combinatorial targeted cellular therapy.
Supplementary Material
Acknowledgments
This work was funded by a research agreement with Novartis Pharmaceuticals, a Leukemia and Lymphoma Society Specialized Centers of Research grant to CHJ, a National Institutes of Health T32 award to MK (T32-GM008076) and an American Society of Hematology Scholar award to SG. We thank Fang Chen and Simon Lacey for performing Luminex assay. Imaging was performed at the University of Pennsylvania Small Animal Imaging Facility (SAIF) Optical/Bioluminescence Core, supported by NIH grant CA016520.
Footnotes
Conflict of Interest: CHJ and DLP have filed patent applications related to CAR technology and could potentially receive licensing royalties from Novartis corporation. The other authors declare no conflict of interest.
Author Contributions: SSK designed and performed research, analyzed data and wrote the paper; MR and SG designed, performed research and wrote the paper; OS performed research, MK, VA, JM and DS performed research; JS contributed reagents and performed research; DLP designed research and wrote the paper; MC designed research and contributed reagents; and CHJ designed research, contributed reagents and wrote the paper. All authors read and approved the manuscript.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Thein MS, Ershler WB, Jemal A, Yates JW, Baer MR. Outcome of older patients with acute myeloid leukemia: an analysis of SEER data over 3 decades. Cancer. 2013;119:2720–2727. doi: 10.1002/cncr.28129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamadani M, Awan FT, Copelan EA. Hematopoietic stem cell transplantation in adults with acute myeloid leukemia. Biol Blood Marrow Transplant. 2008;14:556–567. doi: 10.1016/j.bbmt.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 4.Duval M, Klein JP, He W, Cahn JY, Cairo M, Camitta BM, et al. Hematopoietic stem-cell transplantation for acute leukemia in relapse or primary induction failure. J Clin Oncol. 2010;28:3730–3738. doi: 10.1200/JCO.2010.28.8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Estey EH. Acute myeloid leukemia: 2013 update on risk-stratification and management. Am J Hematol. 2013;88:318–327. doi: 10.1002/ajh.23404. [DOI] [PubMed] [Google Scholar]
- 6.Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39:49–60. doi: 10.1016/j.immuni.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA. 1989;86:10024–10028. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter D, Rheingold S, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2014;33:540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cruz CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013;122:2965–2973. doi: 10.1182/blood-2013-06-506741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walter RB, Gooley TA, van der Velden VH, Loken MR, van Dongen JJ, Flowers DA, et al. CD33 expression and P-glycoprotein-mediated drug efflux inversely correlate and predict clinical outcome in patients with acute myeloid leukemia treated with gemtuzumab ozogamicin monotherapy. Blood. 2007;109:4168–4170. doi: 10.1182/blood-2006-09-047399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griffin JD, Linch D, Sabbath K, Larcom P, Schlossman SF. A monoclonal antibody reactive with normal and leukemic human myeloid progenitor cells. Leuk Res. 1984;8:521–534. doi: 10.1016/0145-2126(84)90001-8. [DOI] [PubMed] [Google Scholar]
- 18.Dinndorf PA, Andrews RG, Benjamin D, Ridgway D, Wolff L, Bernstein ID. Expression of normal myeloid-associated antigens by acute leukemia cells. Blood. 1986;67:1048–1053. [PubMed] [Google Scholar]
- 19.Schwonzen M, Diehl V, Dellanna M, Staib P. Immunophenotyping of surface antigens in acute myeloid leukemia by flow cytometry after red blood cell lysis. Leuk Res. 2007;31:113–116. doi: 10.1016/j.leukres.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 20.Hoyer JD, Grogg KL, Hanson CA, Gamez JD, Dogan A. CD33 detection by immunohistochemistry in paraffin-embedded tissues: a new antibody shows excellent specificity and sensitivity for cells of myelomonocytic lineage. Am J Clin Pathol. 2008;129:316–323. doi: 10.1309/E36008Y2H08Q1AYY. [DOI] [PubMed] [Google Scholar]
- 21.Hills RK, Castaigne S, Appelbaum FR, Delaunay J, Petersdorf S, Othus M, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15:986–996. doi: 10.1016/S1470-2045(14)70281-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123:2343–2354. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther. 2013;24:717–727. doi: 10.1089/hum.2013.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M, et al. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther. 2011;22:1575–1586. doi: 10.1089/hum.2011.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, et al. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther. 2006;13:151–159. doi: 10.1016/j.ymthe.2005.07.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Betts MR, Koup RA. Detection of T-cell degranulation: CD107a and b. Methods Cell Biol. 2004;75:497–512. doi: 10.1016/s0091-679x(04)75020-7. [DOI] [PubMed] [Google Scholar]
- 28.Cao LF, Krymskaya L, Tran V, Mi S, Jensen MC, Blanchard S, et al. Development and application of a multiplexable flow cytometry-based assay to quantify cell-mediated cytolysis. Cytometry A. 2010;77:534–545. doi: 10.1002/cyto.a.20887. [DOI] [PubMed] [Google Scholar]
- 29.Pollard JA, Alonzo TA, Loken M, Gerbing RB, Ho PA, Bernstein ID, et al. Correlation of CD33 expression level with disease characteristics and response to gemtuzumab ozogamicin containing chemotherapy in childhood AML. Blood. 2012;119:3705–3711. doi: 10.1182/blood-2011-12-398370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walter RB, Raden BW, Kamikura DM, Cooper JA, Bernstein ID. Influence of CD33 expression levels and ITIM-dependent internalization on gemtuzumab ozogamicin-induced cytotoxicity. Blood. 2005;105:1295–1302. doi: 10.1182/blood-2004-07-2784. [DOI] [PubMed] [Google Scholar]
- 31.Ehninger A, Kramer M, Rollig C, Thiede C, Bornhauser M, von Bonin M, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014;4:e218. doi: 10.1038/bcj.2014.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wunderlich M, Chou FS, Link KA, Mizukawa B, Perry RL, Carroll M, et al. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia. 2010;24:1785–1788. doi: 10.1038/leu.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Casucci M, Nicolis di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122:3461–3472. doi: 10.1182/blood-2013-04-493361. [DOI] [PubMed] [Google Scholar]
- 35.Mardiros A, Dos Santos C, McDonald T, Brown CE, Wang X, Budde LE, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122:3138–3148. doi: 10.1182/blood-2012-12-474056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, Spinelli O, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014;28:1596–1605. doi: 10.1038/leu.2014.62. [DOI] [PubMed] [Google Scholar]
- 37.Dutour A, Marin V, Pizzitola I, Valsesia-Wittmann S, Lee D, Yvon E, et al. In vitro and in vivo antitumor effect of anti-CD33 chimeric receptor-expressing EBV-CTL against CD33 acute myeloid leukemia. Adv Hematol. 2012;2012:683065. doi: 10.1155/2012/683065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The non-signaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2014;3:125–135. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lympho-blastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larson RA, Sievers EL, Stadtmauer EA, Lowenberg B, Estey EH, Dombret H, et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer. 2005;104:1442–1452. doi: 10.1002/cncr.21326. [DOI] [PubMed] [Google Scholar]
- 41.Kantarjian H, Thomas D, Jorgensen J, Kebriaei P, Jabbour E, Rytting M, et al. Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer. 2013;119:2728–2736. doi: 10.1002/cncr.28136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kantarjian H, Thomas D, Jorgensen J, Jabbour E, Kebriaei P, Rytting M, et al. Inotuzumab ozogamicin, an anti-CD22-calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: a phase 2 study. Lancet Oncol. 2012;13:403–411. doi: 10.1016/S1470-2045(11)70386-2. [DOI] [PubMed] [Google Scholar]
- 43.Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K, et al. Persistence and efficacy of second generation CAR-T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 2013;21:2122–2129. doi: 10.1038/mt.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2014;23:184–191. doi: 10.1038/mt.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonini C, Bondanza A, Perna SK, Kaneko S, Traversari C, Ciceri F, et al. The suicide gene therapy challenge: how to improve a successful gene therapy approach. Mol Ther. 2007;15:1248–1252. doi: 10.1038/sj.mt.6300190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.