

ABSTRACT
The production of glycolytic end products, such as lactate, usually evokes a cellular shift from aerobic to anaerobic ATP generation and O2 insufficiency. In the classical view, muscle lactate must be exported to the liver for clearance. However, lactate also forms under well-oxygenated conditions, and this has led investigators to postulate lactate shuttling from non-oxidative to oxidative muscle fiber, where it can serve as a precursor. Indeed, the intracellular lactate shuttle and the glycogen shunt hypotheses expand the vision to include a dynamic mobilization and utilization of lactate during a muscle contraction cycle. Testing the tenability of these provocative ideas during a rapid contraction cycle has posed a technical challenge. The present study reports the use of hyperpolarized [1-13C]lactate and [2-13C]pyruvate in dynamic nuclear polarization (DNP) NMR experiments to measure the rapid pyruvate and lactate kinetics in rat muscle. With a 3 s temporal resolution, 13C DNP NMR detects both [1-13C]lactate and [2-13C]pyruvate kinetics in muscle. Infusion of dichloroacetate stimulates pyruvate dehydrogenase activity and shifts the kinetics toward oxidative metabolism. Bicarbonate formation from [1-13C]lactate increases sharply and acetyl-l-carnitine, acetoacetate and glutamate levels also rise. Such a quick mobilization of pyruvate and lactate toward oxidative metabolism supports the postulated role of lactate in the glycogen shunt and the intracellular lactate shuttle models. The study thus introduces an innovative DNP approach to measure metabolite transients, which will help delineate the cellular and physiological role of lactate and glycolytic end products.
KEY WORDS: Muscle metabolism, Lactate bioenergetics, Hyperpolarized 13C, Pyruvate
Summary: Hyperpolarized 13C NMR is used to measure the kinetics of lactate with 3 s resolution in skeletal muscle to validate the tenability of the intracellular lactate shuttle and glycogen shunt models.
INTRODUCTION
Biologists often view lactate as a hypoxia indicator and a dead-end product of glycolysis, which muscle must export to liver for clearance in the Cori cycle (Brooks et al., 2000). From this perspective, cellular bioenergetics demarcates conveniently into non-oxidative and oxidative pathways. Without O2, glycolysis can continue to produce ATP by rege`nerating oxidizing equivalent through lactate formation. For many, lactate still implicates an O2 insufficiency (Kreutzer and Jue, 1995).
But lactate also forms in muscle under well-oxygenated conditions (Brooks, 1986a). Such observations have led investigators to question a strict correspondence of the appearance of lactate with an anaerobic threshold (Brooks, 1986b; Gladden, 2004; Wasserman, 1987). Indeed, radioactive tracer experiments have detected the steady state disappearance of lactate in muscle and the appearance of labeled CO2. CO2 formation signals the oxidative metabolism of lactate by pyruvate dehydrogenase (PDH) and tricarboxylic acid (TCA) enzymes (Stanley and Brooks, 1987; Stanley et al., 1985, 1986). 13C NMR studies have also detected the oxidation of [3-13C]lactate to 13C-labeled glutamate in the muscle of a perfused hindquarter model (Bertocci et al., 1997; Bertocci and Lujan, 1999). The collective evidence has helped to spawn the hypothesis that lactate produced in glycolytic fibers can shuttle to oxidative muscles and become a metabolic precursor. In fact, the detection of lactate dehydrogenase (LDH) in the mitochondria has expanded the notion to include a lactate shuttle between the cytosol and the mitochondria (Brooks, 2000; Brooks and Hashimoto, 2007; Passarella et al., 2014; Sahlin et al., 2002; Yoshida et al., 2007).
Nevertheless, the biochemical mechanism underpinning the accumulation of lactate under aerobic conditions still requires clarification. In 2001, a novel hypothesis emerged to rationalize aerobic lactate formation as part of a dynamic glycogen flux during a muscle twitch (Shulman and Rothman, 2001). Because NMR has detected a large energy fluctuation during each contraction, the cell must dynamically restore glycogen in order to sustain any extended period of contractions (Chung et al., 1998). The glycogen shunt model envisions glycogen providing the immediate energy source during the millisecond contraction phase. A rapid resynthesis using glucose and lactate during the extended relaxation phase replenishes the glycogen pool. Because glucose must shunt to glycogen to create the immediate energy precursor, it introduces inefficiency in ATP generation, which would lead to a lactate build-up, even under aerobic conditions. Consequently, during the relatively long relaxation phase of each contraction cycle, the accumulated lactate can serve as a precursor for acetyl CoA formation and oxidative ATP production (Shulman, 2005). In essence, the cell uses the accumulated lactate during aerobic muscle contraction to buffer energy utilization and restore the glycogen pool.
However, both the glycogen shunt and intracellular lactate shuttle models require a rapid mobilization and utilization of lactate. Within seconds, PDH must compete effectively with LDH and alanine aminotransferase (ALT) to direct pyruvate toward lactate transport into the mitochondria or oxidative metabolism. If lactate cannot mobilize rapidly and serve as a metabolic precursor in seconds, then both hypotheses immediately become invalidated.
Testing the tenability of these provocative models has posed many technical challenges because innovative methodological approaches must track in seconds the lactate kinetics in the in situ muscle. We report here the use of hyperpolarized [1-13C]lactate and [2-13C]pyruvate in dynamic nuclear polarization (DNP) NMR experiments to measure the rapid lactate kinetics in rat muscle (Ardenkjaer-Larsen et al., 2003). With a 3 s temporal resolution, 13C DNP NMR has detected both the dynamics of [1-13C]lactate and [2-13C]pyruvate in muscle. Indeed, in situ muscle enzymes can use either pyruvate or lactate as a substrate. The [1-13C] label of lactate appears rapidly in pyruvate, alanine and bicarbonate (HCO3−). HCO3− formation reflects the PDH activity, which leads to acetyl CoA production and oxidative ATP generation. Using dichloroacetate (DCA) to mimic the PDH activation during muscle contraction shifts dramatically the lactate substrate toward oxidative metabolism. With [2-13C]pyruvate as the precursor, DCA increases the formation of acetyl-l-carnitine (ALCAR), acetoacetate (AcAc) and glutamate (Howlett et al., 1999; Stacpoole, 1989).
List of symbols and abbreviations
- AcAc
acetoacetate
- ALCAR
acetyl-l-carnitine
- ALT
alanine aminotransaminase
- DCA
dichloroacetate
- DNP
dynamic nuclear polarization
- LDH
lactate dehydrogenase
- MCT
monocarboxylate transporter
- PDH
pyruvate dehydrogenase
- PDHa
pyruvate dehydrogenase activity
- PDK
pyruvate dehydrogenase kinase
- r
metabolite production rate
- tC
total [13C] signal
- T1
spin lattice relaxation time
- VO2
oxygen consumption
- τ
time to half-maximum
Such rapid mobilization of pyruvate and lactate toward oxidative metabolism supports the postulated role of lactate in the glycogen shunt and the intracellular lactate shuttle model. The Cori cycle does not present the exclusive disposal pathway for lactate formed in muscle. The report then introduces an innovative approach to measure metabolite transients, to delineate the dynamic role of lactate in muscle, to provide evidence supporting two provocative models and to stimulate a discussion about the commonly accepted function of glycolytic end products (Grieshaber et al., 1994; Kreutzer and Jue, 1997).
RESULTS
Fig. 1 shows the time-averaged 13C signals in rat leg muscle without and with the addition of DCA following an injection of hyperpolarized [1-13C]lactate. The spectra reflect the averaging of all transients between 0 and 2 min. The peak assignments were as follows: [1-13C]lactate (185 ppm), [1-13C]alanine (179 ppm), [1-13C]pyruvate (173 ppm), H13CO3− (163 ppm) and 13CO2 (127 ppm). The spectra were normalized to the [1-13C]lactate peak to correct for the varying extent of nuclear polarization. In the presence of DCA, alanine and pyruvate signals decreased, whereas HCO3− and CO2 signals increased (Table 1).
Fig. 1.

Time-averaged 13C signals from rat skeletal muscle after injection of 40 mmol l−1 hyperpolarized [1-13C]lactate. (A) Control. (B) 1 h after DCA infusion. Each spectrum is normalized to the corresponding [1-13C]lactate peak intensity and time averaged for 0–2 min. Relative to the signal intensity of [1-13C]lactate, the DCA stimulation of PDH produces a marked change in 13C spectra, as noted in intensity changes of the alanine, pyruvate, HCO3− and CO2 signals. Scaling all signals to the initial lactate signal intensity corrects for variation in 13C polarization.
Table 1.
Dynamics of [1-13C]lactate
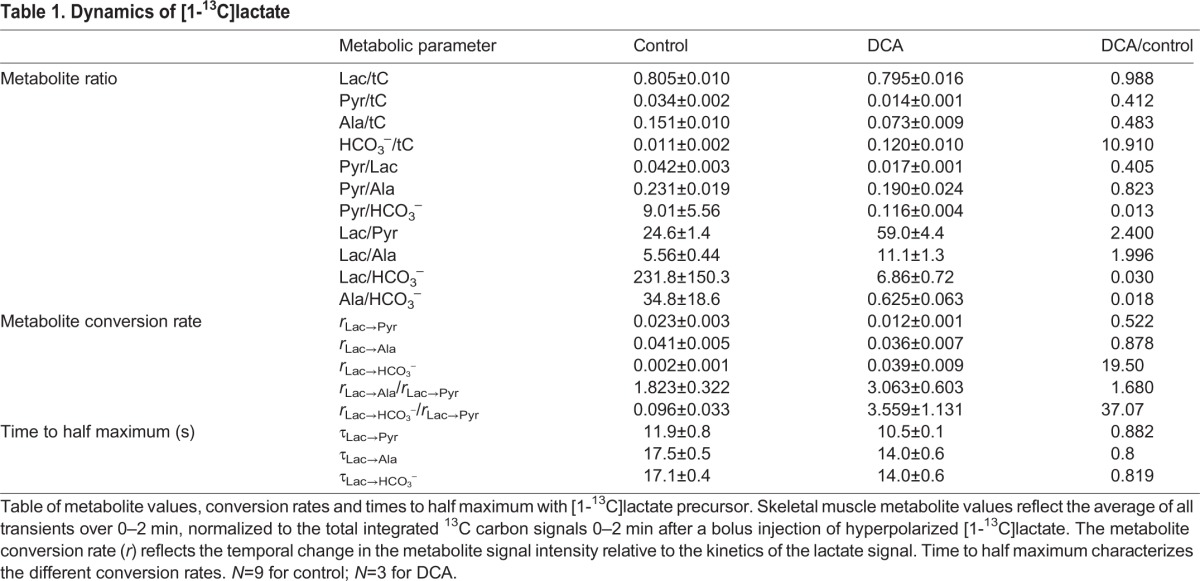
Fig. 2 shows the kinetics after injection of hyperpolarized [1-13C]lactate in rat leg muscle with and without the addition of DCA. Without DCA, the composite metabolism and exchange rates (rx→y) relative to the time-dependent change of the lactate signal were as follows: rLac→Pyr=0.023, rLac→Ala=0.041, rLac→HCO3−=0.002. With DCA, the pyruvate and HCO3− rates changed significantly: rLac→Pyr=0.012, rLac→Ala=0.036 and rLac→HCO3−=0.039. With DCA, rLac→HCO3− increased by almost a factor of 10, rLac→Pyr decreased by a factor of 2 and rLac→Ala decreased slightly (Table 1).
Fig. 2.

Dynamic changes in the 13C signals from rat skeletal muscle after injection of 40 mmol l−1 hyperpolarized [1-13C]lactate. (A) Control. (B) 1 h after DCA infusion. Each time course (3 s resolution) is normalized by the peak value of the corresponding [1-13C]lactate curve. Relative to the signal intensity of [1-13C]lactate, the DCA stimulation of PDH produces a rapid time-dependent change in 13C spectra, as noted in dynamics of the alanine, pyruvate and HCO3− signals.
The integrated area of all the carbon signals (tC) served to normalize the 13C signals to yield insight into the metabolite distribution. In control muscle, the incoming [1-13C]lactate presented the most intense signal lactate/tC=0.805. The 13C label distributed as follows: pyruvate, 0.034; alanine, 0.151; HCO3−, 0.011. The pyruvate/alanine ratio was 0.231. After DCA injection, which stimulates PDH activity, the lactate/tC signal did not change significantly (0.795). However, the 13C label distribution changed as follows: pyruvate, 0.014; alanine, 0.073; HCO3−, 0.120. The pyruvate/alanine ratio now stood as 0.190. DCA increased the HCO3−/tC ratio almost 11 times. The pyruvate/alanine ratio decreased by 18%, whereas the pyruvate/lactate ratio fell by 60% (Table 1).
Fig. 3 shows a typical time-averaged spectra (0–2 min) of the metabolite signals in muscle generated from the precursor [2-13C]pyruvate (208 ppm). [2-13C]pyruvate converted to [2-13C]lactate (70 ppm), [2-13C]alanine (52 ppm) and [2-13C]pyruvate hydrate (PyH; 96 ppm) (Fig. 3A).
Fig. 3.

Time-averaged 13C signals from rat skeletal muscle after injection of 80 mmol l−1 hyperpolarized [2-13C]pyruvate. (A) Control. (B) 1 h after DCA infusion. Each spectrum is normalized by the corresponding [2-13C]pyruvate peak intensity and time averaged for 0–2 min. Relative to the signal intensity of [2-13C]pyruvate, the DCA stimulation of PDH produces a marked change in 13C spectra, as noted in intensity changes of the ALCAR, AcAc and glutamate signals. Scaling all signals to the initial pyruvate signal intensity corrects for variation in 13C polarization.
In control muscle, the DNP experiments could not detect any conversion of [2-13C]pyruvate to glutamate, ALCAR or AcAc. However, with DCA, DNP NMR could detect the conversion of [2-13C]pyruvate to [5-13C]glutamate (184 ppm), [1-13C]ALCAR (175 ppm), [1-13C]AcAc (177 ppm) and [3-13C]AcAc (213 ppm) (Fig. 3B).
Fig. 4 shows the dynamic metabolic response to injected hyperpolarized [2-13C]pyruvate. In control muscle, only lactate and pyruvate had sufficient signal-to-noise ratio to map the dynamic response confidently. With DCA, the ALCAR signal appeared prominently. The time to half maximum for [2-13C]lactate (τPyr→Lac) decreased from 13.1 s in control to 10.6 s after DCA. However, the lactate production rate (rPyr→Lac) did not change significantly between the control (0.053) and DCA-treated (0.048) muscle. Normalized to the lactate kinetics, DCA stimulated the production of ALCAR at rPyr→Alcar=0.026 with a τPyr→Alcar=13.6, Table 2.
Fig. 4.

Dynamic changes in the 13C signal from rat skeletal muscle after injection of 80 mmol l−1 hyperpolarized [2-13C]pyruvate. (A) Control. (B) 1 h after DCA infusion. Each time course (3 s resolution) is normalized by the peak value of the corresponding [2-13C]pyruvate curve. Relative to the signal intensity of [2-13C]pyruvate, the DCA stimulation of PDH produces a rapid time-dependent change in 13C spectra, as noted in a rapid time-dependent changes of the lactate and ALCAR signals.
Table 2.
Dynamics of [2-13C]pyruvate
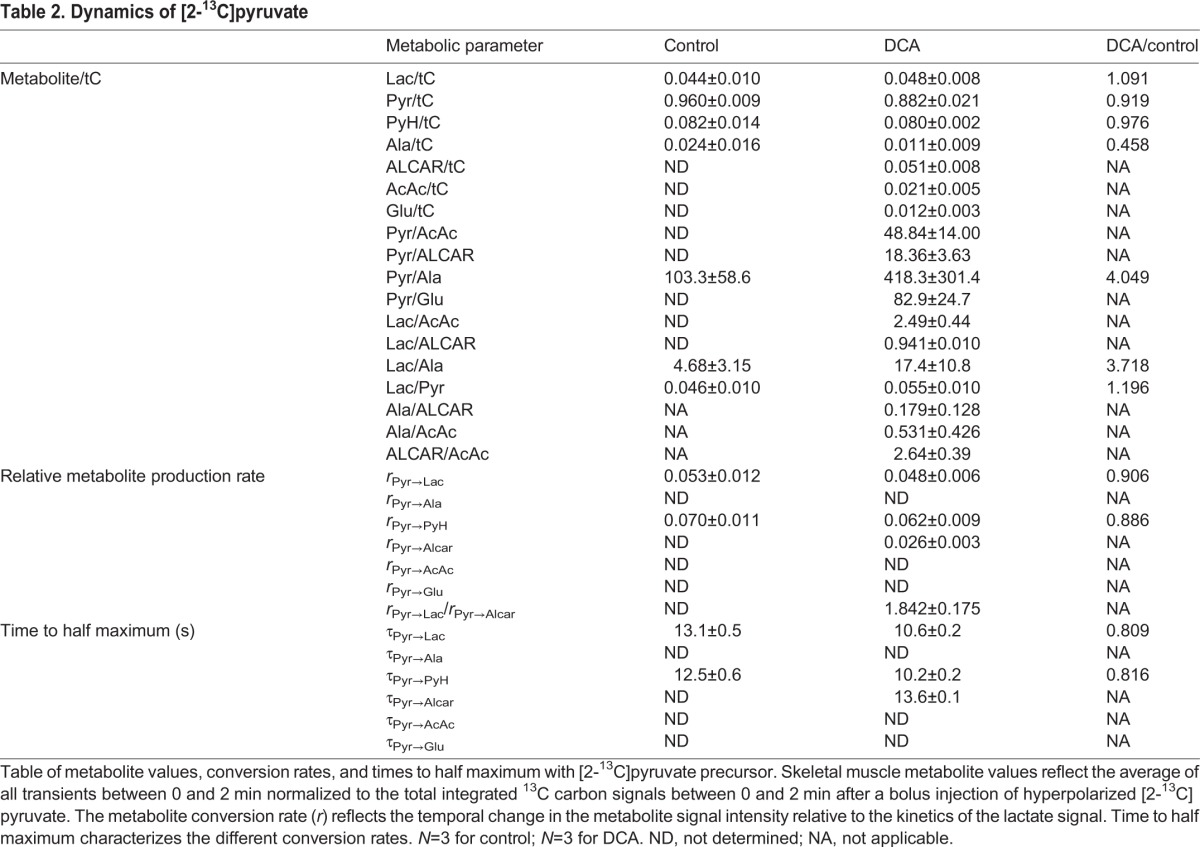
Normalized to tC, the 13C label distributed to the metabolites in control muscle as follows: pyruvate/tC=0.960, lactate/tC=0.044, PyH/tC=0.082 and alanine/tC=0.024. With DCA, pyruvate/tC=0.882, lactate/tC=0.048, PyH/tC=0.080 and alanine/tC=0.011. In addition, other metabolite signals now appeared: ALCAR/tC=0.051, AcAc/tC=0.021, glutamate/tC=0.012 (Table 2).
3D metabolite maps of lactate, alanine, pyruvate and HCO3− were reconstructed from the 2.5 h post-DCA chemical shift imaging (CSI) data (Fig. 5). Normalized metabolite maps (lactate/tC, alanine/tC, HCO3−/tC, pyruvate/tC) and a ratio map of products (alanine/HCO3−) are shown in axial and coronal planes. Despite the large point spread function and partial volume effects, each metabolite map in both axial and coronal planes showed distinct distribution from the other metabolite maps.
Fig. 5.

Overlays of anatomical and metabolite images of rat leg muscle acquired using hyperpolarized [1-13C]lactate. (A) Axial images. (B) Coronal images. 3D CSI image was collected 2.5 h post DCA and overlaid on a 3D anatomical image. 13C MR metabolite maps (lactate, alanine, HCO3−, pyruvate) and metabolite-ratio maps (lactate/tC, alanine/tC, HCO3−/tC, pyruvate/tC, alanine/HCO3−), acquired 25 s after the injection of 40 mmol l−1 hyperpolarized [1-13C]lactate. Although different biodistributions of alanine/tC, HCO3−/tC and pyruvate/tC were observed, a large amount of metabolic products was produced from skeletal muscle tissues (HCO3−/tC). The lactate image would accentuate the vasculature if the vascular volume had a significant contribution. The yellow numbers next to color bars represent relative scale of each metabolite map to the maximum lactate signal or metabolite ratios. The white line in A and B denotes the slice used to construct the respective images, as denoted by the blue arrows. The white line in A and B denotes the slice used to construct the respective images, as denoted by the blue arrows. FOV, field of view.
Figs 6 and 7 show the graphical representations of the 13C label distribution in various metabolites and the production rates during infusion of [1-13C]lactate and [2-13C]pyruvate, respectively.
Fig. 6.

Different metabolite enzyme activity ratios in rat skeletal muscle after the injection of 40 mmol l−1 hyperpolarized [1-13C]lactate. (A) Metabolite ratios calculated from time-averaged (0–2 min) spectra. (B) Metabolite production rates (r). (C) Time to half-maximum (τ). Asterisk indicates statistically significant difference between pre-/post-DCA measurements (*P<0.05).
Fig. 7.

Different metabolite enzyme activity ratios in rat skeletal muscle after the injection of 80 mmol l−1 hyperpolarized [2-13C]pyruvate. (A) Metabolite ratios calculated from time-averaged (0–2 min) spectra. (B) Metabolite production rates (r). (C) Time to half-maximum (τ). Glutamate, ALCAR and AcAc are detected post-DCA only. Asterisk indicates statistically significant difference between pre-/post-DCA measurements (*P<0.05). Double asterisks indicate that the metabolite was detected only post-DCA.
DISCUSSION
Biodistribution of lactate in muscle
In resting muscle cells and normoxic rat blood, pyruvate and lactate concentrations hover around 50 μmol l−1 and 700 μmol l−1, respectively (Goodman et al., 1978). In blood, recent experiments have detected 60 μmol l−1 pyruvate in the basal state (Atherton et al., 2011). After an injection of pH buffered and osmotically balanced 80 mmol l−1 pyruvate, as described in the Materials and methods, the pyruvate level reached 250 μmol l−1 in blood after 1 min. It remained well above 150 μmol l−1 even after 10 min. Other metabolite levels (glucose, insulin, lactate, TAG and NEFA) and physiological parameters, however, exhibit no significant perturbation (Atherton et al., 2011). Injected lactate will presumably reach a similar concentration.
After addition of the hyperpolarized precursors, pyruvate and lactate distribute first in the vasculature and in the interstitial volume, before they enter the cell via a family of monocarboxylate transporters (MCTs) or by free diffusion at high concentrations (>10 mmol l−1) (Juel, 1991; Poole and Halestrap, 1993). The precursor and metabolite product signals in the CSI images show different spatial distributions, resulting in inhomogeneous metabolite ratio maps (lactate/tC, alanine/tC, HCO3−/tC, alanine/HCO3−). While the volumetric CSI maps also suggest their potential utility to assess spatial information of metabolic function, the images confirm that all metabolite signals do not have a predominant contribution from the vasculature. Therefore, the observed 13C DNP signals originate primarily from the 13C-labeled molecules in the muscle/fat tissue. In particular, HCO3− production from large muscle regions, as shown in the HCO3−/tC ratio map, indicates that the DCA-modulated PDH activation globally affects the metabolism in leg muscle.
In fact, the precursor signal in the vasculature will probably not contribute significantly to a non-localized spectrum, since blood constitutes <5% of the total muscle volume. However, the interstitial and intracellular space constitutes, respectively, 20% and 35% of the total muscle volume (Boron and Boulpaep, 2003). So, the hyperpolarized signal of the precursor predominantly reflects signals from both the interstitial and intracellular space. The hyperpolarized lactate signal will increase as it distributes into the surface coil detection volume and begins to decrease as the polarization decays with apparent spin lattice relaxation time (T1) of ∼40 s (Day et al., 2007).
In muscle, MCT1 and MCT4 transporters carry the lactate and pyruvate into the cell with a Km of 13–40 mmol l−1 for l-lactate and >50 mmol l−1 for pyruvate. MCT4 distributes evenly in all muscle fibers, but MCT1 localizes predominantly in oxidative fibers of skeletal muscle and heart muscle. With high-intensity or endurance training, MCT1 expression increases along with a 30–100% rise in maximal rate of lactate transport (McDermott and Bonen, 1993; Pilegaard et al., 1993). The association of MCT1 expression, distribution and increased lactate transport in oxidative muscle has implicated a role for MCT1 in the shuttle of lactate between glycolytic and oxidative muscle fibers (Gladden, 2004; Juel and Halestrap, 1999).
The MCT transporter uses an ordered sequential mechanism in which a proton binds first to the transporter and then the lactate anion binds (Roth and Brooks, 1990a,b). Both lactate and protons translocate across the membrane followed by a sequential release. Because the cycling of the free carrier across the membrane constitutes the rate-limiting step, monocarboxylate exchange proceeds substantially faster than net transport (Carpenter and Halestrap, 1994; Poole and Halestrap, 1993).
Given concentrations of ∼0.1 mmol l−1 pyruvate and ∼1 mmol l−1 lactate in resting myocyte, the sudden arrival of 0.25 mmol l−1 hyperpolarized pyruvate or lactate and the rate-limiting step of the MCT transport, the observed hyperpolarized [13C]pyruvate and [13C]lactate signals in muscle will have significant contributions from both exchange and net transport.
Biodistribution of the 13C label in the cell
The rapid 13C labeling of the lactate pool establishes a basis to map the relative enzymatic activity in the cell. Specifically, the [13C]lactate signal initially reflects the distribution kinetics of lactate into the interstitial and intracellular volume. In muscle the near-equilibrium enzyme, LDH, catalyzes usually the conversion of pyruvate to lactate to regenerate the requisite oxidizing equivalent (NAD+) to sustain glycolysis. The DNP experiments show that the LDH in muscle can also rapidly catalyze the reverse reaction from lactate to pyruvate (Bastiaansen et al., 2014). Under normal physiological conditions, the cell maintains a lactate/pyruvate ratio of approximately 10:1. The observed [13C]lactate signal and its time-dependent change can serve as a reference for analysis of other metabolite pools and kinetics.
Integrated over 2 min, the lactate signal comprises 0.805 of the total 13C signal intensities in the spectra. LDH converts lactate to pyruvate and transfers 0.034 of the total 13C label to pyruvate, assuming the same T1 as the 13C-labeled substrates. ALT catalyzes the conversion of pyruvate to alanine, which appears to have more 13C labeling (0.151) than pyruvate.
The apparent higher 13C label flow into alanine than into pyruvate reflects a significant contribution from exchange and does not necessarily indicate any difference in the flux through the transaminase versus LDH. Since myocytes contain 1.3–1.5 mmol l−1 of endogenous [12C]alanine and only 0.1 mmol l−1 [12C]pyruvate, the non-steady-state 13C exchange between alanine and pyruvate will initially increase the pool of [12C]alanine faster than [13C]pyruvate and lead to a higher signal intensity and apparent conversion rate for [13C]alanine. Studies have already confirmed a significant contribution of exchange in the LDH reaction. However, an overall model has yet to emerge to factor out the exchange versus metabolic contributions in the LDH- and ALT-catalyzed reactions, as observed in the DNP NMR experiments (Kettunen et al., 2010).
In contrast, the HCO3− signal has no contribution from exchange and reflects the unidirectional flux from pyruvate to HCO3− via PDH (Day et al., 2007; Kettunen et al., 2010). In the presence of DCA, PDH activity increases markedly and redistributes the 13C labels toward oxidative metabolism. Since DCA inhibits a pyruvate dehydrogenase kinase (PDK), which in turn stimulates PDH activity, the increased unidirectional flux from pyruvate to HCO3− mimics the biochemical adaptation during muscle contraction, when energy demand and respiration increase suddenly (Howlett et al., 1999). In contrast to the reduction in lactate observed under normal physiological conditions, DCA in the presence of infused lactate produces no significant change in the integrated lactate signal as fraction of the total carbon signal (0.795). But HCO3−/tC increases 11 times over the control level. Moreover, the relative lactate/pyruvate ratio shifts from 24.6 to 59.0 because of the decreasing pyruvate/tC ratio, which falls from 0.034 to 0.014. The pyruvate/alanine ratio also decreases from 0.231 to 0.190 and also reflects a decreasing pool of pyruvate available in the conversion to alanine. The observations confirm that an activated PDH can compete immediately and effectively with the near-equilibrium LDH and ALT enzymes for the precursor pyruvate.
NAD+/NADH
The LDH enzymatic rate depends upon the turnover of a smaller NAD+ and NADH nucleotide pool. Given that the cytosol and mitochondria constitute 90% and 10% of the cell volume, respectively, studies have estimated the NAD+ and NADH concentration in skeletal muscle as follows: total, 450 and 50 µmol kg−1 cell wet wt; cytosol, 150 and 0.28 µmol kg−1 cytosolic wet wt; mitochondria, 3150 and 500 µmol kg−1 mitochondrial wet wt (Li et al., 2009; White and Schenk, 2012). The NAD+/NADH ratio in the cytosol (∼540) stands at a higher oxidization level than the NAD+/NADH ratio in the mitochondria (∼6.3). However, the mitochondrion has a larger pool of NAD+ and NADH. More than 95% of cellular NADH localizes in the mitochondrial compartment. The NAD and NADH in the nucleus contributes slightly. It comprises only about 1% of muscle cell volume. Still, many studies consider the nuclear NAD+ and NADH concentrations in equilibrium with cytosol. In contrast to myocytes, other cells have even lower NAD+ (10–100 µmol l−1) and NADH (130 nmol l−1) values (Fjeld et al., 2003).
The rapid chemical exchange/metabolism of hyperpolarized lactate and pyruvate in muscle indicates the NAD+/NADH turnover occurs rapidly and reacts with LDH to catalyze the exchange of the 13C label and/or the conversion of substrate to net product. In skeletal muscle, LDH has no apparent limitation in catalyzing the back reaction from lactate to pyruvate, even though the reaction produces NADH, which would limit glycolysis and glycogenolysis. Infusing hyperpolarized 13C lactate leads to the rapid appearance of [13C]pyruvate and [13C]alanine. In essence, the DNP experiments find evidence that MCT can rapidly transport lactate into the myocyte, that the LDH reaction can run rapidly in either the forward direction (pyruvate to lactate) or the backward direction (lactate to pyruvate) and NAD+/NADH turnover can proceed rapidly in either direction. These observations support the feasibility of key tenets in the intracellular lactate shuttle model.
Pyruvate dehydrogenase
The relative change in the pyruvate/tC, alanine/tC, HCO3−/tC levels in muscle with DCA reveals a shift in the metabolic partitioning with PDH activation. The HCO3−/lactate and the HCO3−/pyruvate ratios increase to 34 and 78 times control levels, respectively. Relative to the observed lactate kinetics, the relative rate for the reaction from lactate to pyruvate (rlac→pyr) without and with DCA decreases about 2 times. The reaction from lactate to alanine (rLac→Ala) without and with DCA also significantly decreases. In contrast, the reaction from lactate to HCO3− (rLac→HCO3−) increases sharply to 19.5 times that of the control. A study using hyperpolarized [1-13C]pyruvate as the precursor has demonstrated an excellent linear correlation between HCO3−/pyruvate, HCO3−/alanine and rPyr→HCO3−, with PDH activity determined by conventional biochemical assays (Atherton et al., 2011). Indeed, the rLac→HCO3−/rLac→Pyr ratio in the presence of DCA shows the PDH activity (PDHa) increasing 37.1 times from 0.096 to 3.559. Human muscle biopsy experiments show that 30 s after the onset of contraction, DCA induces PDHa to increase only about 4 times (Bangsbo et al., 2002; Howlett et al., 1999). The DNP experiments show a much faster and higher dynamic rise of DCA-induced PDH activation, which implies a large capacity for PDH to activate and accommodate a significant surge in energy demand at the beginning of a contraction cycle.
The DNP experiments also indicate that PDH activity has a high control coefficient for directing pyruvate derived from lactate, glycolysis, glycogenolysis and alanine toward oxidative metabolism via formation of acetyl CoA. Once activated by DCA, PDH can shift the metabolic flux toward oxidative phosphorylation. As envisioned by the glycogen shunt theory (see below), any accumulated lactate during aerobic muscle contraction can act quickly to buffer energy loss and react immediately with PDH to form acetyl CoA, a precursor for the TCA cycle and oxidative phosphorylation. Already, physiological studies on humans have observed a similar shift in the PDHa at the onset of contraction (Bangsbo et al., 2002; Howlett et al., 1999). In the steady state, 13C NMR and tracer studies have also shown lactate to be the precursor for PDH and TCA (Bertocci et al., 1997; Bertocci and Lujan, 1999; Stanley et al., 1985, 1986).
PDH and TCA
Whether the activated PDH controls TCA and respiration still poses an open question. Some researchers have noted that an enhanced PDHa leads directly to an increased oxidative phosphorylation and to reduced lactate formation (Howlett and Hogan, 2002). In essence, PDHa controls respiration. Other researchers, however, have observed a delay in muscle O2 uptake at the onset of exercise and have postulated a limitation in O2 utilization rather than in O2 supply (Grassi et al., 1998; Tschakovsy and Hughson, 1999). They posit a metabolic inertia in PDHa, which has stirred up much controversy. Even though pulmonary oxygen consumption (VO2) measurement during muscle contraction has deconvoluted an initial slow phase followed by a rapid phase, 1H NMR measurements of the intracellular VO2 based on the myoglobin signal do not detect any delay in VO2 or metabolic inertia (Chung et al., 2005; Whipp, 1994). Other muscle studies have also found no delay in PDH activation and cannot discern any relationship between respiration and DCA-activated PDH and rising TCA intermediates (Bangsbo et al., 2006, 2002).
The DNP experiments with [2-13C]pyruvate also do not detect any kinetic delay or inertia in the step from pyruvate to acetyl CoA upon DCA activation. ALCAR forms immediately upon DCA activation, which reflects a rising acetyl CoA pool and a decreasing pyruvate level, maintained, in part, by the influx of hyperpolarized pyruvate and the LDH reaction. Given that DNP NMR could readily detect peaks with one-tenth of the ALCAR signal level observed in the DCA-treated muscle, it would suggest that ALCAR concentration level has increased at least 10 times above the control level. A similar reasoning would suggest that glutamate and AcAc increase to at least 5 times the control level. DCA activation of PDH elicits a sudden and a large flux from pyruvate into acetylCoA and does not appear consistent with any metabolic inertia.
The DNP observation appears to be consistent with findings reported for human muscle. In these studies, within 30 s of DCA infusion, the ALCAR level increases 5.2-fold from 2500 to 13,000 μmol kg−1 dry muscle, whereas the acetyl CoA level rises correspondingly 4.1-fold from 11 to 45 μmol kg−1 dry muscle (Bangsbo et al., 2002; Howlett et al., 1999). However, the ALCAR/acetyl CoA ratio shifts modestly from 227 to 289. Given the previously reported constant ratio of ALCAR/acetyl CoA, the interpretation of the ALCAR signal observed in the DNP NMR would favor an increased flux into the acetyl CoA rather than a different chemical exchange with a significantly altered metabolite pools (Schroeder et al., 2013a,b).
With an increase in the acetyl CoA level during DCA activation, TCA intermediates should also rise, as indicated by the AcAc and [5-13C]glutamate signal. For AcAc, previous studies have shown that DCA increases the flux through β-hydroxybutyrate dehydrogenase to produce a higher level of β-hydroxybutyrate in skeletal muscle, consistent with an elevated AcAc level (Bock and Fleischer, 1975; Kark et al., 1970; Schneider et al., 1981). Since α-ketoglutarate dehydrogenase maintains glutamate and α-ketoglutarate in near equilibrium, the non-detectable glutamate/tC signal in control muscle versus a detectable glutamate/tC signal in DCA-treated muscle also supports a rising level for TCA intermediates.
Yet, in the present experiments with resting muscle, the increase in glutamate and PDHa does not accompany any rise in oxygen consumption or ATP demand. DCA-treated and control resting muscles still have the same energy requirements. Consequently, in the DNP experiments, the rate of glutamate production may not accurately reflect the TCA activity or O2 consumption, because any build-up in α-ketoglutarate does not necessarily indicate a continuing flux towards succinate. Instead, it could shuttle via the α-ketoglutarate malate shuttle to the cytosol to produce glutamate (LaNoue et al., 1973; Lewandowski, 1992). Nevertheless, DNP techniques present an opportunity to delineate the different fluxes at the onset of muscle contraction and the responses to a rapidly changing energy demand.
Metabolic inertia, PDH and VO2
At the onset of muscle contraction, the sudden surge in energy demand activates sharply PDHa (Bangsbo et al., 2002; Howlett et al., 1999). Previous studies have shown that DCA increases the initial PDHa 4-fold over control muscle PDHa. In the DNP experiments, the relative PDHa (as reflected in the rLac→HCO3−/rLac→Pyr) with DCA, rises immediately to 37 times the control level. Such a sharp rise in PDHa reflects the cell's capacity to adapt to a rapid change in energy demand and tends to militate against the notion of a metabolic impediment in PDHa, which would slow down O2 utilization.
Some researchers have argued that the VO2 measured by arterial/venous blood oxygen content and thermodilution do not differ in control versus DCA-treated muscle, despite the difference in the PDH activation kinetics. With that perspective, VO2 appears to be uncoupled from PDH activity (Bangsbo et al., 2006, 2002). However, the arterial/venous-based measurements do not consider the role of myoglobin at the onset of contraction. Indeed, 1H NMR measurements of intracellular myoglobin show an immediate desaturation with a t1/2=24 s at the onset of contraction (Chung et al., 2005). Based on the myoglobin desaturation kinetics, the intracellular VO2 again shows no sign of delay and correlates with sudden and significant activation of PDH observed in the present DNP experiments. Further study, however, must explore the relationship.
Lactate shuttle and glycogen shunt
Conventionally, glycogenolytic ATP generation during the transient mismatch between O2-dependent energy supply and demand rationalizes the increased production of lactate observed during muscle contraction. Brooks et al. (2009) have shown that muscle produces lactate in the presence of abundant O2 and have proposed a lactate shuttle from glycolytic white fibers to oxidative red fibers to redistribute the energy supply. The detection of a mitochondrial LDH has expanded the idea to include a potential shuttle from the cytosol to the mitochondria.
A biochemical mechanism underlying the lactate production under aerobic conditions has also emerged. On the basis of triggered 31P NMR studies that show a rapid and significant millisecond consumption of ATP during a contraction cycle, Shulman and Rothman proposed a glycogen shunt, which posits glycogen supplying the ATP needs in milliseconds and serving as a temporal energy buffer (Chung et al., 1998; Shulman and Rothman, 2001). The limited supply of glycogen in the cell and the significant energy demand during a millisecond contraction cycle require dynamic glycogen replenishment during the relaxation phase, which can last for seconds. Otherwise, glycogen would deplete completely, and no sustained period of contraction can occur. But the glycogen shunt introduces an energy inefficiency, even under aerobic conditions. In essence, lactate accumulates during muscle contraction until its export equals net production. The accumulated lactate can then serve as a precursor for acetyl CoA and oxidative ATP generation, which, in turn, fuels the dynamic restoration of the glycogen pool (Shulman, 2005). Lactate can also serve as a carbon precursor for glyconeogenesis (Bonen et al., 1990; Johnson and Bagby, 1988; McDermott and Bonen, 1992; McLane and Holloszy, 1979).
Both the intracellular lactate shuttle and glycogen shunt models, however, require that muscle must mobilize and rapidly utilize lactate and can convert it quickly to acetyl CoA for oxidative metabolism in the mitochondria. Indeed, the present DNP studies have established that muscle can rapidly mobilize and use lactate. PDH poses no metabolic inertia and can compete readily with LDH to divert accumulated lactate to acetyl CoA. As a consequence, the DNP experiment results provide support for a critical underpinning of both the glycogen shunt model and the intracellular lactate shuttle hypothesis.
Perspectives
The DNP study reaffirms that simply measuring the steady level of lactate overlooks its dynamic role in maintaining homeostasis and belies the system's capacity to switch from net lactate release to lactate consumption in response to changing nutritive state, power output, duration of activity and arterial lactate concentration (Bergman et al., 1999; Brooks et al., 1998). The dynamic nature of lactate metabolism provokes a reconsideration of many key concepts in biology.
In particular, lactate metabolism provides unique insight into the treatment of traumatic brain injury (TBI). TBI induces initially an acute rise in glucose level and cerebral metabolic rates of glucose (CMRG) followed by a more prolonged period of reduced CMRG and a marked decline in the CMRG/oxygen consumption ratio (Brooks and Martin, 2014; Glenn et al., 2003, 2015). However, TBI does not suppress lactate metabolism. The contrasting effect on glucose versus lactate metabolism has led to bold ideas that take advantage of the lactate shuttle mechanism to supply energy, which then bypasses the restriction in glycolytic flux and spares the consumption of limited glucose reserves (Brooks and Martin, 2014; Glenn et al., 2003, 2015). Augmenting the TBI treatment with lactate supplementation has improved the recovery outcome.
Our DNP NMR experiments have specifically aimed to test a key premise in both the intracellular lactate shuttle and glycogen shunt hypotheses, which require a rapid mobilization and metabolism of lactate during a muscle contraction cycle. If experiments cannot detect a rapid lactate dynamics, they would falsify the premise. According to Karl Popper's theory of scientific discovery, these hypotheses will fail immediately. However, the results cannot falsify the premise and appear consistent with the hypothesis that lactate can serve as a transient energy buffer and as a precursor for mitochondrial LDH in muscle. Given the rapid kinetics of lactate in muscle, further studies must now investigate the role in regulating metabolism. Consequently, our study provides many perspectives on the commonly accepted function of pyruvate and lactate, especially in organsims facing diurnal anaerobiosis and physiological challenges, and cautions against an overly simpliistic view of glycolytic end products as merely hypoxia biomarkers.
MATERIALS AND METHODS
Substrates and polarization procedure
HyperSense DNP system (Oxford Instruments Molecular Biotools, Oxford, UK) operating at 1.4 K temperature and 25 mW microwave power in a 3.35 T magnet polarized the substrate samples. For lactate, 90 mg of 2.1 mol l−1 [1-13C]-labeled sodium lactate in 37.5:62.5 w/w water:glycerol with 15 mmol l−1 OX063 trityl radical was mixed with 10 μl of a 1:50 dilution of Dotarem (Guerbet, France) prior to polarization. After the polarization, the sample was dissolved in 5 g of 40 mmol l−1 Tris buffer containing 100 mg l−1 EDTA-Na2 to yield a final solution of 40 mmol l−1 lactate. For pyruvate, a mixture of 25 μl of 14 mol l−1 [2-13C]pyruvic acid and 15 mmol l−1 OX063 trityl radical with 3 μl of diluted Dotarem was prepared. After polarization, the pyruvate sample was dissolved using ∼4.5 g of 80 mmol l−1 NaOH solution with 40 mmol l−1 Tris buffer and 100 mg l−1 EDTA-Na2 to yield an 80 mmol l−1 solution of hyperpolarized pyruvate at pH ∼7.5.
The build-up time constants for the solid-state polarization were ∼2800 s and ∼1100 s for lactate and pyruvate, respectively, and the dissolutions were performed when the solid polarization reached at least 95% of the saturated level. The liquid polarization and t1 of the samples were measured by separate in vitro experiments (Park et al., 2013a). The polarization levels were 27.8±2.6% for [1-13C]lactate (N=3) and 28.3±0.6% for [2-13C]pyruvate (N=5). t1 values were estimated as 41.2±2.1 s for [1-13C]lactate and 47.1±1.7 s for [2-13C]pyruvate.
Animal preparation
Twelve healthy male Sprague-Dawley rats (517–681 g) were prepared for the following two studies. For the first study (N=9), non-localized metabolic kinetics were measured using the hyperpolarized [1-13C]lactate (0.78 mmol kg−1 body weight). Three rats received DCA (Sigma-Aldrich, St Louis, MO, USA) for PDH activation. One hour after DCA injection, non-localized metabolic kinetics was measured again with hyperpolarized [1-13C]lactate. To measure the spatial distribution of the detected metabolites in skeletal muscle, metabolite maps were acquired using CSI from one of the DCA-injected rats (2.5 h post-DCA) immediately after an additional injection of hyperpolarized [1-13C]lactate. For the second study (N=3), non-localized metabolic kinetics was measured using hyperpolarized [2-13C]pyruvate (1.56 mmol kg−1) before and 1 h after DCA injection.
The hyperpolarized solutions were injected at a rate of 0.25 ml s−1. DCA (200 mg kg−1 body weight dissolved in 30 g ml−1 of saline) was administered initially as a 0.5 ml bolus. The rest of the DCA was infused at a rate of 0.1 ml per 3 min.
A custom-made 13C surface coil (internal diameter=28 mm) was placed on top of right rectus femoris of each animal, which was placed inside a proton birdcage quadrature coil (internal diameter=70 mm). Each animal was anesthetized with 1–3% isoflurane in oxygen (∼1.5 l min−1) and catheterized in the tail vein before placing on a clinical 3 T Signa® MR scanner (GE Healthcare, Waukesha, WI, USA). Vital signs such as respiration, heart rate and oxygen saturation were monitored throughout the experiment and body temperature was maintained at ∼37°C using a warm water blanket. All procedures were approved by the local Institutional Animal Care and Use Committee.
NMR protocols
After acquiring anatomical references using the 1H birdcage coil, the homogeneity of the B0 field over the region of leg muscle was optimized by minimizing the line width of the unsuppressed water signal using the linear shim currents and a point-resolved spectroscopy sequence. The surface coil was used for the 13C experiments. For the measurements of metabolic kinetics using the dynamic free induction decay (FID), the NMR signal acquisition used the following parameters: 10 deg radiofrequency (RF) hard pulse, temporal resolution=3 s, spectral width/points=10 kHz/4096, acquisition time=4 min. A volumetric single time-point spiral CSI sequence (field of view=80×80×60 mm3, matrix size=16×16×12, spectral bandwidth/points=972.7 Hz/96, acquisition time=4 s, 20 deg hard pulse) with a circularly reduced k-space sampling scheme for the shortened scan time was used to acquire the CSI (Park et al., 2013b). To obtain the maximum signal of 13C-labeled metabolic products, the CSI scan was started 25 s after the start of the injection. The center frequency was set to the chemical shift position half way between H13CO3− and [1-13C]lactate signals. The reconstructed 13C metabolite maps were overlaid on top of proton MRI for anatomical reference, acquired using a 3D spoiled gradient echo sequence.
Data processing
All 13C data were processed using Matlab (MathWorks, Natick, MA, USA). After the k-space data of each time point from the FID dynamics were apodized by a Gaussian filter (5 Hz) and zero-filled by a factor of four in the spectral dimension, a fast Fourier transform (FFT) and zeroth order phase correction were applied in the frequency domain. Metabolites were quantified by integrating the respective peak in the absorption mode from time-averaged spectra (0–2 min), followed by normalization to total carbon signal, which is the sum of all the 13C-labeled signals. For display purposes, the baseline was subtracted from the time-averaged spectra as described previously (Mayer et al., 2009).
Temporal change of each metabolite signal was estimated from the peak integrals of the time-resolved spectra using two parameters: τ and r. Time point (τ) where the half-maximum of each metabolite signal was achieved was compared to assess the metabolite production rate. The production rate of each metabolite was also estimated from the mean slope of the first four time points (r) following its appearance in the dynamic curve by linearly fitting the metabolite signal, which is then normalized to the initial mean slope of the injected substrate (Schroeder et al., 2013a,b).
For CSI reconstruction, k-space data were apodized by a 30 Hz Gaussian filter and zero-filled by a factor of four spectrally. It was followed by an additional zero-padding by a factor of two in spatial dimension and a 4D fast-Fourier transform (FFT) in both spatial and spectral domains and gridding on to a Cartesian coordinate. For compensating the inhomogeneous coil sensitivity and substrate perfusion, metabolite-ratio images were obtained by normalizing the metabolite maps to the tC map. Voxels with low signal in the tC map (<20%) were masked. Metabolite ratios between products (e.g. alanine/HCO3−, pyruvate/HCO3−, pyruvate/alanine) were also obtained similarly.
To evaluate the statistical significance of the effect of DCA on metabolite levels, only the DCA-injected rats (three pre/post paired datasets per study) were compared using a paired Student's t-test in both metabolite ratios and dynamic analysis. All the values are reported as the means±s.e.
Acknowledgements
The authors acknowledge the technical assistance of James Graham in animal preparation and Arif Wibowo in [13C]lactate sample preparation.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
J.M.P., S.J., D.M., R.E.H., Y.C., D.B., D.M.S. and T.J. contributed to the conception and design of research; J.M.P., S.J., D.M., Y.C., D.B. and T.J. performed experiments; J.M.P., S.J., D.M., R.E.H., Y.C., D.B., D.M.S. and T.J. discussed, analyzed, and interpreted experiments; J.M.P. and T.J. prepared figures, established the framework of the data analysis, incorporated comments to shape the final analysis, and drafted the manuscript; J.M.P., S.J., D.M., R.E.H., Y.C., D.B., D.M.S. and T.J. reviewed and approved the final version of the manuscript.
Funding
The project received funding support from the National Institutes of Health [P41 EB015891, AA05965, AA018681, AA13521-INIA, CA176836, OD012283 and EB009070], The Department of Defense [PC100427], The Lucas Foundation, France Berkeley Fund and GE Healthcare. Deposited in PMC for release after 12 months.
References
- Ardenkjaer-Larsen J. H., Fridlund B., Gram A., Hansson G., Hansson L., Lerche M. H., Servin R., Thaning M. and Golman K. (2003). Increase in signal-to-noise ratio of >10,000 times in liquid-state NMR. Proc. Natl. Acad. Sci. USA 100, 10158-10163. 10.1073/pnas.1733835100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton H. J., Schroeder M. A., Dodd M. S., Heather L. C., Carter E. E., Cochlin L. E., Nagel S., Sibson N. R., Radda G. K., Clarke K. et al. (2011). Validation of the in vivo assessment of pyruvate dehydrogenase activity using hyperpolarised 13C MRS. NMR Biomed. 24, 201-208. 10.1002/nbm.1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangsbo J., Gibala M. J., Krustrup P., Gonzalez-Alonso J. and Saltin B. (2002). Enhanced pyruvate dehydrogenase activity does not affect muscle O2 uptake at onset of intense exercise in humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 282, R273-R280. [DOI] [PubMed] [Google Scholar]
- Bangsbo J., Gibala M. J., Howarth K. R. and Krustrup P. (2006). Tricarboxylic acid cycle intermediates accumulate at the onset of intense exercise in man but are not essential for the increase in muscle oxygen uptake. Pflugers Arch. 452, 737-743. 10.1007/s00424-006-0075-4 [DOI] [PubMed] [Google Scholar]
- Bastiaansen J. A. M., Yoshihara H. A. L., Takado Y., Gruetter R. and Comment A. (2014). Hyperpolarized 13C lactate as a substrate for in vivo metabolic studies in skeletal muscle. Metabolomics 10, 986-994. 10.1007/s11306-014-0630-5 [DOI] [Google Scholar]
- Bergman B. C., Wolfel E. E., Butterfield G. E., Lopaschuk G. D., Casazza G. A., Horning M. A. and Brooks G. A. (1999). Active muscle and whole body lactate kinetics after endurance training in men. J. Appl. Physiol. (1985) 87, 1684-1696. [DOI] [PubMed] [Google Scholar]
- Bertocci L. A. and Lujan B. F. (1999). Incorporation and utilization of [3-13C]lactate and [1,2-13C]acetate by rat skeletal muscle. J. Appl. Physiol. 86, 2077-2089. [DOI] [PubMed] [Google Scholar]
- Bertocci L. A., Jones J. G., Malloy C. R., Victor R. G. and Thomas G. D. (1997). Oxidation of lactate and acetate in rat skeletal muscle: analysis by 13C-nuclear magnetic resonance spectroscopy. J. Appl. Physiol. 83, 32-39. [DOI] [PubMed] [Google Scholar]
- Bock H. and Fleischer S. (1975). Preparation of a homogeneous soluble D-beta-hydroxybutyrate apodehydrogenase from mitochondria. J. Biol. Chem. 250, 5774-5761. [PubMed] [Google Scholar]
- Bonen A., McDermott J. C. and Tan M. H. (1990). Glycogenesis and glyconeogenesis in skeletal muscle: effects of pH and hormones. Am. J. Physiol. 258, E693-E700. [DOI] [PubMed] [Google Scholar]
- Boron W. F. and Boulpaep E. L. (2003). Medical Physiology: A Cellular and Molecular Approach. Philadelphia: Elsevier. [Google Scholar]
- Brooks G. A. (1986a). Lactate production under fully aerobic conditions: the lactate shuttle during rest and exercise. Fed. Proc. 45, 2924-2929. [PubMed] [Google Scholar]
- Brooks G. A. (1986b). The lactate shuttle during exercise and recovery. Med. Sci. Sports Exerc. 18, 360-368. 10.1249/00005768-198606000-00019 [DOI] [PubMed] [Google Scholar]
- Brooks G. A. (2000). Intra- and extra-cellular lactate shuttles. Med. Sci. Sports Exerc. 32, 790-799. 10.1097/00005768-200004000-00011 [DOI] [PubMed] [Google Scholar]
- Brooks G. A. (2009). Cell-cell and intracellular lactate shuttles. J. Physiol. 587, 5591-5600. 10.1113/jphysiol.2009.178350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks G. A. and Hashimoto T. (2007). Investigation of the lactate shuttle in skeletal muscle mitochondria. J. Physiol. 584, 705-706; author reply 707-8 10.1113/jphysiol.2007.142992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks G. A. and Martin N. A. (2014). Cerebral metabolism following traumatic brain injury: new discoveries with implications for treatment. Front. Neurosci. 8, 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks G. A., Wolfel E. E., Butterfield G. E., Cymerman A., Roberts A. C., Mazzeo R. S. and Reeves J. T. (1998). Poor relationship between arterial [lactate] and leg net release during exercise at 4,300 m altitude. Am. J. Physiol. 275, R1192-R1201. [DOI] [PubMed] [Google Scholar]
- Brooks G. A., Fahey T. D., White T. P. and Baldwin K. M. (2000). Exercise Physiology: Human Bioenergetics and Its Application. Mountain View: Mayfield Publishing. [Google Scholar]
- Carpenter L. and Halestrap A. P. (1994). The kinetics, substrate and inhibitor specificity of the lactate transporter of Ehrlich-Lettre tumour cells studied with the intracellular pH indicator BCECF. Biochem. J. 304, 751-760. 10.1042/bj3040751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y., Sharman R., Carlsen R., Unger S. W., Larson D. and Jue T. (1998). Metabolic fluctuation during a muscle contraction cycle. Am. J. Physiol. Cell Physiol. 274, C846-C852. [DOI] [PubMed] [Google Scholar]
- Chung Y., Molé P. A., Sailasuta N., Tran T. K., Hurd R. and Jue T. (2005). Control of respiration and bioenergetics during muscle contraction. Am. J. Physiol. Cell Physiol. 288, C730-C738. 10.1152/ajpcell.00138.2004 [DOI] [PubMed] [Google Scholar]
- Day S. E., Kettunen M. I., Gallagher F. A., Hu D.-E., Lerche M., Wolber J., Golman K., Ardenkjaer-Larsen J. H. and Brindle K. M. (2007). Detecting tumor response to treatment using hyperpolarized 13C magnetic resonance imaging and spectroscopy. Nat. Med. 13, 1382-1387. 10.1038/nm1650 [DOI] [PubMed] [Google Scholar]
- Fjeld C. C., Birdsong W. T. and Goodman R. H. (2003). Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc. Natl. Acad. Sci. USA 100, 9202-9207. 10.1073/pnas.1633591100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladden L. B. (2004). Lactate metabolism: a new paradigm for the third millennium. J. Physiol. 558, 5-30. 10.1113/jphysiol.2003.058701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn T. C., Kelly D. F., Boscardin W. J., McArthur D. L., Vespa P., Oertel M., Hovda D. A., Bergsneider M., Hillered L. and Martin N. A. (2003). Energy dysfunction as a predictor of outcome after moderate or severe head injury: indices of oxygen, glucose, and lactate metabolism. J. Cereb. Blood Flow Metab. 23, 1239-1250. 10.1097/01.WCB.0000089833.23606.7F [DOI] [PubMed] [Google Scholar]
- Glenn T. C., Martin N. A., McArthur D. L., Hovda D. A., Vespa P., Johnson M. L., Horning M. A. and Brooks G. A. (2015). Endogenous nutritive support after traumatic brain injury: peripheral lactate production for glucose supply via gluconeogenesis. J. Neurotrauma 32, 811-819. 10.1089/neu.2014.3482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman M. N., Ruderman N. B. and Aoki T. T. (1978). Glucose and amino acid metabolism in perfused skeletal muscle: effect of dichloroacetate. Diabetes 27, 1065-1074. 10.2337/diab.27.11.1065 [DOI] [PubMed] [Google Scholar]
- Grassi B., Gladden L. B., Samaja M., Stary C. M. and Hogan M. C. (1998). Faster adjustment of O2 delivery does not affect V(O2) on-kinetics in isolated in situ canine muscle. J. Appl. Physiol. 85, 1394-1403. [DOI] [PubMed] [Google Scholar]
- Grieshaber M. K., Hardewig I., Kreutzer U. and Pörtner H. O. (1994). Physiological and metabolic responses to hypoxia in invertebrates. Rev. Physiol. Biochem. Pharmacol. 125, 43-147. 10.1007/BFb0030909 [DOI] [PubMed] [Google Scholar]
- Howlett R. A. and Hogan M. C. (2002). Dichloroacetate accelerates the fall in intracellular PO2 at onset of contractions in Xenopus single muscle fibers. Am. J. Physiol. 284, R481-R485. [DOI] [PubMed] [Google Scholar]
- Howlett R. A., Heigenhauser G. J., Hultman E., Hollidge-Horvat M. G. and Spriet L. L. (1999). Effects of dichloroacetate infusion on human skeletal muscle metabolism at the onset of exercise. Am. J. Physiol. 277, E18-E25. [DOI] [PubMed] [Google Scholar]
- Johnson J. L. and Bagby G. J. (1988). Gluconeogenic pathway in liver and muscle glycogen synthesis after exercise. J. Appl. Physiol. (1985) 64, 1591-1599. [DOI] [PubMed] [Google Scholar]
- Juel C. (1991). Muscle lactate transport studied in sarcolemmal giant vesicles. Biochim. Biophys. Acta 1065, 15-20. 10.1016/0005-2736(91)90004-R [DOI] [PubMed] [Google Scholar]
- Juel C. and Halestrap A. P. (1999). Lactate transport in skeletal muscle - role and regulation of the monocarboxylate transporter. J. Physiol. 517, 633-642. 10.1111/j.1469-7793.1999.0633s.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kark R. A., Blass J. P., Avigan J. and Engel W. K. (1970). Measurement of metabolic activities in small samples of muscle. Neurology 20, 404. [PubMed] [Google Scholar]
- Kettunen M. I., Hu D.-e., Witney T. H., McLaughlin R., Gallagher F. A., Bohndiek S. E., Day S. E. and Brindle K. M. (2010). Magnetization transfer measurements of exchange between hyperpolarized [1-13C]pyruvate and [1-13C]lactate in a murine lymphoma. Magn. Reson. Med. 63, 872-880. 10.1002/mrm.22276 [DOI] [PubMed] [Google Scholar]
- Kreutzer U. and Jue T. (1995). Critical intracellular oxygen in the myocardium as determined with the 1H NMR signal of myoglobin. Am. J. Physiol. 268, H1675-H1681. [DOI] [PubMed] [Google Scholar]
- Kreutzer U. and Jue T. (1997). Metabolic response in Arenicola marina to limiting oxygen as reflected in the 1H-NMR oxymyoglobin signal. Eur. J. Biochem. 243, 233-239. 10.1111/j.1432-1033.1997.0233a.x [DOI] [PubMed] [Google Scholar]
- LaNoue K. F., Walajtys E. I. and Williamson J. R. (1973). Regulation of glutamate metabolism and interactions with the citric acid cycle in rat heart mitochondria. J. Biol. Chem. 248, 7171-7183. [PubMed] [Google Scholar]
- Lewandowski E. D. (1992). Metabolic heterogeneity of carbon substrate utilization in mammalian heart: NMR determinations of mitochondrial versus cytosolic compartmentation. Biochemistry 31, 8916-8923. 10.1021/bi00152a031 [DOI] [PubMed] [Google Scholar]
- Li Y., Dash R. K., Kim J., Saidel G. M. and Cabrera M. E. (2009). Role of NADH/NAD+ transport activity and glycogen store on skeletal muscle energy metabolism during exercise: in silico studies. Am. J. Physiol. Cell Physiol. 296, C25-C46. 10.1152/ajpcell.00094.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer D., Yen Y.-F., Tropp J., Pfefferbaum A., Hurd R. E. and Spielman D. M. (2009). Application of subsecond spiral chemical shift imaging to real-time multislice metabolic imaging of the rat in vivo after injection of hyperpolarized 13C1-pyruvate. Magn. Reson. Med. 62, 557-564. 10.1002/mrm.22041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott J. C. and Bonen A. (1992). Glyconeogenic and oxidative lactate utilization in skeletal muscle. Can. J. Physiol. Pharmacol. 70, 142-149. 10.1139/y92-021 [DOI] [PubMed] [Google Scholar]
- McDermott J. C. and Bonen A. (1993). Lactate transport by skeletal muscle sarcolemmal vesicles. Mol. Cell Biochem. 122, 113-121. 10.1007/BF01076095 [DOI] [PubMed] [Google Scholar]
- McLane J. A. and Holloszy J. O. (1979). Glycogen synthesis from lactate in the three types of skeletal muscle. J. Biol. Chem. 254, 6548-6553. [PubMed] [Google Scholar]
- Park J. M., Josan S., Grafendorfer T., Yen Y.-F., Hurd R. E., Spielman D. M. and Mayer D. (2013a). Measuring mitochondrial metabolism in rat brain in vivo using MR Spectroscopy of hyperpolarized [2-13C]pyruvate. NMR Biomed. 26, 1197-1203. 10.1002/nbm.2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. M., Recht L. D., Josan S., Merchant M., Jang T., Yen Y.-F., Hurd R. E., Spielman D. M. and Mayer D. (2013b). Metabolic response of glioma to dichloroacetate measured in vivo by hyperpolarized 13C magnetic resonance spectroscopic imaging. Neuro Oncol. 15, 433-441. 10.1093/neuonc/nos319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passarella S., Paventi G. and Pizzuto R. (2014). The mitochondrial L-lactate dehydrogenase affair. Front. Neurosci. 8, 407 10.3389/fnins.2014.00407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilegaard H., Juel C. and Wibrand F. (1993). Lactate transport studied in sarcolemmal giant vesicles from rats: effect of training. Am. J. Physiol. 264, E156-E160. [DOI] [PubMed] [Google Scholar]
- Poole R. C. and Halestrap A. P. (1993). Transport of lactate and other monocarboxylates across mammalian plasma membranes. Am. J. Physiol. 264, C761-C782. [DOI] [PubMed] [Google Scholar]
- Roth D. A. and Brooks G. A. (1990a). Lactate and pyruvate transport is dominated by a pH gradient-sensitive carrier in rat skeletal muscle sarcolemmal vesicles. Arch. Biochem. Biophys. 279, 386-394. 10.1016/0003-9861(90)90506-T [DOI] [PubMed] [Google Scholar]
- Roth D. A. and Brooks G. A. (1990b). Lactate transport is mediated by a membrane-bound carrier in rat skeletal muscle sarcolemmal vesicles. Arch. Biochem. Biophys. 279, 377-385. 10.1016/0003-9861(90)90505-S [DOI] [PubMed] [Google Scholar]
- Sahlin K., Fernström M., Svensson M. and Tonkonogi M. (2002). No evidence of an intracellular lactate shuttle in rat skeletal muscle. J. Physiol. 541, 569-574. 10.1113/jphysiol.2002.016683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider S. H., Komanicky P. M., Goodman M. N. and Ruderman N. B. (1981). Dichloroacetate: effects on exercise endurance in untrained rats. Metabolism 30, 590-595. 10.1016/0026-0495(81)90137-2 [DOI] [PubMed] [Google Scholar]
- Schroeder M. A., Ali M. A., Hulikova A., Supuran C. T., Clarke K., Vaughan-Jones R. D., Tyler D. J. and Swietach P. (2013a). Extramitochondrial domain rich in carbonic anhydrase activity improves myocardial energetics. Proc. Natl. Acad. Sci. USA 110, E958-E967. 10.1073/pnas.1213471110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder M. A., Lau A. Z., Chen A. P., Gu Y., Nagendran J., Barry J., Hu X., Dyck J. R. B., Tyler D. J., Clarke K. et al. (2013b). Hyperpolarized 13C magnetic resonance reveals early- and late-onset changes to in vivo pyruvate metabolism in the failing heart. Eur. J. Heart Fail. 15, 130-140. 10.1093/eurjhf/hfs192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman R. G. (2005). Glycogen turnover forms lactate during exercise. Exerc. Sport Sci. Rev. 33, 157-162. 10.1097/00003677-200510000-00002 [DOI] [PubMed] [Google Scholar]
- Shulman R. G. and Rothman D. L. (2001). The “glycogen shunt” in exercising muscle: a role for glycogen in muscle energetics and fatigue. Proc. Natl. Acad. Sci. USA 98, 457-461. 10.1073/pnas.98.2.457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacpoole P. W. (1989). The pharmacology of dichloroacetate. Metabolism 38, 1124-1144. 10.1016/0026-0495(89)90051-6 [DOI] [PubMed] [Google Scholar]
- Stanley W. C. and Brooks G. A. (1987). Measuring lactate production. Am. J. Physiol. 253, E472-E473. [DOI] [PubMed] [Google Scholar]
- Stanley W. C., Gertz E. W., Wisneski J. A., Morris D. L., Neese R. A. and Brooks G. A. (1985). Systemic lactate kinetics during graded exercise in man. Am. J. Physiol. 249, E595-E602. [DOI] [PubMed] [Google Scholar]
- Stanley W. C., Gertz E. W., Wisneski J. A., Neese R. A., Morris D. L. and Brooks G. A. (1986). Lactate extraction during net lactate release in legs of humans during exercise. J. Appl. Physiol. 60, 1116-1120. [DOI] [PubMed] [Google Scholar]
- Tschakovsy M. E. and Hughson R. L. (1999). Interaction of factors determining oxygen uptake at the onset of exercise. J. Appl. Physiol. 86, 1101-1113. [DOI] [PubMed] [Google Scholar]
- Wasserman K. (1987). Determinants and detection of anaerobic threshold and consequences of exercise above it. Circulation 76, VI29-VI39. [PubMed] [Google Scholar]
- Whipp B. J. (1994). The slow component of O2 uptake kinetics during heavy exercise. Med. Sci. Sports Exerc. 26, 1297-1423. 10.1249/00005768-199411000-00005 [DOI] [PubMed] [Google Scholar]
- White A. T. and Schenk S. (2012). NAD+/NADH and skeletal muscle mitochondrial adaptations to exercise. Am. J. Physiol. Endocrinol. Metab. 303, E308-E321. 10.1152/ajpendo.00054.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y., Holloway G. P., Ljubicic V., Hatta H., Spriet L. L., Hood D. A. and Bonen A. (2007). Negligible direct lactate oxidation in subsarcolemmal and intermyofibrillar mitochondria obtained from red and white rat skeletal muscle. J. Physiol. 582, 1317-1335. 10.1113/jphysiol.2007.135095 [DOI] [PMC free article] [PubMed] [Google Scholar]