

Abstract
Three genome-wide association studies (GWAS) have been conducted on the genetic susceptibility of hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC), two of which consistently identified tagging single nucleotide polymorphisms (SNPs) around HLA-DQ/DR. In contrast, large multi-centre association studies between HBV genotype, mutations and the risk of HCC are relatively rare, and their interactions with host variants are even less. We performed a multi-centre study of 1,507 HBV-related HCC cases and 1,560 HBV persistent carriers as controls to evaluate the effects of HBV genotype, mutations, GWAS-identified HLA-DQ/DR SNPs (rs9272105 and rs9275319) and their interactions on HCC risk. We found HBV genotype C was more frequent in HBV-related HCC. And 11 HBV hotspot mutations were independently and significantly associated with HCC risk. We also detected significant interactions of rs9272105 with both the HBV genotype and mutations. Through stepwise regression analysis, HBV genotype, the 11 mutations, HLA-DQ/DR SNPs, and the interaction of rs9272105 with mutation A1752G were all entered into the HCC prediction model, and the area under the curve for the panel including the HLA-DQ/DR SNPs, HBV genotype and mutations was 0.840. The HBV genotype, the mutations and the HLA-DQ/DR SNPs may serve as biomarkers for the surveillance of HBV persistent carriers.
Hepatocellular carcinoma (HCC) is the second leading cause of cancer-related deaths worldwide, with the incidence on the rise both in developed and developing countries1,2. HCC development is influenced by complex factors including viral infection, environmental factors3, and genetic makeup, with most studies having identified susceptibility loci at the human leukocyte antigen (HLA) class II region at 6p214,5,6.
Currently, three genome-wide association studies (GWAS), all from China, have been conducted on hepatitis B virus (HBV)-related HCC, two of which consistently identified HLA-DQ/DR as susceptibility loci5,6. Of the two independent GWAS, one study with 1,538 cases and 1,465 controls for GWA scan identified the single nucleotide polymorphism (SNP) rs9272105 (located between HLA-DQA1 and HLA-DRB1)5, while the other study with 1,161 HCC cases and 1,353 controls for GWA scan identified the SNP rs9275319 at HLA-DQ6. These findings highlight the importance of HLA-DQ/DR molecules in the development of HBV-related HCC.
In contrast, large multi-centre studies on the association between HBV genotype, mutations and HCC risk are relatively rare, especially regarding their interactions with host genetic variants. HBV has been classified into different genotypes according to a sequence divergence of >8% in the entire genome, and the genotypes are further separated into subgenotypes if the divergence in the nucleotide sequence is between 4 and 8%7,8. The HBV genotypes and subgenotypes have distinct geographic distributions and have been implicated to differ with regard to clinical liver diseases, disease outcomes, and responses to interferon treatment9,10,11,12. In East Asia, although HBV genotypes B and C are endemic, the HBV genotype frequency varies in these areas13. Because of the relatively small study sample sizes, the low success rates of HBV typing and the different study designs, the effect of HBV genotype on the outcomes of HBV persistent infection also varied greatly14,15,16,17,18,19. The basal core promoter, which is regulated by the enhancer II to a great extent, controls the transcription of precore mRNA20. The precore protein is processed to produce the secreted hepatitis B e antigen (HBeAg), which indicates active viral replication and is associated with an increased risk of HCC21,22,23. Three small-scale longitudinal studies with less than 50 cases demonstrated that some of the mutations in the enhancer II/basal corepromoter/precore (EnhII/BCP/PC) region would occur years before a diagnosis of HCC is made and gradually accumulate during the development of HCC24,25,26.
HBV mutations are most likely selected via virus-immune interactions in the inflammatory microenvironment. Therefore, we performed a large multi-centre study to evaluate the effects of HBV genotype, mutations in the EnhII/BCP/PC region, GWAS-identified HLA-DQ/DR SNPs (rs9272105 and rs9275319) and their interactions on HCC risk. Furthermore, we evaluated the risk prediction effects of these factors in HBV-related HCC.
Results
Baseline characteristics of the participants
Selected characteristics of the 1,507 HBV-related HCC patients and the 1,560 HBV persistent carriers are described in Supplementary Table S2. As expected, there were similar distributions of age and gender between the HCC patients and the HBV persistent carriers (P = 0.835 and 0.687, respectively). In addition, similar distributions of age and gender were also seen between the cases and controls of the additional sample set from Xi’an (Supplementary Table S3).
Distribution of HBV genotypes and subgenotypes
Among the participants of this study, the HBV genotypes B, C, BC (coinfection) and D and the subgenotypes B2, C1, C2, B2C1, B2C2, B2C1C2 and C1C2 were identified through nested multiplex PCR and sequencing. All the amplicons were of the size expected for each genotype and subgenotype, as shown in Supplementary Fig. S1. In case where the genotype was identified but the subgenotype was not, the HBV subgenotype was identified by sequencing and aligning, especially for C3 and C4. The sequencing peak chart of all the genotypes and subgenotypes is shown in Supplementary Fig. S2.
As shown in Table 1, we found 629 (41.0%) subjects infected with HBV genotype B, 375 subjects (24.4%) with genotype BC, 529 subjects (34.4%) with genotype C, and 3 subjects (0.2%) with genotype D among the HBV persistent carriers, whereas there were 159 subjects (10.8%) with HBV genotype B, 121 subjects (8.2%) with genotype BC, 1,186 subjects (80.6%) with genotype C, and 5 subjects (0.3%) with genotype D among the HCC patients. Although the frequencies of the HBV genotypes B and BC (coinfection) were varied among the studied areas (Supplementary Table S4), the frequency of genotype C was consistently higher in HBV-related HCC subjects (80.6% among HCC vs. 34.4% among HBV persistent carriers, Table 1). Compared with subjects infected with HBV B-related genotypes (B and BC), the subjects infected with non-B genotypes (C and D) had an 8.18-fold increased HCC risk (95% CI = 6.91–9.68). Among the HBV subgenotypes, B2, C2 and B2C2 were predominant. The proportions of subgenotype C2 in HCC patients and HBV persistent carriers were 67.0% and 29.3%, respectively. Similarly, the genotype and subgenotype frequencies also suggested that HBV genotype C and subgenotype C2 were the risk factors for HCC in the Xi’an sample set (Supplementary Table S5).
Table 1. Distribution of HBV genotypes and subgenotypes in HCC and HBV persistent carriers.
| Variable | HCC patients(n = 1507)N (%) | HBV persistentcarriers (n = 1560)N (%) | P |
|---|---|---|---|
| No. of HBV typed | 1471 (97.6) | 1536 (98.5) | 0.089 |
| Genotype | <0.001 | ||
| C | 1186 (80.6) | 529 (34.4) | |
| B | 159 (10.8) | 629 (41.0) | |
| BC | 121 (8.2) | 375 (24.4) | |
| D | 5 (0.3) | 3 (0.2) | |
| Subgenotype | <0.001 | ||
| C2 | 985 (67.0) | 450 (29.3) | |
| C1C2 | 69 (4.7) | 11 (0.7) | |
| C1 | 10 (0.7) | 5 (0.3) | |
| C3 | 111 (7.5) | 61 (4.0) | |
| C4 | 11 (0.7) | 2 (0.1) | |
| B2 | 159 (10.8) | 629 (41.0) | |
| B2C1 | 2 (0.1) | 2 (0.1) | |
| B2C1C2 | 8 (0.5) | 3 (0.2) | |
| B2C2 | 107 (7.3) | 366 (23.8) | |
| B2C3 | 4 (0.3) | 4 (0.3) | |
| D | 5 (0.3) | 3 (0.2) |
Associations of HBV mutations and HBV-related HCC
We successfully amplified and sequenced the EnhII/BCP/PC region from 1,313 (89.3%) HCC patients and 1,413 (92.0%) HBV persistent carriers with HBV genotype results. The wild type nucleotide and hotspot mutations in the EnhII/BCP/PC region are listed in Supplementary Table S6. The sequences of the hotspot mutations are shown in Supplementary Fig.S3. Of those 19 hotspot mutations, C1653T, T1674C/G, A1703G, G1719T, T1727A/G, T1753C, A1762T, G1764A, G1799C, G1899A, G1915A/C and C1969T were significantly associated with an increased risk of HCC, whereas C1673T, A1726C, C1730G and A1752G were significantly associated with a reduced risk of HCC, when adjusted for age and gender (Supplementary Table S7). We then used a conditional logistic regression analysis to test the independence of these hotspot mutations and found that the effects of C1653T, C1673T, T1674C/G, C1730G, A1752G, T1753C, A1762T, G1764A, G1899A, G1915A/C and C1969T on HCC development remained in existence after being conditioned on the other mutations (Supplementary Table S7). After Bonferroni correction, HBV mutations C1653T, T1674C/G, A1752G, T1753C, A1762T, G1764A, G1899A, and C1969T were still significant associated with HCC risk.
Supplementary Table S8 shows the correlations between the hotspot mutations in the EnhII/BCP/PC region of HBV. The results indicated that the HBV mutation C1730G was correlated with T1727A/G and G1799C (r2 > 0.800).
Associations of the HLA SNPs with HBV-related HCC and their interactions with HBV genotype and mutations in HCC
The genotype distributions of the SNPs rs9272105 and rs9275319 in the HCC patients and the HBV persistent carriers are described in Supplementary Table S9. The observed genotype frequencies for the two SNPs in the HBV persistent carriers were all in Hardy-Weinberg equilibrium (P = 0.641 for rs9272105 and P = 0.274 for rs9275319). The logistic regression analyses in the additive genetic model showed that a variant allele of rs9272105 increased the host HCC risk compared to the HBV persistent carriers (adjusted OR = 1.31, 95% CI = 1.18–1.45), whereas a variant allele of rs9275319 was associated with a decreased HCC risk (adjusted OR = 0.66, 95% CI = 0.56–0.78). In addition, the allele frequencies of the two SNPs were similar between the case subjects and the controls from different areas.
Then, subgroup analyses stratified on the HBV genotype model 1 (B, BC and C) and model 2 (B-related and non-B) were conducted on the associations of the HLA SNPs with HCC risk (Supplementary Table S10). Similar association strengths were shown between all the subgroups for rs9275319 (P > 0.05 for heterogeneity test). Interestingly, a stronger effect of rs9272105 was observed among the subjects infected with B-related genotypes (adjusted OR = 1.61, 95% CI = 1.33–1.95) compared to those infected with non-B genotypes (adjusted OR = 1.26, 95% CI = 1.09–1.46) (P = 0.046 for heterogeneity test). A further interaction analysis detected a significant multiplicative interaction between rs9272105 and HBV genotype on HCC risk (P = 0.014 for model 1 and P = 0.046 for model 2). The crossover analysis suggested that subjects infected with HBV genotype C and carrying the rs9272105 AA genotype had a 17.05-fold increased HCC risk (95% CI = 10.99–26.44) compared with those infected with HBV genotype B and carrying the rs9272105 GG genotype. Additionally, the subjects infected with HBV non-B genotypes and carrying the rs9272105 AA genotype had a more prominent risk effect (adjusted OR = 17.70, 95% CI = 12.21–25.64) compared to those infected with HBV B-related genotypes and carrying the rs9272105 GG genotype (Fig. 1).
Figure 1. Crossover analysis of the SNP rs9272105-HBV genotype interaction on HCC susceptibility, including stratification model 1 (B, BC and C) and model 2 (B-related and non-B).
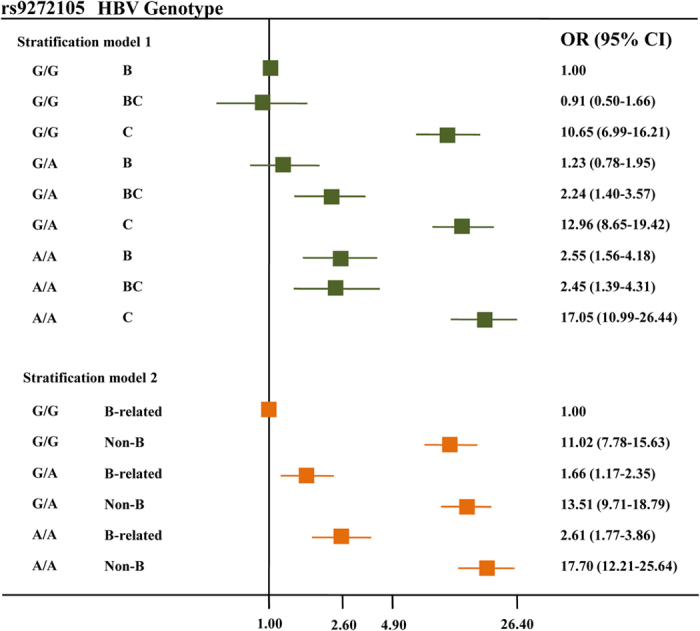
The B-related genotypes included B and BC, whereas the non-B genotypes included C and D.
Multiplicative interactions of rs9272105 and rs9275319 with all the HBV hotspot mutations were also evaluated (Supplementary Table S11). We detected significant interactions between rs9272105 and the HBV mutations C1673T, G1719T, A1726C, C1730G, A1752G and G1799C on HCC risk (P < 0.05). As shown in Fig. 2, rs9272105 multiplicatively interacted with C1673T, G1719T, A1726C, C1730G, A1752G and G1799C. Thus, the effects of the HBV mutations on HCC risk depended on the HLA genetic background. However, we did not observe any significant interaction between rs9275319 and HBV genotype or mutations on HCC risk.
Figure 2. Crossover analysis of the SNP rs9272105-HBV mutations interactions on HCC susceptibility.
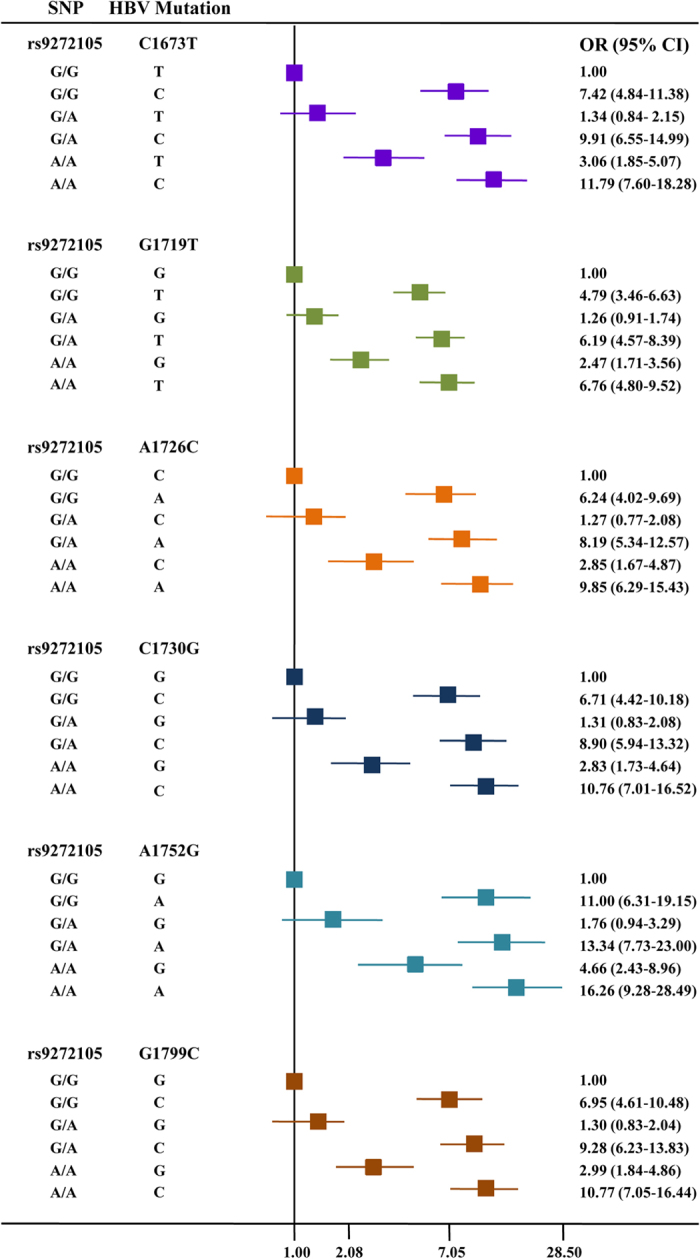
From top to bottom, the HBV mutations are C1673T, G1719T, A1726C, C1730G, A1752G and G1799C, respectively.
Performance of the panels of the HLA SNPs, HBV genotype and mutations in the prediction of HCC risk
After a stepwise regression analysis, the HBV genotype, the 11 independent HCC-related mutations, the HLA SNPs (rs9272105 and rs9275319) and the interaction of rs9272105 with A1752G were entered into the HCC prediction model (Table 2). Then, we constructed the ROC curve to evaluate the risk prediction performance of the HLA SNPs, HBV genotype and mutations for the HCC patients (Fig. 3). The AUC of the panels of the two SNPs, HBV genotype and mutations were 0.565, 0.736 and 0.821, respectively. In fact, the AUC of the panel that combined the two SNPs, HBV genotypes and the 11 mutations was even better than the above panels at 0.840 (sensitivity = 81.3%, specificity = 74.8%). The difference between the AUC of the panel 3 (HBV mutations) and panel 4 (two SNPs, HBV genotypes and mutations) was statistically significant (Z = −5.08, P = 3.82 × 10−7). For the panel including two SNPs, HBV genotype and mutations, the Hosmer-Lemeshow χ2 (8 degrees of freedom) was 11.64 (P = 0.168), giving no cause for concern over model fit or calibration.
Table 2. Results of full model (including age, sex as adjustment) after stepwise regression analysis.
| Variables | SE | Z | OR (95% CI) | P |
|---|---|---|---|---|
| Age | 0.00 | −1.34 | 0.99 (0.98–1.00) | 0.180 |
| Gender | 0.11 | −1.58 | 0.82 (0.63–1.05) | 0.115 |
| HBV genotype | 0.24 | 9.52 | 2.48 (2.06–2.99) | <0.001 |
| C1653T | 0.28 | 5.33 | 2.06 (1.58–2.68) | <0.001 |
| C1673T | 0.44 | 2.48 | 1.82 (1.13–2.93) | 0.013 |
| T1674C/G | 0.21 | 4.30 | 1.71 (1.34–2.18) | <0.001 |
| C1730G | 1.02 | 4.33 | 3.52 (1.99–6.23) | <0.001 |
| A1752G | 1.82 | 5.33 | 5.63 (2.98–10.62) | <0.001 |
| T1753C | 0.21 | 3.52 | 1.60 (1.23–2.08) | <0.001 |
| A1762T | 0.32 | 3.09 | 1.75 (1.23–2.49) | 0.002 |
| G1764A | 0.48 | 4.67 | 2.47 (1.69–3.60) | <0.001 |
| G1899A | 0.21 | 3.32 | 1.57 (1.20–2.05) | 0.001 |
| G1915A/C | 0.24 | 2.54 | 1.49 (1.10–2.04) | 0.011 |
| C1969T | 0.31 | 4.00 | 1.92 (1.40–2.65) | <0.001 |
| rs9275319 | 0.19 | 3.09 | 1.48 (1.15–1.89) | 0.002 |
| rs9272105 | 0.41 | 4.17 | 2.19 (1.52–3.17) | <0.001 |
| A1752G * rs9272105 | 0.11 | −3.21 | 0.52 (0.35–0.78) | 0.001 |
Note: The references of the prediction factors were reset for consistent risk effect.
Abbreviation: SE, standard error; Z, Z value.
Figure 3. The discriminative ability of four panels between the HBV-related HCC patients and the HBV persistent carriers was evaluated by a ROC curve analysis.
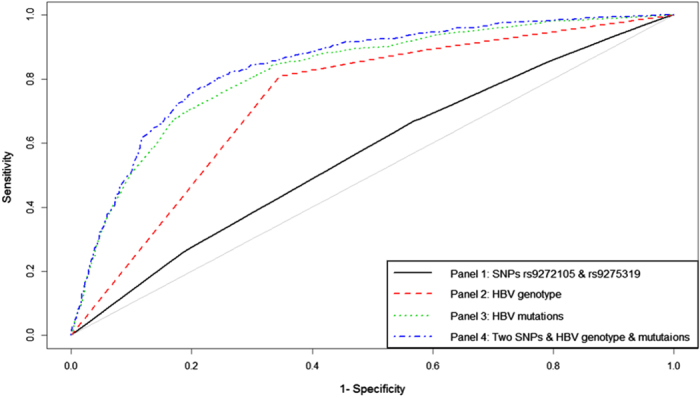
Black line, panel of the SNPs rs9272105 and rs9275319 (AUC = 0.565, sensitivity = 67.0%, specificity = 43.1%); red line, HBV genotype panel (AUC = 0.736, sensitivity = 80.9%, specificity = 65.5%); green line, HBV mutations panel (AUC = 0.821, sensitivity = 70.9%, specificity = 80.1%); blue line, the panel including the two SNPs, HBV genotype and mutations (AUC = 0.840, sensitivity = 81.3%, specificity = 74.8%).
Discussion
In this large multi-centre study, we found that HBV genotype C and subgenotype C2 were the risk factors for HCC, and 11 HBV mutations were found to be significantly associated with HCC risk. We also detected significant interactions of HLA-DQ/DR rs9272105 with both HBV genotype and mutations, which may imply a potential biological significance forrs9272105. Excitingly, the panel that combined the HLA-DQ/DR SNPs, HBV genotypes and mutations provided a high sensitivity and specificity to discriminate the HCC patients from the controls. The HBV carriers who infected with HBV genotype C and carrying the rs9272105 AA genotype, rs9275319 AA genotype and risky nucleotide of the 11 HBV mutations had a relatively high HCC risk, which was useful in screening of high-risk groups of HCC and needed to be validated in further prospective studies.
The development of HCC is a multistage process, and most HCCs arise from chronic hepatitis induced by HBV infection, particularly in China27. With the progression of chronic infection, HBV mutations gradually occur28,29. HBV reverse transcriptase lacks proofreading activity, resulting in an estimated mutation rate of 4.57 × 10−5 nucleotide (nt) substitutions per site per year30. Inflammatory factors could also promote HBV mutations, and the insufficient immune responses elicited by the HBV antigens select the disease-related HBV mutations during the long-term evolutionary process31,32. Only the HBV strains/variants best adapted to the host immune system will survive and thrive in liver33. The HLA system is the name of the locus of genes that encodes the major histocompatibility complex (MHC) in humans. This super-locus contains a large number of genes related to immune system function in humans. HLA class II molecules include three isotypes: HLA-DR, HLA-DQ, and HLA-DP, which have been reported on extensively for their association with HBV infection and hepatocarcinogenesis5,6,34,35,36. In this study, we detected significant interactions of HLA-DQA1/DRB1 rs9272105with HBV genotype and mutations on HCC risk. Thus, HLA- DQ/DR genetic polymorphisms might affect the outcomes of chronic HBV infection via regulating the immune selection of HBV mutations, thereby affecting the risk of HCC caused by the HBV mutations.
In this study, the HBV mutations A1846T and G1896A, which have been reported to be associated with an increased risk of HCC by several studies33,37,38, were not significantly associated with HCC. This may be due to different study areas, the sample sizes and the adjustment for other HBV mutations. There were also novel HBV mutations, including A1752G, G1915A/C and C1969T, which were found to be associated with HBV-related HCC. In addition, the effect of C1730G was reversed from a protective effect (adjusted OR = 0.18, 95% CI = 0.15–0.22) to a risk effect with borderline significance (adjusted OR = 2.07, 95% CI = 1.02–4.20) after being conditionally adjusted by the other mutations. Thus, the functional effects of these HBV mutations on HCC risk deserve further investigation.
We successfully validated the associations between HLA SNPs (rs9272105 and rs9275319) and HCC risk. SNP rs9272105 is located between HLA-DQA1 and HLA-DRB1, and rs9275319 is located between HLA-DQB1 and HLA-DQA2. HLA-DQ and -DR proteins make up the HLA class II complex, an α-β heterodimeric membrane glycoprotein that is expressed on the surface of antigen-presenting cells, such as B lymphocytes, macrophages and den-dritic cells. HLA class II glycoproteins present viral peptides to CD4+ T cells and influence the immune responses. Therefore, SNPs in HLA-DQ and -DR genes may have important roles in immune-mediated diseases, including liver diseases and HCC.
Recently, Cao et al. conducted a case-control study (1,108 HCC patients and 1,628 HBV-positive subjects without HCC) to evaluate the effects of HLA-DP polymorphisms (four SNPs reported by a GWAS of HBV persistent infection), HBV EnhII/BCP/PC region mutations and their interactions on HCC risk in subjects infected with HBV genotype B or genotype C. They sequenced the HBV EnhII/BCP/PC region successfully from 1,429 (52.2%) of the HBV-infected subjects and found that the interactions of rs9277535 AA with the T1674C/G or G1719T mutation in genotype C significantly decreased HCC risk33. The same group also evaluated the effect of the STAT3 SNPs and their interactions with HBV mutations on HCC risk with a sample size of 1,021 HCC patients and 990 HBV-positive subjects without HCC. In this study, they genotyped three SNPs of STAT3and sequenced the HBV EnhII/BCP/PC region successfully from 1,160 (57.7%) of the HBV-infected subjects. Finally, they found the interaction of rs1053004 with T1674C/G significantly increased the HCC risk37. Lately, the group performed a case-control study again with a larger sample size of 1,531 HCC patients and 2,489 HBV-positive subjects without HCC to evaluate the impacts of HLA-DQ SNPs (rs2856718 reported by a GWAS of chronic hepatitis B and rs9275319 reported by a HCC GWAS) and their interactions with HBV mutations on the risk of HCC. They sequenced the HBV EnhII/BCP/PC region successfully from 1,450 (36.11%) of all the HBV-infected subjects. And they found rs2856718 variant genotypes significantly decreased HCC risk and the variant genotypes of rs2856718 were significantly associated with an increased frequency of HBV A1726C mutation in genotype C. However, the association betweenrs9275319 and HCC risk was not observed (adjusted OR = 0.99, 95% CI = 0.83–1.18, P = 0.876), which failed to validate the results of the HCC GWAS (P = 2.72 × 10−17), and was different from our results (P = 1.008 × 10−6)6,39.The findings were exciting; however, the different study design (different sample size and matching criteria) and varying methodology (different models of nest multiplex PCR and primers, the detection rate and definition for hotspot mutations) could influence the reproducible of the results13,15,18,19,33,37,39,40.
To the best of our knowledge, this is the first large multi-centre study revealing that HBV genotype and mutations could affect HCC risk via interacting with HLA-DQ/DR genetic variants, and this is the first study constructing prediction models and estimating sensitivity and specificity. There are four advantages of our study. First, the multiple centres used included a large sample size matched by area, age and gender, and the rigid quality control provided sufficient statistical power and more convincing data. Second, because the HBV genotypes were varied among the controls, the controls from an ongoing large-scale, population-based cohort can be helpful for quality control. Third, the success rates of the determination of the HBV genotypes, subgenotypes and mutations were much higher than in previous studies, and the distribution of the HBV genotypes and subgenotypes between cases and controls was validated successfully in an additional independent sample set. Fourth, the diagnostic performance of the panel including the HLA SNPs, HBV genotype and mutations was relatively high (sensitivity = 81.3%, specificity = 74.8%), and the detection of these factors was non-invasive. However, this study had limitations as well. The sample size of the additional sample set was too small to validate the association of the HBV mutations or HLA-DQ/DR SNPs with HCC risk and the interaction of rs9272105 with the HBV genotype and mutations. Further studies with large sample size in diverse populations are warranted to validate and extend our findings. And HBV DNA levels, HBeAg status and ALT level were not detected in our case-control study because some participants had received antiviral treatments, especially for the HCC patients. In addition, since the controls were from a large-scale, population-based cohort, the sensitivity and specificity of the cirrhosis diagnosis may be relatively low, and the antiviral treatment scheme was not obtained. Therefore, cirrhosis status and antiviral therapy were also not shown. Further rigorous prospective studies were also needed to dynamically monitor the HBV persistent carriers. Since HBV EnhII/BCP/PC region is one of important regulatory regions for HBV replication and is less sensitive to antiviral treatments, we only amplified this region for HBV mutation analysis. The detection methods of HBV genotype and mutations were based on nested PCR and sequencing in this study, thus there might be a small number of HBV carriers who failed to detect HBV genotype and mutations for low viral loads. Moreover, we only focused on the HCC GWAS-identified HLA SNPs in terms of the host genetic polymorphism. Therefore, well-designed studies involving genome-wide genetic factors (at least immune-related genes), mutations of the entire HBV genome, and HBV DNA levels may improve the prediction accuracy.
Methods
The methods were carried out in accordance with the approved guidelines. And all experimental protocols were approved by the institutional review board of Nanjing Medical University.
Participants
The HCC patients were consecutively recruited between January 2006 and May 2014 at the First Affiliated Hospital of Nanjing Medical University (Nanjing, China), the Nantong Tumour Hospital (Nantong, China) and the Qidong Liver Cancer Institute (Qidong, China) from central and southern Jiangsu Province. The diagnosis of HCC was confirmed by a pathological examination and/or an alpha-fetoprotein elevation (>400 ng/ml) combined with an imaging examination (i.e., magnetic resonance imaging and/or computerised tomography). Because HCV infection is rare in Chinese populations, we excluded HCC patients with HCV infection. As a result, 1,507 HBV-related HCC cases consented to participate in the study and provided blood samples (535 cases from Nanjing, 522 cases from Nantong and 450 cases from Qidong). Only the 450 cases from Qidong were shared with the published GWAS6.
Because the cases were all from central and southern Jiangsu Province, the controls from three cities in central and southern Jiangsu Province were screened for being HBV persistent carriers in 2009 (48,417 subjects from Zhangjiagang, 43,563 subjects from Taixing and 57,192 subjects from Danyang) and were confirmed in 2010. All the participants were self-reported Han Chinese. The HBV persistent carriers were those subjects who were positive for both hepatitis B surface antigen (HBsAg) and antibodies against the hepatitis B core antigen (anti-HBc) but were negative for the HCV antibody (anti-HCV) at the two visits. Approximately 2,475 (5.11%), 3,413 (7.83%) and 5,587 (9.77%) HBV persistent carriers were identified from Zhangjiagang, Taixing and Danyang, respectively. Then, we selected 1,560 HBV persistent carriers from these three cities and matched them to the HCC cases by age and gender (510 controls from Zhangjiagang, 500 controls from Taixing and 550 controls from Danyang). These selected controls had no history of cancer by self-report and B ultrasonic scanning, and their demographic information, such as age and gender, was collected by face-to-face interviews. All the controls included in the current study were independent from the two published GWAS5,6.
An additional small sample set for validation of the HBV genotype main effect was consecutively recruited between December 2013 and April 2014 at the Tangdu and Xijing Hospital of the Fourth Military Medical University (Xi’an, China)in Shaanxi Province, including 107 HBV-related HCC cases and 204 HBV persistent carriers, in which age and gender were matched between the cases and controls.
After written informed consent was obtained, each participant was scheduled for an interview by using a structured questionnaire to collect demographic and exposure information. After interview, a venous blood sample of approximately 5 ml was collected from each participant.
Serological testing
HBsAg, anti-HBs, anti-HBc, and anti-HCV were detected by an enzyme-linked immunosorbent assay (Kehua Bio-Engineering Co., Ltd., Shanghai, China) in the serum, following the manufacturer’s instructions as described previously4.
Determination of HBV genotypes and subgenotypes
HBV DNA was extracted from 200 μl of serum of the HBV-positive samples using High Pure Viral Nucleic Acid kits (Roche Diagnostics GmbH, Mannheim, Germany) according to the manufacturer’s instructions. The HBV genotypes and subgenotypes were identified by nested multiplex PCR, which was modified from Cao et al.13.The first round of amplification with rTaq DNA polymerase (TaKaRa, Dalian, China) was carried out as followed: 1) P1-S and P1-AS primers were used for the amplification of the first round of the HBV genotypes A, B, C and D, 2) P2-S and P2-AS primers were used for the amplification of the HBV subgenotypes B1 and B2, and 3) P3-S and P3-AS primers were used for the amplification of the HBV genotypes E and F and the subgenotypes C1and C2. The product (2 μl) of the first round of PCR served as a template for the routine multiplex PCR. The primers used are listed in Supplementary Table S1. The PCR-amplified products were electrophoresed on a 2% agarose gel, stained with ethidium bromide and visualised under UV light. If the HBV genotype was determined successfully but the subgenotype was not, the genotype-specific product was sequenced and aligned with the reference sequences (http://www.ncbi.nlm.nih.gov; http://hbv.geno2pheno.org). All the genotyping assays were performed at the same time at the same lab without knowing the subjects’ case and control statuses; two blank (i.e., water) controls in each 96-well format were used for quality control, and more than 10% of the samples were randomly selected to be confirmed by DNA sequencing, yielding a 100% concordant. The success rates of determining the HBV genotypes and subgenotypes for each area were all above 96%.
HBV mutation analysis
The HBV EnhII/BCP/PC region was amplified in the subjects with a successfully detected HBV genotype using nested PCR13. The primers for the first and second round of the nested PCR are listed in Supplementary Table S1. Direct DNA sequencing was carried out by using ABI PRISM BigDye sequencing kits and an ABI 3730 Genetic Analyser (Applied Biosystems, Foster City, CA). The HBV sequences were aligned and analysed using MEGA 5.0 software. After the alignment, the nucleotide with the highest frequency at each site in the HBV EnhII/BCP/PC region from the HBV persistent carriers was termed as the wild type nucleotide. Nucleotide substitutions with the other three nucleotides and deletions at each site were termed as mutations. A site with combined mutation frequencies >10% from all the participants was termed a hotspot.
Genotyping of the HLA-DP/DQ SNPs
The genomic DNA was extracted from the leukocyte pellets by traditional proteinase K digestion, followed by phenol-chloroform extraction and ethanol precipitation. The SNPs rs9272105 and rs9275319 were genotyped by a TaqMan allelic discrimination assay on an ABI 7900 system (Applied Biosystems, La Jolla, CA). The information on the primers and probes is shown in Supplementary Table S1. All the genotyping assays were performed without knowing the subjects’ case and control statuses; two blank (i.e., water) controls in each 384-well format were used for quality control, and more than 10% of the samples were randomly selected to repeat, yielding a 100% concordant. The success rates of genotyping for the rs9272105 and rs9275319 SNPs were 98.73 and 98.79%, respectively.
Statistical analysis
The differences of the demographic characteristics, HBV genotypes, subgenotypes and mutations frequencies, in addition to the genotype frequencies of the SNPs between the cases and controls, were calculated by Student’s t-tests or a one-way analysis of variance (for the continuous variables) and a χ2 test (for the categorical variables). The associations of the HBV genotype, mutations and the genotypes of the SNPs with HCC risk were estimated by computing odds ratios (ORs) and their 95% confidence intervals (CIs) from logistic regression analyses. The heterogeneity of the associations between subgroups was assessed by the χ2 -based Q test. The linkage disequilibrium (LD) information for the mutation hotspots was evaluated by a pairwise r2. A risk prediction model to classify the HCC cases and controls was constructed according to the following steps: (1) Prediction factor selection: HBV genotype, the 11 independent HCC-related mutations (C1653T, C1673T, T1674C/G, C1730G, A1752G, T1753C, A1762T, G1764A, G1899A, G1915A/C and C1969T), the HLA SNPs (rs9272105 and rs9275319), the HCC-related multiplicative interactions (rs9272105 with the HBV genotype, the HBV mutations C1673T, G1719T, A1726C, C1730G, A1752G and G1799C) and the main effect of the interactions were considered to be the predictive factors by conducting a backwards stepwise logistic regression model, with a significance level of 0.05 for removing the respective variables. (2) Risk model construction: the variables that remained in the backwards stepwise model were included, and the risk prediction model was constructed using a logistic regression model. (3) Risk model evaluation: the model performance was evaluated by receiver-operator characteristic (ROC) curves, and the area under the curve (AUC) was used to classify the HCC cases and controls. The sensitivity and specificity were calculated to illustrate the model effects using the “best threshold” criteria of the ROC curve. The difference in the area under two correlated ROC curves was evaluated by DeLong’s test. The model’s calibration was assessed by Hosmer-Lemeshow χ2 test. All the statistical analyses were performed with R software (version 2.13.0; The R Foundation for Statistical Computing), and P ≤ 0.05 in a two-sided test was considered statistically significant.
Additional Information
How to cite this article: Wen, J. et al. Hepatitis B virus genotype, mutations, human leukocyte antigen polymorphisms and their interactions in hepatocellular carcinoma: a multi-centre case-control study. Sci. Rep. 5, 16489; doi: 10.1038/srep16489 (2015).
Supplementary Material
Acknowledgments
This work was supported in part by the National Science Foundation for Distinguished Young Scholars of China (81225020), the National Key Basic Research Program For Youth (2013CB911400), the National Program for Support of Top-notch Young Professionals, the National Major S&T Projects (2009ZX10004-904; 2011ZX10004-902; 2013ZX10004-905), the Foundation for the Program for New Century Excellent Talents in University (NCET-10-0178), the Foundation for the Author of National Excellent Doctoral Dissertation (201081), the Foundation of Jiangsu Province for Distinguished Young Scholars (BK2012042), Jiangsu Specially-Appointed Professor project, the National Natural Science Foundation of China (31100895), the Outstanding Young Scholar Project of Fudan University, the Jiangsu Province Clinical Science and Technology Projects (BL2012008) and the Priority Academic Program for the Development of Jiangsu Higher Education Institutions (Public Health and Preventive Medicine).
Footnotes
Author Contributions Z.B.H. and H.B.S. designed the study. J.W., C.S., D.K.J., T.B.J., L.G.Z., J.Z.A., Y.L., S.J.M., N.Q., C.L., J.P.C., Y.J., L.L.Y., L.L., T.T.G., J.B.L., J.J., F.C.Z., Y.F.Z., L.Y., J.G.C. and C.C. collected the data. Z.B.H., J.C.D. and J.W. conducted the statistical analysis and interpretation. J.W. and Z.B.H. wrote the report. Z.B.H., X.J.Z., J.F.X. and H.B.S. revised the report. All the authors reviewed the report and approved the final version.
References
- Jemal A. et al. Global cancer statistics. CA Cancer J Clin 61, 69–90 (2011). [DOI] [PubMed] [Google Scholar]
- Siegel R., Naishadham D. & Jemal A. Cancer statistics, 2012. CA Cancer J Clin 62, 10–29 (2012). [DOI] [PubMed] [Google Scholar]
- El-Serag H. B. & Rudolph K. L. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 132, 2557–76 (2007). [DOI] [PubMed] [Google Scholar]
- Hu L. et al. Genetic variants in human leukocyte antigen/DP-DQ influence both hepatitis B virus clearance and hepatocellular carcinoma development. Hepatology 55, 1426–31 (2012). [DOI] [PubMed] [Google Scholar]
- Li S. et al. GWAS identifies novel susceptibility loci on 6p21.32 and 21q21.3 for hepatocellular carcinoma in chronic hepatitis B virus carriers. PLoS Genet 8, e1002791 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D. K. et al. Genetic variants in STAT4 and HLA-DQ genes confer risk of hepatitis B virus-related hepatocellular carcinoma. Nat Genet 45, 72–5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norder H. et al. Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology 47, 289–309 (2004). [DOI] [PubMed] [Google Scholar]
- Kramvis A., Kew M. & Francois G. Hepatitis B virus genotypes. Vaccine 23, 2409–23 (2005). [DOI] [PubMed] [Google Scholar]
- Cao G. W. Clinical relevance and public health significance of hepatitis B virus genomic variations. World J Gastroenterol 15, 5761–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan H. L. et al. High viral load and hepatitis B virus subgenotype ce are associated with increased risk of hepatocellular carcinoma. J Clin Oncol 26, 177–82 (2008). [DOI] [PubMed] [Google Scholar]
- Zhang H. W. et al. Risk factors for acute hepatitis B and its progression to chronic hepatitis in Shanghai, China. Gut 57, 1713–20 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai C. T., Chu C. J., Hussain M. & Lok A. S. HBV genotype B is associated with better response to interferon therapy in HBeAg(+) chronic hepatitis than genotype C. Hepatology 36, 1425–30 (2002). [DOI] [PubMed] [Google Scholar]
- Yin J. et al. Distribution and hepatocellular carcinoma-related viral properties of hepatitis B virus genotypes in Mainland China: a community-based study. Cancer Epidemiol Biomarkers Prev 19, 777–86 (2010). [DOI] [PubMed] [Google Scholar]
- Yang H. I. et al. Associations between hepatitis B virus genotype and mutants and the risk of hepatocellular carcinoma. J Natl Cancer Inst 100, 1134–43 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J. et al. Role of hepatitis B virus genotype mixture, subgenotypes C2 and B2 on hepatocellular carcinoma: compared with chronic hepatitis B and asymptomatic carrier state in the same area. Carcinogenesis 29, 1685–91 (2008). [DOI] [PubMed] [Google Scholar]
- Hung C. H. et al. Role of viral genotypes and hepatitis B viral mutants in the risk of hepatocellular carcinoma associated with hepatitis B and C dual infection. Intervirology 56, 316–24 (2013). [DOI] [PubMed] [Google Scholar]
- McMahon B. J. The influence of hepatitis B virus genotype and subgenotype on the natural history of chronic hepatitis B. Hepatol Int 3, 334–42 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S. et al. A matched case-control study of hepatitis B virus mutations in the preS and core promoter regions associated independently with hepatocellular carcinoma. J Med Virol 83, 45–53 (2011). [DOI] [PubMed] [Google Scholar]
- Yin J. et al. Significant association of different preS mutations with hepatitis B-related cirrhosis or hepatocellular carcinoma. J Gastroenterol 45, 1063–71 (2010). [DOI] [PubMed] [Google Scholar]
- Kay A. & Zoulim F. Hepatitis B virus genetic variability and evolution. Virus Res 127, 164–76 (2007). [DOI] [PubMed] [Google Scholar]
- Yang H. I. et al. Hepatitis B e antigen and the risk of hepatocellular carcinoma. N Engl J Med 347, 168–74 (2002). [DOI] [PubMed] [Google Scholar]
- Chen C. J. et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 295, 65–73 (2006). [DOI] [PubMed] [Google Scholar]
- Wu C. F. et al. Long-term tracking of hepatitis B viral load and the relationship with risk for hepatocellular carcinoma in men. Carcinogenesis 29, 106–12 (2008). [DOI] [PubMed] [Google Scholar]
- Qu L. S. et al. Effect of combined mutations in the enhancer II and basal core promoter of hepatitis B virus on development of hepatocellular carcinoma in Qidong, China. Hepatol Res 44, 1186–95(2014). [DOI] [PubMed] [Google Scholar]
- Bai X. et al. Temporal acquisition of sequential mutations in the enhancer II and basal core promoter of HBV in individuals at high risk for hepatocellular carcinoma. Carcinogenesis 32, 63–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen M. F. et al. Independent risk factors and predictive score for the development of hepatocellular carcinoma in chronic hepatitis B. J Hepatol 50, 80–8 (2009). [DOI] [PubMed] [Google Scholar]
- Hernandez-Gea V., Toffanin S., Friedman S. L. & Llovet J. M. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 144, 512–27 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran A. et al. Emergence of and takeover by hepatitis B virus (HBV) with rearrangements in the pre-S/S and pre-C/C genes during chronic HBV infection. J Virol 65, 3566–74 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X., Jin Y., Qian G. & Tu H. Sequential accumulation of the mutations in core promoter of hepatitis B virus is associated with the development of hepatocellular carcinoma in Qidong, China. J Hepatol 49, 718–25 (2008). [DOI] [PubMed] [Google Scholar]
- Orito E. et al. Host-independent evolution and a genetic classification of the hepadnavirus family based on nucleotide sequences. Proc Natl Acad Sci USA 86, 7059–62 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian J. P. et al. Massive APOBEC3 editing of hepatitis B viral DNA in cirrhosis. PLoS Pathog 6, e1000928 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R. et al. Association of human APOBEC3 cytidine deaminases with the generation of hepatitis virus B x antigen mutants and hepatocellular carcinoma. Hepatology 46, 1810–20 (2007). [DOI] [PubMed] [Google Scholar]
- Zhang Q. et al. HLA-DP polymorphisms affect the outcomes of chronic hepatitis B virus infections, possibly through interacting with viral mutations. J Virol 87, 12176–86 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbarek H. et al. A genome-wide association study of chronic hepatitis B identified novel risk locus in a Japanese population. Hum Mol Genet 20, 3884–92 (2011). [DOI] [PubMed] [Google Scholar]
- Hu Z. et al. New loci associated with chronic hepatitis B virus infection in Han Chinese. Nat Genet 45, 1499–503 (2013). [DOI] [PubMed] [Google Scholar]
- Kamatani Y. et al. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat Genet 41, 591–5 (2009). [DOI] [PubMed] [Google Scholar]
- Xie J. et al. Interaction of signal transducer and activator of transcription 3 polymorphisms with hepatitis B virus mutations in hepatocellular carcinoma. Hepatology 57, 2369–77 (2013). [DOI] [PubMed] [Google Scholar]
- Park Y. M. et al. Combinations of eight key mutations in the X/preC region and genomic activity of hepatitis B virus are associated with hepatocellular carcinoma. J Viral Hepat 21, 171–7 (2014). [DOI] [PubMed] [Google Scholar]
- Ji X. et al. Impacts of human leukocyte antigen DQ geneticpolymorphisms and their interactions with hepatitis B virus mutations on the risks of viral persistence, liver cirrhosis, and hepatocellular carcinoma.Infect Genet Evol 28, 201–9 (2014). [DOI] [PubMed] [Google Scholar]
- Yin J. et al. Association between the various mutations in viral core promoter region to different stages of hepatitis B, ranging of asymptomatic carrier state to hepatocellular carcinoma. Am J Gastroenterol 106, 81–92 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.