

A 5:1 mixture of 4-hydroxypyridine with benzene 1,3,5-tricarboxylic acid in methanol yields the title hydrogen-bonded framework compound. This compound crystallizes in the orthorhombic space group Pna21 and is a polymorph of the same stoichiometric species, reported in Cc.
Keywords: crystal structure, hydrogen-bond framework, polymorph
Abstract
Slow co-crystallization of a solution of benzene-1,3,5-tricarboxylic acid with a large excess of 4-hydroxypyridine produces an interpenetrating, three-dimensional, hydrogen-bonded framework consisting of three 4-pyridone and one benzene-1,3,5-tricarboxylic acid molecules, C9H6O6·3C5H5NO. This structure represents an orthorhombic polymorph of the previously reported C-centered, monoclinic structure [Campos-Gaxiola et al. (2014 ▸). Acta Cryst. E70, o453–o454].
Chemical context
We have been interested in the co-crystallization properties of benzene carboxylic acid derivatives (namely: benzene-1,4-dicarboxylic acid and benzene-1,3,5-tricarboxylic acid) with 3- and 4-hydroxypyridines (Staun & Oliver, 2012 ▸, 2015 ▸; Bhogala et al., 2005 ▸). A variety of 3-hydroxypyridine co-crystallants with benzene carboxylic acids have already been reported and we discontinued pursuit of those materials (Shattock et al., 2008 ▸). Both 4-hydroxypyridine and benzene-1,3,5-tricarboxylic acid have been used extensively in both metal-organic frameworks as well as suitable donor/acceptor species in crystal engineering (see for example: Castillo et al., 2001 ▸; Qian et al., 2014 ▸). Recently we reported the characterization of the 1:1 co-crystallant 4-hydroxypyridinium 3,5-dicarboxybenzoate (Staun & Oliver, 2015 ▸). We also discovered that from similar preparative conditions (slow evaporation from methanol) with a larger molar ratio of 4-hydroxypyridine to benzene-1,3,5-tricarboxylic acid (BTC) a new species could be obtained; reported herein. A comparison of the structure with the Cambridge Structure Database revealed an identical structural motif, albeit in a different crystal system (Campos-Gaxiola et al., 2014 ▸). Thus, we report the orthorhombic polymorph of benzene-1,3,5-tricarboxylic acid–4-pyridone (1/3).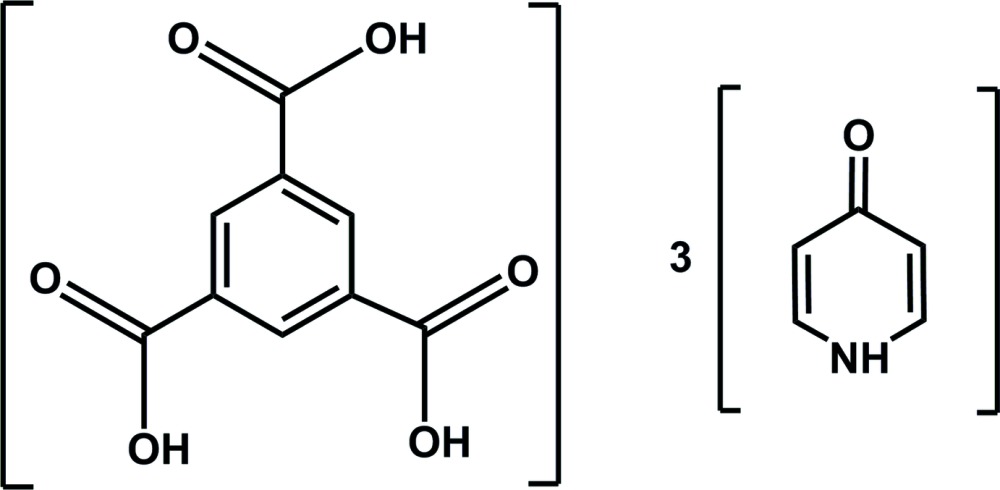
Structural commentary
The dihedral angles formed by the carboxylic acid moieties with respect to the benzene ring are 2.95 (16), 6.23 (10) and 10.28 (18)°. These are comparable with those for the previously reported polymorph of this compound [3.9 (2), 9.3 (2), and 13.3 (2)°; Campos-Gaxiola et al., 2014 ▸]. It should be noted that the 4-hydroxypyridine has undergone rearrangement from a hydroxypyridine to the pyridone form of the molecule as previously observed (Tyl et al., 2008 ▸). The 4-pyridone C—O bond distances range from 1.280 (8) to 1.295 (8) Å. These distances are comparable with previously reported examples of this molecule (Staun & Oliver, 2012 ▸; Tyl et al., 2008 ▸). Inspection of the bond distances about each pyridone ring shows a slight tendency for the C—C bonds α to the nitrogen [1.347 (12) to 1.371 (11) Å] to be shorter than those to the carbonyl carbon [1.410 (11) to 1.421 (10) Å]. This supports the proposed formal, localized double bond along the ‘edges’ of the pyridone ring.
Two of the three 4-pyridone rings are co-planar with the benzene tricarboxylic acid moiety, similar to that of the previously reported structure (Campos-Gaxiola et al., 2014 ▸). The remaining 4-pyridone is essentially perpendicular to this plane, also similar to the Campos-Gaxiola structure (Table 1 ▸).
Table 1. Pyridone / BTC interplanar angles ().
| Pyridone ring | This work | Campos-Gaxiola |
|---|---|---|
| N1 | 7.3(2) | 12.9 |
| N2 | 8.5(2) | 13.2 |
| N3 | 87.5(3) | 87.1 |
Supramolecular features
Each of the pyridone molecules forms a hydrogen-bonded chain of symmetry-related molecules. N1 and N2 form hydrogen bonds to O1i and O2ii, respectively, related by the crystallographic n-glide [symmetry codes: (i) x −  , −y + , z; (ii) x + , −y +
, −y + , z; (ii) x + , −y +  , z]. N3 forms hydrogen bonds to O3iii and O6iv related by translation along the crystallographic c-axis and the [
, z]. N3 forms hydrogen bonds to O3iii and O6iv related by translation along the crystallographic c-axis and the [ 01] direction, respectively [symmetry codes: (iii) x, y, z + 1; (iv) x − 1, y, z + 1). Thus N3 forms a bifurcated hydrogen bond. These chains of hydrogen-bonded pyridone molecules are bridged by the BTC molecule. Each carboxylic acid moiety on BTC donates a hydrogen bond to a nearby pyridone carbonyl oxygen (Fig. 1 ▸, Table 2 ▸). These OCOOH⋯Opy contacts are short for O—H⋯O contacts indicating strong intermolecular hydrogen bonding. As a result of the N3 pyridone being oriented almost perpendicular to the plane of the other three molecules, the resulting architecture is a three-dimensional hydrogen-bonded network. The BTC, N1 and N2 pyridone molecules form a graph-set
01] direction, respectively [symmetry codes: (iii) x, y, z + 1; (iv) x − 1, y, z + 1). Thus N3 forms a bifurcated hydrogen bond. These chains of hydrogen-bonded pyridone molecules are bridged by the BTC molecule. Each carboxylic acid moiety on BTC donates a hydrogen bond to a nearby pyridone carbonyl oxygen (Fig. 1 ▸, Table 2 ▸). These OCOOH⋯Opy contacts are short for O—H⋯O contacts indicating strong intermolecular hydrogen bonding. As a result of the N3 pyridone being oriented almost perpendicular to the plane of the other three molecules, the resulting architecture is a three-dimensional hydrogen-bonded network. The BTC, N1 and N2 pyridone molecules form a graph-set  (44) ring that is parallel with the ab plane (Macrae et al., 2008 ▸). This corresponds with that observed by Campos-Gaxiola et al. The BTC and N3 pyridone form an
(44) ring that is parallel with the ab plane (Macrae et al., 2008 ▸). This corresponds with that observed by Campos-Gaxiola et al. The BTC and N3 pyridone form an  (30) ring that is perpendicular to the previous ring. Further inspection of this network reveals that there are two independent, interpenetrating networks (Fig. 2 ▸). The BTC molecules in the two networks form typical slipped π–π-stacks [Cg⋯Cg = 3.592 (5) Å, Cg⋯perp = 3.302 (4) Å; Cg represents the center of gravity of the ring, perp is the shortest perpendicular distance; Spek, 2009 ▸]. Other potential π–π contacts are beyond 4 Å. Due to the efficient packing of these molecules there is a significant number of close C—H⋯O contacts, primarily between pyridone carbon atoms and carboxylic acid oxygen atoms, with one notable example being a contact from C9 to O3v [symmetry code: (v) x + 1, y, z].
(30) ring that is perpendicular to the previous ring. Further inspection of this network reveals that there are two independent, interpenetrating networks (Fig. 2 ▸). The BTC molecules in the two networks form typical slipped π–π-stacks [Cg⋯Cg = 3.592 (5) Å, Cg⋯perp = 3.302 (4) Å; Cg represents the center of gravity of the ring, perp is the shortest perpendicular distance; Spek, 2009 ▸]. Other potential π–π contacts are beyond 4 Å. Due to the efficient packing of these molecules there is a significant number of close C—H⋯O contacts, primarily between pyridone carbon atoms and carboxylic acid oxygen atoms, with one notable example being a contact from C9 to O3v [symmetry code: (v) x + 1, y, z].
Figure 1.

Labeling scheme for title compound. Atomic displacement ellipsoids are depicted at the 50% probability level. Dashed lines represent hydrogen bonds within the asymmetric unit.
Table 2. Hydrogen-bond geometry (, ).
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| N1H1NO1i | 0.88 | 1.89 | 2.762(8) | 169 |
| N2H2NO2ii | 0.88 | 1.90 | 2.711(8) | 152 |
| N3H3NO3iii | 0.88 | 2.01 | 2.773(10) | 144 |
| N3H3NO6iv | 0.88 | 2.59 | 3.124(9) | 120 |
| O5H5OO1 | 0.84 | 1.75 | 2.555(7) | 161 |
| O7H7OO2 | 0.84 | 1.73 | 2.463(7) | 145 |
| O9H9OO3 | 0.84 | 1.70 | 2.526(7) | 167 |
| C1H1O4i | 0.95 | 2.38 | 3.227(10) | 148 |
| C4H4O5 | 0.95 | 2.53 | 3.174(9) | 126 |
| C6H6O7ii | 0.95 | 2.26 | 3.051(9) | 140 |
| C7H7O8ii | 0.95 | 2.66 | 3.530(9) | 153 |
| C9H9O3v | 0.95 | 2.58 | 3.227(9) | 126 |
| C11H11O6iv | 0.95 | 2.46 | 3.076(11) | 123 |
| C11H11O9vi | 0.95 | 2.55 | 3.159(9) | 122 |
| C12H12O6vii | 0.95 | 2.49 | 3.302(11) | 143 |
| C14H13O4viii | 0.95 | 2.60 | 3.405(10) | 143 |
| C15H15O8iii | 0.95 | 2.66 | 3.608(10) | 178 |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  ; (iv)
; (iv)  ; (v)
; (v)  ; (vi)
; (vi)  ; (vii)
; (vii)  ; (viii)
; (viii)  .
.
Figure 2.

Space-filling views displaying the interpenetrating networks (a) along the a axis; (b) along the c axis.
Database survey
A search in the Cambridge Structural Database (CSD, Version 5.36 plus 3 updates; Groom & Allen, 2014 ▸) for 4-hydroxypyridine with benzene-1,3,5-tricarboxylic acid produced only one hit. The compound is closely related to the title compound, namely: benzene-1,3,5-tricarboxylic acid-pyridinium-2-olate (1/3) (Campos-Gaxiola et al., 2014 ▸). However, the structure is reported to be in the monoclinic space group Cc.
Comparison with the structure of the monoclinic polymorph
Inspection of an overlay of the two structures reveals some differences between the two polymorphs (Fig. 3 ▸). The orientation of the carboxylic acid groups of the BTC in the title compound has one ‘reversed’ with respect to the others, while the Campos-Gaxiola structure has all three oriented in the same direction, forming a propeller-like motif about the BTC. This results in a change in the hydrogen-bonding motif, reversing the orientations of the pyridone moieties. Perhaps the most prominent structural change is the orientation of the pyridone perpendicular to the plane of the BTC. In the title compound the pyridone rings are oriented with planes that are parallel to each other along the channels they occupy and are related by the screw axis parallel to the c axis. The perpendicular pyridone rings in the Campos-Gaxiola structure alternate their orientation along the channel, related by the c-glide. The change in hydrogen-bonding directionality is propagated to the orientation of the N1 and N2 pyridone chains. Examining the orientation of the carbonyl of the pyridone in these two chains reveals that the Campos-Gaxiola structure has the N1 and N2 chains oriented with the carbonyl along the a-axis forming a ‘parallel‘ alignment of the adjacent pyridone chains; again the c-glide is the cause for this arrangement. The N1 and N2 chains in the title compound adopt an ‘anti-parallel’ orientation with carbonyls in one chain being oriented in the opposite direction to the next chain, again a function of the screw axis. This is highlighted in Fig. 3 ▸ with the pyridone chain on the left of the figure showing an overlap of the pyridone rings between the two structures and the chain on the right of the figure showing the opposite orientation of the pyridone rings.
Figure 3.

Overlay of the title compound (red) with the Campos-Gaxiola (light green) structure. The BTC moiety is used as the target for overlay. The view is along the c axis of both structures. Non-H atoms depicted as arbitrary spheres, H atoms as short sticks.
Synthesis and crystallization
The compound was formed by dissolving 4-hydroxypyridine (0.112 g, 1.18 mmol) in methanol (3 mL) and benzene 1,3,5-tricarboxylic acid (0.052 g, 0.24 mmol) in methanol (3 mL). The two solutions were combined and allowed to evaporate over 5 d yielding crystals suitable for diffraction studies. The crystallization process yields crystals of both the previously reported 1:1 co-crystal (Staun & Oliver, 2015 ▸) and those of the title compound. Presumably the differences in solvent composition and time for crystallization can yield one polymorph over the other. Several crystallization attempts were made using the methodology described herein (slow evaporation from methanol) and all yielded mixtures of the 1:1 and the 3:1 co-crystals reported herein. No evidence of the Campos-Gaxiola structure was observed within the crystals examined (reported as colorless rectangular prisms).
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 3 ▸. Where possible, hydrogen atoms were initially located from a difference Fourier map and were subsequently refined using a riding model with C—H = 0.95 Å, N—H = 0.88 Å and O—H = 0.84 Å. U iso(H) was set to 1.2U eq(C/N) and 1.5U eq(O). The reliability for the correct enantiomorph of the space group is low, due to the use of Mo Kα radiation with a light atom structure. Analysis of the Flack x [0.1 (10); Flack, 1983 ▸], Hooft y [0.2 (10); Hooft et al., 2008 ▸] and Parsons z [−0.2 (12); Parsons et al., 2013 ▸] parameters tends to indicate that the correct enantiomorph of the space group and absolute structure has been determined (Flack & Bernardinelli, 1999 ▸). Since these values are not close to zero the model could be refined as a racemic twin. However, this does not yield new or useful information and we have retained the standard model.
Table 3. Experimental details.
| Crystal data | |
| Chemical formula | C9H6O63C5H5NO |
| M r | 495.44 |
| Crystal system, space group | Orthorhombic, P n a21 |
| Temperature (K) | 120 |
| a, b, c () | 12.699(3), 26.498(6), 6.6591(14) |
| V (3) | 2240.9(8) |
| Z | 4 |
| Radiation type | Mo K |
| (mm1) | 0.11 |
| Crystal size (mm) | 0.11 0.07 0.05 |
| Data collection | |
| Diffractometer | Bruker APEXII |
| Absorption correction | Multi-scan (SADABS; Krause et al., 2015 ▸) |
| T min, T max | 0.647, 0.745 |
| No. of measured, independent and observed [I > 2(I)] reflections | 19034, 3257, 2418 |
| R int | 0.109 |
| max () | 23.4 |
| (sin /)max (1) | 0.558 |
| Refinement | |
| R[F 2 > 2(F 2)], wR(F 2), S | 0.069, 0.171, 1.04 |
| No. of reflections | 3257 |
| No. of parameters | 328 |
| No. of restraints | 1 |
| H-atom treatment | H-atom parameters constrained |
| max, min (e 3) | 0.43, 0.43 |
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2056989015017867/zl2644sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015017867/zl2644Isup2.hkl
CCDC reference: 1427116
Additional supporting information: crystallographic information; 3D view; checkCIF report
supplementary crystallographic information
Crystal data
| C9H6O6·3C5H5NO | Dx = 1.469 Mg m−3 |
| Mr = 495.44 | Mo Kα radiation, λ = 0.71073 Å |
| Orthorhombic, Pna21 | Cell parameters from 1078 reflections |
| a = 12.699 (3) Å | θ = 3.1–19.2° |
| b = 26.498 (6) Å | µ = 0.11 mm−1 |
| c = 6.6591 (14) Å | T = 120 K |
| V = 2240.9 (8) Å3 | Rod, colorless |
| Z = 4 | 0.11 × 0.07 × 0.05 mm |
| F(000) = 1032 |
Data collection
| Bruker APEXII diffractometer | 3257 independent reflections |
| Radiation source: fine-focus sealed tube | 2418 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.109 |
| Detector resolution: 8.33 pixels mm-1 | θmax = 23.4°, θmin = 1.5° |
| combination of ω and φ–scans | h = −14→14 |
| Absorption correction: multi-scan (SADABS; Krause et al., 2015) | k = −29→29 |
| Tmin = 0.647, Tmax = 0.745 | l = −7→7 |
| 19034 measured reflections |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.069 | H-atom parameters constrained |
| wR(F2) = 0.171 | w = 1/[σ2(Fo2) + (0.1002P)2] where P = (Fo2 + 2Fc2)/3 |
| S = 1.04 | (Δ/σ)max < 0.001 |
| 3257 reflections | Δρmax = 0.43 e Å−3 |
| 328 parameters | Δρmin = −0.43 e Å−3 |
| 1 restraint | Absolute structure: Flack x determined using 801 quotients [(I+)-(I-)]/[(I+)+(I-)] (Parsons et al., 2013). |
| Primary atom site location: structure-invariant direct methods | Absolute structure parameter: 0.1 (10) |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.4424 (4) | 0.29680 (18) | 0.8670 (10) | 0.0258 (14) | |
| N1 | 0.1387 (5) | 0.2479 (3) | 0.8807 (11) | 0.0282 (18) | |
| H1N | 0.0730 | 0.2374 | 0.8841 | 0.034* | |
| C1 | 0.2175 (6) | 0.2147 (3) | 0.9013 (15) | 0.029 (2) | |
| H1 | 0.2017 | 0.1799 | 0.9202 | 0.035* | |
| C2 | 0.3205 (6) | 0.2300 (3) | 0.8955 (14) | 0.028 (2) | |
| H2 | 0.3754 | 0.2059 | 0.9094 | 0.034* | |
| C3 | 0.3455 (5) | 0.2814 (3) | 0.8692 (14) | 0.023 (2) | |
| C4 | 0.2598 (6) | 0.3152 (3) | 0.8499 (14) | 0.029 (2) | |
| H4 | 0.2720 | 0.3503 | 0.8335 | 0.035* | |
| C5 | 0.1594 (6) | 0.2968 (3) | 0.8551 (15) | 0.030 (2) | |
| H5 | 0.1023 | 0.3197 | 0.8400 | 0.036* | |
| O2 | 0.7873 (4) | 0.71124 (18) | 0.8498 (10) | 0.0258 (14) | |
| N2 | 1.1005 (5) | 0.7401 (2) | 0.8856 (11) | 0.0268 (18) | |
| H2N | 1.1684 | 0.7465 | 0.8913 | 0.032* | |
| C6 | 1.0301 (6) | 0.7782 (3) | 0.9018 (15) | 0.029 (2) | |
| H6 | 1.0550 | 0.8116 | 0.9216 | 0.034* | |
| C7 | 0.9253 (6) | 0.7700 (3) | 0.8907 (14) | 0.025 (2) | |
| H7 | 0.8779 | 0.7976 | 0.9023 | 0.030* | |
| C8 | 0.8859 (5) | 0.7208 (3) | 0.8619 (14) | 0.0216 (19) | |
| C9 | 0.9618 (5) | 0.6815 (3) | 0.8493 (14) | 0.026 (2) | |
| H9 | 0.9392 | 0.6476 | 0.8330 | 0.031* | |
| C10 | 1.0663 (6) | 0.6920 (3) | 0.8605 (14) | 0.028 (2) | |
| H10 | 1.1161 | 0.6654 | 0.8507 | 0.034* | |
| O3 | 0.0529 (4) | 0.56842 (19) | 0.8945 (9) | 0.0217 (14) | |
| N3 | −0.0040 (5) | 0.5697 (3) | 1.4925 (12) | 0.0283 (19) | |
| H3N | −0.0161 | 0.5691 | 1.6226 | 0.034* | |
| C11 | −0.0812 (6) | 0.5564 (3) | 1.3647 (15) | 0.023 (2) | |
| H11 | −0.1479 | 0.5468 | 1.4167 | 0.028* | |
| C12 | −0.0659 (7) | 0.5566 (3) | 1.1636 (14) | 0.025 (2) | |
| H12 | −0.1218 | 0.5476 | 1.0756 | 0.030* | |
| C13 | 0.0335 (6) | 0.5702 (3) | 1.0850 (13) | 0.020 (2) | |
| C14 | 0.1109 (6) | 0.5847 (3) | 1.2259 (13) | 0.023 (2) | |
| H13 | 0.1780 | 0.5954 | 1.1794 | 0.027* | |
| C15 | 0.0916 (7) | 0.5838 (3) | 1.4248 (13) | 0.022 (2) | |
| H15 | 0.1453 | 0.5931 | 1.5172 | 0.026* | |
| O4 | 0.6262 (4) | 0.39539 (18) | 0.7880 (11) | 0.0327 (16) | |
| O5 | 0.4504 (3) | 0.39158 (18) | 0.7983 (10) | 0.0270 (14) | |
| H5O | 0.4625 | 0.3607 | 0.8143 | 0.040* | |
| O6 | 0.7915 (4) | 0.57133 (19) | 0.7531 (10) | 0.0257 (14) | |
| O7 | 0.6792 (4) | 0.63488 (19) | 0.8051 (13) | 0.046 (2) | |
| H7O | 0.7346 | 0.6513 | 0.8273 | 0.069* | |
| O8 | 0.2964 (4) | 0.61537 (19) | 0.7789 (11) | 0.0293 (14) | |
| O9 | 0.2359 (4) | 0.53621 (19) | 0.8195 (9) | 0.0261 (15) | |
| H9O | 0.1794 | 0.5512 | 0.8474 | 0.039* | |
| C16 | 0.5281 (5) | 0.4719 (3) | 0.7808 (14) | 0.0161 (17) | |
| C17 | 0.6164 (5) | 0.5025 (3) | 0.7756 (13) | 0.0196 (19) | |
| H17 | 0.6845 | 0.4878 | 0.7736 | 0.023* | |
| C18 | 0.6062 (5) | 0.5544 (3) | 0.7733 (13) | 0.0163 (17) | |
| C19 | 0.5061 (6) | 0.5761 (3) | 0.7777 (14) | 0.0200 (18) | |
| H19 | 0.4990 | 0.6118 | 0.7774 | 0.024* | |
| C20 | 0.4175 (5) | 0.5460 (3) | 0.7823 (13) | 0.0177 (18) | |
| C21 | 0.4275 (6) | 0.4933 (3) | 0.7853 (14) | 0.0195 (18) | |
| H21 | 0.3667 | 0.4725 | 0.7903 | 0.023* | |
| C22 | 0.5408 (6) | 0.4164 (3) | 0.7873 (14) | 0.0229 (19) | |
| C23 | 0.3122 (5) | 0.5701 (3) | 0.7927 (13) | 0.021 (2) | |
| C24 | 0.7028 (6) | 0.5873 (3) | 0.7757 (14) | 0.0204 (18) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.011 (3) | 0.021 (3) | 0.046 (4) | −0.002 (2) | 0.004 (3) | 0.002 (3) |
| N1 | 0.013 (3) | 0.029 (4) | 0.043 (5) | −0.010 (3) | 0.004 (4) | −0.003 (4) |
| C1 | 0.024 (5) | 0.025 (5) | 0.040 (6) | −0.008 (4) | −0.001 (4) | −0.001 (5) |
| C2 | 0.024 (5) | 0.024 (5) | 0.037 (6) | 0.002 (4) | 0.005 (4) | 0.001 (4) |
| C3 | 0.016 (4) | 0.024 (4) | 0.028 (5) | 0.001 (4) | −0.001 (4) | −0.008 (4) |
| C4 | 0.021 (4) | 0.026 (5) | 0.039 (6) | −0.005 (4) | 0.000 (5) | 0.001 (5) |
| C5 | 0.021 (4) | 0.030 (5) | 0.038 (6) | 0.002 (4) | −0.002 (4) | 0.001 (5) |
| O2 | 0.013 (3) | 0.022 (3) | 0.042 (4) | 0.000 (2) | −0.004 (3) | −0.003 (3) |
| N2 | 0.012 (3) | 0.030 (4) | 0.038 (5) | −0.003 (3) | −0.003 (4) | 0.002 (4) |
| C6 | 0.018 (5) | 0.021 (5) | 0.046 (6) | −0.006 (4) | 0.000 (4) | 0.003 (5) |
| C7 | 0.023 (5) | 0.014 (4) | 0.037 (5) | 0.001 (3) | 0.001 (4) | 0.006 (4) |
| C8 | 0.011 (4) | 0.028 (5) | 0.026 (5) | −0.005 (3) | −0.006 (4) | 0.002 (4) |
| C9 | 0.020 (4) | 0.014 (4) | 0.044 (6) | −0.003 (3) | 0.002 (4) | 0.001 (4) |
| C10 | 0.021 (4) | 0.025 (5) | 0.040 (6) | 0.000 (4) | 0.004 (4) | 0.001 (5) |
| O3 | 0.018 (3) | 0.025 (3) | 0.023 (4) | 0.004 (2) | 0.001 (3) | 0.000 (3) |
| N3 | 0.042 (5) | 0.019 (4) | 0.024 (4) | 0.003 (4) | 0.001 (4) | −0.005 (3) |
| C11 | 0.012 (4) | 0.022 (5) | 0.036 (6) | 0.000 (3) | 0.003 (4) | 0.001 (5) |
| C12 | 0.024 (5) | 0.017 (5) | 0.033 (6) | 0.003 (4) | −0.003 (4) | −0.007 (4) |
| C13 | 0.024 (5) | 0.006 (4) | 0.028 (6) | 0.005 (4) | 0.001 (4) | 0.000 (4) |
| C14 | 0.011 (4) | 0.026 (5) | 0.031 (6) | 0.002 (4) | −0.002 (4) | 0.000 (4) |
| C15 | 0.018 (5) | 0.024 (5) | 0.022 (5) | 0.001 (4) | −0.007 (4) | 0.001 (4) |
| O4 | 0.012 (3) | 0.023 (3) | 0.063 (5) | 0.005 (2) | 0.006 (3) | 0.004 (4) |
| O5 | 0.012 (3) | 0.018 (3) | 0.050 (4) | −0.003 (2) | 0.000 (3) | 0.006 (4) |
| O6 | 0.009 (3) | 0.025 (3) | 0.043 (4) | 0.000 (2) | 0.001 (3) | −0.003 (3) |
| O7 | 0.013 (3) | 0.021 (3) | 0.106 (6) | −0.004 (2) | 0.011 (4) | −0.011 (4) |
| O8 | 0.016 (3) | 0.019 (3) | 0.053 (4) | 0.001 (2) | 0.000 (3) | −0.004 (3) |
| O9 | 0.013 (3) | 0.022 (3) | 0.043 (4) | −0.001 (2) | 0.006 (3) | 0.003 (3) |
| C16 | 0.010 (4) | 0.016 (4) | 0.022 (4) | 0.004 (3) | −0.003 (4) | −0.007 (4) |
| C17 | 0.010 (4) | 0.027 (4) | 0.021 (5) | 0.003 (3) | 0.000 (4) | −0.003 (4) |
| C18 | 0.011 (4) | 0.015 (4) | 0.022 (4) | 0.002 (3) | 0.002 (4) | −0.006 (4) |
| C19 | 0.016 (4) | 0.019 (4) | 0.025 (5) | 0.000 (3) | 0.000 (4) | 0.003 (4) |
| C20 | 0.011 (4) | 0.014 (4) | 0.028 (5) | 0.000 (3) | −0.003 (4) | −0.002 (4) |
| C21 | 0.013 (4) | 0.022 (4) | 0.024 (5) | −0.003 (3) | 0.001 (4) | 0.004 (4) |
| C22 | 0.021 (5) | 0.023 (4) | 0.025 (5) | 0.000 (4) | 0.003 (4) | −0.005 (5) |
| C23 | 0.011 (4) | 0.021 (5) | 0.030 (6) | −0.005 (3) | −0.003 (4) | 0.000 (4) |
| C24 | 0.022 (5) | 0.016 (4) | 0.023 (5) | −0.001 (3) | 0.002 (4) | 0.002 (4) |
Geometric parameters (Å, º)
| O1—C3 | 1.295 (8) | C11—H11 | 0.9500 |
| N1—C5 | 1.334 (10) | C12—C13 | 1.413 (12) |
| N1—C1 | 1.340 (10) | C12—H12 | 0.9500 |
| N1—H1N | 0.8800 | C13—C14 | 1.413 (12) |
| C1—C2 | 1.371 (11) | C14—C15 | 1.347 (12) |
| C1—H1 | 0.9500 | C14—H13 | 0.9500 |
| C2—C3 | 1.410 (11) | C15—H15 | 0.9500 |
| C2—H2 | 0.9500 | O4—C22 | 1.220 (8) |
| C3—C4 | 1.414 (10) | O5—C22 | 1.324 (8) |
| C4—C5 | 1.365 (10) | O5—H5O | 0.8400 |
| C4—H4 | 0.9500 | O6—C24 | 1.213 (9) |
| C5—H5 | 0.9500 | O7—C24 | 1.310 (9) |
| O2—C8 | 1.280 (8) | O7—H7O | 0.8400 |
| N2—C6 | 1.353 (10) | O8—C23 | 1.221 (8) |
| N2—C10 | 1.356 (10) | O9—C23 | 1.332 (8) |
| N2—H2N | 0.8800 | O9—H9O | 0.8400 |
| C6—C7 | 1.350 (11) | C16—C17 | 1.385 (9) |
| C6—H6 | 0.9500 | C16—C21 | 1.398 (9) |
| C7—C8 | 1.410 (10) | C16—C22 | 1.481 (10) |
| C7—H7 | 0.9500 | C17—C18 | 1.380 (9) |
| C8—C9 | 1.421 (10) | C17—H17 | 0.9500 |
| C9—C10 | 1.358 (10) | C18—C19 | 1.395 (10) |
| C9—H9 | 0.9500 | C18—C24 | 1.506 (10) |
| C10—H10 | 0.9500 | C19—C20 | 1.380 (10) |
| O3—C13 | 1.293 (10) | C19—H19 | 0.9500 |
| N3—C11 | 1.345 (10) | C20—C21 | 1.401 (10) |
| N3—C15 | 1.348 (10) | C20—C23 | 1.484 (10) |
| N3—H3N | 0.8800 | C21—H21 | 0.9500 |
| C11—C12 | 1.353 (12) | ||
| C5—N1—C1 | 120.3 (7) | C11—C12—C13 | 119.7 (8) |
| C5—N1—H1N | 119.8 | C11—C12—H12 | 120.2 |
| C1—N1—H1N | 119.8 | C13—C12—H12 | 120.2 |
| N1—C1—C2 | 121.0 (8) | O3—C13—C12 | 121.6 (8) |
| N1—C1—H1 | 119.5 | O3—C13—C14 | 121.9 (8) |
| C2—C1—H1 | 119.5 | C12—C13—C14 | 116.4 (8) |
| C1—C2—C3 | 120.4 (7) | C15—C14—C13 | 121.4 (9) |
| C1—C2—H2 | 119.8 | C15—C14—H13 | 119.3 |
| C3—C2—H2 | 119.8 | C13—C14—H13 | 119.3 |
| O1—C3—C2 | 121.3 (7) | C14—C15—N3 | 119.8 (8) |
| O1—C3—C4 | 122.0 (7) | C14—C15—H15 | 120.1 |
| C2—C3—C4 | 116.7 (7) | N3—C15—H15 | 120.1 |
| C5—C4—C3 | 119.4 (7) | C22—O5—H5O | 109.5 |
| C5—C4—H4 | 120.3 | C24—O7—H7O | 109.5 |
| C3—C4—H4 | 120.3 | C23—O9—H9O | 109.5 |
| N1—C5—C4 | 122.2 (7) | C17—C16—C21 | 120.2 (6) |
| N1—C5—H5 | 118.9 | C17—C16—C22 | 119.7 (6) |
| C4—C5—H5 | 118.9 | C21—C16—C22 | 120.1 (6) |
| C6—N2—C10 | 120.0 (6) | C18—C17—C16 | 120.5 (7) |
| C6—N2—H2N | 120.0 | C18—C17—H17 | 119.7 |
| C10—N2—H2N | 120.0 | C16—C17—H17 | 119.7 |
| C7—C6—N2 | 121.8 (8) | C17—C18—C19 | 119.7 (7) |
| C7—C6—H6 | 119.1 | C17—C18—C24 | 120.0 (6) |
| N2—C6—H6 | 119.1 | C19—C18—C24 | 120.2 (6) |
| C6—C7—C8 | 120.4 (7) | C20—C19—C18 | 120.3 (7) |
| C6—C7—H7 | 119.8 | C20—C19—H19 | 119.8 |
| C8—C7—H7 | 119.8 | C18—C19—H19 | 119.8 |
| O2—C8—C7 | 122.6 (7) | C19—C20—C21 | 120.2 (6) |
| O2—C8—C9 | 121.0 (7) | C19—C20—C23 | 119.2 (6) |
| C7—C8—C9 | 116.4 (7) | C21—C20—C23 | 120.6 (6) |
| C10—C9—C8 | 120.6 (7) | C16—C21—C20 | 119.1 (6) |
| C10—C9—H9 | 119.7 | C16—C21—H21 | 120.4 |
| C8—C9—H9 | 119.7 | C20—C21—H21 | 120.4 |
| N2—C10—C9 | 120.8 (7) | O4—C22—O5 | 123.0 (7) |
| N2—C10—H10 | 119.6 | O4—C22—C16 | 123.4 (7) |
| C9—C10—H10 | 119.6 | O5—C22—C16 | 113.6 (6) |
| C11—N3—C15 | 121.2 (8) | O8—C23—O9 | 123.6 (6) |
| C11—N3—H3N | 119.4 | O8—C23—C20 | 124.6 (6) |
| C15—N3—H3N | 119.4 | O9—C23—C20 | 111.8 (6) |
| N3—C11—C12 | 121.4 (8) | O6—C24—O7 | 124.5 (7) |
| N3—C11—H11 | 119.3 | O6—C24—C18 | 123.6 (7) |
| C12—C11—H11 | 119.3 | O7—C24—C18 | 111.9 (6) |
| C5—N1—C1—C2 | −0.3 (15) | C21—C16—C17—C18 | −0.5 (14) |
| N1—C1—C2—C3 | 0.5 (15) | C22—C16—C17—C18 | −178.8 (8) |
| C1—C2—C3—O1 | 178.7 (9) | C16—C17—C18—C19 | 0.4 (13) |
| C1—C2—C3—C4 | 0.1 (14) | C16—C17—C18—C24 | 177.7 (8) |
| O1—C3—C4—C5 | −179.4 (9) | C17—C18—C19—C20 | −0.6 (14) |
| C2—C3—C4—C5 | −0.8 (14) | C24—C18—C19—C20 | −177.8 (8) |
| C1—N1—C5—C4 | −0.4 (15) | C18—C19—C20—C21 | 0.8 (14) |
| C3—C4—C5—N1 | 1.0 (15) | C18—C19—C20—C23 | 178.3 (8) |
| C10—N2—C6—C7 | −1.1 (14) | C17—C16—C21—C20 | 0.7 (13) |
| N2—C6—C7—C8 | 0.2 (15) | C22—C16—C21—C20 | 179.0 (9) |
| C6—C7—C8—O2 | −179.8 (9) | C19—C20—C21—C16 | −0.9 (13) |
| C6—C7—C8—C9 | 1.0 (14) | C23—C20—C21—C16 | −178.3 (8) |
| O2—C8—C9—C10 | 179.4 (9) | C17—C16—C22—O4 | −0.2 (15) |
| C7—C8—C9—C10 | −1.4 (14) | C21—C16—C22—O4 | −178.4 (9) |
| C6—N2—C10—C9 | 0.7 (14) | C17—C16—C22—O5 | 177.9 (7) |
| C8—C9—C10—N2 | 0.6 (15) | C21—C16—C22—O5 | −0.3 (13) |
| C15—N3—C11—C12 | 0.2 (13) | C19—C20—C23—O8 | 7.1 (15) |
| N3—C11—C12—C13 | 1.0 (14) | C21—C20—C23—O8 | −175.5 (9) |
| C11—C12—C13—O3 | 176.6 (8) | C19—C20—C23—O9 | −172.7 (8) |
| C11—C12—C13—C14 | −2.2 (13) | C21—C20—C23—O9 | 4.7 (12) |
| O3—C13—C14—C15 | −176.3 (8) | C17—C18—C24—O6 | 10.1 (14) |
| C12—C13—C14—C15 | 2.4 (13) | C19—C18—C24—O6 | −172.7 (9) |
| C13—C14—C15—N3 | −1.3 (14) | C17—C18—C24—O7 | −169.9 (8) |
| C11—N3—C15—C14 | −0.1 (13) | C19—C18—C24—O7 | 7.3 (12) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N···O1i | 0.88 | 1.89 | 2.762 (8) | 169 |
| N2—H2N···O2ii | 0.88 | 1.90 | 2.711 (8) | 152 |
| N3—H3N···O3iii | 0.88 | 2.01 | 2.773 (10) | 144 |
| N3—H3N···O6iv | 0.88 | 2.59 | 3.124 (9) | 120 |
| O5—H5O···O1 | 0.84 | 1.75 | 2.555 (7) | 161 |
| O7—H7O···O2 | 0.84 | 1.73 | 2.463 (7) | 145 |
| O9—H9O···O3 | 0.84 | 1.70 | 2.526 (7) | 167 |
| C1—H1···O4i | 0.95 | 2.38 | 3.227 (10) | 148 |
| C4—H4···O5 | 0.95 | 2.53 | 3.174 (9) | 126 |
| C6—H6···O7ii | 0.95 | 2.26 | 3.051 (9) | 140 |
| C7—H7···O8ii | 0.95 | 2.66 | 3.530 (9) | 153 |
| C9—H9···O3v | 0.95 | 2.58 | 3.227 (9) | 126 |
| C11—H11···O6iv | 0.95 | 2.46 | 3.076 (11) | 123 |
| C11—H11···O9vi | 0.95 | 2.55 | 3.159 (9) | 122 |
| C12—H12···O6vii | 0.95 | 2.49 | 3.302 (11) | 143 |
| C14—H13···O4viii | 0.95 | 2.60 | 3.405 (10) | 143 |
| C15—H15···O8iii | 0.95 | 2.66 | 3.608 (10) | 178 |
Symmetry codes: (i) x−1/2, −y+1/2, z; (ii) x+1/2, −y+3/2, z; (iii) x, y, z+1; (iv) x−1, y, z+1; (v) x+1, y, z; (vi) −x, −y+1, z+1/2; (vii) x−1, y, z; (viii) −x+1, −y+1, z+1/2.
References
- Bhogala, B. R., Basavoju, S. & Nangia, A. (2005). CrystEngComm, 7, 551–562.
- Bruker (2012). APEX2 and SAINT. Bruker AXS Inc., Madison, Wisconsin, USA.
- Campos-Gaxiola, J. J., Zamora Falcon, F., Corral Higuera, R., Höpfl, H. & Cruz-Enríquez, A. (2014). Acta Cryst. E70, o453–o454. [DOI] [PMC free article] [PubMed]
- Castillo, O., Luque, A., Lloret, F. & Román, P. (2001). Inorg. Chim. Acta, 324, 141–149.
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Flack, H. D. (1983). Acta Cryst. A39, 876–881.
- Flack, H. D. & Bernardinelli, G. (1999). Acta Cryst. A55, 908–915. [DOI] [PubMed]
- Groom, C. R. & Allen, F. H. (2014). Angew. Chem. Int. Ed. 53, 662–671. [DOI] [PubMed]
- Hooft, R. W. W., Straver, L. H. & Spek, A. L. (2008). J. Appl. Cryst. 41, 96–103. [DOI] [PMC free article] [PubMed]
- Krause, L., Herbst-Irmer, R., Sheldrick, G. M. & Stalke, D. (2015). J. Appl. Cryst. 48, 3–10. [DOI] [PMC free article] [PubMed]
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst. 41, 466–470.
- Parsons, S., Flack, H. D. & Wagner, T. (2013). Acta Cryst. B69, 249–259. [DOI] [PMC free article] [PubMed]
- Qian, J., Jiang, F., Zhang, L., Su, K., Pan, J., Li, Q., Yuan, D. & Hong, M. (2014). Chem. Commun. 50, 1678–1681. [DOI] [PubMed]
- Shattock, T. R., Arora, K. K., Vishweshwar, P. & Zaworotko, M. J. (2008). Cryst. Growth Des. 8, 4533–4545.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Staun, S. L. & Oliver, A. G. (2012). Acta Cryst. C68, o84–o87. [DOI] [PubMed]
- Staun, S. L. & Oliver, A. G. (2015). Acta Cryst. E71, 861–863. [DOI] [PMC free article] [PubMed]
- Tyl, A., Nowak, M. & Kusz, J. (2008). Acta Cryst. C64, o661–o664. [DOI] [PubMed]
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2056989015017867/zl2644sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015017867/zl2644Isup2.hkl
CCDC reference: 1427116
Additional supporting information: crystallographic information; 3D view; checkCIF report