

Abstract
Objective:
To determine the clinical features and presence in CSF of antineuronal antibodies in patients with pathologically proven autoimmune encephalitis derived from a cohort of patients with suspected Creutzfeldt-Jakob disease (CJD).
Methods:
The Dutch Surveillance Centre for Prion Diseases performed 384 autopsies on patients with suspected CJD over a 14-year period (1998–2011). Clinical information was collected from treating physicians. Antineuronal antibodies were tested in CSF obtained postmortem by immunohistochemistry on fresh frozen rat brain sections, by Luminex assay for the presence of well-characterized onconeural antibodies, and by cell-based assays for antibodies against NMDAR, GABABR1/2, GABAAR GLUR1/2, LGI1, Caspr2, and DPPX.
Results:
In 203 patients, a diagnosis of definite CJD was made, while in 181 a variety of other conditions were diagnosed, mainly neurodegenerative. In 22 of these 181, the neuropathologist diagnosed autoimmune encephalitis. One patient was excluded because of lack of clinical information. Inflammatory infiltrates were predominantly perivascular and consisted mainly of T cells. The predominant locations were basal ganglia and thalamus (90%) and temporal lobes and hippocampus (81%). In 6 patients (29%), antineuronal antibodies were detected in postmortem CSF, directed against Hu, NMDAR, GABABR1/2, Caspr2, and an unidentified synaptic antigen in 2. The most frequent symptoms were dementia (90%), gait disturbance (86%), cerebellar signs (67%), and neuropsychiatric symptoms (67%). Immunopathologic and clinical findings did not differ between autoantibody-negative patients and patients with antineuronal antibodies.
Conclusions:
It is important to consider immune-mediated disorders in the differential diagnosis of rapidly progressive neurologic deficits.
Autoimmune encephalitis and sporadic Creutzfeldt-Jakob disease (sCJD) may present with similar clinical, radiologic, electrophysiologic, and laboratory findings.1–4 In sCJD, the course of disease is fatal, while autoimmune encephalitis associated with antibodies directed against neuronal surface antigens (NSA-Abs) usually responds well to treatment.5,6
We encountered several patients with rapidly progressive dementia diagnosed as possible or probable sCJD, who at autopsy at the Dutch Surveillance Centre for Prion Diseases (DSCPD) turned out to have had potentially treatable autoimmune encephalitis. We retrospectively reviewed all prion-negative autopsy brain reports from the DSCPD to determine the number of pathologically confirmed cases of autoimmune encephalitis in a cohort of patients with suspected sCJD. In addition, we determined the clinical, radiologic, and neurophysiologic characteristics of patients with pathologically confirmed autoimmune encephalitis and examined the CSF for the presence of antineuronal autoantibodies.
METHODS
Patients.
The DSCPD performed 384 autopsies on patients with suspected Creutzfeldt-Jakob disease (CJD) over a 14-year period (1998–2011).7 At brain autopsy, definite CJD was diagnosed in 203 patients based on the presence of spongiosis and positive prion staining. In 181 patients, neuropathology showed a variety of other conditions, mainly neurodegenerative disorders (table e-1 at Neurology.org/nn). In 22 patients, the pathologist identified inflammatory infiltrates consistent with a diagnosis of autoimmune encephalitis. Of these 22 patients, we collected clinical information from treating physicians, including discharge notes, laboratory, neurophysiology, and radiology results. One patient lacked sufficient clinical information and was excluded from the study.
Based on the clinical information, the 21 patients with pathologically proven autoimmune encephalitis were retrospectively classified as probable, possible, or no CJD according to the CDC Diagnostic Criteria for Creutzfeldt-Jakob Disease, 2010 (http://www.cdc.gov/ncidod/dvrd/cjd/resources/CDCs_Diagnostic_Criteria_for_CJD-2010.pdf).
Standard protocol approvals, registrations, and patient consents.
Local ethical committee approval was obtained for research on retained tissues after written informed consent was given by the patients during life or their next of kin after death (Medical Ethics Committee of the University Medical Centre Utrecht 11-531/C). All information was analyzed anonymously.
Antineuronal antibody testing.
Postmortem derived CSF samples from all cases were stored at −80°C until testing. As sampling was done postmortem, no serum samples were available at the DSCPD. All CSF samples were tested for autoimmune encephalitis–associated antineuronal antibodies in several assays at a dilution of 1:2. CSF samples were tested on fresh frozen rat brain sections for autoantibodies against neuronal cell surface antigens, as previously described.8 Antibodies against the onconeural antigens HuD, CDR62 (Yo), Tr (DNER), amphiphysin, CRMP-5 (CV2), NOVA-1 (Ri), Ma1/2, and Zic4 were assayed, using another immunohistochemistry protocol9 combined with a recently described multiplex bead-based assay.10 In addition, CSF samples were tested on cultured hippocampal neurons and on HeLa cells overexpressing NMDAR1, GABABR1/2, GABAAR, GLUR1/2, and DPPX as previously described.9,11 Finally, samples were tested in a commercial assay (EUROIMMUN, Lübeck, Germany) for anti-LGI1 and anti-Caspr2 antibodies (previously tested as voltage-gated potassium channel complex antibodies). The sensitive assays on frozen rat brain sections and cultured hippocampal neurons detect clinically relevant (high serum titer) GAD65 and LGI1 antibodies in CSF, rendering underreporting of antibodies by the lack of serum availability unlikely.12
Neuropathology.
Brains were removed at autopsy and selected tissue samples were immediately frozen and stored at −80°C while the rest of the brain was fixed in formalin and used for histologic and immunologic purposes. Histopathologic examination was performed as described before.7,13 In short, relevant sections were stained for α-synuclein, ubiquitin, amyloid-β, and tau in all patients. In a subset of patients, additional immune stainings were carried out for B cells (CD20), T cells (CD3, CD4, and CD8), and macrophages (CD68).
RESULTS
Pathology consistent with autoimmune encephalitis was found at autopsy in 22 out of 384 (6%) patients with clinically suspected CJD, constituting 12% (22/181) of the patients who turned out at autopsy not to have had CJD. Because one patient lacked sufficient clinical information, we present the clinical, pathologic, and serologic findings in 21 patients with pathologically confirmed autoimmune encephalitis. Detailed information on the patients is summarized in tables e-2 and e-3.
Neuropathology.
In all 21 patients, inflammatory infiltrates were present (figure e-1). Gliosis was observed in 10 (48%) patients while vasculitis was seen in none. The infiltrates were predominantly perivascular in 15 (71%), predominantly parenchymal in 5 (24%), and both perivascular and parenchymal in 1 (5%) patient. The infiltrates consisted of CD3-positive T cells in 14/15 (93%) patients tested. CD8-positive cells were observed in 7/8 (88%) and CD4-positive cells in 4/5 (80%) patients examined. In 8/12 (67%) patients, CD20-positive B cells were present, mainly in the perivascular infiltrates. CD68-positive cells were seen in 11/13 (85%) patients.
Infiltrates were mainly located in the basal ganglia and thalamus in 19 (90%), temporal lobes and hippocampus in 17 (81%), cerebellum in 12 (57%), frontal lobes in 11 (52%), other cortical areas in 12 (57%), brainstem in 7 (33%), and periventricular white matter in 4 (19%). Neuropathologic changes were far more extensive than MRI changes.
Postmortem CSF testing for antineuronal antibodies.
Antineuronal antibodies were detected in 6/21 (29%) of the postmortem collected CSF samples. Five of these antibodies were directed against NSA. One each was directed against GABAB1/B2, NMDAR, and Caspr2, while 2 CSF samples harbored antibodies against an unidentified synaptic antigen (figure 1). One additional sample contained antibodies against the intracellular onconeural Hu antigen. Of the 4 known antibodies detected in CSF, the Hu antibody had been described long before the time of the patient's illness, while GABAB1/B2, NMDAR, and Caspr2 antibodies were first reported after the death of the respective patients. Three of the 6 patients with antineuronal antibodies presented with limbic encephalitis, 2 with encephalomyelitis, and 1 with Morvan syndrome (associated with anti-Caspr2 antibodies). Further clinical findings in the 6 patients with pathologically confirmed autoimmune encephalitis and antineuronal antibodies are summarized in table 1 and supplementary information (1). The phenotypes associated with the known antibodies are classical for these disorders.14 The localization and cellular composition of the inflammatory infiltrates found at autopsy did not differ between the antibody-positive and antibody-negative patients (table e-4).
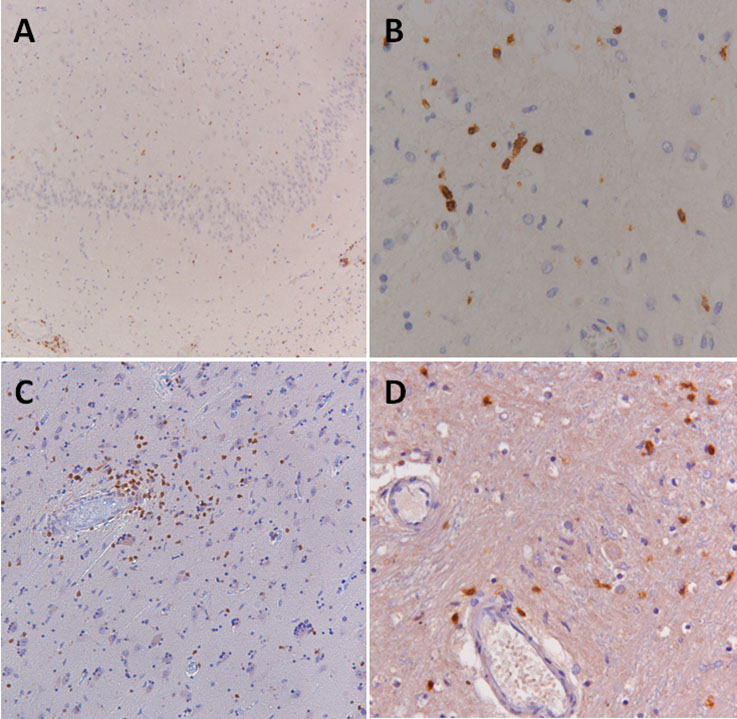
Figure 1. Unidentified synaptic antigen in CSF of 2 patients with autoimmune encephalitis.

In 2 patients, a synaptic staining pattern in the hippocampus was observed not corresponding to any of the known antigens tested. One patient was a 73-year-old man with limbic encephalitis (A, B; patient 11) and the other a 77-year-old man with encephalomyelitis (C, D; patient 18). Immunohistochemistry was performed as described in the Methods (A, C). To confirm the synaptic staining pattern in these 2 patients, CSF samples were incubated (1:2 dilution) live with primary hippocampal cultures (B, D) as previously described.9
Table 1.
Clinical summary of 6 patients with pathologically confirmed autoimmune encephalitis and associated antineuronal autoantibodies
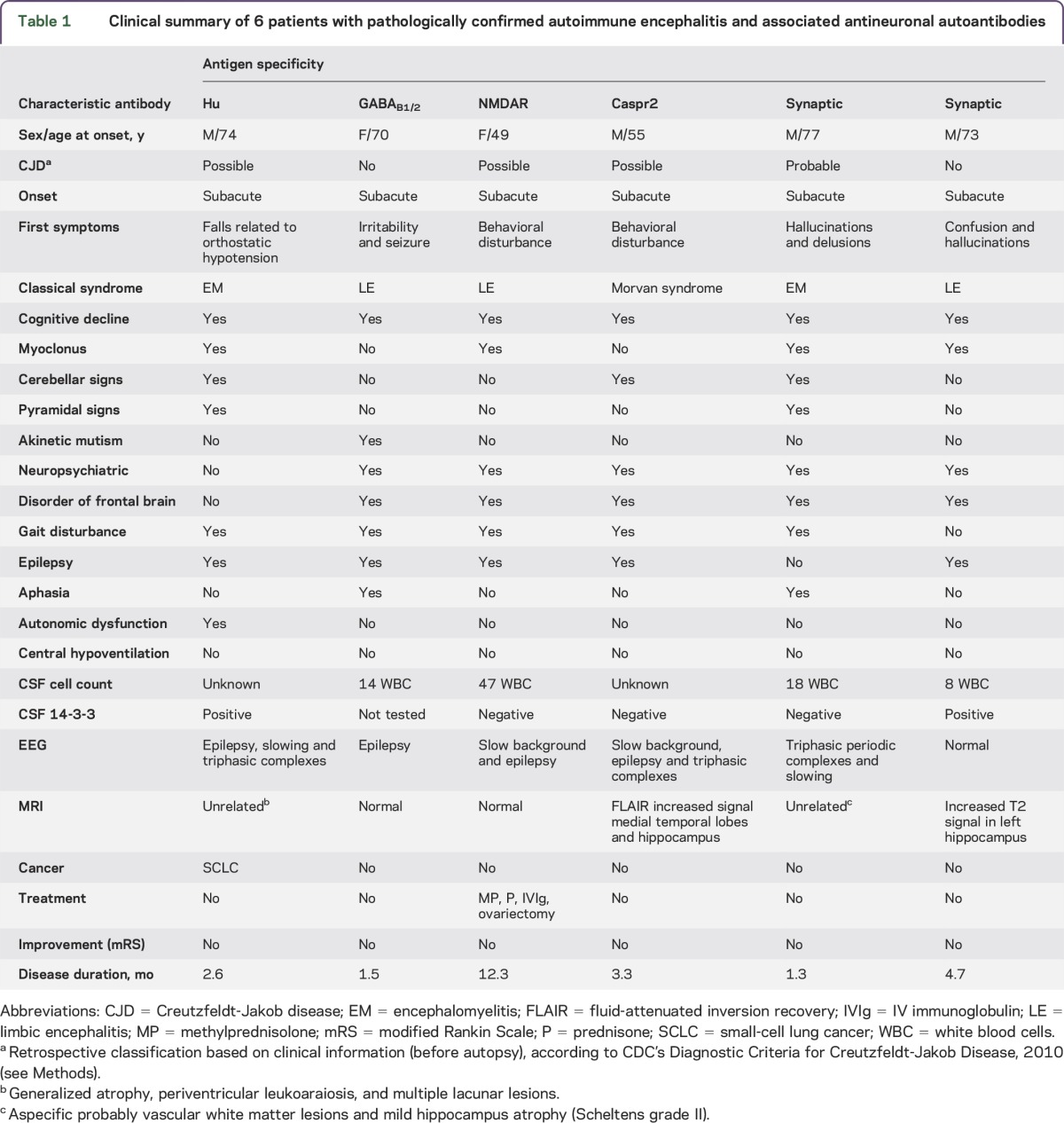
Summarizing, 5/21 (24%) patients with pathologically confirmed autoimmune encephalitis had CSF autoantibodies directed against NSA (3 against well-defined and 2 against unknown antigens).
Clinical characteristics.
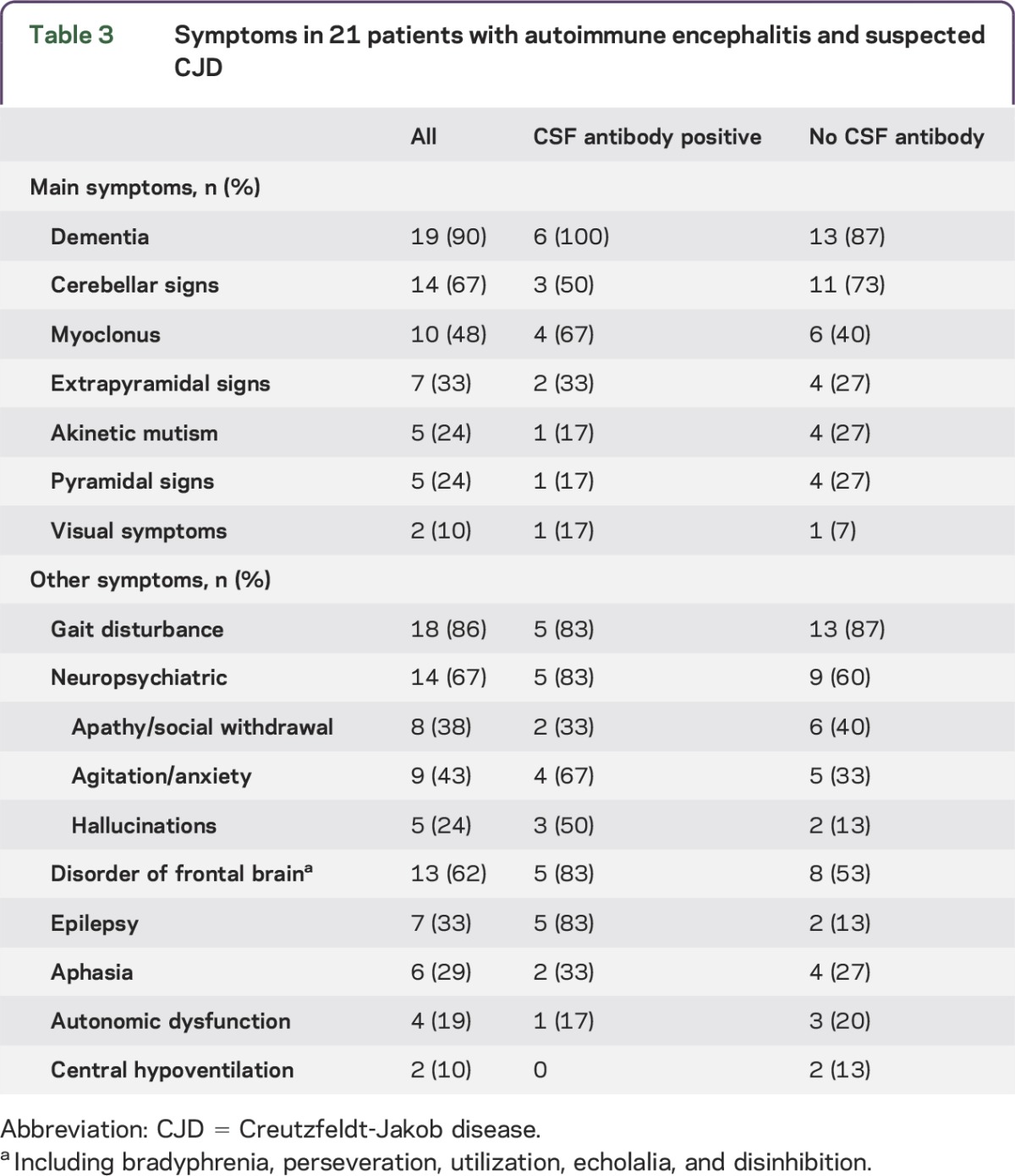
All 21 patients with autoimmune encephalitis had a rapidly progressive illness with a median duration of 3.3 months and died within 13 months of first symptoms (table 2, tables e-2 and e-3). In retrospect, 14 of the 21 (67%) patients with autoimmune encephalitis fulfilled the clinical 2010 CDC criteria for possible or probable sCJD while 7 (33%) did not. In the subgroup of 6 patients with antineuronal antibody–associated autoimmune encephalitis, 67% (4 patients) also fulfilled CDC criteria for possible or probable sCJD (table 2). The most frequent main symptoms included dementia (90%), cerebellar signs (67%), myoclonus (48%), and extrapyramidal signs (33%) (table 3). Other symptoms that were frequently present included gait disturbance (86%), neuropsychiatric symptoms (67%), and frontal lobe symptoms (62%). There were no clear differences in clinical presentation of autoimmune encephalitis with or without detectable antineuronal antibodies in CSF (tables 2 and 3). Only 4 patients received immunotherapy, consisting of steroids in 2, IV immunoglobulins (IVIg) in 1, and both IVIg and steroids in 1. None of the patients showed substantial improvement and immunotherapy was discontinued in all 4.
Table 2.
Clinical characteristics in 21 patients with autoimmune encephalitis and suspected CJD
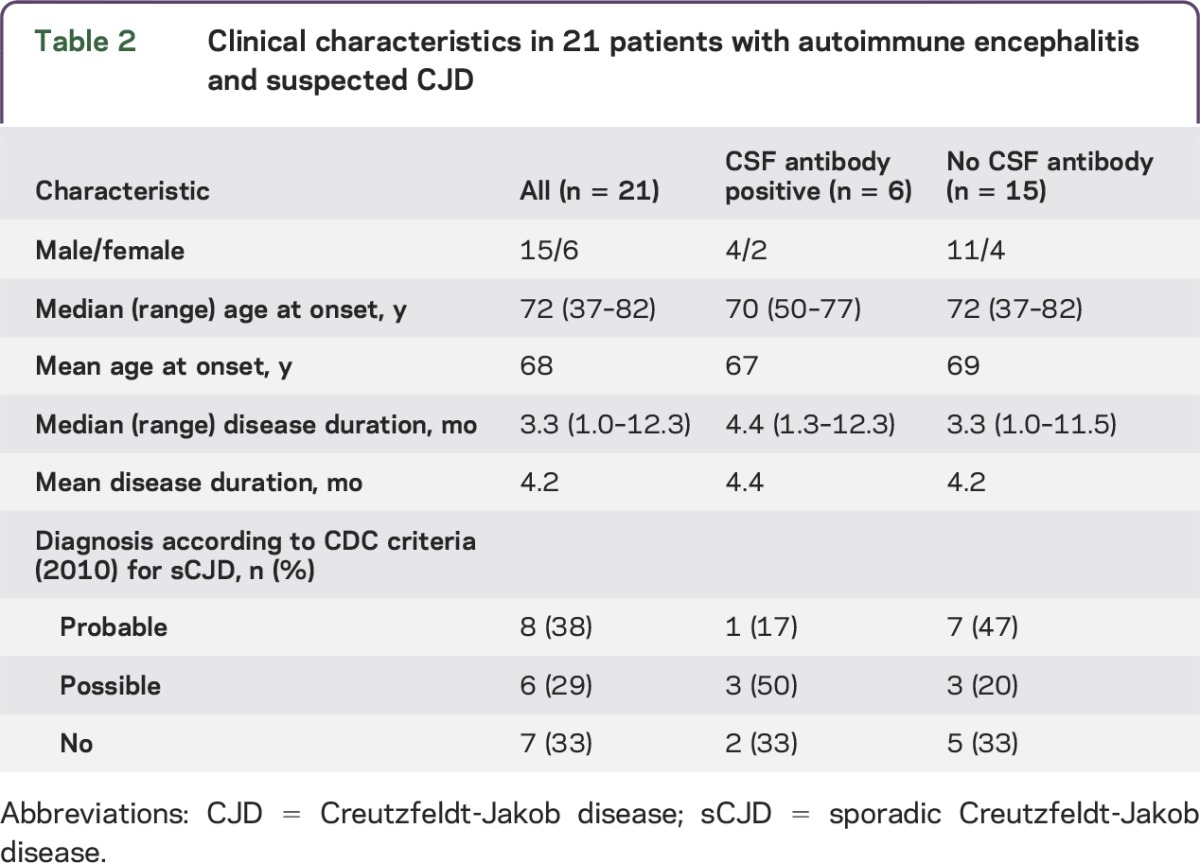
Table 3.
Symptoms in 21 patients with autoimmune encephalitis and suspected CJD
Additional investigations.
The CSF had been examined in 18 of the 21 patients and was abnormal in 14/18 (78%), showing increased protein in 14 (78%), pleiocytosis in 10 (56%), and positive 14-3-3 protein in 9 (50%) (table 4).
Table 4.
CSF, EEG, and MRI findings in patients with autoimmune encephalitis
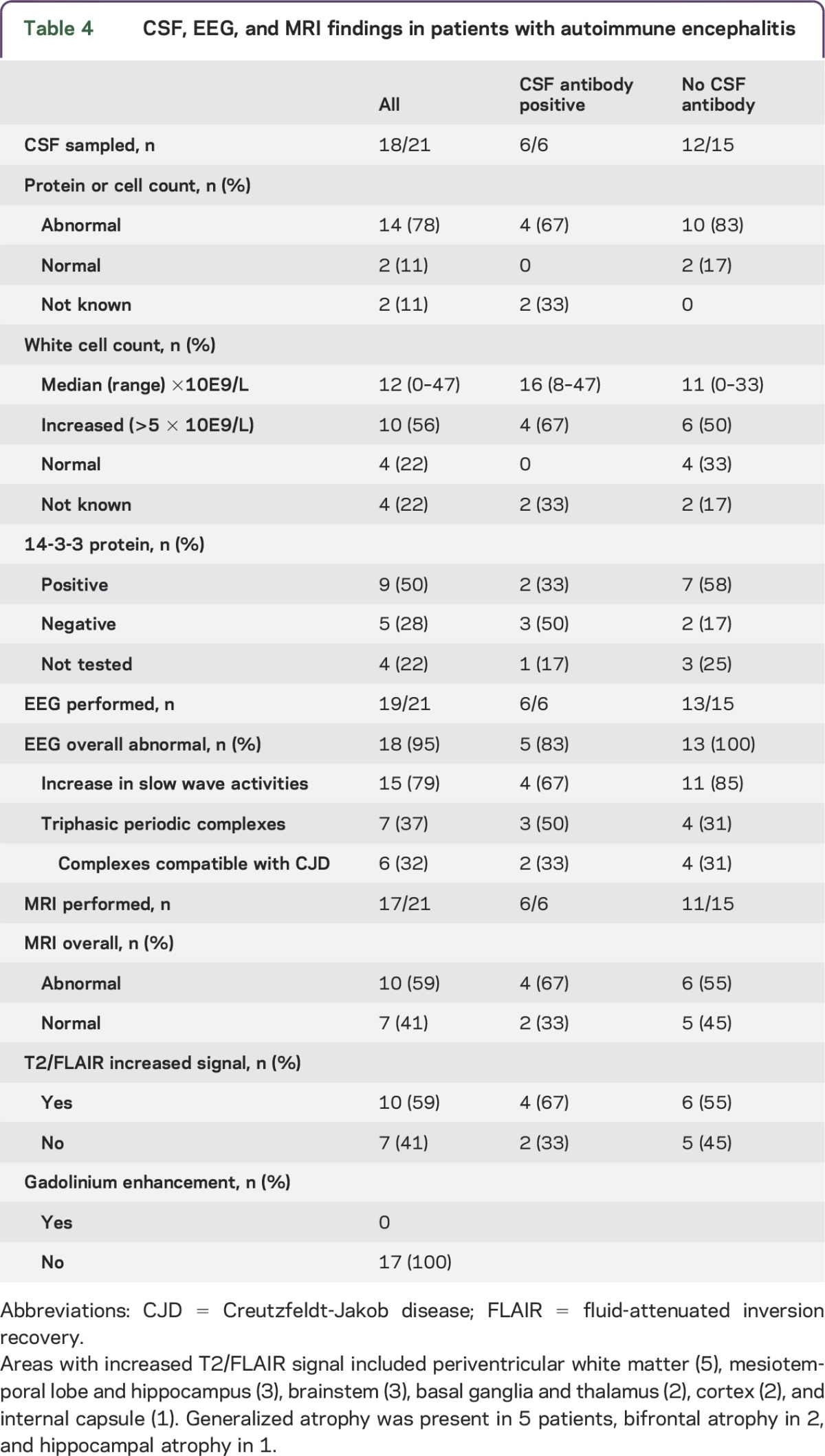
EEG was performed at least once in 19 of the patients and was abnormal in 18/19 (95%) with an increase of slow wave activities in 15/19 (79%). Seven of the patients showed triphasic periodic complexes that were considered compatible with CJD in 6 (32%). Epileptiform abnormalities were registered in 7 (37%) of the patients (table 4).
MRI was abnormal in 10/17 (59%) patients, showing increased signal on T2-weighted or fluid-attenuated inversion recovery images (table 4). Imaging abnormalities were located in the medial temporal lobes in 3, periventricular in 5 (most likely vascular lesions in 3), basal ganglia and thalamus in 2, and brainstem in 1. None of the patients had MRI changes typical of sCJD, according to 2010 CDC criteria, although most investigations lacked diffusion-weighted imaging (DWI) sequences.
The patients with autoimmune encephalitis with antineuronal autoantibodies in their CSF did not clearly differ from the antibody-negative patients in CSF, EEG, or MRI findings (table 4).
Associated tumors.
In 9 patients (43%), a tumor was present. Based on criteria published by an EU consortium, the etiology of the neurologic syndrome could be classified as definitely paraneoplastic in 5 and possibly paraneoplastic in 2.15 The tumor was probably unrelated in 2 of the patients, precluding a paraneoplastic etiology. Detailed clinical and oncologic findings are presented in table e-3 and supplementary information (2).
DISCUSSION
The first finding of our study is that approximately 7% of patients with suspected CJD have autoimmune encephalitis at autopsy. Secondly, antineuronal autoantibodies could be demonstrated in the CSF from 6 of 21 (29%) patients with pathologically confirmed autoimmune encephalitis. One case each of anti-Hu, GABAB1/B2, NMDAR, and Caspr2 was detected, while 2 CSF samples had antibodies against an unidentified NSA. Third, in retrospect, 67% of the patients met the 2010 CDC criteria for possible or probable CJD, both in the antibody-positive and antibody-negative group.
Several other studies have addressed the clinical overlap between prion disorders and autoimmune encephalitis. Chitravas et al.3 studied prion-negative brain autopsy cases referred to the US National Prion Disease Pathology Surveillance Center. Neuropathology diagnosed an immune-mediated disorder in 26/304 (8.5%) of the prion-negative cases with histology, including primary angiitis of the CNS (7), acute disseminated encephalomyelitis (6), limbic encephalitis (6), neurosarcoidosis (4), paraneoplastic cerebellar degeneration (2), and granulomatosis with polyangiitis (Wegener) (1). This study did not report the antineuronal antibody status of the patients. In contrast to our study, most of the misdiagnosed patients did not meet the WHO criteria for CJD.
Grau-Rivera et al.1 examined the presence of NSA-Abs in the CSF of patients with suspected or pathologically confirmed CJD. NSA-Abs were found in 6 of 346 patients (1.7%) with a rapidly progressive neurologic disorder suggestive of CJD during life. In contrast to our study, none of these 6 patients met the diagnostic criteria for probable or possible CJD. The target antigens included NMDAR, LGI1, Caspr2, aquaporin 4, Tr (DNER), and an unknown protein. All patients stabilized or improved after appropriate treatment. None of the 49 patients with definite CJD had NSA-abs. Another study reported serum NSA-Abs in <5% of patients with sCJD, all at very low titers.2 In this study, we tested CSF of all patients identified with autoimmune encephalitis at autopsy. CSF is proven more sensitive and specific to detect NSA-Abs as shown for NMDR,16 AMPAR,17 and GABABR.18
In 50% of our patients with autoimmune encephalitis tested, 14-3-3 protein was elevated in CSF (part of the CDC's 2010 diagnostic criteria), casting further doubt on the usefulness of this test in diagnosing CJD.19 EEG was also not useful as triphasic waves were present in 32% of our autoimmune patients. None of our patients with autoimmune encephalitis showed sCJD-specific MRI abnormalities, supporting the higher specificity of sCJD MRI criteria over 14-3-3.19,20 However, DWI was not performed in most of our patients, and we do not know whether DWI would have retained its specificity in this series. On the other hand, 56% of patients had an increased leukocyte count (>5 × 10E9/L) in their CSF. Although the pleiocytosis was usually mild, it provides a clue to an inflammatory etiology.
Other studies describing clinical overlap between prion and autoimmune disorders focused primarily on rapidly progressive dementia, detecting a 2%–9% prevalence of immune-mediated disorders.21–23 In a series of patients with suspected autoimmune dementia and good response to immunotherapy, almost 9% of the disorders had initially been diagnosed as sCJD.24 In particular, patients with encephalitis associated with LGI1-Abs may present with myoclonus-like movements and other symptoms that can be mistaken for CJD.4
In general, the prognosis of autoimmune encephalitis appears to be determined by the involved immunopathogenic mechanisms.25 Disorders accompanied by antibodies directed against intracellular antigens are probably T-cell-mediated and generally respond poorly to immunotherapy. Most paraneoplastic neurologic disorders with onconeural antibodies belong to this category. On the other hand, in disorders associated with NSA-Abs, the antibodies are probably pathogenic and these disorders respond favorably to first- and second-line immunotherapy. Examples include NMDAR,5 LGI1,26,27 and Caspr227,28 encephalitis. The immunopathology of NSA-Abs-associated encephalitis shows mild to moderate, mainly perivascular, lymphocytic infiltrations containing both B and T cells, antibody-producing cells, and immunoglobulin G deposits.29–31 It is possible that we missed some cases with burned out or with bland neuropathology and that the 22 cases that we identified represent an underestimation. The encephalitis associated with Hu or Ma2 antibodies is characterized by extensive B and T cell lymphocytic infiltrations, containing T cells with a cytotoxic phenotype.31–33 In our series, immunopathologic findings did not differ between autoantibody-negative patients and the patients with NSA-Ab (table e-4), nor did clinical, CSF, radiologic, or electrophysiologic characteristics (tables 2–4). The immunopathologic mechanism in the 15 patients with autoimmune encephalitis without detectable autoantibodies remains unclear. It cannot be excluded that some of these patients harbored NSA-Abs that were degraded by postmortem interval or during storage at −80°C in low protein CSF. The lack of serum availability may also have contributed to underreporting of NSA-Abs.
Limitations based on the retrospective nature of the study are applicable. In addition, bias was introduced by the fact that all patients had suspected CJD and had come to autopsy. In particular, the poor outcome with a median survival of 3.3 months (range 1.0–12.3 months) in this study of autoimmune encephalitis was largely determined by this inclusion bias.
It is important to consider both prion and immune-mediated disorders in the differential diagnosis of rapidly progressive neurologic deficits, knowing that EEG and 14-3-3 in CSF do not discriminate sufficiently to distinguish the two. Detection in serum or CSF of specific antineuronal antibodies can establish a diagnosis of autoimmune encephalitis and guide appropriate treatment.
Supplementary Material
GLOSSARY
- CJD
Creutzfeldt-Jakob disease
- DSCPD
Dutch Surveillance Centre for Prion Diseases
- DWI
diffusion-weighted imaging
- IVIg
IV immunoglobulin
- NSA-Abs
antibodies directed against neuronal surface antigens
- sCJD
sporadic Creutzfeldt-Jakob disease
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
Study conception: P.M., A.J.R., P.S.S. Study design: P.M., A.J.R., P.S.S. Sample acquisition: C.J., M.S., C.M.v.D., A.J.R. Acquisition of laboratory data: P.M., C.J., M.H.v.C., E.d.G., M.T., A.J.R. Acquisition of clinical data: P.M., J.W.d.B., C.J., M.S., A.J.R., P.S.S. Study supervision, analysis and interpretation of data: M.T., P.S.S. Writing of first draft: P.M., J.W.d.B., P.S.S. Editing and revising final draft for intellectual content: all authors.
STUDY FUNDING
No targeted funding.
DISCLOSURE
P. Maat, J. de Beukelaar, C. Jansen, M. Schuur, C. van Duijn, and M. van Coevorden report no disclosures. E. de Graaf holds a patent for the detection of anti-DNER antibodies. M. Titulaer served on the scientific advisory board for MedImmne LLC, received travel funding from Sun Pharma, is on the editorial board for Neurology: Neuroimmunology & Neuroinflammation, and received research support from Netherlands Organiszation for Scientific Research, ErasmusMC, and Dutch Epilepsy Foundations. A. Rozemuller is on the scientific advisory board for Netherlands Brain Bank and received research support from Neuroscience Campus Amsterdam, Erasmus Mundo. P.S. Smitt holds a patent for the detection of anti-DNER and received research support from Euroimmun. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Grau-Rivera O, Sanchez-Valle R, Saiz A, et al. Determination of neuronal antibodies in suspected and definite Creutzfeldt-Jakob disease. JAMA Neurol 2014;71:74–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rossi M, Mead S, Collinge J, Rudge P, Vincent A. Neuronal antibodies in patients with suspected or confirmed sporadic Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 2015;86:692–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chitravas N, Jung RS, Kofskey DM, et al. Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease. Ann Neurol 2011;70:437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geschwind MD, Tan KM, Lennon VA, et al. Voltage-gated potassium channel autoimmunity mimicking Creutzfeldt-Jakob disease. Arch Neurol 2008;65:1341–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004;127:701–712. [DOI] [PubMed] [Google Scholar]
- 7.Jansen C, Parchi P, Capellari S, et al. Human prion diseases in the Netherlands (1998-2009): clinical, genetic and molecular aspects. PLoS One 2012;7:e36333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petit-Pedrol M, Armangue T, Peng X, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol 2014;13:276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Graaff E, Maat P, Hulsenboom E, et al. Identification of delta/notch-like epidermal growth factor-related receptor as the Tr antigen in paraneoplastic cerebellar degeneration. Ann Neurol 2012;71:815–824. [DOI] [PubMed] [Google Scholar]
- 10.Maat P, Brouwer E, Hulsenboom E, et al. Multiplex serology of paraneoplastic antineuronal antibodies. J Immunol Methods 2013;391:125–132. [DOI] [PubMed] [Google Scholar]
- 11.Maat P, de Graaff E, van Beveren NM, et al. Psychiatric phenomena as initial manifestation of encephalitis by anti-NMDA receptor antibodies. Acta Neuropsychiatr 2013;25:128–136. [DOI] [PubMed] [Google Scholar]
- 12.Gresa-Arribas N, Arino H, Martinez-Hernandez E, et al. Antibodies to inhibitory synaptic proteins in neurological syndromes associated with glutamic acid decarboxylase autoimmunity. PLoS One 2015;10:e0121364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jansen C, Parchi P, Capellari S, et al. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol 2010;119:189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Coevorden-Hameete MH, de Graaff E, Titulaer MJ, Hoogenraad CC, Sillevis Smitt PA. Molecular and cellular mechanisms underlying anti-neuronal antibody mediated disorders of the central nervous system. Autoimmun Rev 2014;13:299–312. [DOI] [PubMed] [Google Scholar]
- 15.Graus F, Delattre JY, Antoine JC, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry 2004;75:1135–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol 2014;13:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoftberger R, van Sonderen A, Leypoldt F, et al. Encephalitis and AMPA receptor antibodies: novel findings in a case series of 22 patients. Neurology 2015;84:2403–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoftberger R, Titulaer MJ, Sabater L, et al. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology 2013;81:1500–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paterson RW, Torres-Chae CC, Kuo AL, et al. Differential diagnosis of Jakob-Creutzfeldt disease. Arch Neurol 2012;69:1578–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vitali P, Maccagnano E, Caverzasi E, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology 2011;76:1711–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL. Rapidly progressive dementia. Ann Neurol 2008;64:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papageorgiou SG, Kontaxis T, Bonakis A, Karahalios G, Kalfakis N, Vassilopoulos D. Rapidly progressive dementia: causes found in a Greek tertiary referral center in Athens. Alzheimer Dis Assoc Disord 2009;23:337–346. [DOI] [PubMed] [Google Scholar]
- 23.Sala I, Marquie M, Sanchez-Saudinos MB, et al. Rapidly progressive dementia: experience in a tertiary care medical center. Alzheimer Dis Assoc Disord 2012;26:267–271. [DOI] [PubMed] [Google Scholar]
- 24.Flanagan EP, McKeon A, Lennon VA, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc 2010;85:881–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lancaster E, Dalmau J. Neuronal autoantigens: pathogenesis, associated disorders and antibody testing. Nat Rev Neurol 2012;8:380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain 2010;133:2734–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lancaster E, Huijbers MG, Bar V, et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol 2011;69:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tuzun E, Zhou L, Baehring JM, Bannykh S, Rosenfeld MR, Dalmau J. Evidence for antibody-mediated pathogenesis in anti-NMDAR encephalitis associated with ovarian teratoma. Acta Neuropathol 2009;118:737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y, Sangha N, Martinez-Lage M, Dalmau J. Analysis of complement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology 2011;77:589–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bien CG, Vincent A, Barnett MH, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 2012;135:1622–1638. [DOI] [PubMed] [Google Scholar]
- 32.Bernal F, Graus F, Pifarre A, Saiz A, Benyahia B, Ribalta T. Immunohistochemical analysis of anti-Hu-associated paraneoplastic encephalomyelitis. Acta Neuropathol 2002;103:509–515. [DOI] [PubMed] [Google Scholar]
- 33.Blumenthal DT, Salzman KL, Digre KB, Jensen RL, Dunson WA, Dalmau J. Early pathologic findings and long-term improvement in anti-Ma2-associated encephalitis. Neurology 2006;67:146–149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}