

Abstract
Peptides that translocate spontaneously across cell membranes could transform the field of drug delivery by enabling the transport of otherwise membrane-impermeant molecules into cells. In this regard, a 9-aminoacid-long motif (representative sequence: PLIYLRLLR, hereafter Translocating Motif 9, TM9) that spontaneously translocates across membranes while carrying a polar dye was recently identified by high-throughput screening. Here we investigate its transport properties by a combination of in cuvette physico-chemical assays, rational mutagenesis, live-cell confocal imaging and fluorescence correlation spectroscopy measurements. We unveil TM9 ability to self-aggregate in a concentration-dependent manner and demonstrate that peptide self-aggregation is a necessary –yet not sufficient– step for effective membrane translocation. Furthermore we show that membrane crossing can occur with apolar payloads while it is completely inhibited by polar ones. These findings are discussed and compared to previous reports. The present results impose a careful rethinking of this class of sequences as direct-translocation vectors suitable for delivery purposes.
The ability to cross the plasma-membrane and gain access to the cell interior, often even to a specific subcellular compartment, is a requirement for any intracellular drug-delivery protocol. To this end, direct membrane crossing is a particularly attractive approach to cytoplasmic delivery since it avoids the complications of vesicle-mediated internalization pathways1,2,3,4,5,6. This is still an unsolved issue, however, but today, peptide-based strategies appear to be particularly promising. Indeed, high-delivery yields, low toxicity, wide range of target cells, and the possibility to easily modify their sequence/function make peptides an excellent platform for the engineering of efficient transporters. Of note in the field, Marks and co-workers recently developed a straightforward combinatorial chemistry approach capable of achieving the high-throughput screening of thousands of sequences at the same time7. These authors selected out of several thousand sequences 18 Spontaneous Membrane-Translocating Peptides (SMTPs) with a characteristic length of 12 amino acids and demonstrated their ability to deliver low-molecular-weight polar cargo-molecules across synthetic lipid bilayers and into live cells at relatively low peptide concentration (0.5–2 μM). Importantly these peptides were shown not to irreversibly affect membrane integrity. A number of later studies (both theoretical and experimental) were performed on one of these representative peptides (known as Translocating Peptide 2, TP2; sequence: PLIYLRLLR-G-QF) that provide detailed knowledge on the mechanism yielding its spontaneous membrane translocation8,9,10,11,12,13,14. Notably, available data clearly ascribe TP2 translocating activity to the very first 9 residues of its sequence (highlighted in bold above), while the additional 3 residues were only functional to specific in-cuvette translocation assays (for details see Ref. 7). Yet, no detailed reports are available so far on the properties of the putative 9-aa translocating motif of TP2. Here we address this matter by a combination of in cuvette physico-chemical assays, live-cell confocal imaging, and fluorescence correlation spectroscopy measurements on fluorescently-labeled variants of this 9-aa peptide, hereafter named Translocating Motif 9 (TM9). We use ATTO 425 and Tetramethylrhodamine-5-maleimide (TAMRA) as representative apolar and polar low-molecular-weight cargoes, respectively. By means of the standard “pyrene 1:3 ratio method”15, we demonstrate that TM9 is able to self-aggregate in a concentration-dependent manner, regardless of the polarity of the attached cargo. In particular, we identify the Critical Micelle Concentration (CMC)16 above which the peptide appears in an aggregated form. We confirm this result by Dynamic Light Scattering (DLS) measurements and find that the aggregated form corresponds to a nanoparticle with a hydrodynamic radius of about 100–150-nm. Concentration-dependent aggregation and cargo-polarity together determine the peptide translocation properties in live cells. In particular, while TAMRA-labeled TM9 enters cells by a vesicle-mediated process at any tested concentration, its ATTO 425-labeled counterpart is able to effectively relocate into cells by direct plasma-membrane translocation. In this latter case, however, we additionally prove that direct translocation switches-on only above the peptide characteristic CMC, while endocytosis dominates below the CMC. Furthermore, as revealed by quantitative fluorescence microscopy on live cells, translocation of the TM9-ATTO 425-based nanoparticle occurs preferentially across selected plasma membrane regions, analogously to what already observed for several cell-penetrating peptides17. In addition, our data suggest a critical role of integer extracellular glycosaminoglycans (GAGs) for this localized process to be effective, as GAGs enzymatic digestion leads to peptide homogeneous translocation throughout the plasma membrane. Based on this knowledge, we finally propose a direct comparison of TM9 with its precursor sequence, TP2, widely used in the literature. We show that TP2 is able to form aggregates in a concentration-dependent manner, similarly to TM9. This behavior, however, does not correspond to the expected concentration-dependent translocation of TP2 into cells, since endocytosis is shown to be the dominant mechanism of entry under the experimental conditions tested here, regardless of the specific fluorophore-cargo chosen. Importantly, we discuss this apparent contradiction and relate it to the specific experimental conditions that significantly affect the peptide physicochemical behavior and, as a consequence, its membrane translocation ability. Collectively, our results impose a careful rethinking of this class of sequences as direct-translocation vectors for delivery purposes. We believe that peptide propensity to self-aggregate, exact peptide sequence, and nature of the attached cargo need further attention and study in an effort to rationally engineer a new generation of spontaneously-translocating sequences.
Results and Discussion
TM9 direct translocation across the plasma membrane depends on the polarity of the cargo and on peptide concentration
Marks and co-workers recently introduced a straightforward combinatorial peptide chemistry and orthogonal high-throughput screening to select peptides that spontaneously translocate across synthetic lipid bilayer membranes without permeabilization effects, while carrying a small polar cargo (TAMRA)7. A family of Spontaneous Membrane Translocating Peptide (SMTP) sequences with the general structure reported in Fig. 1a was identified. In particular, the first 9 residues represent the screened segment, with putative translocating properties (example: 1PLIYLRLLR9, as in Fig. 1a). A Glycine in position 10 constitutes a flexible linker, while Glutamate in position 11 and Phenylalanine in position 12 are functional to the in vitro test of membrane permeability used7. A C-terminal Cysteine residue was also added for labeling purposes. Following this general strategy, and in order to better evaluate the properties of the putative 9-aa-long translocating motif, we removed the Glutamate and Phenylalanine residues, while preserving the Glycine as a flexible linker and Cysteine to chemically attach a cargo moiety (overall sequence: PLIYLRLLR-G-C, hereafter named Translocating Motif 9, TM9). The purified peptide was labeled on the Cys residue with either ATTO 425 (as model of apolar cargo) or TAMRA (as a model of polar cargo, in analogy with previous studies7,13,14) (Fig. 1a). Labeled peptides were tested on live CHO-K1 cells by confocal fluorescence microscopy. As a first test, we performed experiments on cells exposed to increasing concentrations of either TM9-ATTO 425 or TM9-TAMRA. As detailed in the Experimental Procedures section, purified dye-labeled peptides were pre-dissolved in water solution, sonicated, quantified by UV-Vis absorbance, and added to the cell medium at the desired concentration (in both cases, fluorescent peptides are incubated with cells for 20 minutes before washing). As reported in Fig. 1b, we observed that for TM9-ATTO 425 concentrations up to 3 μM only fluorescent vesicles (bright spots in Fig. 1b-0.5 and 3 μM) are detected, indicating that endocytosis is the dominant mechanism responsible of peptide uptake. On the contrary, an intense and diffuse fluorescence signal is present at higher peptide concentrations (e.g. 5 and 10 μM), suggesting that a vesicle-independent direct translocation process across the plasma membrane becomes dominant in this regime (Fig. 1b-5 and 10 μM). The observed fluorescence staining (labeled cytoplasm and apparently dark nucleus) is consistent with the peculiar spectral properties of ATTO 425 (Fig. S1): just like many coumarin derivatives18, in fact, this fluorophore markedly increases its fluorescence quantum yield in apolar, lipophilic environments of the cell (cytoplasm) compared to aqueous environments (nucleus and surrounding medium). By contrast, under the same experimental conditions, TM9-TAMRA clearly shows punctuate fluorescence throughout the complete range of tested concentrations, with no apparent direct translocation into the intracellular medium (Fig. 1b). To reinforce our conclusion that the massive cytoplasmic staining observed for TM9-ATTO 425 at high concentrations depends on direct plasma membrane translocation we performed an uptake experiment at 4 °C, a well-known strategy to block active endocytosis while preserving direct permeation5. As can be seen in Fig. 1c, endocytosis of TM9-ATTO 425 at 3 μM is almost completely inhibited (i.e. only accumulation of the peptide at the level of the plasma membrane can be detected, Fig. 1c, left panels). On the contrary, under the same experimental conditions, TM9-ATTO 425 at 10 μM still shows diffuse cytoplasmic staining, thus corroborating the hypothesis of a temperature-independent direct translocation process across the membrane. To exclude any possible toxic effect of the labeled peptides on cell viability, we incubated cells for 2 hours in presence of varying peptide concentrations and performed the standard WST-8 assay 24 hours after removal of the peptide solution (Fig. S2).
Figure 1. SMTP general structure, TP2 and derived TM9 peptide sequences, fluorophores studied here and cell uptake of dye-labeled TM9.

(a) SMTP structural composition, TP2 precursor sequence as representative example and TM9 peptide sequence with ATTO 425 and TAMRA structures. (b) Confocal images of cells treated with increasing concentrations of TM9-ATTO 425 and TM9-TAMRA in free serum medium. Concerning the former, endocytic bright spots are detectable at 0.5 and 3 μM, while direct translocation is prevalent at 5 and 10 μM. Concerning the latter, only endocytosis is present at all the concentrations tested. Scale bars: 10 μm. (c) Confocal images of cells treated with 3 μM (left) and 10 μM (right) of TM9-ATTO 425 at 4 °C. While in the former case only cell plasma membranes are labelled (i.e. endocytosis is blocked), in the latter one diffuse cytoplasmic staining is still evident (i.e. direct translocation occurs).
TM9 is able to self-aggregate in a concentration-dependent manner
The translocation properties observed for TM9-ATTO 425 prompted us to speculate that peptide physicochemical properties (e.g. aggregation state) may change in a concentration-dependent manner, thus affecting transport properties in cells. As a first test, we dissolved either unlabeled or dye-labeled peptides in water solutions and performed a standard pyrene 1:3 ratio experiment, as a means to test their aggregation propensity15. Notably (Fig. 2), a significant decrease of the 1:3 ratio is detected going from 0.5 μM to 100 μM peptide concentration in all cases: a clear flex at a threshold CMC value is identified, analogously to previous measurements on short peptide sequences19,20. The CMC values for TM9, TM9-ATTO 425 and TM9-TAMRA are shown in Table 1. It is worth noting that the presence of a fluorophore, regardless of its polarity, lowers the CMC value of about one order of magnitude.We measured 58 ± 4 μM for CMC in the case of unlabeled TM9 and 5.7 ± 0.8 μM and 3.8 ± 0.5 μM for its ATTO 425- and TAMRA-labeled variants, respectively. This effect prompted us to test the intrinsic propensity of isolated fluorophores to form aggregates: we found that both TAMRA and ATTO 425 have a characteristic CMC in the μM range, even if at higher values (∼80 μM for TAMRA and ~25 μM for ATTO 425, see Fig. S3), in line with available data21. In order to evaluate the dimension of the peptide aggregates we performed Dynamic Light Scattering (DLS) measurements at concentrations above the CMC threshold. Table 1 shows the measured hydrodynamic radii for unlabeled and dye-labeled TM9 nanoparticles.
Figure 2. Variation of the pyrene 1:3 ratio at increasing concentrations of unlabeled, ATTO 425- and TAMRA- labeled TM9 peptide.
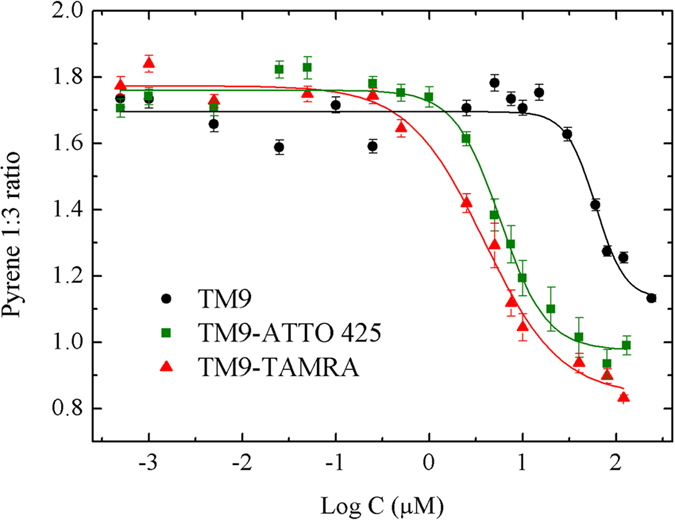
Vertical bars: standard errors.
Table 1. Critical Micelle Concentration (CMC) from pyrene 1:3 ratio method and hydrodynamic radius (RH) from DLS for unlabeled and dye-labeled TM9 and TP2 peptides.
| Peptide | TM9 | TM9-ATTO 425 | TM9-TAMRA | TP2 | TP2-ATTO 425 | TP2-TAMRA |
|---|---|---|---|---|---|---|
| CMC (μM) | 58 ± 4 | 5.7 ± 0.8 | 3.8 ± 0.5 | 1.0 ± 0.2 | 7.1 ± 0.9 | 3.7 ± 0.5 |
| RH (nm) | 112 ± 14 | 156 ± 8 | 135 ± 14 | 163 ± 6 | 114 ± 11 | 113 ± 12 |
Addressing the spatiotemporal regulation of TM9 translocation in live cells
In order to get further insight on peptide translocation into cells we incubated CHO-K1 cells in serum-free medium, added the selected concentration of TM9-ATTO 425 (see also the Experimental Procedures section) and performed a time-lapse, real-time imaging of peptide internalization process, this time without replacing the cell medium (Supporting Information, Movie 1). This latter procedure, made possible by the intrinsic spectral properties of ATTO 425 (as discussed above), allowed us to image the otherwise elusive process of peptide entry into cells. Interestingly, in fact, at concentrations above the CMC, few micrometric zones (typically one or two per cell) in which the fluorescence signal suddenly increases (i.e. the peptide locally concentrates) are detected on the plasma membrane shortly after peptide administration. Then fluorescence rapidly diffuses into the rest of the cell (Fig. 3a). Accordingly, if the average fluorescence intensity is measured in time at different locations within the cell (e.g. arrows in Fig. 3a) a quantitative picture of the timing of peptide intracellular spreading can be achieved. As reported in the plot of Fig. 3b, a nearly-constant intensity value is obtained if the ROI is placed at the peptide entry point, while a time lag of null fluorescence intensity is measured if the ROI is far from the entry point (with time lag proportional to the distance from the entry point). Overall, about 50% of the cells are interested by the translocation process. Of note, in a number of reports similar translocation processes originating from spatially-restricted sites of the plasma membrane (also known as “Nucleation Zones”, NZs) were related to peptide binding to selected extracellular-matrix components and subsequent translocation into the intracellular medium17,22,23,24. Here we test the hypothesis that TM9 uptake and translocation might be regulated also at the extracellular level. In this regard, a classical molecular target is represented by glycosaminoglycans (GAGs) that, together with proteins, constitute the “glycocalyx” that covers cell surface. To test the possible role of GAGs we performed a confocal microscopy experiment using PgsA-745 cells. This cell line, derived from CHO-K1 cells, does not produce GAGs, as it does not express xylosyltransferase (a required enzyme in GAG synthesis)25,26. We administered TM9-ATTO 425 at 10 μM to PgsA-745 and performed a time-lapse acquisition (see also Supporting Information, Movie 2). We observed that, differently from wild type CHO-K1 cells, the peptide enters nearly 100% of the cells by a translocation process that is homogeneously distributed throughout the plasmatic membrane (i.e. no NZs are detected, Fig. 3c). This is confirmed by the spatial analysis of peptide uptake in different locations within the cell (i.e. a linear increase of fluorescence in all the selected ROIs is detected, with no time lag, Fig. 3d). As a last control experiment, we analyzed peptide entry into CHO-K1 cells in which the GAGs were enzymatically digested by chondroitinase ABC (ChABC, Fig. S4). Upon administration of TM9-ATTO 425 at 10 μM concentration in serum-free medium, we monitored internalization by real-time imaging (Supporting Information, Movie 3). We detected homogeneous spreading of the fluorescence signal in 100% of the cells (Fig. 3e); fluorescence increased steadily during the observation time, with no time lag (Fig. 3f). Collectively, these results point at a significant role of integer GAGs in re-directing the peptide entry process towards spatially-restricted sites of the plasma membrane or NZs. Based on the present data and on the well-recognized role of GAGs as endocytosis effectors27,28,29, we cannot exclude that NZs are sites of GAG absence (or marked spatial inhomogeneity) rather than GAG accumulation.
Figure 3. Cell uptake and diffusion kinetics of direct translocating TM9-ATTO 425 in GAGs rich and deficient cells.

(a,c,e) diffusion of TM9-ATTO 425 solutions 12 μM in CHO-K1, 10 μM in PgsA-745 and 10 μM in GAGs cleaved CHO-K1, respectively. A NZ-dependent uptake is clearly detectable in the former case, while a homogeneous diffusion of peptide is shown for GAG-deficient and GAG-cleaved cells. The colored arrows indicate the ROIs used to evaluate peptide diffusion. (b,d,f) plot of the average fluorescence (AU) with respect to time (s) in the selected ROIs. In the case of CHO-K1 a different trend is shown with respect to the location within the cell, while, a linear increase occurs in all the locations within PgsA-745 and GAG-cleaved cells. Scale bars: 10 μm.
In order to further probe the spatiotemporal regulation of TM9-ATTO 425 translocation, we analyzed its extra- and intracellular diffusion properties by Raster Image Correlation Spectroscopy (RICS)30 measurements. These measurements were performed at a peptide concentration above the CMC (10 μM in the case reported in Fig. 4). Concerning the external solution, in some of the collected images we were able to identify bright diffusing spots blurred by the scanning process (Fig. 4a): these bright spots likely correspond to peptide nano-aggregates. By applying a single-particle RICS protocol (see Experimental Procedures) we retrieved a diffusion coefficient of 2.6 ± 0.5 μm2/s (Fig. 4d), compatible with aggregates with a hydrodynamic radius of about 100 nm (in good agreement with DLS results) (Fig. 4c). In order to identify the aggregation state of TM9-ATTO 425 within the cell, we performed the same RICS measurement in the cytoplasm and in the nucleus (Fig. 4b). Here, we were not able to identify isolated bright diffusing spots and we found a diffusion coefficient of 5.5 ± 0.3 μm2/s in the cytoplasm and of 13.1 ± 0.8 μm2/s in the nucleus (Fig. 4d). This suggests a disruption of peptide aggregates during or after cell permeation. In fact, the measured diffusion coefficient in live cell is in agreement with previous intracellular measurements on analogous monomeric peptides31,32. Moreover, the twofold lower diffusion coefficient measured in the cytoplasm compared to the nucleus indicates a higher propensity of the peptide to interact with the cytoplasmic environment. Please note that this result is somewhat expected given the presence of membranous structures within the cytoplasm only.
Figure 4. RICS and single-particle RICS analysis and results for TM9-ATTO 425.
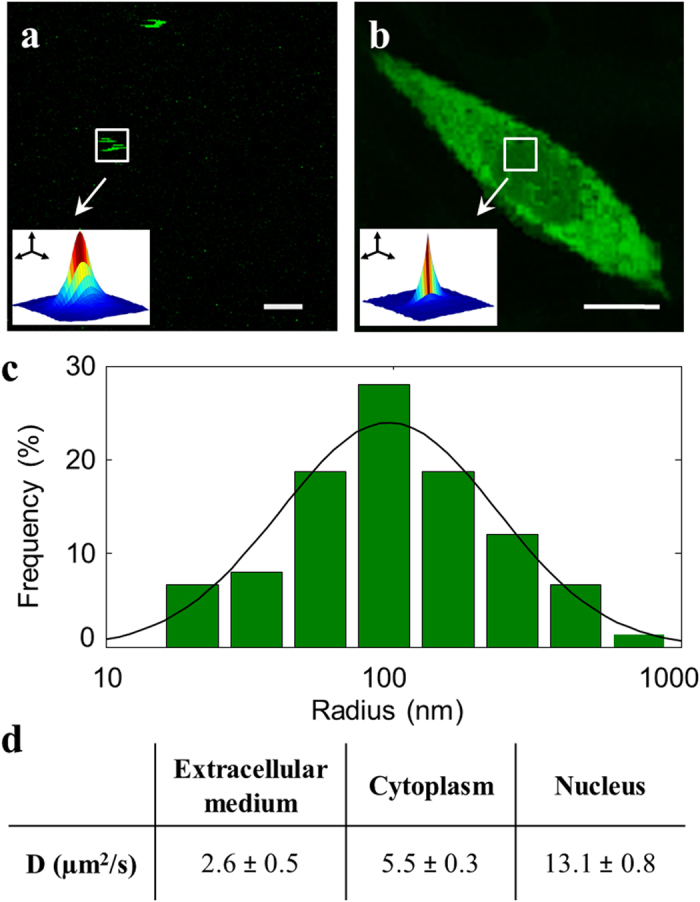
(a) Nanoparticle aggregates found in the external solution, for which RICS measurement was repeated overall 30 times. Scale bar: 1.5 μm. (b) A cell region selected for RICS experiments. Scale bar: 10 μm. (c) Size distribution and gaussian fitting for nanoparticles found in the external solution. The mean radius is 100 ± 30 nm. (d) TM9-ATTO 425 diffusion coefficients (μm2/s) obtained from RICS for nanoparticles and for internalized peptide.
Comparison of TM9 with the TP2 precursor sequence
At this point, one must check whether the properties unveiled so far for TM9 are shared by its precursor sequence, TP2. To this end, we performed the pyrene 1:3 ratio assay on TP2 dissolved in water at varying concentrations, ranging from 0.5 μM to 100 μM (Fig. 5a). Notably, the calculated I1/I3 parameter varies non-linearly with peptide concentration, as expected for a concentration-dependent aggregation process. In this case, and in contrast to TM9, the characteristic CMC value of the unlabeled peptide (1.0 ± 0.2 μM) increases upon addition of the fluorophore, regardless of fluorophore polarity (CMC = 7.1 ± 0.9 μM in the case of ATTO 425, and CMC = 3.7 ± 0.5 μM in the case of TAMRA, see also Table 1). As expected, DLS measurements confirm the presence of peptide-based aggregates at concentrations above the CMC for both unlabeled and labeled TP2 and yield an estimate of the aggregate size (about 160 nm for TP2 and about 110 nm for both types of dye-labeled TP2). We then investigated cell uptake of the two fluorescent variants of TP2 at concentrations both below and above their respective CMC values (Fig. 5b). Quite surprisingly, no direct translocation was observed for TP2, regardless of the selected concentration and of the chosen fluorescent cargo (polar or apolar). In fact, as shown by confocal images in Fig. 5b, only punctuate fluorescence staining was detected within live cells after peptide administration, suggesting endocytosis as the dominant uptake mechanism for these TP2 adducts.These results do not appear to be consistent with previous reports on TP27,14 and this, in turn, suggests the need for a careful comparison of the experimental protocols adopted. For what concerns in vitro cell studies, in previous reports dye-labeled TP2 was added from stock DMSO solutions directly to the cell medium up to the desired final concentration7,14. Indeed, if we adopt this same procedure, we do obtain direct translocation for both TP2-ATTO 425 and TP2-TAMRA even below the CMC value (see, for instance, the behavior of both TP2-ATTO 425 and TP2-TAMRA at 2 μM reported in Fig. 5c). Unfortunately, although not affecting cell viability (its final concentration in the cell medium is always around 1% or even less, as in the literature), DMSO can influence TP2 solubilization by altering peptide/water miscibility kinetics and lead to the formation of precipitates (see in cuvette absorption measurements reported in Fig. S5). As a consequence, this type of protocol does not appear to be transferable from these in vitro tests to more complex situations like tissues or entire organisms.
Figure 5. Decreasing of pyrene 1:3 ratio at TP2 increasing concentrations, TP2 cell uptake and cell uptake before and after the homogenization of TP2 solution (1% DMSO).
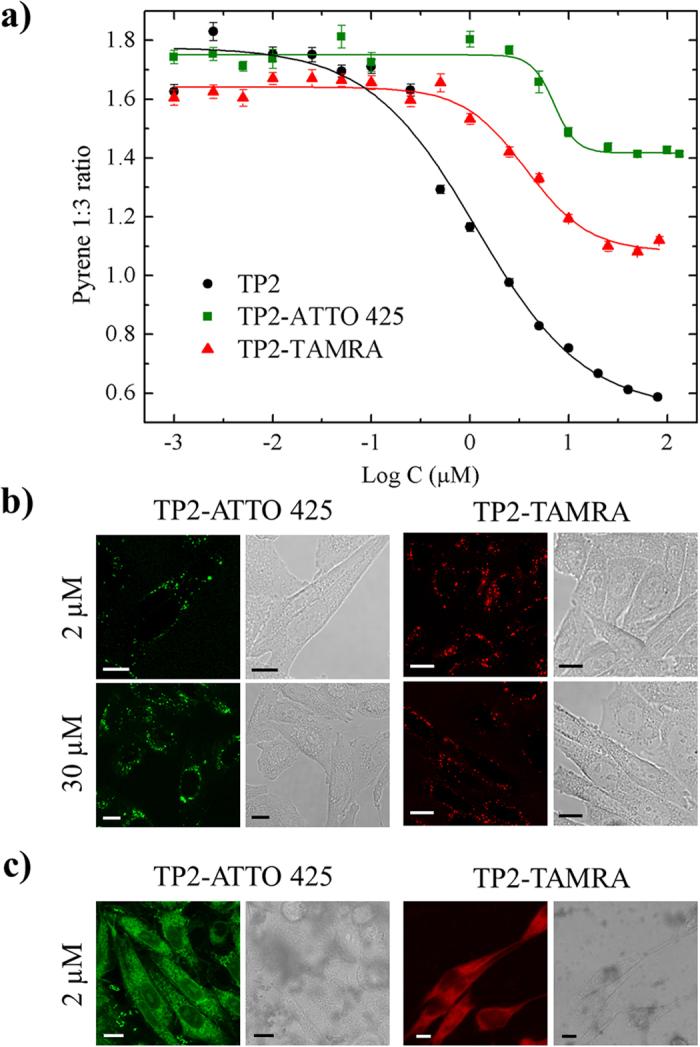
(a) Plot of the pyrene 1:3 ratio of free and dye-labeled TP2 peptide. Vertical bars: standard errors. (b) Confocal images of cells treated with increasing concentrations of TP2-ATTO 425 and TP2-TAMRA in free serum medium. Scale bars: 10 μm. (c) Confocal images of cells treated with TP2-ATTO 425 and TP2-TAMRA at a final concentration of 2 μM (peptide added to the cell medium directly from the DMSO stock solution). Scale bars: 10 μm.
Conclusions
In this work we addressed the spontaneous membrane-translocation properties of a recently-introduced class of short peptidic sequences (representative translocating motif: PLIYLRLLR, TM9). Thanks to a combination of in cuvette physico-chemical assays, rational mutagenesis, and live-cell fluorescence-based measurements we demonstrated that TM9 self-aggregates in a concentration-dependent manner.Moreover we showed that self-aggregation is a necessary –but not sufficient– step for effective membrane translocation. Indeed, membrane crossing is enabled by apolar payloads while it is inhibited by polar ones. These new results on this interesting class of sequences show the need of further studies for their exploitation as direct-translocation vectors suitable for drug-delivery purposes. In particular, peptide propensity to self-aggregate, peptide sequence and influence of the attached cargo must be considered as key parameters in the effort of rationally engineer new spontaneously-translocating sequences, better tailored to delivery applications both in vitro and in vivo.
Experimental procedures
Peptide synthesis, purification and labeling
All peptides were prepared by solid-phase synthesis using Fmoc chemistry on an automatic Liberty Blue Peptide Synthesizer with an integrated microwave system (CEM, North Carolina, USA). HPLC analysis and purification were performed on a Dionex Ultimate 3000 PLC system with autosampler. Crude peptides were purified by RP-HPLC on a Jupiter 4 μm Proteo 90 Å column (250 × 10 mm; Phenomenex) using these solvents: water:TFA 100:0.01 v/v (eluent A)/acetonitrile:water:TFA 95:5:0.01 v/v (eluent B), flux 5 ml/min. The identity of purified product was confirmed by electrospray mass spectroscopy, using an API3200QTRAP Hybrid Triple Quadrupole/Linear Ion Trap (ABSciex, Foster City, California, USA). Labeling of purified peptides was performed as follows: a suitable amount of peptide (10 μmol) was dissolved in 0.2 mL of freshly degassed 10 mM PBS buffer (pH 7.4). 1.5 equivalents of fluorophore, ATTO 425-maleimide (ATTO-TEC GmbH, Germany) or Tetramethylrhodamine-5-maleimide (TAMRA, Sigma Aldrich), dissolved in DMF were added to the solution. The mixture was reacted for 2 h at 25 °C. Crude product was purified by HPLC and the identity of labeled peptides was confirmed by LC-MS. The purified product was freeze-dried and stored at −80 °C. When needed, it was dissolved in water and the concentration of each peptide stock solution was verified by UV–vis absorbance (Jasco 550 spectrometer, Jasco, Tokyo, Japan).
Preparation of peptide solution for translocation experiments
Homogeneous peptide solutions used for translocation experiments were always freshly prepared. Purified dye-labeled peptides were pre-dissolved in 100–200 μl of water and sonicated for 15 minutes at 25 °C. Then they were quantified by UV-Vis absorbance (concentration ranging between 100 and 200 μM; for ATTO 425 Maleimide: ε436(water) = 45000 cm−1M−1, for TAMRA: ε543(water) = 80000 cm−1M−1). An appropriate amount from this stock water-solution was diluted in 200–300 μl of serum-free DMEM F-12 growth medium and then administered to cells. Ionic strength was adjusted to physiological values adding the required amount of 10X PBS buffer.
Cell culture and translocation experiments
Chinese Hamster Ovary (CHO-K1) and PgsA-745 cells were purchased from ATCC (reference numbers: CCL-61 and CRL-2242, respectively) and grown in DMEM F-12 medium supplemented with 10% of Fetal Bovine Serum (FBS) at 37 °C with 5% CO2, according to manufacturer’s instructions. In order to perform translocation experiments, cells were plated onto 35-mm glass-bottom petri dishes (WillCo-dish GWSt-3512) 24 h before the experiment. The day of the experiment, cells were washed with PBS at room temperature. At this point the peptide solution was homogeneously added, and cells incubated for 20 minutes at 37 °C. In standard uptake experiments, after incubation, cells were washed with PBS and fresh complete medium was added before confocal imaging. For the experiment of real-time imaging of peptide direct translocation through nucleation zones (limited to TM9-ATTO 425 at high concentration) we did not wash the peptide solution before imaging, for the reasons explained in the Results section.
Confocal microscopy and Raster Image Correlation Spectroscopy (RICS)
Confocal imaging experiments were performed with a Leica TCS SP5 SMD inverted confocal microscope (Leica Microsystems) interfaced with a diode laser (Picoquant) for excitation at 405 nm. Glass-bottom Petri dishes containing plated cells were mounted in a temperature-controlled chamber at 37 °C and 5% CO2 (Leica Microsystems) and viewed with a 63 × 1.2 numerical aperture (NA) water immersion objective (Leica Microsystems). The excitation wavelengths were 405 nm and 561 nm for ATTO 425 and TAMRA, respectively. The collection range adopted was 450–550 nm and 580–680 nm for acquiring ATTO 425 and TAMRA, respectively. The diameter of the detection pinhole was set to the size of 1 Airy disk. All images and videos collected were analyzed by ImageJ software version 1.440 (NIH Image; http://rsbweb.nih.gov/ij/). RICS measurements were performed with an Olympus FluoView 1000-ASW-2.0 confocal laser scanning microscope using a 60× (NA 1.2) planApo water-immersion objective. The diameter of the detection pinhole was set to the size of 1 Airy disk. All experiments were carried out at 37 °C and 5% CO2 using an incubation chamber enclosing the microscope stage and body. Sequential image series at 12 bits were collected at a fixed pixel size of 50 nm and with a pixel dwell time of 10 μs for measurements in solution and in cells. Typically, a ROI of 256 × 256 pixels (corresponding to an area of 6.4 × 6.4 μm) was selected and 200 total frames were collected. RICS correlation function was calculated by the SimFCS software, developed at the Laboratory for Fluorescence Dynamics (www.lfd.uci.edu). Fitting of RICS function to extract the diffusion coefficient (D, μm2/s) of the dye-labeled peptide was performed by SimFCS software. In order to measure the diffusion coefficient of the peptide nanoparticles directly in the media above the cells we applied the following protocol (also named ‘single-particle RICS’ in the main text in order to distinguish it from the classical RICS analysis). Peptide nanoparticles appear as bright isolated spots during the imaging process. Since they are diffusing, their image is blurred by the raster scan process. The RICS principle can be thus used to estimate their diffusivity. In this case, the signal-to-noise ratio is so high that averaging over many frames is not required. Thus, we calculate the RICS correlation function in a ROI containing each selected particle separately and measure the diffusion coefficient one particle at a time by fitting with a free diffusion model33,34. This protocol allows obtaining an estimate of the hydrodynamic radius of the diffusing particle, analogously to the DLS measurement but through direct observation of a single particle.
GAGs labeling and cleavage in CHO-K1 cells
Fluorescein-labeled Wisteria Fluribunda Lectin (2 mg/ml, Sigma Aldrich, catalog number: FL-1351) was used as a well-recognized marker of GAGs. It was diluted 1:100 in DMEM F-12 (10% FBS) and added to CHO-K1 cells, which were then incubated for 2 hours at 37 °C with 5% CO2. Chondroitinase ABC (Sigma Aldrich) was then added at a final concentration of 0.2 U/ml obtaining an efficient cleavage of surface proteoglycans. After 4 hours of incubation, translocation experiments were performed, as described above.
WST-8 Cell Viability Assay
Cytotoxicity of unlabeled peptides and dye-labeled peptides was evaluated by the WST-8 assay. CHO-K1 cell proliferation was evaluated by plating 5 × 103 cells per well in 96-well plates. After 24 hours in serum-containing DMEM-F12 medium, the cells were incubated with a serum-free DMEM-F12 solution of unlabeled or dye-labeled peptides for 2 hours at 37 °C. After the incubation, the medium was removed, cells were washed with PBS 1X and incubated for 2 hours at 37 °C with 100 μl of a 10%-solution of WST-8 dissolved in serum-containing DMEM-F12. Absorbance at 450 nm was measured using a multiplate reader (Microplate Reader, GloMax®-Discover and Explorer Systems, Promega). Cell viability was quantitatively determined by comparing peptide-treated cells with untreated cells (as a reference of 100% viability) and cells treated with dimethyl sulfoxide (DMSO) 20% v/v. Reported data represent the average of three independent experiments, in which three wells for each concentration were measured. Error bars represent the standard errors.
Critical Micelle Concentration (CMC) determination
All the peptide solutions were always freshly prepared and dye-labeled peptides were quantified by UV-Vis absorbance. Unlabeled peptides were quantified by amino acidic digestion obtained by the mineralization of a known amount of peptide (5–50 μg) dissolved in 200–500 μl of HCl 6 M, with the following conditions: temperature: 170 °C; time: 20 minutes. Finally, derivatized amino acids were detected by RP-HPLC (Jupiter 4 μm Proteo 90 Å column (250 × 4.6 mm; Phenomenex) with these solvents: water:TFA 100:0.01 v/v (eluent A)/acetonitrile:water:TFA 95:5:0.01 v/v (eluent B), flux 1 ml/min, and their identity was confirmed by electrospray mass spectroscopy. CMC values of peptide and labeled-peptide systems were then determined by the pyrene 1:3 ratio method15. Fluorescence emission spectra at 25 °C of unlabeled and labeled peptide water solutions containing 5 × 10−7 μM pyrene were recorded using an excitation wavelength of 335 nm. The intensities I1 and I3 were measured at the wavelengths corresponding to the first and third vibronic bands of pyrene (373 nm, emission range: 368–378 nm, and 385 nm, emission range: 380–390 nm, respectively). According to literature15, the plot of the pyrene 1:3 ratio index as a function of the concentration of the surfactant can be described by a decreasing sigmoid of the Boltzmann type and indicates the transition from the monomeric to the aggregated form of the peptide. Experimental raw data were fitted using the OriginPro8 software, and the center of the sigmoid is identified as the characteristic CMC.
Dynamic Light Scattering (DLS) measurements
DLS measurements were performed at 25 °C in a 50-μL quartz cuvette on a Zetasizernano ZS DLS (Malvern Instrument) following the manufacturer’s instructions. Water solutions of labeled-peptide at 100 μM were analyzed with a single scattering angle of 90°. Each value reported is the average of five consecutive measurements.
Additional Information
How to cite this article: Macchi, S. et al. Spontaneous membrane-translocating peptides: influence of peptide self-aggregation and cargo polarity. Sci. Rep. 5, 16914; doi: 10.1038/srep16914 (2015).
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Silvia Landi and Dr. Gerardo Abbandonato for useful discussions. Work partly supported by “Borsa Pegaso” (POR-FSE Regione Toscana) to S.M.
Footnotes
Author Contributions S.M. performed the experiments, analyzed the data, wrote the manuscript; G.S. performed experiments, analyzed the data; C.D.R. performed experiments, analyzed the data; C.B. synthetized the peptides; F.B. designed the research, wrote the manuscript; F.C. designed the research, analyzed the data, wrote the manuscript.
References
- Salomone F. et al. A novel chimeric cell-penetrating peptide with membrane-disruptive properties for efficient endosomal escape. J. Control. Release 163, 293–303 (2012). [DOI] [PubMed] [Google Scholar]
- Salomone F., Cardarelli F., Signore G., Boccardi C. & Beltram F. In Vitro Efficient Transfection by CM18-Tat11 Hybrid Peptide: A New Tool for Gene-Delivery Applications. PLoS One 8, e70108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara C. & Sagan S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 587, 1693–702 (2013). [DOI] [PubMed] [Google Scholar]
- Wang F. et al. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J. Control. Release 174, 126–36 (2014). [DOI] [PubMed] [Google Scholar]
- Copolovici D. M., Langel K., Eriste E. & Langel Ü. Cell-penetrating peptides: design, synthesis, and applications. ACS Nano 8, 1972–94 (2014). [DOI] [PubMed] [Google Scholar]
- Di Pisa M., Chassaing G. & Swiecicki J.-M. Translocation Mechanism(s) of Cell-Penetrating Peptides: Biophysical Studies Using Artificial Membrane Bilayers. Biochemistry 54, 194–207 (2015). [DOI] [PubMed] [Google Scholar]
- Marks J. R., Placone J., Hristova K. & Wimley W. C. Spontaneous membrane-translocating peptides by orthogonal high-throughput screening. J. Am. Chem. Soc. 133, 8995–9004 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis T., Leveritt J. M. & PeBenito L. Implicit membrane treatment of buried charged groups: application to peptide translocation across lipid bilayers. Biochim. Biophys. Acta 1838, 2149–59 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y. & Patel S. Structural and Thermodynamic Insight into Spontaneous Membrane-Translocating Peptides Across Model PC/PG Lipid Bilayers. J. Membr. Biol. 248, 505–515 (2015). [DOI] [PubMed] [Google Scholar]
- Lin J. et al. Interactions of membrane active peptides with planar supported bilayers: an impedance spectroscopy study. Langmuir 28, 6088–96 (2012). [DOI] [PubMed] [Google Scholar]
- Cruz J. et al. A membrane-translocating peptide penetrates into bilayers without significant bilayer perturbations. Biophys. J. 104, 2419–28 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecicki J.-M. et al. The efficacies of cell-penetrating peptides in accumulating in large unilamellar vesicles depend on their ability to form inverted micelles. Chembiochem 15, 884–91 (2014). [DOI] [PubMed] [Google Scholar]
- He J., Hristova K. & Wimley W. C. A highly charged voltage-sensor helix spontaneously translocates across membranes. Angew. Chem. Int. Ed. Engl. 51, 7150–3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J. et al. Direct cytosolic delivery of polar cargo to cells by spontaneous membrane-translocating peptides. J. Biol. Chem. 288, 29974–29986 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguiar J., Carpena P., Molina-Bolívar J. A. & Carnero Ruiz C. On the determination of the critical micelle concentration by the pyrene 1:3 ratio method. J. Colloid Interface Sci. 258, 116–122 (2003). [Google Scholar]
- Ruckenstein. E. & Nagarajan R.. Critical Micelle Concentration. A Transition Point for Micellar Size Distribution. J. Phys. Chem. 79, 2622–2626 (1975). [Google Scholar]
- Duchardt F., Fotin-Mleczek M., Schwarz H., Fischer R. & Brock R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 8, 848–66 (2007). [DOI] [PubMed] [Google Scholar]
- Signore G., Nifosì R., Albertazzi L., Storti B. & Bizzarri R. Polarity-sensitive coumarins tailored to live cell imaging. J. Am. Chem. Soc. 132, 1276–88 (2010). [DOI] [PubMed] [Google Scholar]
- Cao M., Cao C., Zhang L., Xia D. & Xu H. Tuning of peptide assembly through force balance adjustment. J. Colloid Interface Sci. 407, 287–295 (2013). [DOI] [PubMed] [Google Scholar]
- Qiu F., Chen Y. & Zhao X. Comparative studies on the self-assembling behaviors of cationic and catanionic surfactant-like peptides. J. Colloid Interface Sci. 336, 477–484 (2009). [DOI] [PubMed] [Google Scholar]
- Moreno-Villoslada I. et al. Comparative Study of the Self-Aggregation of Rhodamine 6G in the Presence of Poly (sodium 4-styrenesulfonate), Poly (N -phenylmaleimide- co -acrylic acid), Poly (styrene- alt -maleic acid), and Poly (sodium acrylate). J Phys Chem B 114, 11983–11992 (2010). [DOI] [PubMed] [Google Scholar]
- Verdurmen W. P. R., Thanos M., Ruttekolk I. R., Gulbins E. & Brock R. Cationic cell-penetrating peptides induce ceramide formation via acid sphingomyelinase: implications for uptake. J. Control. Release 147, 171–9 (2010). [DOI] [PubMed] [Google Scholar]
- Favretto M. E., Wallbrecher R., Schmidt S., van de Putte R. & Brock R. Glycosaminoglycans in the cellular uptake of drug delivery vectors-bystanders or active players? J. Control. Release 180, 81–90 (2014). [DOI] [PubMed] [Google Scholar]
- Brock R. The uptake of arginine-rich cell-penetrating peptides: putting the puzzle together. Bioconjug. Chem. 25, 863–8 (2014). [DOI] [PubMed] [Google Scholar]
- Esko J. D. et al. Inhibition of chondroitin and heparan sulfate biosynthesis in Chinese Hamster Ovary cell mutants defective in galactosyltransferase I. J. Biol. Chem. 262, 12189–95 (1987). [PubMed] [Google Scholar]
- Brinkmann T., Weilke C. & Kleesiek K. Recognition of acceptor proteins by UDP-D-xylose proteoglycan core protein β-D-xylosyltransferase. J. Biol. Chem. (1997). doi: 10.1074/jbc.272.17.11171 [DOI] [PubMed] [Google Scholar]
- Wallbrecher R. et al. The stoichiometry of peptide-heparan sulfate binding as a determinant of uptake efficiency of cell-penetrating peptides. Cell. Mol. Life Sci. 71, 2717–29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson H. C. & Belting M. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol. 35, 51–55 (2014). [DOI] [PubMed] [Google Scholar]
- Bechara C. et al. Massive glycosaminoglycan-dependent entry of Trp-containing cell-penetrating peptides induced by exogenous sphingomyelinase or cholesterol depletion. Cell. Mol. Life Sci. 72, 809–820 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digman M. A. & Gratton E. Analysis of diffusion and binding in cells using the RICS approach. Microsc. Res. Tech. 72, 323–32 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardarelli F., Serresi M., Bizzarri R., Giacca M. & Beltram F. In vivo study of HIV-1 Tat arginine-rich motif unveils its transport properties. Mol. Ther. 15, 1313–1322 (2007). [DOI] [PubMed] [Google Scholar]
- Cardarelli F., Serresi M., Bizzarri R. & Beltram F. Tuning the transport properties of HIV-1 Tat arginine-rich motif in living cells. Traffic 9, 528–539 (2008). [DOI] [PubMed] [Google Scholar]
- Digman M. A. et al. Measuring fast dynamics in solutions and cells with a laser scanning microscope. Biophys. J. 89, 1317–1327 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzo C., Piazza V., Gratton E., Beltram F. & Cardarelli F. Probing short-range protein Brownian motion in the cytoplasm of living cells. Nat. Commun. 5, 5891 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.