

Abstract

Recent studies in adipose tissue, pancreas, muscle, and macrophages suggest that MAP4K4, a serine/threonine protein kinase may be a viable target for antidiabetic drugs. As part of the evaluation of MAP4K4 as a novel antidiabetic target, a tool compound, 16 (PF-6260933) and a lead 17 possessing excellent kinome selectivity and suitable properties were delivered to establish proof of concept in vivo. The medicinal chemistry effort that led to the discovery of these lead compounds is described herein together with in vivo pharmacokinetic properties and activity in a model of insulin resistance.
Keywords: MAP4K4, HGK, ZC1, kinase inhibitor, MAP4K4 inhibitor, P-loop, PF-6260933
Mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4), also called HGK or ZC1, is a serine/threonine protein kinase that belongs to the mammalian STE20/MAP4K family.1 MAP4K4 is overexpressed in human cancers and cancer cell lines and appears to play an important role in cell transformation, invasiveness, adhesion, and migration.2 More recently, studies in adipose tissue, pancreas, muscle, and macrophages suggest that MAP4K4 could also be a potential target for antidiabetic drugs.3,4 For example, suppression of the expression of MAP4K4 using small interfering RNA in human skeletal muscle cells from both healthy subjects and type II diabetic patients prevented TNF-α induced insulin resistance of glucose uptake.5 Furthermore, a recent genetic analysis suggests that common genetic variants in the MAP4K4 locus are associated with type II diabetes underlying pathomechanisms, insulin resistance, and β-cell failure.6 As part of the evaluation of MAP4K4 as a novel antidiabetic target, it was desirable to deliver a tool compound with greater kinome selectivity than the existing chemical matter (unpublished results) and reasonable properties to establish proof of concept in a mouse diabetes model. Herein we describe the medicinal chemistry that led to the discovery of such a compound. Since our work was conducted, alternative series of MAP4K4 inhibitors for oncology and neurodegenerative indications have recently been described.7−9
A virtual screening effort of the internal Pfizer file using an in-house crystal structure of the kinase domain of MAP4K4, identified compound 1 as a potent MAP4K4 inhibitor (Table 1). It was selective in a standard kinase panel and upon cocrystallization the complex between 1 and MAP4K4 revealed that the P-loop adopts a rare folded conformation state (Figure 1).10 The P-loop, which normally exists in an extended conformation, is a stretch of nine amino acids that in MAP4K4 is delimited by residues Val31 to Val39. The binding site adopts a tunnel-like shape that provides a significant hydrophobic enclosure for the inhibitor. There are potential π–π interactions between the MAP4K4 P-loop Tyr36 residue and the three aromatic rings of compound 1. The primary amide is the only group that escapes the hydrophobic tunnel, being partially exposed to solvent. The structure suggests that it forms a hydrogen bond with Asp115.
Table 1. MM-GB/SA Score, Enzyme and Cell Potencies, and Permeability Data of Analogues of Compound 1.

MM-GB/SA relative score in kcal/mol. The best scoring analogue according to the MM-GB/SA method is used as reference (0.0 kcal/mol).
Arithmetic mean and standard error of ≥3 measurements (unless noted otherwise).
Less than three independent replicates.
Passive permeability (Papp in 10–6 cm/s) measured using low-efflux MDCKII cells.13
Figure 1.
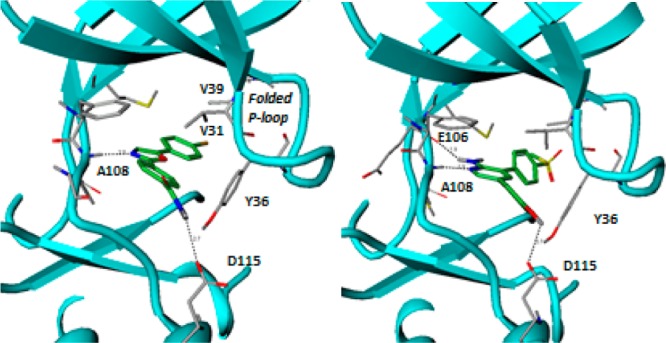
Crystal structure of the complex between MAP4K4 and (a) compound 1 (in green) (PDB code: 4ZP5) and (b) compound 13 (in green) (PDB code: 5DI1).
Compound 1 had low molecular weight (MW = 299) and a measured LogD of 3.6. It was a weak inhibitor of MAP4K4 (Table 1) but with good ligand efficiency (LE = 0.48). The kinase activity is measured using a FRET-based assay that utilizes catalytic domain of human recombinant MAP4K4. The cell assay is designed to identify compounds inhibiting MAP4K4’s phosphorylation of serine/threonine residues on traf2 (see Supporting Information for full details). In an attempt to improve the enzyme and cell potencies of 1, several heterocyclic alternatives to the oxazole hinge binding group were evaluated by the MM-GB/SA scoring procedure.11 The MM-GB/SA method involves simulations of a ligand in its bound and unbound states and computes its free energy of binding from the energy differences between these two states. The energy terms include intramolecular and intermolecular interactions, solvation energy, and conformational entropy. The most promising analogues from this analysis were synthesized and evaluated for MAP4K4 inhibitory activity (Table 1). The best scoring compounds, 1–4, were the most potent in the enzyme assay, whereas heterocycles 5–7, which had higher scores (and hence predicted to be less potent), displayed weaker potencies. The pyridine analogue 2 was found to be the most potent inhibitor in the enzyme assay but displayed weak inhibition in the cell-based assay. After the identification of pyridine as a competent hinge binder, the WaterMap methodology12 was employed to characterize unstable water molecules that could lead to incremental increase in potency when liberated to the bulk upon ligand binding. The hydration sites obtained from a simulation using the crystal structure of the complex between MAP4K4 and 1 are illustrated in Figure 2; the compound is omitted in the simulation (compounds 1 and 2 are included in the figure for comparison). The overlap between the map and compounds 1 and 2 suggests that they replace the favorable waters (colored green) with the primary amide and displace most of the unfavorable waters (colored red), except for three moderately unstable waters that sit in a deep pocket close to the methionine gatekeeper residue.
Figure 2.
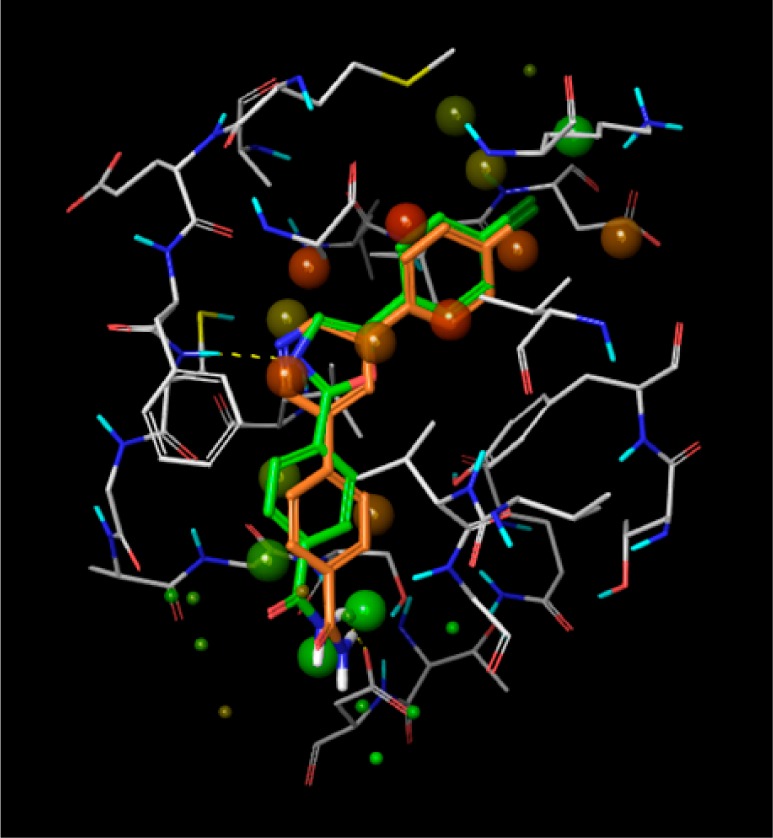
Hydration sites obtained using WaterMap simulations. The crystal structure of the complex between MAP4K4 and compound 1 was used. The compound is omitted in the simulations (compounds 1 and 2 were added to the figure above for comparison). The most favorable and unfavorable water molecules with respect to a water molecule with bulk-like properties are colored green and red, respectively.
This map inspired the design of analogues 8 and 9 (Table 1). The methoxy and cyclopropoxy groups were expected to lead to gains in potency due to partial or full displacement of one or two of the hydration sites highlighted in Figure 2. These modifications unfortunately did not result in improved enzyme potency when compared to compound 2. It is possible that the gain resulting from displacing the unstable waters is offset by an effect that opposes binding, such as a greater desolvation penalty for the ether oxygen or that the WaterMap estimates were not accurate.
Despite reasonable enzyme potency for some compounds (1–4, 8–9), they all have weak or no activity in a cell assay. As expected, enzyme potency is necessary but not sufficient to ensure cell activity. Impaired membrane permeability, differences in protein constructs between enzyme and cell assays (kinase domain vs full length), and competition with cellular ATP concentrations may be some of the determining factors in this enzyme-whole cell potency disconnect.. Passive permeability data, obtained using the MDCKII-LE cell line, which displays low expression of active transporters,13 indicated that most of the compounds have moderate permeability (Table 1) suggesting that it is not an impeding factor. Measured PgP efflux ratios for many of these compounds showed no evidence of efflux. To assess the impact of physiological ATP concentrations on compound activity, 1, 3, and 9 were tested in the enzyme assay using [ATP] = 1 mM instead of [ATP] = Km, (∼10 μM for MAP4K4). The respective IC50 values under these conditions were >10 μM for 1 and 3 and 9.8 μM for 9, which are similar to their cell activities. It is plausible that this chemotype relies heavily on the single hinge contact with the protein for activity and, therefore, is less capable of competing with ATP, which establishes two hinge contacts. To test this hypothesis, an amino group was introduced to the C(2) position of the pyridine ring in 2 to provide compound 10 (Table 2). This was accompanied by a modest 4-fold gain in enzyme potency and led to a ∼30-fold gain in cell activity. The H → NH2 change led to a drop of 0.5 units of logD but not much of a change in permeability despite the increased hydrogen bond donors. The efflux ratios were similar (MDR BA/AB efflux ratio of 1.2 for 2 vs 1.4 for 10) and the kinetic solubility was improved (<1.1 μM @ pH 6.5 for 2 vs 299 μM for 10). Several analogues of 10 that preserved the aminopyridine motif as the potential hinge-binder, but with alterations of the substituents on the two phenyl rings, were prepared (Table 2) in an effort to optimize overall properties directed toward identification of an in vivo tool compound.
Table 2. Enzyme and Cell Potencies and Permeability Data of Aminopyridines.

eLogD = measured logD at pH 7.4 [calculated logD (ACD Laboratories program v12) in italics].
Arithmetic mean and standard error of ≥3 measurements (unless noted otherwise).
Less than three independent replicates.
Passive permeability (Papp in 10–6 cm/s) measured using Low-Efflux MDCKII cells as described.13
Compound 11, lacking the amide group, shows loss of kinase potency accentuating the importance of the interactions between the primary amide group of compound 10 and Asp115 for achieving potency. Exchanging the Cl substituent in 10 for a SO2Me group (12) resulted in some loss of potency. Replacement of the amide of 12 by a hydroxy group in 13 resulted in similar potency in the enzyme and cellular assays despite the improved passive permeability. Crystallization of the complex between 13 and MAP4K4 revealed the same folded conformation state for the P-loop as the one adopted in the complex with 1 (Figure 1).10 The crystal structure illustrates the double hinge contact with the aminopyridine ring. It also displays a direct hydrogen bond between the phenol OH and Asp115, reinforcing the role of this interaction in MAP4K4 potency. The crystal structure also shows that the two flanking aromatic rings are not in plane relative to the central aromatic ring, thus mitigating ADME concerns of an otherwise planar triaromatic scaffold. To alleviate metabolic issues of a phenolic group, the pyridone analogue 14 was made, which led to a loss in both the enzyme and cell potencies. The reversed pyridone (15) regained some of the potency lost in 14 presumably due to the interactions with Asp115.
To optimize the hydrogen bond with Asp115, compound 16 was prepared, as modeling indicated that the NH2 group of the newly introduced aminopyridine group interacts with Asp115 in a similar fashion as the phenol OH in 13. Compound 16 (PF-6260933) was very potent against the enzyme with a very good ligand efficiency (LE = 0.57) and was the most potent analogue in the cell assay.
The kinome selectivity of 16 was assessed at a dose of 1 μM in a panel of kinases (Figure 3). Bolstered by the promising selectivity, this compound was advanced to an ActivX screen, which allows for profiling protein and lipid kinase interactions in cell lysates.14 Remarkably, the initial in vitro kinome selectivity profile translated to cell lysates; with 16 demonstrating very good selectivity against more than 150 native kinases that could be quantified in human peripheral blood mononuclear cells; with only MAP4K4 (ZC1, HGK) and closely related GCK family members (ZC2, TNIK and ZC3, MINK) showing >70% inhibition at 1 μM (see Supporting Information). The intrafamily lack of selectivity with an ATP-site inhibitor was expected as the ATP-site similarity is >95% between the three kinases (MAP4K4, TNIK, and MINK). Indeed, compound 16 showed a kinase activity IC50 (tested at Km) of 15 and 8 nM for TNIK and MINK, respectively.
Figure 3.

Heat map obtained from an Invitrogen selectivity panel (% inhibition of kinases tested @1 μM).
Compound 16 demonstrated good physicochemical properties as well as moderate human liver microsomal (HLM) clearance (Table 3). Compound 16 had good solubility at both stomach and intestinal pH15 and a reasonably clean DDI profile. It also showed cytotoxicity (THLE assay) only at levels above 119 μM. Thus, 16 was progressed to pharmacokinetic (PK) evaluation in the mouse. Following oral dosing (10 mg/kg), it displayed a plasma exposure that provided free drug concentrations above the cell IC50 value for about 4–6 h after administration (Figure 4). Based on this profile, compound 16 was selected as a fit-for-purpose tool for in vivo pharmacological characterization.
Table 3. Physicochemical/ADME Properties for 16/17.
| properties | 16 | 17 |
|---|---|---|
| MW/eLogD/TPSAa | 296/3.3/78 | 297/2.8/91 |
| HLM Clu (mL/min/kg)b | 89 | 13 |
| solubility, pH 1.2, mg/mL | 13.0 | 3.96 |
| solubility, pH 6.5, mg/mL | 5.8 | 0.002 |
| MDR BA/AB ratioc | 2.7 | 1.1 |
| drug–drug interactions (DDI) | >30 μMd | >30 μMe |
| CYP inhibition IC50 | 3 μM (1A2) | |
| TDI,f % inhibition (dose) | 54% (30 μM) | 8% (30 μM) |
| 61% (60 μM) | 24% (60 μM) | |
| hERG IC50g | 1.8 μM | >10 μM |
| THLE IC50h | >119 μM | >300 μM |
| AMESi | Positive | Negative |
eLogD = measured logD at pH 7.4.
Free clearance calculated using measured protein binding in human liver microsomes (HLM).
MDR BA/AB efflux ratio using MDCK cell line transfected with human MDR1.
<15% Inhibition at 3 μM for isoforms (3A4, 2D6, 2C8, and 2C9).
<15% Inhibition at 3 μM for isoforms (3A4, 2D6, 1A2, 2C8, and 2C9).
Change in midazolam metabolism after incubation with compound.16
Measurement of potassium current in human embryonic kidney 293 (HEK293) cells stably transfected with the human ether-a-go-go-related gene (hERG) channel.
Transformed human liver epithelial cell assay (THLE).17
Activity in a bacterial mutagenesis assay.18
Figure 4.
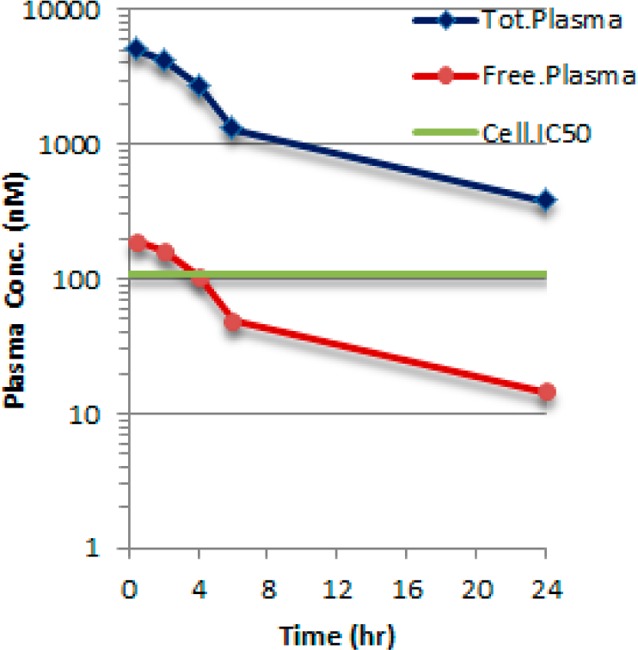
Total and free plasma concentration of 16 after PO dosing (10 mg/kg) to mice. Mouse plasma protein fraction unbound (fu = 0.038).
To recapitulate the in vivo effects demonstrated by MAP4K4-siRNA,4 compound 16 was dosed orally (15 mg/kg) in a mouse model of LPS-induced TNFα levels in blood (compounds were dosed 30 min before an intravenous injection of LPS). Compound 16 showed >90% inhibition (similar to the effect of rosiglitazone) of LPS-induced plasma TNFα levels at 1.5 h with a corresponding plasma concentration of 439 ng/mL (unbound levels of 56.2 nM, which was lower than the cellular IC50).
In order to gain further understanding of the in vivo effect of a MAP4K4 inhibitor, compound 16 and rosiglitazone, a thiazolidinone-based antidiabetic drug, were tested in the ob/ob rodent model of type II diabetes.19,20 Animals were treated with vehicle (n = 12), rosiglitazone (n = 12) at 1 mg/kg, p.o., bid, or 16 (n = 12) at 10 mg/kg, p.o., bid for 4 weeks. Compound 16 (10 mg/kg, bid) provided significant improvement in fasting hyperglycemia (44% reduction in blood glucose, Figure 5), which was comparable to the effect observed with rosiglitazone and also consistent with the profile observed with MAP4K4-siRNA knockdown.5
Figure 5.

(a) Effects of 16 on LPS-induced TNFα levels in C57-BL/6J mice). (b) Effects of 16 on glycemic control in ob/ob mice.
Despite its kinome selectivity and in vivo activity, compound 16 had liabilities that prevented its advancement beyond its utility as a biological tool compound. Compound 16 displayed inhibition in the hERG assay, nonkinase pharmacological promiscuity (Cerep), and demonstrated time-dependent inhibition (TDI)16 of CYP3A4, which led to drug accumulation and toxicity when dosed beyond 10 mg/kg in mice. Toxicity limited dosing to 10 mg/kg bid in chronic pharmacodynamic models, though this was sufficient to provide ∼24 h coverage of cellular IC50 levels. In addition, compound 16 had a mutagenic risk demonstrated by the AMES assay.18
In order to understand the underlying causes of TDI, compound 16 was subjected to incubation with human liver microsomes. These studies showed that oxidation of the distal aminopyridine ring leads to hydroxylation of the ring followed by metabolism to a putative pyridoquinone-imine species that was trapped with glutathione (Supporting Information). Thus, a covalent adduct of the pyridoquinone-imine intermediate with CYP3A4 could result in the observed TDI and may also be contributing to the observed mutagenicity.
Various design strategies to attenuate the observed TDI were explored.21 Addition of substituents ortho to the terminal amine (F, CN CF3) or incorporation of additional nitrogen atoms to the aminopyridine core provided no improvement. However, conversion of the aminopyridyl tail to the corresponding 2-aminopyrimidine (17) retained potency with a good ligand efficiency (LE = 0.53) and kinome selectivity profile (Figure 3), while eliminating the TDI risk (Table 3). Gratifyingly, compound 17 was negative in the AMES assay. Commensurate with the reduced lipophilicity, 17 demonstrated a more desirable physicochemical, metabolic (Table 3), and off-target profile (Supporting Information). Compound 17 thus offered a lead compound for further development of a MAP4K4 inhibitor with an improved overall profile.
Compounds 1–9 were synthesized according to modified literature procedures (Supporting Information). Scheme 1 illustrates the synthesis of the analogues having 3,5-disubstituted-pyridine (or 2-aminopyridine) as the hinge binding group. Commercially available halopyridine 18 underwent sequential Suzuki couplings with a variety of substituted phenylboronic acids or pinacol esters to provide compounds 10–13, 16, and 17. Compounds 14 and 15 were obtained from bromide 19 via Pd-catalyzed borylation to provide a boronate ester 28 that was then coupled with the appropriate halogenated pyridones.
Scheme 1. Synthesis of Pyridines 10–17.

Conditions: (a) 4-ClPhB(OH)2 (21) or (4-SO2Me)PhB(OH)2 (22), Pd(dppf)Cl2, Na2CO3, dioxane, H2O, 110 °C or KOH, PhCH3, EtOH, H2O, 140 °C; (b) (4-CONH2) PhB(OH)2 (23) or PhB(OH)2 (24), (4-OH)PhB(OH)2 (25), or (pyridin-2-NH2)-B2Pin2, (26), or (pyrimidin-2-NH2)Bpin (27); catalyst: Ph(AmPhos)2Cl2, Pd(dppf)Cl2, or Pd(Ph3P)4; base: Na2CO3, K2CO3, Cs2CO3, or KOH; solvent: EtOH, THF, PhCH3, or dioxane, H2O, heat; (c) B2Pin2 (26), Pd(dppf)Cl2, KOAc, dioxane, 60 °C; (d) 5-bromopyridin-2-ol (28) or 4-chloropyridin-2-ol (29), Pd(dppf)Cl2, Na2CO3, dioxane, H2O, 110 °C.
In summary, we describe two 2-aminopyridine leads 16 (PF-6260933)22 and 17 that are potent and selective MAP4K4 inhibitors. Compound 16 had suitable PK properties in mouse to be used as a tool in an in vivo model of diabetes. Compound 17 showed improved overall drug-like properties compared to 16 and thus offered a lead compound for advanced preclinical development. Further studies with these leads will be the subject of future publications.
Acknowledgments
We thank Amit Kalgutkar and Tim Ryder for analysis of the metabolite ID experiments, and John C. Kath, Michael Robbins and Robert G. Linde for early work on this project.
Glossary
ABBREVIATIONS
- MW
molecular weight
- TPSA
topological polar surface area
- HLM
human liver microsomes
- LPS
lipopolysaccharide
- dppf
1,1′-Bis(diphenylphosphino)ferrocene
- B2Pin2
Bis(pinacolato)diboron
- hERG
human ether-a-go-go-related gene
- PK
pharmacokinetic
- TDI
time-dependent inhibition
- MM-GB/SA
molecular mechanics with generalized born/surface area solvation
- ND
not determined
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00215.
Description of synthesis procedures and identification of compounds, experimental protocols for the in vitro and in vivo assays, ActivX results for compound 16 and metabolite ID information, and Cerep promiscuity profiles of compounds 16 and 17 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Yao Z.; Zhou G.; Wang X. S.; Brown A.; Diener K.; Gan H.; Tan T. H. A novel human STE20-related protein kinase, HGK, that specifically activates the c-Jun N-terminal kinase signaling pathway. J. Biol. Chem. 1999, 274, 2118–2225. 10.1074/jbc.274.4.2118. [DOI] [PubMed] [Google Scholar]
- Liang J. J.; Wang H.; Rashid A.; Tan T.-H.; Hwang R. F.; Hamilton S. R.; Abbruzzese J. L.; Evans D. B.; Wang H. Expression of MAP4K4 is associated with worse prognosis in patients with stage II pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2008, 14, 7043–7049. 10.1158/1078-0432.CCR-08-0381. [DOI] [PubMed] [Google Scholar]
- Tang X.; Guilherme A.; Chakladar A.; Powelka A. M.; Konda S.; Virbasius J. V.; Nicoloro S. M. C.; Straubhaar J.; Czech M. P. An RNA interference-based screen identifies MAP4K4/NIK as a negative regulator of PPARγ, adipogenesis, and insulin-responsive hexose transport. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 2087–2092. 10.1073/pnas.0507660103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouadi M.; Tesz G. J.; Nicoloro S. M.; Wang M.; Chouinard M.; Soto E.; Ostroff G. R.; Czech M. P. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature 2009, 458, 1180–1184. 10.1038/nature07774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzakri K.; Zierath J. R. MAP4K4 gene silencing in human skeletal muscle prevents tumor necrosis factor-α-induced insulin resistance. J. Biol. Chem. 2007, 282, 7783–7789. 10.1074/jbc.M608602200. [DOI] [PubMed] [Google Scholar]
- Sartorius T.; Staiger H.; Ketterer C.; Heni M.; Machicao F.; et al. Association of Common Genetic Variants in the MAP4K4 Locus with Prediabetic Traits in Humans. PLoS One 2012, 7, e47647. 10.1371/journal.pone.0047647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndubaku C. O.; Crawford T. D.; Chen H.; Boggs J. W.; Drobnick J.; Harris S. F.; Jesudason R.; McNamara E.; Nonomiya J.; Sambrone A.; Schmidt S.; Smyczek T.; Vitorino P.; Wang L.; Wu P.; Yeung S.; Chen J.; Chen K.; Ding C. Z.; Wang T.; Xu Z.; Gould S. E.; Murray L. J.; Ye W. Structure-Based Design of GNE-495, a Potent and Selective MAP4K4 Inhibitor with Efficacy in Retinal Angiogenesis. ACS Med. Chem. Lett. 2015, 6, 913–918. 10.1021/acsmedchemlett.5b00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Stanley M.; Boggs J. W.; Crawford T. D.; Bravo B. J.; Giannetti A. M.; Harris S. F.; Magnuson S. R.; Nonomiya J.; Schmidt S.; Wu P.; Ye W.; Gould S. E.; Murray L. J.; Ndubaku C. O.; Chen H. Fragment-based identification and optimization of a class of potent pyrrolo[2,1-f][1,2,4]triazine MAP4K4 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 4546–4552. 10.1016/j.bmcl.2014.07.071. [DOI] [PubMed] [Google Scholar]
- Schroder P.; Forster T.; Kleine S.; Becker C.; Richters A.; Ziegler S.; Rauh D.; Kumar K.; Waldmann H. Neuritogenic Militarinone-Inspired 4-Hydroxypyridones Target the Stress Pathway Kinase MAP4K4. Angew. Chem. 2015, 10.1002/ange.201501515. [DOI] [PubMed] [Google Scholar]
- Guimarães C. R. W.; Rai B. K.; Munchhof M. J.; Liu S.; Wang J.; Bhattacharya S. K.; Buckbinder L. Understanding the Impact of the P-loop Conformation on Kinase Selectivity. J. Chem. Inf. Model. 2011, 51, 1199–1204. 10.1021/ci200153c. [DOI] [PubMed] [Google Scholar]
- Guimarães C. R. W.; Cardozo M. MM-GB/SA rescoring of docking poses in structure-based lead optimization. J. Chem. Inf. Model. 2008, 48, 958–970. 10.1021/ci800004w. [DOI] [PubMed] [Google Scholar]
- Abel R.; Young T.; Farid R.; Berne B. J.; Friesner R. A. Role of the Active-Site Solvent in the Thermodynamics of Factor Xa Ligand Binding. J. Am. Chem. Soc. 2008, 130, 2817–2831. 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L.; Whitney-Pickett C.; Umland J. P.; Zhang H.; Zhang X.; Gebhard D. F.; Lai Y.; Federico J. J. 3rd; Davidson R. E.; Smith R.; Reyner E. L.; Lee C.; Feng B.; Rotter C.; Varma M. V.; Kempshall S.; Fenner K.; El-Kattan A. F.; Liston T. E.; Troutman M. D. Development of a new permeability assay using low-efflux MDCKII cells. J. Pharm. Sci. 2011, 100, 4974–4985. 10.1002/jps.22674. [DOI] [PubMed] [Google Scholar]
- Patricelli M. P.; Nomanbhoy T. K.; Wu J.; Brown H.; Zhou D.; Zhang J.; Jagannathan S.; Aban A.; Okerberg E.; Herring C.; Nordin B.; Helge W.; Yang Q.; Lee J. – D.; Gray N.; Kozarich J. W. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol. 2011, 18, 699–710. 10.1016/j.chembiol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma M. V.; Gardner I.; Steyn S. J.; Nkansah P.; Rotter C. J.; Whitney-Pickett C.; Zhang H.; Di L.; Cram M.; Fenner K. S.; El-Kattan A. F. pH-Dependent solubility and permeability criteria for provisional biopharmaceutics classification (BCS and BDDCS) in early drug discovery. Mol. Pharmaceutics 2012, 9, 1199–1212. 10.1021/mp2004912. [DOI] [PubMed] [Google Scholar]
- Orr S. T.; Rip S. L.; Ballard T. E.; Henderson J. L.; Scott D. O.; Obach R. S.; Sun H.; Kalgutkar A. S. Mechanism-Based Inactivation (MBI) of Cytochrome P450 Enzymes: Structure–Activity Relationships and Discovery Strategies To Mitigate Drug–Drug Interaction Risks. J. Med. Chem. 2012, 55, 4896–4933. 10.1021/jm300065h. [DOI] [PubMed] [Google Scholar]
- Benbow J. W.; Aubrecht J.; Banker M. J.; Nettleton D.; Aleo M. D. Predicting safety toleration of pharmaceutical chemical leads: cytotoxicity correlations to exploratory toxicity studies. Toxicol. Lett. 2010, 197, 175–182. 10.1016/j.toxlet.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Aubrecht J.; Osowski J. J.; Persaud P.; Cheung J. R.; Ackerman J.; Lopes S. H.; Ku W. W. Bioluminescent Salmonella reverse mutation assay: a screen for detecting mutagenicity with high throughput attributes. Mutagenesis 2007, 22, 335–342. 10.1093/mutage/gem022. [DOI] [PubMed] [Google Scholar]
- Xu H.; Barnes G. T.; Yang Q.; Tan G.; Yang D.; Chou C. J.; Sole J.; Nichols A.; Ross J. S.; Tartaglia L. A.; Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 2003, 112, 1821–1830. 10.1172/JCI200319451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsusue K.; Haluzik M.; Lambert G.; Yim S.-H.; Gavrilova O.; Ward J. M.; Brewer B. Jr.; Reitman M. L.; Gonzalez F. J. Liver-specific disruption of PPARγ in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Invest. 2003, 111, 737–747. 10.1172/JCI200317223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C.; Pairish M.; Kephart S.; Bouzida D.; Cui J.; Deal J.; Dong L.; Gu D.; Linton A.; McAlpine I.; Yamazaki S.; Smith E.; John-Baptiste A.; Bagrodia S.; Kania R.; Guo C. Structural modifications of a 3-methoxy-2-aminopyridine compound to reduce potential for mutagenicity and time-dependent drug-drug interaction. Bioorg. Med. Chem. Lett. 2012, 22, 7605–7609. 10.1016/j.bmcl.2012.10.011. [DOI] [PubMed] [Google Scholar]
- PF-6260933 is commercially available via Sigma Aldrich (catalog #PZ0272).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.