

Abstract

Oral squamous cell carcinoma (OSCC) is the most common cancer affecting the oral cavity, and US clinics will register about 30,000 new patients in 2015. Current treatment modalities include chemotherapy, surgery, and radiotherapy, which often result in astonishing disfigurement. Cancers of the head and neck display enhanced levels of glucose-regulated proteins and translation initiation factors associated with endoplasmic reticulum (ER) stress and the unfolded protein response (UPR). Previous work demonstrated that chemically enforced UPR could overwhelm these adaptive features and selectively kill malignant cells. The threonyl-tRNA synthetase (ThRS) inhibitor borrelidin and two congeners were discovered in a cell-based chemical genomic screen. Borrelidin increased XBP1 splicing and led to accumulation of phosphorylated eIF2α and UPR-associated genes, prior to death in panel of OSCC cells. Murine embryonic fibroblasts (MEFs) null for GCN2 and PERK were less able to accumulate UPR markers and were resistant to borrelidin. This study demonstrates that UPR induction is a feature of ThRS inhibition and adds to a growing body of literature suggesting ThRS inhibitors might selectively target cancer cells.
Keywords: UPR; borrelidin; Chop; Xbp1, BiP/GRP78; oral cancer; natural products; high throughput screen; ER stress; oral squamous cell carcinoma; protein folding
Patients suffering from oral squamous cell carcinoma (OSCC) continue to have limited treatment options beyond surgery and radiotherapy, and survivors are often left physically disfigured and in of need adjunctive therapy for assistance with basic functions. Our ability to improve disease in these patients chemotherapeutically has not increased substantially since the introduction of cisplatin in 1978. This paucity of treatment options has fueled a worldwide search for therapeutic small molecules and natural products that target a variety of metabolic processes. Recent reports using immunohistochemistry or reverse phase protein arrays have indicated that chaperones, glucose-regulated proteins, and translation factors associated with enhanced endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) are significantly increased and predictive of recurrence in OSCC.1−4
The UPR is a conserved signaling program that facilitates a rapid survival response in the face of cellular stresses that interfere with protein folding or post-translational modification in the ER. PKR-like ER kinase (PERK), inositol-requiring 1-alpha (IRE1α), and activating transcription factor 6 alpha (ATF6α) are ER transmembrane proteins that constantly monitor luminal protein folding. When the demand for folding outpaces the capacity of the ER these sensors initiate the UPR. Activated PERK phosphorylates the alpha subunit of eukaryotic translation initiation factor 2-alpha (eIF2α), which attenuates mRNA translation at the initiation step thereby mediating a halt in general protein synthesis.5−7 The UPR is characterized by this energy-conserving pause in translation and is accompanied by an IRE1α/ATF6-mediated transcriptional increase in the production of chaperones and foldases that return to the ER to improve folding. Most evidence supports the notion that when a stress is robust or protracted, eIF2α phosphorylation induces activating transcription factor 4 (ATF4), which activates transcription of C/EBP homologous protein (CHOP) and directs the cell toward an apoptotic fate.8−10 ATF4 and CHOP appear to function as a heterodimer to activate genes that encode translational machinery and adaptive genes, further increasing protein synthesis and luminal folding burden prior to death.10
Several groups recently reported that many human OSCC cell lines and archived biopsies from head and neck and thyroid cancer patients displayed significantly enhanced levels of the translation factors eIF2α and eIF4e, compared to normal patient controls or adjacent nonmalignant tissue.1,2 These findings support the idea that UPR might be an underlying mechanism by which tumor cells are able to survive harsh microenvironmental stresses (i.e., low oxygen tension and nutrient deprivation).11 The UPR might be an attractive therapeutic target whereby a drug might be delivered systemically and have selective anticancer effects, as it would only be detrimental to cells with increased UPR pressure.
It was previously demonstrated that the proteasome inhibitor Velcade (bortezomib)12 and the natural products patulin,13 celastrol,14 cantharidin,15 and lobophorin16 could induce UPR-dependent cell death in a panel of OSCC cell lines. The UPR activating properties of these natural products was identified using a productive HTS platform that utilized two Chinese hamster ovary (CHO) cell lines that individually reported (luciferase) the activation of Chop or Xbp1 splicing.13 An unique natural product library of organic extracts from marine and terrestrial organisms from biodiverse habitats in Costa Rica was screened at the University of Michigan Center for Chemical Genomics.17 The macrocyclic antibiotic borrelidin was identified from an extract able to activate the Chop reporter. Two congeners of borrelidin that have never been described as natural products were identified in the purification process.
Borrelidin was first isolated in 1949 from Streptomyces rochei(18) and has been evaluated for antibiotic, antimalarial, and anticancer properties of many cell types and animal models. The ability of borrelidin to modulate diverse molecular functions has been described. Endothelial cells cultured with borrelidin underwent caspase-mediated cell death leading to capillary tube collapse;19,20 and yeast and acute lymphoblastic leukemia cells treated with borrelidin experienced amino acid deprivation-induced GCN4 and GCN2 activation, respectively,21,22 prior to cell death. The ability of borrelidin to impair proliferation and modulate translation in bacterial and mammalian cells has been reliably attributed to caspase activation and noncompetitive threonyl-tRNA synthetase (ThRS) inhibition.23 In support of this notion it was reported that borrelidin-resistant CHO cells had 10–20-fold higher ThRS activity than cocultured borrelidin-sensitive cells.24 Having been identified as a molecule that could activate Chop- and Xbp1-luciferase reporters, and predicated on the knowledge that it could perturb protein synthesis, it was hypothesized that activation of the UPR might be a mechanism by which borrelidin exerts its cytotoxic effect.
A cell-based high throughput screen (HTS) that has identified novel UPR-inducing small molecules and natural product hits was previously described.13,25 Iterative bioassay-guided C18 fractionation and RP-18 HPLC purification of previously reported UPR-inducing natural extracts13 identified the known 18-membered macrocyclic polyketide borrelidin 1 and two amide-containing congeners designated CR1 2 and CR2 3 (Figure 1). 2 was previously described.26 Compound 3 was also isolated from the same RP-C18 fraction containing congeners 1 and 2. The HRESIMS of the molecule provided a molecular formula of C30H50N2O6 showing a [M + Na]+ ion peak at m/z 557.1566 (Figure S13). Compound 3 had a high structural similarity to both 1 and 2, as evidenced by nearly identical 1H and 13C NMR chemical shifts when measured in CD3OD (Table S4). However, the chemical formula suggested only seven degrees of unsaturation, compared to the eight in 2, and the loss of δC 120.5 suggested the absence of a nitrile group. Furthermore, COSY and HMBC correlation of H-11 to a carbonyl group at 172.6, as well as presence of a 1H signal at δH 1.91, suggested the substitution of a nitrile with a terminal acetyl amide (Figures S14–S17). The amide being connected to C-12 via a methylene (δH 3.65, δC 39.4) showed HMBC correlation to C-23 and therefore completed the planar structure of 3. The configurations of eight stereocenters in both congeners 2 and 3 were predicted to be the same as 1 based on the very comparable chemical shifts and coupling constants (Table S4).
Figure 1.

Borrelidin congeners.
Treatment of UMSCC1 cultures with each purified borrelidin 1–3 revealed that the nonamide parent molecule 1, and to a lesser extent 2, induced mRNA transcripts for CHOP, ATF3, and ATF4 in OSCC, which are required for stress-mediated cell death, but did not induce cytoprotective BiP/GRP78 mRNA (Figure 2A). Treatment with 1 and 2 led to modest XBP1 splicing in the same cell line (Figure 2 B), which is a hallmark of ER stress. The ability of 1 and 2 to reduce proliferation coincided with the level of UPR induction; 3 could neither activate UPR nor reduce proliferation (Figure 2C−E). The very modest accumulation of BiP/GRP78 and spliced XBP1 observed after 6 h suggests that borrelidin might preferentially activate the cell death arm of the UPR. Normal human epidermal keratinocytes (nHEK) treated with borrelidin demonstrated similar IC50 values (not shown) as OSCC cells; however, concerns about toxicity in nonmalignant cells in culture are attenuated by in vivo studies demonstrating that malaria infected mice treated with 0.25 mg/kg borrelidin daily recovered from disease and developed durable immunity.27 Gene expression and proliferation assays performed with a panel of leukemia cell lines revealed that the ability of borrelidin to increase UPR and cell death mRNA transcripts, activate caspases, and reduce proliferation was not a phenomenon unique to OSCC (Figure S1). A recent study reported that a tyrosine residue at position 313 of ThRS interacts solely with the cyanide moiety of borrelidin for effective binding and inhibition.28 This finding is bolstered by the current observation that as the substitution of a bulky yet flexible N-methylacetylamide group in 3 against the sturdy and polar nitrile group in 2 compromised UPR activation and the antiproliferative activity in OSCC (Figure 2). This finding provides a crucial lead into structure activity modulation of the borrelidin core scaffold for any future medicinal chemistry enhancement to the molecule. The current findings that 3 could not splice XBP1, increase CHOP, nor inhibit OSCC proliferation are a strong indication that ThRS inhibition is a mechanism by which borrelidin upregulated the UPR.
Figure 2.

(A) RT-qPCR analysis of UMSCC1 treated with 5 μM (1–3) 6 h. (B) RT-PCR, same samples to appreciate XBP1. (C–E) OSCC proliferation assays treated with 1–3 for 24 h (two-way ANOVA; p < 0.0001 for dose and interaction).
To address the paucity of mechanistic detail in the literature, UPR, DNA damage, and apoptosis quantitative RT2 Profiler PCR Arrays were performed with cDNA pools generated from UMSCC1 treated with 1, 2, and 3 (10 μM). Increased transcripts were observed in each array for samples treated with 1 but not with 2 or 3 (Tables S1–S3). Two stocks of borrelidin (derived from Streptomyces parvulus, hereafter, referred to as “borrelidin” to distinguish from extract-derived 1) were purchased from Sigma and used for the balance of nongene array studies. Proliferation assays and quantitative reverse transcription (RT-qPCR) analyses with a panel of OSCC cell lines validated each stock reduced proliferation and induced UPR gene expression similar to 1 (Figures S2 and S3). To confirm and extend the array data, RT-qPCR analysis of apoptotic mRNA transcripts was performed. Notably, the UPR-associated cell death genes TRB3, NOXA, PUMA, and to a lesser extent DR5 were induced (Figure S4). Time-course luminescent caspase 3/7 assays demonstrated the presence of active caspase enzymes as early as 4 h after treatment (Figure 3A), and immunoblot analysis revealed an accumulation of the cleaved (active form) caspases 9 and 3 and fragmented PARP (Figure 3B). These results are similar to previous observations demonstrating borrelidin-induced apoptosis in rat aorta cultures and human umbilical vein endothelial cells.19 Electrophoretic resolution of genomic DNA revealed nucleosome-sized DNA fragments, a hallmark of apoptotic cell death, occurred between 16 and 36 h (Figure 3C). Human alveolar basal epithelial cells rendered doubly deficient for BAX and BAK using Zinc Finger Nuclease-mediated genome editing were significantly more resistant than parental controls (Figure 3D). Considered together these findings implicate apoptosis as a major contributor in the ability of borrelidin to reduce cancer cell proliferation. Although the quantitative DNA damage array identified 16 DNA damage-associated genes to be upregulated by 1 (Table S2), DNA damage could not be detected with a COMET assay with doses of borrelidin up to 20 μM between 0 and 48 h (data not shown).
Figure 3.
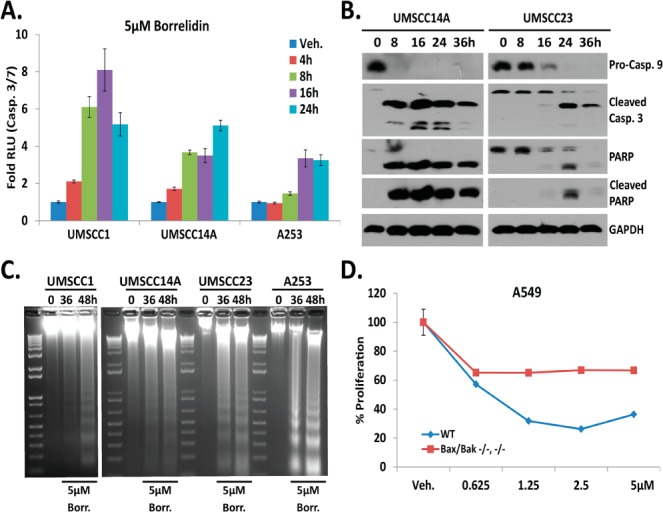
(A) Luminescent caspase 3/7 assay. (B) Immunoblot analysis of 5 μM borrelidin. (C) Electophoretic resolution of genomic DNA. (D) Proliferation assay with BAX(−/–/–/–/) BAK(−/−) A549 cells at 16 h (two-way ANOVA, p value for dose < 0.0001, interaction 0.0016, and between wildtype and knockout <0.0001).
To elucidate the precise role of the UPR in borrelidin-mediated apoptosis, a panel of murine embryonic fibroblasts (MEFs) null for key UPR and stress signaling proteins (Perk, Gcn2, Hri, Pkr, Chop, and Atf4), and wildtype (wt) littermate controls was employed. Proliferation assays revealed no difference in cell growth between Hri- and Pkr-null and wt MEFs (data not shown). Perk-deleted cells were significantly resistant and immunoblot analysis of whole cell lysates revealed similar levels of phosphorylated eIF2α and Chop (Figure 4A). General control nonrepressed 2 kinase (Gcn2)-null cells were similarly protected; however, neither phosphorylation of eIF2α nor accumulation of Chop occurred in the absence of Gcn2 (Figure 4B), consistent with previous findings.29 Borrelidin-resistant Perk-null cells accumulated significantly fewer Noxa transcripts, and Gcn2-null cells were significantly less able to accumulate Noxa and Puma, and the UPR-associated death genes Gadd45β, Trb3, and Dr5 (Figure 4C). Phosphorylation of eIF2α by Perk and Gcn2 occurs during stress to attenuate protein synthesis and conserve energy to afford the cell an opportunity for recovery. MEFs with a Ser51Ala mutation at the phosphorylation site in eIF2α cannot undergo this critical translational pause and are exquisitely sensitive to ER stress. Consistent with this notion, A/A MEFs were significantly more sensitive to borrelidin than wt (S/S) (Figure 4D). Chop-null MEFs were also more resistant than wt cells (Figure 4 E), consistent with the hypothesis that Chop accumulation is required for UPR-mediated cell death. Although Atf4-null cells were also resistant to borrelidin and demonstrated reduced expression of cell death genes (Figure S5), we could not appreciate ATF4 protein accumulation in any MEF or OSCC cells, for reasons that are not clear.
Figure 4.

(A, left) Proliferation assay with wildtype (wt) and Perk-null MEF treated with borrelidin (Bor) (two-way ANOVA, p values for dose <0.0001, interaction 0.0088, and between wt and Perk-null <0.0001); (right) immunoblot analysis 5 μM Bor. (B, left) proliferation assay with wt and Gcn2-null MEF treated with Bor 16 h (two-way ANOVA p values for dose <0.0001, interaction 0.0009, and between wt and Gcn2-null <0.0001); (right) immunoblot analysis of 5 μM Bor. (C) RT-qPCR of cell death transcripts in wt and Perk-null MEF (left) and wt and Gcn2-null MEF (right). (D) Proliferation assays with eIF2α wt (S/S) and mutant (A/A) MEF 16 h. (E) Proliferation assays with wt and Chop-null MEF 16 h (two-way ANOVA p values for dose, interaction, and between wildtype and Chop-null <0.0001).
While our studies were underway it was reported that borrelidin could induce eIF2α phosphorylation, CHOP accumulation, and death in lymphoblastic leukemia cells via the GCN2 stress pathway.29 The current work represents the first stepwise approach to determine the mechanism of eIF2α phosphorylation and CHOP activation by borrelidin. Four kinases, PERK, PKR, GCN2, and HRI, serve as stress sensors and initiate signaling through eIF2α phosphorylation.30,31 Using a MEF model system the data demonstrated Perk- and Gcn2-null cells are less sensitive to borrelidin than wildtype MEFs and that the rate of death in Pkr- and Hri-null cells is indistinguishable from controls. Considered with the current finding that borrelidin led to the splicing of XBP1 mRNA, a feature unique to the induction of ER stress, this work establishes for the first time that borrelidin-induced CHOP accumulation and cell death can operate through both Gcn2 and Perk signaling. The fact that eIF2α phosphorylation was only attenuated in PERK-deficient cells (vis à vis being absent in Gcn2 null cells) might be an indication that amino acid deprivation is a predominant mechanism or that Perk deficient MEFs have acquired a compensatory mechanism (i.e., increased Gcn2 levels) during selection. ATF4 accumulation could not be detected in whole cell lysates or cytosolic and nuclear extracts of borrelidin-treated cells, consistent with a previous report.22 The absence of ATF4 protein suggests an unknown transcription factor (e.g., ATF5 or ER-resident ATF6) might be driving CHOP expression. This work provides the first demonstration that transcriptional activation of CHOP, downstream of eIF2α phosphorylation, is required for borrelidin to efficiently exert a cytotoxic affect.
A recent study reported a possible binding site for borrelidin on ThRS and suggested the tyrosine residue 313 in the binding pocket is critical for borrelidin binding. The study demonstrated that Y313 interacted solely with the cyanide moiety of borrelidin for effective binding in the pocket.28 Another study reported that borrelidin sits deep within a highly conserved region of the binding pocket and interacts with ThRS from multiple directions. The absence of van der Waals contacts between borrelidin and Q566, L567, S386, and 12′ cyano groups in Archaeal ThRS could contribute to borrelidin-resistance.32 These studies support our finding that the replacement of a cyano group with N-methylacetylamide could lead to intramolecular hydrogen bonding and profoundly affect the hydrophobic interaction. Furthermore, intramolecular hydrogen bonding in CR2 would also disturb the hydrogen bond interaction between OH-11 and D564 and perturb a key borrelidin-ThRS interaction and causing a loss of activity. In summary, the novel substitutions identified in borrelidin CR 2 3 at key interacting ThRS residues, and its loss of UPR-inducing activity, provides an important clue for any future medicinal chemistry enhancement of the molecule.
Although studies have suggested that borrelidin might possess value as an anticancer agent, more target-specific and less-toxic derivatives will need to be identified.33 While borrelidin may not be a tractable drug lead, our work supports the notion that ThRS inhibition and UPR induction might be a productive approach to cancer therapy. In this comparative study of three related borrelidin structures, dramatic loss of biological activity was observed in the amide congeners, corresponding with their reported ability to inhibit ThRS. This indicates that modest functional group modifications can dramatically influence biological responses to borrelidin, and provides further motivation to explore detailed SAR.
Experimental Procedures
Isolation of borrelidin and congeners. The natural product extracts from which 1, 2, and 3 were identified are from a collection of cultivated marine microorganisms as part of the Costa Rica International Cooperative Biodiversity Group. For isolation procedure including culture maintenance, fermentation, and spectral characterization, see Supporting Information.
Acknowledgments
We thank the Technical Office, CONAGEBIO, Ministry of the Environment and Telecommunications, Costa Rica for providing sample collection permits.
Glossary
ABBREVIATIONS
- ATF4
activating transcription factor 4
- CHOP
C/EBP homologous protein
- eIF2α
eukaryotic initiation factor 2-α
- Noxa
Phorbol-12-myristate-13-acetate-induced protein 1
- Puma
p53 upregulated modulator of apoptosis
- Trb3
tribbles homologue three
- XBP1
X-box binding protein 1
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00133.
Experimental procedures for cell-based assays, including statistical analyses; and the methods used for isolation, purification, and analytic characterization of 1–3 (PDF)
Author Contributions
¶ These authors contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Funding was provided by NIH DE019678, Children’s Research Foundation of Michigan (to A.M.F.), Hyundai Hope on Wheels, Detroit Country Day Men’s Lacrosse Team (to A.M.F. and M.U.C), International Cooperative Biodiversity Groups initiative (U01 TW007404) at the Fogarty International Center (to G.T.-C. and D.H.S.), Hans W. Vahlteich Professorship (to D.H.S.), and DK042394, DK088227, and HL052173 (to R.J.K.).
The authors declare no competing financial interest.
Supplementary Material
References
- Nathan C. O.; Liu L.; Li B. D.; Abreo F. W.; Nandy I.; De Benedetti A. Detection of the proto-oncogene eIF4E in surgical margins may predict recurrence in head and neck cancer. Oncogene 1997, 15, 579–584. 10.1038/sj.onc.1201216. [DOI] [PubMed] [Google Scholar]
- Chandy B.; Abreo F.; Nassar R.; Stucker F. J.; Nathan C. O. Expression of the proto-oncogene eIF4E in inflammation of the oral cavity. Otolaryngol.--Head Neck Surg. 2002, 126, 290–295. 10.1067/mhn.2002.123104. [DOI] [PubMed] [Google Scholar]
- Slotta-Huspenina J.; Berg D.; Bauer K.; Wolff C.; Malinowsky K.; Bauer L.; Drecoll E.; Bettstetter M.; Feith M.; Walch A.; Hofler H.; Becker K. F.; Langer R. Evidence of prognostic relevant expression profiles of heat-shock proteins and glucose-regulated proteins in oesophageal adenocarcinomas. PLoS One 2012, 7, e41420. 10.1371/journal.pone.0041420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C. Y.; Chen W. H.; Liao C. T.; Chen I. H.; Chiu C. C.; Wang H. M.; Yen T. C.; Lee L. Y.; Chang J. T.; Cheng A. J. Positive association of glucose-regulated protein 78 during oral cancer progression and the prognostic value in oral precancerous lesions. Head Neck 2010, 32, 1028–1039. 10.1002/hed.21287. [DOI] [PubMed] [Google Scholar]
- Prostko C. R.; Brostrom M. A.; Brostrom C. O. Reversible phosphorylation of eukaryotic initiation factor 2 alpha in response to endoplasmic reticular signaling. Mol. Cell. Biochem. 1993, 127–128, 255–265. 10.1007/BF01076776. [DOI] [PubMed] [Google Scholar]
- Brostrom C. O.; Prostko C. R.; Kaufman R. J.; Brostrom M. A. Inhibition of translational initiation by activators of the glucose-regulated stress protein and heat shock protein stress response systems. Role of the interferon-inducible double-stranded RNA-activated eukaryotic initiation factor 2alpha kinase. J. Biol. Chem. 1996, 271, 24995–25002. 10.1074/jbc.271.40.24995. [DOI] [PubMed] [Google Scholar]
- Scheuner D.; Song B.; McEwen E.; Liu C.; Laybutt R.; Gillespie P.; Saunders T.; Bonner-Weir S.; Kaufman R. J. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 2001, 7, 1165–1176. 10.1016/S1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Harding H. P.; Zhang Y.; Zeng H.; Novoa I.; Lu P. D.; Calfon M.; Sadri N.; Yun C.; Popko B.; Paules R.; Stojdl D. F.; Bell J. C.; Hettmann T.; Leiden J. M.; Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. 10.1016/S1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Rutkowski D. T.; Arnold S. M.; Miller C. N.; Wu J.; Li J.; Gunnison K. M.; Mori K.; Sadighi Akha A. A.; Raden D.; Kaufman R. J. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006, 4, e374. 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J.; Back S. H.; Hur J.; Lin Y. H.; Gildersleeve R.; Shan J.; Yuan C. L.; Krokowski D.; Wang S.; Hatzoglou M.; Kilberg M. S.; Sartor M. A.; Kaufman R. J. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Kaufman R. J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- Fribley A.; Zeng Q.; Wang C. Y. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol. Cell. Biol. 2004, 24, 9695–9704. 10.1128/MCB.24.22.9695-9704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fribley A. M.; Cruz P. G.; Miller J. R.; Callaghan M. U.; Cai P.; Narula N.; Neubig R. R.; Showalter H. D.; Larsen S. D.; Kirchhoff P. D.; Larsen M. J.; Burr D. A.; Schultz P. J.; Jacobs R. R.; Tamayo-Castillo G.; Ron D.; Sherman D. H.; Kaufman R. J. Complementary cell-based high-throughput screens identify novel modulators of the unfolded protein response. J. Biomol. Screening 2011, 16, 825–835. 10.1177/1087057111414893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fribley A. M.; Miller J. R.; Brownell A. L.; Garshott D. M.; Zeng Q.; Reist T. E.; Narula N.; Cai P.; Xi Y.; Callaghan M. U.; Kodali V.; Kaufman R. J. Celastrol induces unfolded protein response-dependent cell death in head and neck cancer. Exp. Cell Res. 2015, 330, 412–422. 10.1016/j.yexcr.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y.; Garshott D. M.; Brownell A. L.; Yoo G. H.; Lin H. S.; Freeburg T. L.; Yoo N. G.; Kaufman R. J.; Callaghan M. U.; Fribley A. M. Cantharidins Induce ER Stress and a Terminal Unfolded Protein Response in OSCC. J. Dent. Res. 2015, 94, 320. 10.1177/0022034514559376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz P. G.; Fribley A. M.; Miller J. R.; Larsen M. J.; Schultz P. J.; Jacob R. T.; Tamayo-Castillo G.; Kaufman R. J.; Sherman D. H. Novel Lobophorins Inhibit Oral Cancer Cell Growth and Induce Atf4- and Chop-Dependent Cell Death in Murine Fibroblasts. ACS Med. Chem. Lett. 2015, 6, 877–881. 10.1021/acsmedchemlett.5b00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M. J.; Larsen S. D.; Fribley A.; Grembecka J.; Homan K.; Mapp A.; Haak A.; Nikolovska-Coleska Z.; Stuckey J. A.; Sun D.; Sherman D. H. The role of HTS in drug discovery at the University of Michigan. Comb. Chem. High Throughput Screening 2014, 17, 210–230. 10.2174/1386207317666140109121546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger J.; Jampolsky L. M.; Goldberg M. W. Borrelidin, a new antibiotic with antiborrelia activity and penicillin enhancement properties. Arch Biochem 1949, 22, 476–478. [PubMed] [Google Scholar]
- Kawamura T.; Liu D.; Towle M. J.; Kageyama R.; Tsukahara N.; Wakabayashi T.; Littlefield B. A. Anti-angiogenesis effects of borrelidin are mediated through distinct pathways: threonyl-tRNA synthetase and caspases are independently involved in suppression of proliferation and induction of apoptosis in endothelial cells. J. Antibiot. 2003, 56, 709–715. 10.7164/antibiotics.56.709. [DOI] [PubMed] [Google Scholar]
- Wakabayashi T.; Kageyama R.; Naruse N.; Tsukahara N.; Funahashi Y.; Kitoh K.; Watanabe Y. Borrelidin is an angiogenesis inhibitor; disruption of angiogenic capillary vessels in a rat aorta matrix culture model. J. Antibiot. 1997, 50, 671–676. 10.7164/antibiotics.50.671. [DOI] [PubMed] [Google Scholar]
- Eastwood E. L.; Schaus S. E. Borrelidin induces the transcription of amino acid biosynthetic enzymes via a GCN4-dependent pathway. Bioorg. Med. Chem. Lett. 2003, 13, 2235–2237. 10.1016/S0960-894X(03)00406-2. [DOI] [PubMed] [Google Scholar]
- Schulze C. J.; Bray W. M.; Loganzo F.; Lam M. H.; Szal T.; Villalobos A.; Koehn F. E.; Linington R. G. Borrelidin B: isolation, biological activity, and implications for nitrile biosynthesis. J. Nat. Prod. 2014, 77, 2570–2574. 10.1021/np500727g. [DOI] [PubMed] [Google Scholar]
- Hutter R.; Poralla K.; Zachau H. G.; Zahner H. [Metabolic products of microorganisms. 5l. On the mechanism of action of borrelidin-inhibition of the threonine incorporation in sRNA]. Biochem Z. 1966, 344, 190–196. [PubMed] [Google Scholar]
- Gerken S. C.; Arfin S. M. Chinese hamster ovary cells resistant to borrelidin overproduce threonyl-tRNA synthetase. J. Biol. Chem. 1984, 259, 9202–9206. [PubMed] [Google Scholar]
- Flaherty D. P.; Golden J. E.; Liu C.; Hedrick M.; Gosalia P.; Li Y.; Milewski M.; Sugarman E.; Suyama E.; Nguyen K.; Vasile S.; Salaniwal S.; Stonich D.; Su Y.; Mangravita-Novo A.; Vicchiarelli M.; Smith L. H.; Diwan J.; Chung T. D. Y.; Pinkerton A. B.; Aube J.; Miller J. R.; Garshott D. M.; Callaghan M. U.; Fribley A. M.; Kaufman R. J.. Selective Small Molecule Activator of the Apoptotic Arm of the UPR. In Probe Reports from the NIH Molecular Libraries Program; National Institutes of Health: Bethesda, MD, 2010. [PubMed] [Google Scholar]
- Novoa E. M.; Camacho N.; Tor A.; Wilkinson B.; Moss S.; Marin-Garcia P.; Azcarate I. G.; Bautista J. M.; Mirando A. C.; Francklyn C. S.; Varon S.; Royo M.; Cortes A.; Ribas de Pouplana L. Analogs of natural aminoacyl-tRNA synthetase inhibitors clear malaria in vivo. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E5508–5517. 10.1073/pnas.1405994111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azcarate I. G.; Marin-Garcia P.; Camacho N.; Perez-Benavente S.; Puyet A.; Diez A.; Ribas de Pouplana L.; Bautista J. M. Insights into the preclinical treatment of blood-stage malaria by the antibiotic borrelidin. Br. J. Pharmacol. 2013, 169, 645. 10.1111/bph.12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Zhang J.; Liu C.; Fang B.; Wang X.; Xiang W. Identification of borrelidin binding site on threonyl-tRNA synthetase. Biochem. Biophys. Res. Commun. 2014, 451, 485–490. 10.1016/j.bbrc.2014.07.100. [DOI] [PubMed] [Google Scholar]
- Habibi D.; Ogloff N.; Jalili R. B.; Yost A.; Weng A. P.; Ghahary A.; Ong C. J. Borrelidin, a small molecule nitrile-containing macrolide inhibitor of threonyl-tRNA synthetase, is a potent inducer of apoptosis in acute lymphoblastic leukemia. Invest. New Drugs 2012, 30, 1361–1370. 10.1007/s10637-011-9700-y. [DOI] [PubMed] [Google Scholar]
- Ron D.; Zhang Y.; Harding H. P. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Wek R. C.; Jiang H. Y.; Anthony T. G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. 10.1042/BST0340007. [DOI] [PubMed] [Google Scholar]
- Fang P.; Yu X.; Jeong S. J.; Mirando A.; Chen K.; Chen X.; Kim S.; Francklyn C. S.; Guo M. Structural basis for full-spectrum inhibition of translational functions on a tRNA synthetase. Nat. Commun. 2015, 6, 6402. 10.1038/ncomms7402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhikshapathi D. V. R. N.; Kumar Y. S.; Rao Y. M.; Kishan V. Borrelidin: A prospective drug. Indian Journal of Biotechnology 2010, 9, 18–23. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.