

Abstract

Several new mercaptoacetamides were synthesized and studied as HDAC6 inhibitors. One compound, 2b, bearing an aminoquinoline cap group, was found to show 1.3 nM potency at HDAC6, with >3000-fold selectivity over HDAC1. 2b also showed excellent efficacy at increasing tubulin acetylation in rat primary cortical cultures, inducing a 10-fold increase in acetylated tubulin at 1 μM. To assess possible therapeutic effects, compounds were assayed for their ability to increase T-regulatory (Treg) suppressive function. Some but not all of the compounds increased Treg function, and thereby decreased conventional T cell activation and proliferation in vitro.
Keywords: HDAC6-selective inhibitors; mercaptoacetamides; 8-aminoquinoline; 1,2,3,4-tetrahydroquinoline; Treg
Post-translational modifications of histones, the proteins around which DNA is wrapped, play a key role in regulating chromatin structure and thereby control biological events such as gene expression and DNA repair. The covalent modifications that occur on histone tails include acetylation, methylation, ubiquitinylation, phosphorylation, sumoylation, and ADP ribosylation.1 These epigenetic modifications have led to significant interest in the discovery of small molecules able to modulate the responsible enzymes. We are interested in histone acetylation and deacetylation, which are reversible processes regulated by histone/protein acetyltransferases (HATs) and histone/protein deacetylases (HDACs).2 HDACs are a family of 11 zinc-dependent enzymes that are divided into 4 groups. Class I includes HDACs 1, 2, 3, and 8; class IIa includes HDACs 4, 5, 7, and 9; class IIb includes HDACs 6 and 10; and class IV contains only HDAC 11.3 HDACs specifically remove acetyl groups from lysine residues on target proteins, thereby regulating their function. Lysine acetylation is implicated in many cellular processes, such as cell cycle regulation, cytoskeleton dynamics, and cell motility.
Much attention has focused in recent years on the important roles that HDACs play in carcinogenesis, including regulation of the expression of genes involved in cell proliferation, cell-cycle regulation, and apoptosis.4−6 Several HDACs are overexpressed in various tumor types, further validating them as important therapeutic targets.4,5 In addition to their application in oncology, histone deacetylase inhibitors (HDACIs) can exhibit neuroprotective effects in cellular and animal models of acute and chronic neurodegenerative injury and disease,7,8 including traumatic brain injury, stroke, and Alzheimer’s disease (AD). Furthermore, valproic acid, a weak HDAC inhibitor, is widely used clinically in the control of bipolar disorder and epilepsy.9
At present, over 20 HDACIs have entered clinical studies, and several are already on the market. The latter include a hydroxamic acid derivative, suberoylanilide hydroxamic acid (SAHA, vorinostat, Zolinza), and a depsipeptide, FK228 (romidepsin, Istodax), that were approved for the treatment of cutaneous T-cell lymphoma; chidamide (Epidaza) which was developed in China for use in refractory peripheral T-cell lymphoma; and LBH-589 (panobinostat, Farydak), which was recently approved for use in patients with multiple myeloma. In addition, ACY-1215 (ricolinostat) is an HDAC6 selective inhibitor currently being evaluated in clinical trials (Figure 1).10,11
Figure 1.

Structures of several known pan-HDACIs and ricolinostat, an HDAC6-selective inhibitor in clinical trials.
As a result of this clinical potential, the design of novel HDACIs continues to attract the interest of medicinal chemists. The pharmacophore of a typical HDACI involves a cap group that interacts with the surface of the enzyme, a linker that occupies a hydrophobic channel, and a metal chelator that coordinates with the zinc ion at the bottom of the catalytic pocket.12,13 Many first generation HDACIs, like SAHA, are pan-inhibitory, i.e., they have little, if any, isoform selectivity. Additionally, many HDACIs, such as trichostatin A (TSA, Figure 1) and SAHA, contain a hydroxamic acid function as the zinc-binding group (ZBG). Unfortunately, hydroxamates are metabolically unstable (SAHA has a half-life of 1.5–2 h in humans when administered orally), and their potent metal-chelating ability can lead to off-target activity at other zinc-containing enzymes.14,15 In addition, many hydroxamic acid based inhibitors have been shown to be Ames-positive and to cause chromosomal aberrations, thus linking them to the potential for genotoxicity.16 Accordingly, alternative ZBGs, such as mercaptoacetamides, may be preferable depending on the therapeutic goal.17,18 Certainly, the application of an HDACI to chronic disorders such as certain CNS diseases would require that the compound does not cause genotoxicity.
In addition to their effects on histones, many HDACs, including HDAC6, act on non-histone proteins. Thus, HDAC6 participates in the deacetylation of α-tubulin, cortactin, and HSP90, and thereby regulates important biological processes, including microtubule stability and function, and cell motility.19 HDAC6 has emerged as an attractive target for drug development, as its inhibition is believed to offer potential therapy for cancer and many neurodegenerative conditions, including spinal cord injury.20 Only a few highly selective HDAC6 inhibitors have been reported to date. For instance, tubacin (Figure 1) was developed over 10 years ago and has been extensively used in various disease models to validate HDAC6 as a therapeutic target.21 However, its high lipophilicity and tedious synthesis limits its use as a drug. For this reason, we developed tubastatin A (Figure 1), a more potent hydroxamic acid based HDAC6 inhibitor.22 Despite the fact that these small molecules are of great interest as chemical tools for probing the biological function of HDAC6,22,23 it is now clear that, in order to minimize undesirable side effects, there is a need to identify druggable, isoform-selective inhibitors that are free of certain safety concerns associated with the hydroxamate class of HDACIs.12
Within this context, we considered it worthwhile to further investigate the potency, isoform selectivity, and biological effects of additional mercaptoacetamide-based inhibitors. We and others have previously reported on mercaptoacetamide-based HDACIs that exhibit HDAC6 selectivity combined with promising therapeutic profiles compared to their respective hydroxamic acid analogues.7,24 In particular, compound 1 (Figure 2) was found to regulate cell surface levels of the amyloid precursor protein (APP) and to modulate the levels of Aβ synthesis and Aβ degradation enzymes. Aβ peptides are neurotoxic products, mainly related to AD, that are released following the cleavage of APP by β- and γ-secretases. AD mice treated with compound 1 had decreased brain Aβ levels, decreased tau phosphorylation, and improved learning and memory.24 Additionally, a related mercaptoacetamide was found to decrease the inflammatory response in the brain of animals subjected to traumatic brain injury.25
Figure 2.
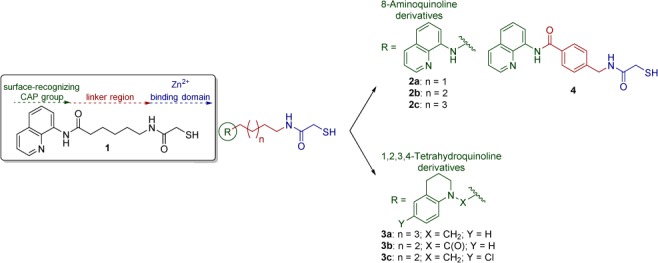
Mercaptoacetamide-based HDACIs presented in this work, containing 8-aminoquinoline and 1,2,3,4-tetrahydroquinoline as the cap groups.
One possible drawback of thiol-containing HDACIs is their ability to undergo oxidative dimerization to form disulfides. Thus, the rate of dimerization of the thiol-based compounds must be considered when profiling the activity of these inhibitors against the purified HDAC proteins. It is furthermore possible that the disulfide bond of an oxidized HDACI could be reduced by the high levels of glutathione present inside of cells to restore the parent thiol.
Given these considerations, we carried out additional chemistry and biology on mercaptoacetamides structurally related to compound 1 (Figure 2). Herein we describe the synthesis, HDAC inhibitory potency, and selectivity of these compounds.
All new ligands prepared contain four to seven CH2 units within the linker region, as this appears to be the optimal length for small molecules to fit into the binding pocket of HDAC6.26 The choice of cap groups was based on the structure of compound 1 (Figure 2) and of other HDACIs previously explored by us.
The mercaptoacetamides 2a–c were synthesized starting from N-protected amino alcohols of varying lengths (5a–c, Scheme 1). Their conversion into the corresponding alkyl halides (6a–c) took place under mild conditions in the presence of triphenylphosphine (PPh3) and tetrabromomethane (CBr4). 8-Aminoquinoline was alkylated with 6a–c under microwave (μW) irradiation to give intermediates 7a–c, respectively. The Boc protecting groups were removed with trifluoroacetic acid (TFA), and then the free amines were subjected to a coupling reaction with 2-(tritylthio)acetic acid in the presence of benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP). Removal of the trityl protecting groups in the presence of TFA and triethylsilane (Et3SiH) afforded the final mercaptoacetamides 2a–c.
Scheme 1. Synthesis of Compounds 2a–c and 3a,b.

Reagents and conditions: (a) PPh3, CBr4, THF, 0 °C to rt, 4 h, 78–95% yield; (b) 8-aminoquinoline, Cs2CO3, DMF, μW, 120 °C, 40 min, 14–17% yield; (c) TFA, CH2Cl2, rt, 2 h, 54–98% yield; (d) 2-(tritylthio)acetic acid, PyBOP, Et3N, CH2Cl2, rt, 20 h, 56–89% yield; (e) TFA, Et3SiH, CH2Cl2, 0 °C to rt, 2 h, 51–88% yield; (f) N,O-dimethylhydroxylamine hydrochloride, EDC, DMAP, Et3N, rt, overnight, 90% yield; (g) LiAlH4, THF, −78 to 0 °C, 10 min, 94% yield; (h) 1,2,3,4-tetrahydroquinoline, NaBH(OAc)3, CH3CO2H, 1,2-dichloroethane, rt, overnight, 21% yield; (i) 1,2,3,4-tetrahydroquinoline, PyBOP, DIPEA, DMF, rt, overnight, 61% yield.
The mercaptoacetamide 3a, containing a tetrahydroquinoline (THQ) cap, was obtained from coupling of N-Boc-7-aminoheptanoic acid (10, Scheme 1) with N,O-dimethylhydroxylamine hydrochloride to give the Weinreb amide 11, which was reduced to aldehyde 12 with LiAlH4. Subsequently, reductive amination of intermediate 12 with the THQ cap afforded 13. Next, Boc deprotection, amide formation in the presence of PyBOP, and removal of the trityl group gave mercaptoacetamide 3a.
Compound 3b was synthesized through coupling of the THQ cap moiety with N-Boc-6-aminohexanoic acid (16). The resulting amide was then taken through the same three final steps described above for analogue 3a.
The same synthetic path used for compound 3a led to analogue 3c, introducing the 6-chloro-1,2,3,4-tetrahydroquinoline core in the cap-linker reductive amination step (Scheme 2).
Scheme 2. Synthesis of Compounds 3c and 4.

Reagents and conditions: (a) N,O-dimethylhydroxylamine hydrochloride, EDC, DMAP, Et3N, rt, overnight, quantitative yield; (b) LiAlH4, THF, −78 °C, 1.5 h, 50% yield; (c) 6-chloro-1,2,3,4-tetrahydroquinoline, NaBH(OAc)3, 1,2-dichloroethane, rt, overnight, 66% yield; (d) TFA, CH2Cl2, rt, 2 h, 86–93% yield; (e) 2-(tritylthio)acetic acid, PyBOP, Et3N, CH2Cl2, rt, 20 h, 25–59% yield; (f) TFA, Et3SiH, CH2Cl2, 0 °C to rt, 2 h, 51–73% yield; (g) 8-aminoquinoline, EDC, DMAP, Et3N, DMF, rt, overnight, 88% yield.
To synthesize compound 4, 4-[((tert-butoxycarbonyl)amino)methyl]benzoic acid (25, Scheme 2) and 8-aminoquinoline were reacted in the presence of 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC) and 4-dimethylaminopyridine (DMAP). The final N-deprotection, amide coupling, and S-deprotection steps were carried out as previously described.
With the desired compounds in hand, we then assayed the activity of each at HDAC1 and HDAC6 using a fluorescence-based assay. IC50 values were determined in duplicate. Results are listed in Table 1. TSA and compound 1 were used as positive controls. In evaluating the HDAC inhibitory effects of compounds 2–4, the assays were performed in the presence of tris(2-carboxyethyl)phosphine (TCEP), following the procedure described by Baud and co-workers (see details in the Supporting Information).27 Thus, TCEP was added to stock solutions of mercaptoacetamides 2–4 in order to avoid dimerization of these compounds when diluted in DMSO and water.
Table 1. HDAC Inhibitory Activity of Mercaptoacetamidesa.

Results were determined by Reaction Biology Corp. (Malvern, PA, USA); unless otherwise stated, IC50 values displayed are the mean of two experiments ± standard deviation obtained from curve fitting of a 10-point enzyme assay, starting from 30 μM HDACI with 3-fold serial dilution. Values are extracted from fitting dose–response curves to the data points.
CLogP values were calculated using ChemBioOffice-ChemDraw V12.
Reference (24).
No inhibitory activity.
Result of a single experiment.
Trichostatin A.
Prior SAR studies suggested that the chain length for HDAC6 inhibitors is optimal when n = 2 or 3 (Figure 2).26 Our findings corroborate these results, and also suggest that compounds with a shorter alkyl chain, n = 1, may still exhibit very good activity (2a, HDAC6 IC50 = 7.9 nM).
Previously, our research had determined that a benzyl linker was optimal for potent HDAC6-selective hydroxamic acid derivatives.23 Having this in mind, we explored an aromatic linker together with the mercaptoacetamide ZBG; however, analogue 4 presented very low activity. This result indicates that while the benzyl linker is appropriate for hydroxamic acids, this may not be the case for thiols. Of course, this comparison is not entirely accurate, as the length of the linker region of compound 4 is larger than that of the linker region found in tubastatin A.
Both THQ and 8-aminoquinoline caps generated potent and selective analogues, with the exception of 3c, but it is important to note that the THQ cap is more lipophilic, suggesting that compounds such as 3a (cLogP = 4.21) are more likely to penetrate the blood–brain barrier for the treatment of CNS diseases. On the other hand, replacing the aminoquinoline cap in compound 2c (n = 3) with the THQ cap to arrive at compound 3a (n = 3) resulted in a slight decrease in potency. This difference in potency may reflect the ability of the 8-aminoquinoline cap to establish an additional hydrogen bonding interaction with the surface of the enzyme, due to the presence of a second nitrogen atom.
A decrease in potency is observed when the amide linker of compound 1 is replaced with a secondary amine as in compound 2c, although the same pattern is not maintained for compounds with a shorter alkyl chain (2a and 2b).
To assess HDAC6 selectivity in a more biologically relevant context, we studied the ability of compounds 1, 2a, 2b, and 3a to induce acetylation of α-tubulin, the primary substrate of HDAC6, in neurons. Low micromolar concentrations of the test compounds applied to primary cultures of rat cortical neurons (E17) led to a dose-dependent increase in acetylated α-tubulin (AcTub) levels. Consistent with a selective inhibition of HDAC6, the same concentrations of compounds did not increase the acetylation levels of histone H3, which is a target of class I HDACs (Figure 3). All tested compounds induced tubulin acetylation at 10 μM or less, with compound 2b showing the greatest efficacy (more than 10-fold induction of AcTub at 1 μM). The results at 10 μM are comparable to those obtained with the hydroxamate-containing HDACIs tubastatin A (TubA) and trichostatin A (TSA), which were used as positive controls. At 1 μM, TubA and TSA are significantly more potent than the present compounds. Compound 1 produced a 5-fold induction of tubulin acetylation at 1 μM, while 2a and 3a had weaker activity at the same concentration.
Figure 3.

(a) Western blots of acetylated tubulin compared to α-tubulin in primary cortical neurons. Rat primary cortical cultures (E17) were treated with TSA, tubastatin A, or the thiol analogues 1, 2a,b, and 3a for 6 h at the indicated concentrations, and their effects on acetylated tubulin (AcTub) were compared with those of the DMSO control. (b) Results from an analogous set of experiments in which the increase of acetylated histone H3 (AcH3), compared to histone H3, was evaluated [**p < 0.01 and *p < 0.05, significant increase compared to DMSO control; 1-way ANOVA followed by Dunnett’s post test; (a) n = 4 experiments; (b) n = 3 experiments].
The pharmacological modulation of T-regulatory (Treg) suppression is considered as a possible therapeutic approach to slow or reverse the pathogenesis of autoimmune disorders such as inflammatory bowel disease and rheumatoid arthritis, and to prevent allograft rejection. Pharmacologic inhibition of HDAC6, using hydroxamate-based compounds such as tubastatin A and its analogues, was previously shown to increase the suppressive functions of murine23,28 and human29 Foxp3+ Treg cells.
To examine whether the present compounds might have an anti-inflammatory action, they were tested for their ability to enhance the immunosuppressive function of murine Foxp3+ Treg cells in vitro. Thus, carboxyfluorescein succinimidyl ester (CFSE)-labeled conventional T-effector (Teff) cells were incubated in the presence and absence of Tregs, with or without the addition of selected HDAC6 inhibitors at multiple concentrations. Using the thiols reported herein at 1 μM, varying effects were observed. Direct effects of compounds on Teff cell proliferation in these assays were not seen, as shown by comparable Teff cell proliferation in the absence of Treg cells (0:1 Treg:Teff ratio). Compared to corresponding cells exposed to DMSO alone, all seven compounds, namely 2a–c, 3a–c, and 4, increased Treg function, resulting in decreased Teff cell proliferation at the 1:1 Treg:Teff ratio, but only three compounds, namely 2a–c, were also effective at the 1:2 Treg:Teff ratio, and only compound 2b was also effective at the 1:4 Treg:Teff ratio (Figure 4).
Figure 4.

Effects of thiol compounds (1 μM) on murine Treg suppressive functions in vitro. Residual CFSE + Teff cell proliferation is shown at each ratio of Treg:Teff cell; assays were performed in triplicate and repeated at least once; mean ± SD shown; *p < 0.05 and **p < 0.01 compared to corresponding DMSO-treated cultures.
We have developed a small series of mercaptoacetamides, two of which exhibit single digit nM HDAC6 IC50s. In particular, compound 2b induced a dose-dependent increase in acetylated α-tubulin in primary cortical neurons, apparent at 0.5 μM, and with no significant effect on histone H3 acetylation. Some of these compounds were also found to enhance Treg suppression of Teff proliferation in vitro. Because there is no history of mercaptoacetamides being linked to genotoxicity, the results found herein suggest that these compounds should be explored further with regard to therapy in cancer and in inflammatory and degenerative diseases, in which prolonged drug use will be required. Further efforts to generate analogues with improved penetration of the blood–brain barrier are being made in order to evaluate these compounds in various animal models of neurodegenerative diseases, and the efficacy of these compounds in models of autoimmunity and transplant rejection will be reported in future studies.
Acknowledgments
The authors gratefully acknowledge Drs. Werner Tueckmantel and Jianjun Cheng for reviewing the article and providing comments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00303.
Experimental details (PDF)
Author Contributions
M.C.F.S. and G.P.V. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by NIH Grants NS079183 and U19MH085195, A.P.K.
The authors declare no competing financial interest.
Supplementary Material
References
- Bannister A. J.; Kouzarides T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolden J. E.; Peart M. J.; Johnstone R. W. Anticancer Activities of Histone Deacetylase Inhibitors. Nat. Rev. Drug Discovery 2006, 5, 769–784. 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Miller T. A.; Witter D. J.; Belvedere S. Histone Deacetylase Inhibitors. J. Med. Chem. 2003, 46, 5097–5116. 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]
- Guo S.-Q.; Zhang Y.-Z. Histone Deacetylase Inhibition: an Important Mechanism in the Treatment of Lymphoma. Cancer Biol. Med. 2012, 9, 85–89. 10.3969/j.issn.2095-3941.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai T.; Cui H.; Chen L. Multi-Targeted Histone Deacetylase Inhibitors in Cancer Therapy. Curr. Med. Chem. 2012, 19, 475–487. 10.2174/092986712798918842. [DOI] [PubMed] [Google Scholar]
- Mensah A. A.; Kwee I.; Gaudio E.; Rinaldi A.; Ponzoni M.; Cascione L.; Fossati G.; Stathis A.; Zucca E.; Caprini G.; Bertoni F. Novel HDAC Inhibitors Exhibit Pre-Clinical Efficacy in Lymphoma Models and Point to the Importance of CDKN1A Expression Levels in Mediating Their Anti-Tumor Response. Oncotarget 2014, 6, 5059–5071. 10.18632/oncotarget.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozikowski A. P.; Chen Y.; Gaysin A.; Chen B.; D'Annibale M. A.; Suto C. M.; Langley B. C. Functional Differences in Epigenetic Modulators - Superiority of Mercaptoacetamide-Based Histone Deacetylase Inhibitors Relative to Hydroxamates in Cortical Neuron Neuroprotection Studies. J. Med. Chem. 2007, 50, 3054–3061. 10.1021/jm070178x. [DOI] [PubMed] [Google Scholar]
- Selenica M.-L.; Benner L.; Housley S. B.; Manchec B.; Lee D. C.; Nash K. R.; Kalin J.; Bergman J. A.; Kozikowski A. P.; Gordon M. N.; Morgan D. Histone Deacetylase 6 Inhibition Improves Memory and Reduces Total tau Levels in a Mouse Model of tau Deposition. Alzheimer's Res. Ther. 2014, 6, 12. 10.1186/alzrt241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel C. J.; Zhang F.; Huang E. Y.; Guenther M. G.; Lazar M. A.; Klein P. S. Histone Deacetylase Is a Direct Target of Valproic Acid, a Potent Anticonvulsant, Mood Stabilizer, and Teratogen. J. Biol. Chem. 2001, 276, 36734–36741. 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- Shindoh N.; Mori M.; Terada Y.; Oda K.; Amino N.; Kita A.; Taniguchi M.; Sohda K.-Y.; Nagai K.; Sowa Y.; Masuoka Y.; Orita M.; Sasamata M.; Matsushime H.; Furuichi K.; Sakai T. YM753, a Novel Histone Deacetylase Inhibitor, Exhibits Antitumor Activity with Selective, Sustained Accumulation of Acetylated Histones in Tumors in the WiDr Xenograft Model. Int. J. Oncol. 2008, 32, 545–555. 10.3892/ijo.32.3.545. [DOI] [PubMed] [Google Scholar]
- Thaler F.; Mercurio C. Towards Selective Inhibition of Histone Deacetylase Isoforms: What Has Been Achieved, Where We Are and What Will Be Next. ChemMedChem 2014, 9, 523–526. 10.1002/cmdc.201300413. [DOI] [PubMed] [Google Scholar]
- Kozikowski A. P.; Butler K. V. Chemical Origins of Isoform Selectivity in Histone Deacetylase Inhibitors. Curr. Pharm. Des. 2008, 14, 505–528. 10.2174/138161208783885353. [DOI] [PubMed] [Google Scholar]
- Kalin J. H.; Butler K. V.; Kozikowski A. P. Creating Zinc Monkey Wrenches in the Treatment of Epigenetic Disorders. Curr. Opin. Chem. Biol. 2009, 13, 263–271. 10.1016/j.cbpa.2009.05.007. [DOI] [PubMed] [Google Scholar]
- Patil V.; Sodji Q. H.; Kornacki J. R.; Mrksich M.; Oyelere A. K. 3-Hydroxypyridin-2-thione as Novel Zinc Binding Group for Selective Histone Deacetylase Inhibition. J. Med. Chem. 2013, 56, 3492–3506. 10.1021/jm301769u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Itoh Y.; Miyata N. Isoform-Selective Histone Deacetylase Inhibitors. Curr. Pharm. Des. 2008, 14, 529–544. 10.2174/138161208783885335. [DOI] [PubMed] [Google Scholar]
- Olaharski A. J.; Ji Z.; Woo J.-Y.; Lim S.; Hubbard A. E.; Zhang L.; Smith M. T. The Histone Deacetylase Inhibitor Trichostatin A Has Genotoxic Effects in Human Lymphoblasts In Vitro. Toxicol. Sci. 2006, 93, 341–347. 10.1093/toxsci/kfl068. [DOI] [PubMed] [Google Scholar]
- Kalin J. H.; Zhang H.; Gaudrel-Grosay S.; Vistoli G.; Kozikowski A. P. Chiral Mercaptoacetamides Display Enantioselective Inhibition of Histone Deacetylase 6 and Exhibit Neuroprotection in Cortical Neuron Models of Oxidative Stress. ChemMedChem 2012, 7, 425–439. 10.1002/cmdc.201100522. [DOI] [PubMed] [Google Scholar]
- Kerr J. S.; Galloway S.; Lagrutta A.; Armstrong M.; Miller T.; Richon V. M.; Andrews P. A. Nonclinical Safety Assessment of the Histone Deacetylase Inhibitor Vorinostat. Int. J. Toxicol. 2010, 29, 3–19. 10.1177/1091581809352111. [DOI] [PubMed] [Google Scholar]
- Namdar M.; Perez G.; Ngo L.; Marks P. A. Selective Inhibition of Histone Deacetylase 6 (HDAC6) Induces DNA Damage and Sensitizes Transformed Cells to Anticancer Agents. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20003–20008. 10.1073/pnas.1013754107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivieccio M. A.; Brochier C.; Willis D. E.; Walker B. A.; D’Annibale M. A.; McLaughlin K.; Siddiq A.; Kozikowski A. P.; Jaffrey S. R.; Twiss J. L.; Ratan R. R.; Langley B. HDAC6 Is a Target for Protection and Regeneration Following Injury in the Nervous System. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 19599–19604. 10.1073/pnas.0907935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty S. J.; Koeller K. M.; Wong J. C.; Grozinger C. M.; Schreiber S. L. Domain-Selective Small-Molecule Inhibitor of Histone Deacetylase 6 (HDAC6)-Mediated Tubulin Deacetylation. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 4389–4394. 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler K. V.; Kalin J.; Brochier C.; Vistoli G.; Langley B.; Kozikowski A. P. Rational Design and Simple Chemistry Yield a Superior, Neuroprotective HDAC6 Inhibitor, Tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalin J. H.; Butler K. V.; Akimova T.; Hancock W. W.; Kozikowski A. P. Second-Generation Histone Deacetylase 6 Inhibitors Enhance the Immunosuppressive Effects of Foxp3+ T-Regulatory Cells. J. Med. Chem. 2012, 55, 639–651. 10.1021/jm200773h. [DOI] [PubMed] [Google Scholar]
- Sung Y. M.; Lee T.; Yoon H.; DiBattista A. M.; Song J. M.; Sohn Y.; Moffat E. I.; Turner R. S.; Jung M.; Kim J.; Hoe H.-S. Mercaptoacetamide-Based Class II HDAC Inhibitor Lowers Aβ Levels and Improves Learning and Memory in a Mouse Model of Alzheimer’s Disease. Exp. Neurol. 2013, 239, 192–201. 10.1016/j.expneurol.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; West E. J.; Van K. C.; Gurkoff G. G.; Zhou J.; Zhang X.-M.; Kozikowski A. P.; Lyeth B. G. HDAC Inhibitor Increases Histone H3 Acetylation and Reduces Microglia Inflammatory Response Following Traumatic Brain Injury in Rats. Brain Res. 2008, 1226, 181–191. 10.1016/j.brainres.2008.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Petukhov P. A.; Jung M.; Velena A.; Eliseeva E.; Dritschilo A.; Kozikowski A. P. Chemistry and Biology of Mercaptoacetamides as Novel Histone Deacetylase Inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 1389–1392. 10.1016/j.bmcl.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Baud M. G. J.; Leiser T.; Haus P.; Samlal S.; Wong A. C.; Wood R. J.; Petrucci V.; Gunaratnam M.; Hughes S. M.; Buluwela L.; Turlais F.; Neidle S.; Meyer-Almes F.-J.; White A. J. P.; Fuchter M. J. Defining the Mechanism of Action and Enzymatic Selectivity of Psammaplin A against Its Epigenetic Targets. J. Med. Chem. 2012, 55, 1731–1750. 10.1021/jm2016182. [DOI] [PubMed] [Google Scholar]
- de Zoeten E. F.; Wang L.; Butler K.; Beier U. H.; Akimova T.; Sai H.; Bradner J. E.; Mazitschek R.; Kozikowski A. P.; Matthias P.; Hancock W. W. Histone Deacetylase 6 and Heat Shock Protein 90 Control the Functions of Foxp3+ T-Regulatory Cells. Mol. Cell. Biol. 2011, 31, 2066–2078. 10.1128/MCB.05155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimova T.; Ge G.; Golovina T.; Mikheeva T.; Wang L.; Riley J. L.; Hancock W. W. Histone/Protein Deacetylase Inhibitors Increase Suppressive Functions of Human FOXP3+ Tregs. Clin. Immunol. 2010, 136, 348–363. 10.1016/j.clim.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.