

Abstract
Studies of directed ortho metalation reactions on an aromatic substrate with multiple potential directing groups have identified conditions that favor either of two regioisomers. One of these regioisomers has been converted to an analogue of the stilbene pawhuskin A, and been shown to have high selectivity as an antagonist of the delta opioid receptor. Docking studies have suggested that this compound can adopt a conformation similar to naltrindole, a known delta antagonist.
Keywords: Pawhuskin, Opioid, Delta receptor, Directed ortho metalation, Docking
Graphical Abstract
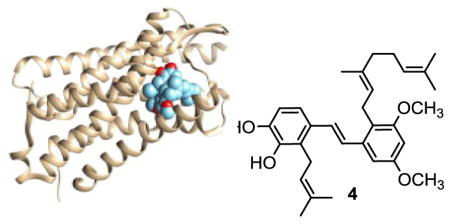
The development of opioid compounds for treatment of pain is one of the triumphs of modern medicine.1 These compounds, however, are associated with numerous negative side effects, most prominently including sensitization to chronic treatment leading to development of addiction and the associated societal problems.2 The canonical opioid receptors kappa (KOP), mu (MOP), and delta (DOP) mediate a variety of key physiological processes, and are involved with the adaptation to chronic opioid analgesic treatment to different degrees.1,3 The primary analgesic response is attributable to activation of the MOP.4 The DOP is much less well studied but appears to play an interesting role in the development of learned habitual responses to chronic treatment with these potent analgesics.5 Because of this role in the addictive effects of the opioid pain medications, selective DOP receptor antagonists are gaining interest in the field of pain management and psychiatry.6–8
Our interest in opioids stems from the reported isolation of the pawhuskin family of natural products.9 These compounds are non-nitrogenous opioid receptor modulators based around a stilbene core, and show significant potential as a scaffold for further exploration aimed at developing novel drug leads. There are several other non-nitrogenous scaffolds that are being studied as leads for opioid receptor modulators with the most prominent being the salvinorins, which have been studied predominantly as KOP agonists.10–12 Our studies of the pawhuskins have led to the synthesis of pawhuskin A (1)13 and C (2)14 (Figure 1) as well as several analogues, and to the demonstration that compound 1 is a moderately selective KOP antagonist. During these explorations we synthesized compound 3,15 with the prenyl group on the “left-half” of the molecule (the portion biochemically derived from shikimate) placed in a different orientation than in the parent pawhuskin A. To our surprise, this regioisomer turned out to be an opioid receptor antagonist with high selectivity for the KOP (δ/κ > 67 and δ/μ > 67) and to be a bit more potent than pawhuskin A (Ke = 0.15 μM vs. 0.20 μM).15
Figure 1.

Structures of Pawhuskins and Analogues.
In our synthesis of pawhuskin A we employed a directed ortho metalation approach (Scheme 1).13 Lithiation of the ring may be directed by the MOM protecting group and presumably the benzylic alcohol anion of the known starting material (5) to afford the intermediate anion. Transmetalation to the copper species followed by treatment with prenyl bromide gave the final product alcohol (6) in modest yields as the only easily isolated product. In attempts to improve the yield use of copper iodide and TMEDA was explored because this had been shown in our prior work with halogen metal exchange reactions in similar systems to afford superior yields.16 The addition of TMEDA and use of copper iodide in ether afforded a mixture of the arene 6 and the isomeric prenylated compound 7 in a 1:1.2 ratio (Table 1, entry 1) and a combined yield of 36%. A more thorough exploration of the conditions showed that either regioisomer could be made with some selectivity. Slightly colder reaction temperatures afforded the best combined yield of products favoring compound 7 (entry 2). Forgoing the transmetalation step improved the ratio of compound 7 to 6 but the overall yield was particularly disappointing (entry 3). Reaction at room temperature in THF with copper bromide but without TMEDA afforded the alternate regioisomer 6 as the predominant product (6:7 2.9:1 entry 4) in a combined yield of 47%. Variation of the reaction temperature and the scale, which also might afford better control of the reaction temperature, did not improve this ratio (entries 5–7).
Scheme 1.

Table 1.
Effect of temperature and other parameters on directed ortho metalation of compound 5.
| Trial | Scale (mmol) | TMEDA (mmol) | n– BuLi (mmol) | CuBr·DMS (mmol) | Prenyl bromide (mmol) | Solvent [conc.] | T | 6 : 7 | % Yield |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 6.51 | 14.01 | 14.25 | 6.53 a | 7.16 | Et2O [0.07 M] | −10 °C to rt | 1.0 : 1.2 | 36 |
| 2 | 7.97 | 16.67 | 17.5 | 7.98a | 11.93 | Et2O [0.06 M] | −20 to 0 °C to rt | 1.0 : 1.9 | 25 |
| 3 | 4.18 | 8.67 | 9.2 | NA | 6.31 | Et2O [0.06 M] | −20 to 0 °C to rt | 1.0 : 4.0 | 10 |
| 4 | 4.46 | NA | 9.52 | 4.91 | 4.94 | THF [0.13 M] | rt | 2.9 : 1.0 | 47 |
| 5 | 0.92 | NA | 1.95 | 1.02 | 1.11 | THF [0.13 M] | rt | 1.1 : 1.0 | 25 |
| 6 | 1.31 | NA | 2.75 | 1.11 | 1.53 | THF [0.13M] | 0 °C | 1.0 : 1.0 | 39 |
| 7 | 4.53 | NA | 9.52 | 4.98 | 4.94 | THF [0.13 M] | 0 °C | 1.8 : 1.0 | 51 |
In these experiments, copper iodide was used.
With a viable route to compound 6 in hand we set about preparation of the pawhuskin A analogue 4. Treatment of the benzylic alcohol 6 with methanesulfonyl chloride in trimethylamine gave the mesylate which was converted into the bromide without isolation. An Arbuzov reaction was carried out by heating the bromide with triethyl phosphite to give the desired phosphonate 8 in moderate yield. Horner-Wadsworth-Emmons coupling of phosphonate 8 and the known aldehyde 915 afforded the protected stilbene 10 (Scheme 2). Global deprotection of the methoxymethyl ether groups by treatment with p-toluenesulfonic acid in methanol gave the desired analogue 4.
Scheme 2.

Analogue 4 was tested for opioid receptor activity by first assessing if intrinsic agonist activity was present. After finding no agonist activity, this compound was tested for antagonist selectivity against the mu, delta and kappa opioid receptors (MOP, DOP, and KOP). To our surprise the analogue 4 displayed strong antagonist activity that was very selective for the DOP (Ke = 25 nM, κ/δ> 400, MOP/DOP μ /δ> 400, Figure 2). This was an intriguing result. In essence, moving the prenyl substituent from a position ortho to the stilbene junction in isomer 4 to a position meta to the central olefin, as in compound 3,15 shifted the activity from highly delta selective to highly kappa selective (Table 2).
Figure 2.

Antagonist activity of pawhuskin analogue 4 at the DOP.
Table 2.
Apparent affinities of pawhuskin A (1), 3 and 4.
| apparent affinity (Ke) of antagonists in μM
|
|||
|---|---|---|---|
| Compound | DOP | KOP | MOP |
| 1 | 2.9 | 0.2 | 570 |
| 3 | >10 | 0.15 | >10 |
| 4 | 0.025 | >10 | >10 |
In order to rationalize this dramatic change in selectivity when the prenyl group is shifted, docking studies were conducted. The structure of the mouse DOP with the bound antagonist naltrindole was solved in 2012 by the Kobilka group.17 We started with this structure and used the Autodock Vina18 software package to perform docking of compounds 3 and 4 into the receptor. Stilbene 4 fits neatly into the DOP receptor binding pocket with the free phenols of the catechol ring predicted to make hydrogen bonds with LYS108, GLN105, TYR109, and TYR308 of the DOP structure (Figure 3A, visualization was conducted using the Chimera software suite).17,19 Interestingly, the hydrophobic isoprenoid groups of isomer 4 overlap quite well with the indole (geranyl group) and cyclopropylmethyl (prenyl group) groups of naltrindole (Figure 3B). When the KOP selective compound 3 is docked using the same procedure, the lowest energy conformation overlaps almost perfectly with that of the predicted lowest energy conformation of compound 4. The prominent exception is the prenyl group which is now directed up and away from the region occupied by the cyclopropylmethyl moiety of naltrindole in the x-ray structure (Figure 3C). This change robs compound 3 of key hydrophobic interactions that presumably support the binding of naltrindole and compound 4 to the DOP. The lowest energy docking pose for compound 4 has a score of −8.9 vs. a −7.3 for the lowest energy pose of stilbene 3. This correlates nicely with the large difference in the functional binding assay.
Figure 3.

Docking studies. A. The structure of compound 4 bound in the DOP looking down into the active site from the top. Shows the key proposed H-bonds in blue. B. Analogue 4 (blue) and naltrindole (in pink, from the crystal structure) shown in the active site. C. Lowest energy docking poses of pawhuskin A (1, gray), 3 (pink) and 4 (blue). D. Key features of the pharmacophore of 4 based on the message and address concept of opioid pharmacology, with message region interactions blue and address region interactions pink. The graphics were rendered using the Chimera software suite.
The overall binding motif of compound 4 can be viewed in the context of the message and address concept of opioid binding in which the geranyl group, like the indole of naltrindole, extends into a region of the receptor that confers selectivity. In contrast the prenyl group and the phenols are the message which allows binding to key parts of the receptor architecture, in this case blocking the ability of ligand to bind and signal (Figure 3D). In contrast, for the docked pose of kappa-selective compound 3 the hydrophobic contributions to the message part of the binding are not possible. This dramatically reduces the overall interaction as depicted by the docking score.
The differences with respect to the docking of the natural product pawhuskin A (1, Figure 3C) are more difficult to rationalize. If one assumes that pawhuskin A adopts an orientation similar to the delta-selective compound 4, it leads to an intermediate docking score of −8.4. In this orientation, the phenols of pawhuskin A are orientated away from the space occupied by the methoxy groups of compound 4. If that orientation improves the hydrophobic interaction between compound 4 and the receptor, it would lead to stronger binding. A favorable orientation of the prenyl group of pawhuskin A may compensate for some of the reduction caused by the absence of the methoxy groups, and allow functional antagonism at the DOP albeit with reduced apparent affinity.
In conclusion, we have synthesized a highly selective delta opioid receptor antagonist (4) based on the stilbene motif of the pawhuskin natural products. Our studies on directed ortho metalation of compound 5 have uncovered conditions which favor prenylation ortho to the benzylic position (i.e. compound 6) or meta to this substituent (compound 7). Incorporation of compound 7 into the final stilbene has yielded the kappa-selective compound 3, while incorporation of the isomeric 6 has given this new delta-selective stilbene 4. Docking studies have shed some light on the potential differences in the binding modes of the stilbene isomers 3 and 4 to the DOP, and provided some rationale for the large difference in selectivity. We are currently undertaking further studies on the activity of compounds 3 and 4, as well as synthesis of other pawhuskin analogues. These efforts will be reported in due course.
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (DA02-6573 to JDN) and the Roy J. Carver Charitable Trust as a Research Program of Excellence (to DFW) is gratefully acknowledged.
Footnotes
Supplementary data including experimental details for the chemical synthesis of novel compounds, 1H and 13C NMR spectra, and details of the functional opioid receptor assay are available.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Pasternak GW. Clin Neuropharmacol. 1993;16:1. doi: 10.1097/00002826-199302000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Benyamin R, Trescot AM, Datta S, Buenaventura R, Adlaka R, Sehgal N, Glaser SE, Vallejo R. Pain Physician. 2008;11:S105. [PubMed] [Google Scholar]
- 3.Raynor K, Kong HY, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T. Mol Pharmacol. 1994;45:330. [PubMed] [Google Scholar]
- 4.Matthes HWD, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, LeMeur M, Dolle P, Tzavara E, Hanoune J, Roques BP, Kieffer BL. Nature. 1996;383:819. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- 5.Gendron L, Mittal N, Beaudry H, Walwyn W. Br J Pharmacol. 2015;172:403. doi: 10.1111/bph.12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abdelhamid EE, Sultana M, Portoghese PS, Takemori AE. J Pharmacol Exp Ther. 1991;258:299. [PubMed] [Google Scholar]
- 7.Burford NT, Livingston KE, Canals M, Ryan MR, Budenholzer LML, Han Y, Shang Y, Herbst JJ, O'Connell J, Banks M, Zhang LT, Filizola M, Bassoni DL, Wehrman TS, Christopoulos A, Traynor JR, Gerritz SW, Alt A. J Med Chem. 2015;58:4220. doi: 10.1021/acs.jmedchem.5b00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nemoto T, Iihara Y, Hirayama S, Iwai T, Higashi E, Fujii H, Nagase H. Bioorg Med Chem Lett. 2015;25:2927. doi: 10.1016/j.bmcl.2015.05.038. [DOI] [PubMed] [Google Scholar]
- 9.Belofsky G, French AN, Wallace DR, Dodson SL. J Nat Prod. 2004;67:26. doi: 10.1021/np030258d. [DOI] [PubMed] [Google Scholar]
- 10.Prisinzano TE. J Med Chem. 2013;56:3435. doi: 10.1021/jm400388u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riley AP, Groer CE, Young D, Ewald AW, Kivell BM, Prisinzano TE. J Med Chem. 2014;57:10464. doi: 10.1021/jm501521d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simonson B, Morani AS, Ewald AWM, Walker L, Kumar N, Simpson D, Miller JH, Prisinzano TE, Kivell BM. Br J Pharmacol. 2015;172:515. doi: 10.1111/bph.12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neighbors JD, Buller MJ, Boss KD, Wiemer DF. J Nat Prod. 2008;71:1949. doi: 10.1021/np800351c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neighbors JD, Salnikova MS, Wiemer DF. Tetrahedron Lett. 2005;46:1321. [Google Scholar]
- 15.Hartung AM, Beutler JA, Navarro HA, Wiemer DF, Neighbors JD. J Nat Prod. 2014;77:311. doi: 10.1021/np4009046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Topczewski JJ, Kodet JG, Wiemer DF. J Org Chem. 2011;76:909. doi: 10.1021/jo1022102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Nature. 2012;485:400. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trott O, Olson AJ. J Computl Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.