

Abstract
Human cytomegalovirus (HCMV) pUL93 is essential for virus growth, but its precise function in the virus life cycle is unknown. Here, we characterize a UL93 stop mutant virus (UL93st-TB40/E-BAC) to demonstrate that the absence of this protein does not restrict viral gene expression; however, cleavage of viral DNA into unit-length genomes as well as genome packaging is abolished. Thus, pUL93 is required for viral genome cleavage and packaging.
TEXT
Human cytomegalovirus (HCMV) protein pUL93, a putative tegument protein, is required for the growth of HCMV (1, 2), but the exact function of this protein is unknown. There is no functional study done to date on HCMV pUL93; however, pUL17, the positional homolog of pUL93 in herpes simplex virus 1 (HSV-1), has been shown to be required for the localization of capsids to the DNA replication compartments in the infected cell nucleus, where viral genome cleavage and packaging take place (3, 4). pUL17 has also been found to interact with pUL25, a homolog of HCMV pUL77, to form the capsid vertex-specific component (CVSC), which is found at the 12 vertices of the icosahedral capsid (4–10). The exact function of the CVSC is currently unknown, but it is thought to aid in capsid stability and nuclear egress (10–13). pUL25-null mutants package viral DNA only transiently and produce mostly empty capsids, while pUL17-null mutants completely abolish DNA packaging (3, 14). It is important to note that pUL17 and pUL93 are positional homologs that share only 1.5% sequence identity, 1.9% sequence similarity, and no distinct conserved domains (based on amino acid ClustalW alignment). Moreover, several HCMV proteins have been found to have very different functions compared to their homologs in other herpesviruses, highlighting the importance of studying HCMV proteins independently. For example, HCMV pUL50 and pUL53 were presumed to recruit host protein kinase C (PKC) for disruption of the nuclear lamina based on data from other herpesviruses but were instead found to recruit viral protein kinase pUL97 for this purpose (15). Targeting of essential structural viral proteins holds great promise for the development of antivirals that would be highly specific and effective and also less susceptible to the development of resistance because the mutation of a structural protein would likely compromise capsid assembly and maturation (16). Characterization of pUL93 will aid in the development of these promising antivirals in addition to advancing our understanding of HCMV maturation events.
Here, we have characterized a UL93 stop mutant virus. We report that although pUL93 is not required for expression of different kinetic classes of viral genes, DNA-containing C-capsids and virions cannot be detected in the nucleus and the cytoplasm, respectively, of infected cells in the absence of pUL93. Upon further investigation, pUL93 was found to be required for the cleavage of viral genomic DNA into unit-length genomes.
pUL93 is dispensable for viral gene expression.
To study the role of pUL93 in the HCMV life cycle, we engineered a UL93 stop mutant (UL93st-TB40/E-BAC) by replacing the initiating codon (ATG) in UL93 with a stop codon (TAG) using two-step bacterial artificial chromosome (BAC) recombineering (Fig. 1A) (17–19). BAC constructs were validated by restriction fragment length polymorphism (RFLP) and PCR sequencing of the region of the BAC genome containing this change (data not shown). Upon transfection in human foreskin fibroblasts (HF), this mutant BAC did not yield any detectable infectious virus particles in cell culture medium. To enable the growth of this virus, we constructed pUL93-complementing cells by transducing HF with a lentiviral vector (pLV-EF1α-MCS-IRES-Puro; cDNA-pLV01 [20]) that expressed FLAG-tagged pUL93. These complementing cells were then transfected with UL93st-TB40/E-BAC to grow this virus. Upon infection in noncomplementing HF, UL93st-TB40/E-BAC virus did not yield any detectable infectious virus particles in cell culture medium when monitored for up to 10 days postinfection (dpi), but in complementing HF, the UL93st-TB40/E-BAC virus grew to high titers (2.24 × 108 PFU/ml) (Fig. 1B). We used the harvested UL93st-TB40/E-BAC virus to infect noncomplementing HF at a multiplicity of infection (MOI) of 3.0. Cells were harvested at 3 dpi, and immunoblot (IB) assays probing for viral immediate early (anti-IE1/2 antibody [CH160]; P1215; Virusys Corporation, Taneytown, MD), delayed early (anti-pUL44 antibody [ICP36]; CA006-100; Virusys Corporation, Taneytown, MD), and late (anti-gB antibody; CA005-100; Virusys Corporation, Taneytown, MD) proteins in whole-cell lysates showed expression of all these classes of viral genes, indicating that pUL93 is not required for viral gene expression (Fig. 2). Nevertheless, levels of gB expression were slightly reduced in UL93st-TB40/E-BAC virus infection compared to the wild-type virus infection, indicating some impact on late gene expression.
FIG 1.
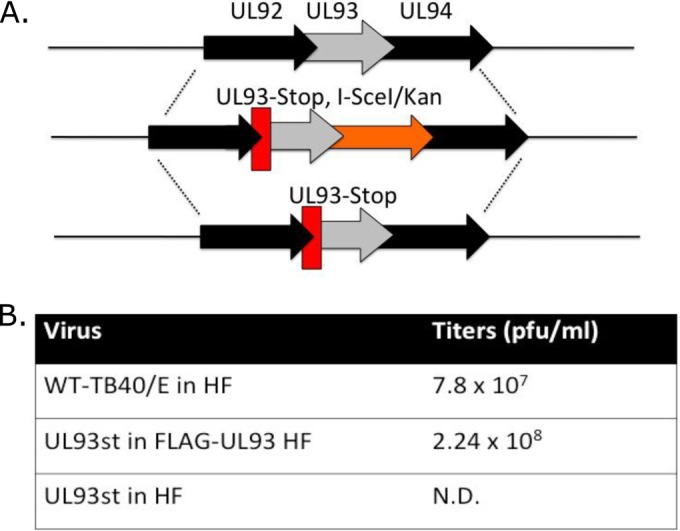
Construction and growth of a UL93 stop mutant virus. (A) Schematic of the UL92-to-UL94 region in TB40/E-BAC (top line) with other bacmid constructs shown below. A UL93 stop mutant virus (UL93st-TB40/E-BAC) was constructed using two-step BAC recombineering (17). In the first step, a kanamycin (Kan) cassette fused to the UL93 stop mutation and an I-SceI site was inserted in the BAC genome using homologous recombination. The Kan cassette was then spliced out utilizing the I-SceI site, leaving only the UL93 stop mutation in the genome. (B) Final virus yields were determined at day 10 postinfection for wild-type TB40/E (WT-TB40/E in HF), UL93st-TB40/E-BAC in complementing cells (UL93st in FLAG-UL93 HF), and UL93st-TB40/E-BAC in noncomplementing cells (UL93st in HF). N.D., not detected, as the number of PFU in this preparation was below the threshold of detection in the assay.
FIG 2.
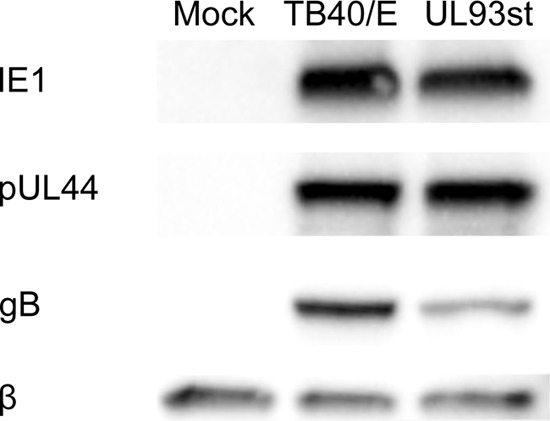
pUL93 is not required for viral gene expression. HF were infected with WT-TB40/E or UL93st-TB40/E-BAC virus (UL93st) at an MOI of 3.0 or mock infected, and cells were harvested at 72 h postinfection for immunoblot assays probing for early (IE1), delayed early (pUL44), and late (gB) viral proteins.
pUL93 is required for nucleocapsid maturation.
To further study the stage of the HCMV life cycle where pUL93 functions are critical, we performed transmission electron microscopy (TEM) of HF infected with either wild-type (WT) TB40/E or UL93st-TB40/E-BAC viruses. HF were infected at an MOI of 3.0 with either of the viruses and fixed for TEM at 4 dpi. Ultrastructural analysis of the nucleus and the cytoplasm of WT-TB40/E-infected cells revealed the presence of empty (A-), scaffold-containing (B-), and genome-containing (C-) capsids in the nucleus, and virions as well as dense bodies in the cytoplasm, typical of a productive infection in fibroblasts (19, 21, 22), while C-capsids and virions were not detected in cells infected with UL93st-TB40/E-BAC virus (Fig. 3A to F). We enumerated mature and immature virus particles in each infection group based on the established morphological characteristics of these particles, which are illustrated in Fig. 3G to K (23, 24). Four individual WT-TB40/E-infected cells showed an average of 10 A-capsids, 54 B-capsids, and 13 C-capsids in the nucleus, while four individual UL93st-TB40/E-BAC virus-infected cells showed an average of less than 1 A-capsid and 45 B-capsids in the nucleus (Fig. 3L). C-capsids were absent from the nuclei of UL93st-TB40/E-BAC virus-infected cells. In the cytoplasm, four WT-TB40/E-infected cells showed an average of 56 virions, 7 dense bodies, and 12 nontypeable particles, compared to no virions, 78 dense bodies, and 8 nontypeable particles in UL93st-TB40/E-BAC virus-infected cells (Fig. 3M). Thus, pUL93 is required for the production of C-capsids and mature virions.
FIG 3.

pUL93 is required for nucleocapsid maturation. (A to F) Transmission electron micrographs (TEM) of HF infected with WT-TB40/E (A, C, and E) or UL93st-TB40/E-BAC (UL93st) (B, D, and F) viruses at an MOI of 3.0 and fixed for processing at 4 days postinfection. (A and B) A single infected cell showing nuclear inclusion (NI) as well as cytoplasmic inclusion (CI). (C and D) Nuclear inclusion illustrating A- (black arrowhead), B- (black-tailed arrow), and C- (white-tailed arrow) capsids. (E and F) Cytoplasmic inclusion illustrating mature virions (white-tailed arrow) and dense bodies (white arrowhead). Bars, 4 μm (A and B), 500 nm (C and D), and 1 μm (E and F). (G to K) Magnified images (not to scale) of A-capsids (G), B-capsids (H), C-capsids (I), virions (J), and dense bodies (K) are shown. (L and M) A-, B-, and C-capsids in the nucleus (L) and virions, dense bodies, and nontypeable particles in the cytoplasm (M) were enumerated in 4 individual infected cells, and the mean values are displayed.
pUL93 is required for viral genome cleavage.
Based on the above data that show that C-capsids and virions are absent during infection with a UL93 stop mutant virus, we hypothesized that this could be due to two reasons: pUL93 is required for viral genome cleavage and packaging and it could also be necessary for maintaining capsid stability after genome cleavage and packaging. Proteolytic digestion of capsid scaffold and packaging of viral DNA are believed to occur simultaneously (25–29), and A-capsids are generated as a result of abortive attempts to package viral DNA (24, 30). Although roles of A- and B-capsids as intermediate or abortive capsid forms in the life cycle of herpesviruses have not been clearly distinguished, we hypothesized that a defect in virus genome packaging would arrest the virus capsid maturation prior to the production of C-capsids (capsids with packaged DNA). A defect after packaging would result in the instability and probable complete degradation of C-capsids, which may not be limited to the ejection of viral DNA, considering that C-capsids sustain much higher internal pressure from the packaged DNA. Thus, A-capsid numbers may not increase despite a reduction in the number of C-capsids. To explore the possibility of a DNA packaging defect, DNA was harvested from WT-TB40/E or UL93st-TB40/E-BAC virus-infected HF and digested with either EcoRI or KpnI, and Southern blot assays were performed to probe for viral terminal DNA. The use of Southern blot assays to probe for terminal and junction fragments in herpesviruses has been well established (31–35). As a control, HF were infected with WT-TB40/E and treated with 2-bromo-5,6-dichloro-1-(β-d-ribofuranosyl) benzimidazole (BDCRB; a viral terminase inhibitor in HCMV) (36, 37) or mock treated. BDCRB selectively inhibits pUL56 and pUL89, thereby blocking viral genome cleavage without affecting viral gene expression or capsid and dense body formation (24, 38–42). The presence of the 4.4- and 4.5-kb terminal fragment in WT-TB40/E digested with EcoRI and KpnI, respectively, indicates the occurrence of genome cleavage. As expected, this band was not detected in the BDCRB-treated control. Importantly, this band was also absent in UL93st-TB40/E-BAC virus (Fig. 4), indicating that pUL93 is required for the cleavage of viral genomic DNA. Based on the TB40/E genome sequence (GenBank accession no. EF999921.1), there can be multiple junction fragments that would bind to a terminal probe in a Southern blot assay. The number of these fragments will depend on the presence or absence of different genomic isomers in virus stock. However, all these junction fragments will be larger than the terminal fragment, as seen in the Southern blot image (Fig. 4).
FIG 4.
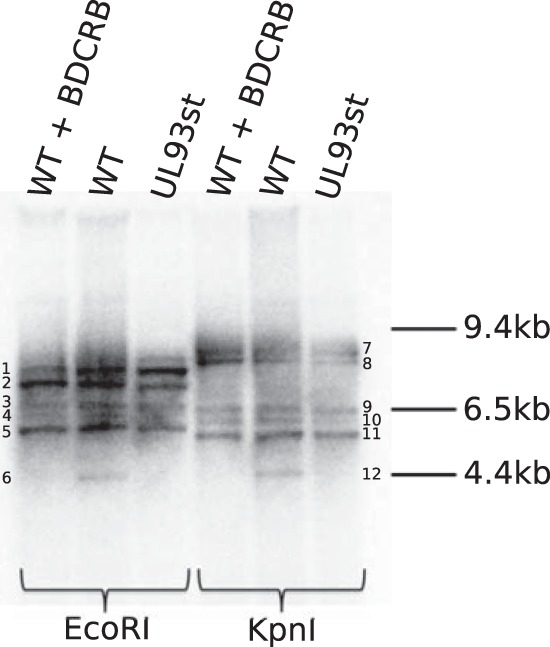
pUL93 is required for viral genome cleavage. DNA extracted from HF infected with WT-TB40/E in the presence of BDCRB, infected with WT-TB40/E in the absence of BDCRB, or infected with UL93st-TB40/E-BAC virus (UL93st) at an MOI of 3.0 was digested with EcoRI or KpnI, size fractionated by gel electrophoresis, and blotted. Hybridization was performed using a terminal DNA probe. Fragments labeled 6 and 12 in the autoradiographic image are terminal fragments that represent cleaved viral genomic DNA. Fragments 1, 2, 3, 4, 5, 7, 8, 9, 10, and 11 are all possible junction fragments.
Together, our data confirm an essential role of pUL93 during HCMV infection and indicate that this protein is critical for viral genome cleavage and packaging. Future studies will examine if pUL93 interacts with pUL77 to form a structure similar to the CVSC in HSV-1 and contributes to capsid stability and/or nuclear egress.
ACKNOWLEDGMENTS
We are thankful to Glenn Hoskins at the Electron Microscopy Facility at the University of Mississippi Medical Center (UMMC) for help with electron microscopy. Melanie Wilson, Eva Bengten, and Cecille Snell at UMMC provided guidance and assistance with Southern blot assays. Christian Sinzger provided the TB40/E-BAC and Klaus Osterrieder provided the Isce1 recombination plasmids. Michael McVoy at Virginia Commonwealth University provided helpful advice about Southern blot probe design and data interpretation. Madeline Archer and Leslie Davis helped with cell culture in the Tandon lab.
An American Heart Association predoctoral fellowship (AHA 15PRE25090135; principal investigator, B. M. DeRussy) and Scientist Development Grant (AHA 14SDG20390009; principal investigator, R. Tandon) supported this research.
REFERENCES
- 1.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A 100:14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci U S A 100:12396–12401. doi: 10.1073/pnas.1635160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taus NS, Salmon B, Baines JD. 1998. The herpes simplex virus 1 UL 17 gene is required for localization of capsids and major and minor capsid proteins to intranuclear sites where viral DNA is cleaved and packaged. Virology 252:115–125. doi: 10.1006/viro.1998.9439. [DOI] [PubMed] [Google Scholar]
- 4.Scholtes L, Baines JD. 2009. Effects of major capsid proteins, capsid assembly, and DNA cleavage/packaging on the pUL17/pUL25 complex of herpes simplex virus 1. J Virol 83:12725–12737. doi: 10.1128/JVI.01658-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thurlow JK, Murphy M, Stow ND, Preston VG. 2006. Herpes simplex virus type 1 DNA-packaging protein UL17 is required for efficient binding of UL25 to capsids. J Virol 80:2118–2126. doi: 10.1128/JVI.80.5.2118-2126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newcomb WW, Homa FL, Brown JC. 2006. Herpes simplex virus capsid structure: DNA packaging protein UL25 is located on the external surface of the capsid near the vertices. J Virol 80:6286–6294. doi: 10.1128/JVI.02648-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trus BL, Newcomb WW, Cheng N, Cardone G, Marekov L, Homa FL, Brown JC, Steven AC. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-filled HSV-1 capsids. Mol Cell 26:479–489. doi: 10.1016/j.molcel.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Preston VG, Murray J, Preston CM, McDougall IM, Stow ND. 2008. The UL25 gene product of herpes simplex virus type 1 is involved in uncoating of the viral genome. J Virol 82:6654–6666. doi: 10.1128/JVI.00257-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cockrell SK, Huffman JB, Toropova K, Conway JF, Homa FL. 2011. Residues of the UL25 protein of herpes simplex virus that are required for its stable interaction with capsids. J Virol 85:4875–4887. doi: 10.1128/JVI.00242-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toropova K, Huffman JB, Homa FL, Conway JF. 2011. The herpes simplex virus 1 UL17 protein is the second constituent of the capsid vertex-specific component required for DNA packaging and retention. J Virol 85:7513–7522. doi: 10.1128/JVI.00837-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klupp BG, Granzow H, Keil GM, Mettenleiter TC. 2006. The capsid-associated UL25 protein of the alphaherpesvirus pseudorabies virus is nonessential for cleavage and encapsidation of genomic DNA but is required for nuclear egress of capsids. J Virol 80:6235–6246. doi: 10.1128/JVI.02662-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang K, Baines JD. 2011. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc Natl Acad Sci U S A 108:14276–14281. doi: 10.1073/pnas.1108564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang K, Wills E, Lim HY, Zhou ZH, Baines JD. 2014. Association of herpes simplex virus pUL31 with capsid vertices and components of the capsid vertex-specific complex. J Virol 88:3815–3825. doi: 10.1128/JVI.03175-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McNab AR, Desai P, Person S, Roof LL, Thomsen DR, Newcomb WW, Brown JC, Homa FL. 1998. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J Virol 72:1060–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma M, Kamil JP, Coughlin M, Reim NI, Coen DM. 2014. Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J Virol 88:249–262. doi: 10.1128/JVI.02358-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanner EJ, Liu HM, Oberste MS, Pallansch M, Collett MS, Kirkegaard K. 2014. Dominant drug targets suppress the emergence of antiviral resistance. eLife 3:e03830. doi: 10.7554/eLife.03830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 18.Thomason L, Court DL, Bubunenko M, Costantino N, Wilson H, Datta S, Oppenheim A. 2007. Recombineering: genetic engineering in bacteria using homologous recombination. Curr Protoc Mol Biol Chapter 1:Unit 1.16. doi: 10.1002/0471142727.mb0116s78. [DOI] [PubMed] [Google Scholar]
- 19.Brechtel TM, Mocarski ES, Tandon R. 2014. Highly acidic C-terminal region of cytomegalovirus pUL96 determines its functions during virus maturation independently of a direct pp150 interaction. J Virol 88:4493–4503. doi: 10.1128/JVI.03784-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Omoto S, Mocarski ES. 2013. Cytomegalovirus UL91 is essential for transcription of viral true late (gamma2) genes. J Virol 87:8651–8664. doi: 10.1128/JVI.01052-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tandon R, Mocarski ES. 2008. Control of cytoplasmic maturation events by cytomegalovirus tegument protein pp150. J Virol 82:9433–9444. doi: 10.1128/JVI.00533-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tandon R, AuCoin DP, Mocarski ES. 2009. Human cytomegalovirus exploits ESCRT machinery in the process of virion maturation. J Virol 83:10797–10807. doi: 10.1128/JVI.01093-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol 20:392–401. doi: 10.1016/j.tim.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tandon R, Mocarski ES, Conway JF. 2015. The A, B, Cs of herpesvirus capsids. Viruses 7:899–914. doi: 10.3390/v7030899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Black LW, Silverman DJ. 1978. Model for DNA packaging into bacteriophage T4 heads. J Virol 28:643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laemmli UK, Favre M. 1973. Maturation of the head of bacteriophage T4. I. DNA packaging events. J Mol Biol 80:575–599. [DOI] [PubMed] [Google Scholar]
- 27.Fuller DN, Raymer DM, Kottadiel VI, Rao VB, Smith DE. 2007. Single phage T4 DNA packaging motors exhibit large force generation, high velocity, and dynamic variability. Proc Natl Acad Sci U S A 104:16868–16873. doi: 10.1073/pnas.0704008104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fuller DN, Raymer DM, Rickgauer JP, Robertson RM, Catalano CE, Anderson DL, Grimes S, Smith DE. 2007. Measurements of single DNA molecule packaging dynamics in bacteriophage lambda reveal high forces, high motor processivity, and capsid transformations. J Mol Biol 373:1113–1122. doi: 10.1016/j.jmb.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rickgauer JP, Fuller DN, Grimes S, Jardine PJ, Anderson DL, Smith DE. 2008. Portal motor velocity and internal force resisting viral DNA packaging in bacteriophage phi29. Biophys J 94:159–167. doi: 10.1529/biophysj.107.104612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mocarski ES Jr, Shenk T, Pass RF. 2006. Cytomegaloviruses, p 2701–2772. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 31.McVoy MA, Nixon DE, Hur JK, Adler SP. 2000. The ends on herpesvirus DNA replicative concatemers contain pac2 cis cleavage/packaging elements and their formation is controlled by terminal cis sequences. J Virol 74:1587–1592. doi: 10.1128/JVI.74.3.1587-1592.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Przech AJ, Yu D, Weller SK. 2003. Point mutations in exon I of the herpes simplex virus putative terminase subunit, UL15, indicate that the most conserved residues are essential for cleavage and packaging. J Virol 77:9613–9621. doi: 10.1128/JVI.77.17.9613-9621.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McVoy MA, Adler SP. 1994. Human cytomegalovirus DNA replicates after early circularization by concatemer formation, and inversion occurs within the concatemer. J Virol 68:1040–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spaete RR, Mocarski ES. 1985. The alpha sequence of the cytomegalovirus genome functions as a cleavage/packaging signal for herpes simplex virus defective genomes. J Virol 54:817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borst EM, Wagner K, Binz A, Sodeik B, Messerle M. 2008. The essential human cytomegalovirus gene UL52 is required for cleavage-packaging of the viral genome. J Virol 82:2065–2078. doi: 10.1128/JVI.01967-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Underwood MR, Harvey RJ, Stanat SC, Hemphill ML, Miller T, Drach JC, Townsend LB, Biron KK. 1998. Inhibition of human cytomegalovirus DNA maturation by a benzimidazole ribonucleoside is mediated through the UL89 gene product. J Virol 72:717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krosky PM, Underwood MR, Turk SR, Feng KW, Jain RK, Ptak RG, Westerman AC, Biron KK, Townsend LB, Drach JC. 1998. Resistance of human cytomegalovirus to benzimidazole ribonucleosides maps to two open reading frames: UL89 and UL56. J Virol 72:4721–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Townsend LB, Devivar RV, Turk SR, Nassiri MR, Drach JC. 1995. Design, synthesis, and antiviral activity of certain 2,5,6-trihalo-1-(beta-d-ribofuranosyl)benzimidazoles. J Med Chem 38:4098–4105. doi: 10.1021/jm00020a025. [DOI] [PubMed] [Google Scholar]
- 39.Cayatte C, Schneider-Ohrum K, Wang Z, Irrinki A, Nguyen N, Lu J, Nelson C, Servat E, Gemmell L, Citkowicz A, Liu Y, Hayes G, Woo J, Van Nest G, Jin H, Duke G, McCormick AL. 2013. Cytomegalovirus vaccine strain Towne-derived dense bodies induce broad cellular immune responses and neutralizing antibodies that prevent infection of fibroblasts and epithelial cells. J Virol 87:11107–11120. doi: 10.1128/JVI.01554-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hwang JS, Kregler O, Schilf R, Bannert N, Drach JC, Townsend LB, Bogner E. 2007. Identification of acetylated, tetrahalogenated benzimidazole d-ribonucleosides with enhanced activity against human cytomegalovirus. J Virol 81:11604–11611. doi: 10.1128/JVI.01130-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams SL, Hartline CB, Kushner NL, Harden EA, Bidanset DJ, Drach JC, Townsend LB, Underwood MR, Biron KK, Kern ER. 2003. In vitro activities of benzimidazole d- and l-ribonucleosides against herpesviruses. Antimicrob Agents Chemother 47:2186–2192. doi: 10.1128/AAC.47.7.2186-2192.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krosky PM, Borysko KZ, Nassiri MR, Devivar RV, Ptak RG, Davis MG, Biron KK, Townsend LB, Drach JC. 2002. Phosphorylation of beta-d-ribosylbenzimidazoles is not required for activity against human cytomegalovirus. Antimicrob Agents Chemother 46:478–486. doi: 10.1128/AAC.46.2.478-486.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]