

ABSTRACT
We previously showed that the simian immunodeficiency virus SIVmac239 is susceptible to human immunodeficiency virus (HIV) integrase (IN) strand transfer inhibitors (INSTIs) and that the same IN drug resistance mutations result in similar phenotypes in both viruses. Now we wished to determine whether tissue culture drug selection studies with SIV would yield the same resistance mutations as in HIV. Tissue culture selection experiments were performed using rhesus macaque peripheral blood mononuclear cells (PBMCs) infected with SIVmac239 viruses in the presence of increasing concentrations of dolutegravir (DTG), elvitegravir (EVG), and raltegravir (RAL). We now show that 22 weeks of selection pressure with DTG yielded a mutation at position R263K in SIV, similar to what has been observed in HIV, and that selections with EVG led to emergence of the E92Q substitution, which is a primary INSTI resistance mutation in HIV associated with EVG treatment failure. To study this at a biochemical level, purified recombinant SIVmac239 wild-type (WT) and E92Q, T97A, G118R, Y143R, Q148R, N155H, R263K, E92Q T97A, E92Q Y143R, R263K H51Y, and G140S Q148R recombinant substitution-containing IN enzymes were produced, and each of the characteristics strand transfer, 3′-processing activity, and INSTI inhibitory constants was assessed in cell-free assays. The results show that the G118R and G140S Q148R substitutions decreased Km′ and Vmax′/Km′ for strand transfer compared to those of the WT. RAL and EVG showed reduced activity against both viruses and against enzymes containing Q148R, E92Q Y143R, and G140S Q148R. Both viruses and enzymes containing Q148R and G140S Q148R showed moderate levels of resistance against DTG. This study further confirms that the same mutations associated with drug resistance in HIV display similar profiles in SIV.
IMPORTANCE Our goal was to definitively establish whether HIV and simian immunodeficiency virus (SIV) share similar resistance pathways under tissue culture drug selection pressure with integrase strand transfer inhibitors and to test the effect of HIV-1 integrase resistance-associated mutations on SIV integrase catalytic activity and resistance to integrase strand transfer inhibitors. Clinically relevant HIV integrase resistance-associated mutations were selected in SIV in our tissue culture experiments. Not only do we report on the characterization of SIV recombinant integrase enzyme catalytic activities, we also provide the first research anywhere on the effect of mutations within recombinant integrase SIV enzymes on drug resistance.
INTRODUCTION
The limitations of current highly active antiretroviral therapy (HAART) include the incidence of drug resistance observed in patients, issues of drug adherence, and numbers of newly acquired infections. The emergence of drug-resistant viruses can lead to increases in viral load and treatment failure, and such viruses can be transmitted to both healthy individuals as well as to untreated HIV-positive individuals, therefore threatening the long-term efficacy of HAART. New classes of drugs and an understanding of pathways to drug resistance are important if we are to curtail the emergence of drug-resistant viruses. The latest class of approved antiretroviral (ARV) drugs for treatment of HIV infection are integrase (IN) strand transfer inhibitors (INSTIs), which include raltegravir (RAL), elvitegravir (EVG), and dolutegravir (DTG).
Integration is a fundamental step in the replication cycle of retroviruses that occurs after nuclear entry and is the last step before an irreversible productive infection of the cell transpires. Integration is a multistep process that is catalyzed by the viral integrase (IN) enzyme. Retroviral integrases catalyze the insertion of proviral DNA that is generated during reverse transcription into the host chromosome. In an initial reaction, also referred to as 3′ processing, dinucleotides from each 3′ end of the viral DNA are cleaved, thereby exposing 3′-reactive hydroxyl ends. Then, the 3′-end processed DNA is covalently linked to the host DNA in a process referred to as strand transfer (1, 2). INSTIs selectively inhibit the strand transfer reaction.
Animal models are an invaluable tool and can provide proof-of-concept data on efficacy, safety, and dosing regimens for pharmaceutical companies, regulators, policy-makers, health care providers, and patients. Animal models have also helped shed light on HIV pathogenesis. Indeed, both simian immunodeficiency virus (SIV) and chimeric simian/human immunodeficiency virus (SHIV) infection models have been used to recapitulate key aspects of the pathogenesis of human HIV-1 infection. Macaque models of rectal and vaginal transmission of SIV and SHIV have provided data that support the use of topical gels with reverse transcriptase (RT) inhibitors such as tenofovir (TFV) and MIV150 (3–5) and integrase inhibitors such as L-870812, an analogue of RAL (6), toward reductions of viral transmission rates. A long-acting form of a new INSTI, cabotegravir (also known as S/GSK-1265744), which is a DTG analogue, was shown to protect macaques against repeated vaginal and rectal challenges using SHIV (7, 8). Prolonged TFV monotherapy of macaques infected with SIV or SHIV resulted in the emergence of viral mutants with the K65R substitution in RT, the same mutation that is associated with TFV treatment failure (9, 10). In another study, macaques treated during preexposure prophylaxis with emtricitabine (FTC) or Truvada (TFV and FTC) and later infected by SIV sometimes developed the M184V substitution in RT, which is associated with resistance to FTC in HIV (11).
Previous reports by our group and others have shown that INSTIs are capable of inhibiting SIV/simian-tropic HIV (stHIV) replication and infectivity in tissue culture (12–14). We and others have also shown that INSTIs exhibit potent antiviral activity against SIV in tissue culture studies and in vivo with inhibitory concentrations in the nanomolar range; moreover, SIV that was mutated in IN displayed resistance profiles similar to those of HIV (12–14).
Now we wished to determine whether HIV and SIV share similar resistance pathways under tissue culture drug selection pressure with INSTIs and in cell-free assays and to test the effects of HIV-1 IN resistance-associated mutations on SIV IN catalytic activities.
Here, we present the biochemical characterization of SIV recombinant IN proteins on the basis of a series of SIV recombinant IN enzymes that carry clinically relevant HIV-1 drug resistance substitutions. This research involved site-directed mutagenesis and the cloning of the SIVmac239 IN open reading frame into a bacterial expression vector. Overall, our findings show similarities in resistance profiles between the IN enzymes of HIV-1 and SIV with regard to EVG and DTG and provide support for the preclinical testing of novel INSTIs in nonhuman primate models in order to assist in predictions of clinical outcomes.
MATERIALS AND METHODS
Cells and reagents.
The HEK293T cell line was obtained from the American Type Culture Collection (CRL-11268). TZM-bl cells were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program. TZM-bl and 293T cells were cultured and propagated as described previously (15). Whole monkey blood (obtained from Primus Bio-Ressources, Inc., Vaudreuil-Dorion, Québec, Canada) was collected from uninfected rhesus macaques (Macaca mulatta) and delivered in BD Vacutainer heparin tubes (Becton, Dickinson and Company). Rhesus macaque peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Hypaque (GE Healthcare) and stimulated with 10 μg/ml phytohemagglutinin A (Invitrogen) and 20 U/ml of human interleukin-2 (IL-2) (Roche). PBMCs were cultured as previously described (12). Merck & Co., Inc., Gilead Sciences, Inc., and ViiV Healthcare, Ltd., kindly supplied raltegravir (RAL), elvitegravir (EVG), and dolutegravir (DTG), respectively. Bovine serum albumin (BSA) and diethylenetriaminepentaacetic acid (DTPA) were purchased from Sigma-Aldrich (St. Louis, MO). DNA-BIND 96-well plates containing N-oxysuccinimide ester groups to bind viral long terminal repeat (LTR) DNA were purchased from Corning (Lowell, MA). Delfia Eu-labeled streptavidin (SA-Eu chelate) enhancement solution was purchased from PerkinElmer (Waltham, MA).
SIV infectivity of TZM-bl cells.
Noncompetitive short-time infectivity assays in TZM-bl cells were used to evaluate SIV infectivity. Single-cycle infectivity was measured through the infection of 30,000 TZM-bl cells using a constant amount (normalized against RT activity) of wild-type (WT) and mutant viruses using serial 1:3 dilutions in a 96-well plate (Corning). After 48 h, luciferase activity was measured using the luciferase assay system (Promega, Madison, WI). The results were analyzed with GraphPad Prism 5.0 software to calculate the 50% effective concentration (EC50) and 95% confidence intervals (95% CI). The RT-normalized infectivity of WT virus was defined as 100%, and relative infectivity was calculated in proportion to induced levels of luciferase activity.
Selection of drug-resistant viruses.
Cell culture selections were performed using WT virus. Selections were performed as previously described using rhesus macaque PBMCs (16). Stimulation of PBMCs was facilitated with RPMI 1640 medium (Gibco/Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen), 2 mM l-glutamine, 50 U of penicillin/ml, 50 g of streptomycin/ml, 5% (vol/vol) IL-2 (Roche), and 10 μg of phytohemagglutinin (Invitrogen) per ml and maintained at 37°C under 5% CO2. Prior to infection, rhesus PBMC cultures were stimulated for 48 to 72 h. Briefly, WT viruses were serially passaged in rhesus PBMCs in the presence of increasing concentrations of DTG, EVG, and RAL for as long as 29 weeks. Potential resistance-associated mutations were identified by sequencing the IN region of pol gene viral RNA. SIV RNA extraction was performed using the QIAamp RNA minikit (Qiagen, Valencia, CA).
Cloning and mutagenesis.
The pET15b vector that carries an N-terminal His tag sequence was used for cloning. The SIV integrase-coding region was amplified using 2 sets of primers (Mac_BamHI and Mac_NdeI) (Table 1) bearing BamHI and NdeI restriction sites for the construction of pET15b. Double digestion of PCR product was carried out simultaneously using BamHI and NdeI restriction enzymes. Digested PCR products were dephosphorylated using calf intestinal alkaline phosphatase (CIP) according to the manufacturer's instructions (New England BioLabs, Inc., Whitby, ON, Canada) and then purified using the QIAquick PCR purification kit (Qiagen, Toronto, ON, Canada). Subsequently, SIVmac239 integrase (GenBank accession no. M33262) was introduced into the pET15b plasmid by ligation using T4 DNA ligase as described in the manufacturer's protocol (New England BioLabs, Inc., Whitby, ON, Canada). The F185H substitution was introduced by site-directed mutagenesis to increase protein solubility as previously described (17). The latter substitution has been shown to increase the solubility of recombinant integrase without changing its activity in vitro (18, 19). The primer pairs used for site-directed mutagenesis to produce the G118R, G140S, Y143R, Q148R, N155H, and R263K mutations have been described previously (12). The primer pairs for other desired mutations in this study (i.e., E92Q, T97A, and F185H) are described in Table 1. Briefly, reaction mixtures were prepared with 10 ng of double-stranded DNA (dsDNA) template, 125 ng of each oligonucleotide primer, 5 μl of 10 × reaction buffer (Invitrogen), 3 μl of QuikSolution, and 2 μl of 5 mM deoxynucleoside triphosphate (dNTP) solution in a final volume of 50 μl. Pfu Turbo DNA polymerase (2.5 U) was added to the reaction mixture just before the start of the denaturation cycle. Double-stranded plasmid DNA was denatured at 95°C, after which oligonucleotide primers were annealed to the plasmid at 60°C. Synthesis and extension of the new DNA strand were catalyzed by DNA polymerase at 72°C. Once the temperature cycles were completed, DpnI (10 U) was added to each amplification reaction mixture, and the mixture was incubated for 2 h at 37°C to digest methylated and hemimethylated parental DNA. Digested products were transformed into Escherichia coli XL10-Gold ultracompetent cells (Stratagene, Agilent Technologies, Mississauga, ON, Canada): Tetr Δ(mcrA)183 Δ(mcrCB-hsdSMR-mrr)173 endA1 supE44 thi-1 recA1 gyrA96 relA1 lac Hte [F′ proAB lacIq ZΔM15 Tn10 (Tetr) Amy Camr].
TABLE 1.
List of the primers and DNA substrates used for cloning, site-directed mutagenesis, strand transfer, and 3′ processing
| Step | Primer | Sequence (5′→3′)a |
|---|---|---|
| Cloning | Mac_NdeI | 5′-GCGGCAGCCATATGTTCTTGGAAAAGATAGAGCCAG-3′ |
| Mac_BamHI | 5′-TTAGCAGCCGGATCCCTATGCCACCTCTCTAGCCTCTC-3′ | |
| Site-directed mutagenesis | E92Q sense | 5′-GCAGAGGTAATTCCACAACAGACAGGAAGACAGAC-3′ |
| E92Q antisense | 5′-GTCTGTCTTCCTGTCTGTTGTGGAATTACCTCTGC-3′ | |
| T97A sense | 5′-CACAAGAGACAGGAAGACAGGCAGCACTATTTCTGTTAAAA-3′ | |
| T97A antisense | 5′-TTTTAACAGAAATAGTGCTGCCTGTCTTCCTGTCTCTTGTG-3′ | |
| F185H sense | 5′-AATGGCAGTTCATTGCATGAATCATAAAAGAAGGGGAGGAATAGGG-3′ | |
| F185H antisense | 5′-CCCTATTCCTCCCCTTCTTTTATGATTCATGCAATGAACTGCCATT-3′ | |
| DNA substrates used for strand transfer assay | Donor LTR DNA sense (primer A) | 5AmMC12-ACCCTTTTAGTCAGTGTGGAAAATCTCTAGCAGT-3′ |
| Donor LTR DNA antisense (primer B) | 5′-ACTGCTAGAGATTTTCCACACTGACTAAAAG-3′ | |
| Target DNA sense (primer C) | 5′-TGACCAAGGGCTAATTCACT-3Bio | |
| Target DNA antisense (primer D) | 5′-AGTGAATTAGCCCTTGGTCA-3Bio | |
| DNA substrates used for 3′ processing | LTR-3′-sense (primer E) | 5AmMC12-ACCCTTTTAGTCAGTGTGGAAAATCTCTAGCAGT-BioTEG |
| LTR-3′-antisense (primer F) | 5′-ACTGCTAGAGATTTTCCACACTGACTAAAAG-3′ |
“5AmMC12” refers to a reactive amino group attached to the 5′ end of the oligonucleotides using a 12-carbon linker, and “BioTEG” refers to a modified 3′ end with a biotin tag (3Bio) attached via a triethylene glycol spacer. BamHI and NdeI restriction sites are in boldface in the sequences of the Mac_NdeI and Mac_BamHI cloning primers. All primers were designed using QuikChange Primer Design software (Agilent Technologies).
Generation of genetically homogenous SIV.
To generate replication-competent homogenous SIVmac239, 12.5 μg of each appropriately mutated SIVmac239 proviral DNA construct was separately transfected into 293T cells using Lipofectamine 2000 (Invitrogen) as recommended by the manufacturer. 293T cells were incubated in Opti-MEM (2% FBS) for 6 h prior to being washed with phosphate-buffered saline (PBS) (GIBCO) and further grown in fresh medium. At 48 h postwash, cell supernatants were harvested, centrifuged, passed through a 0.45-μm-pore filter to remove cellular debris, and treated with Benzonase (EMD), and cell-free viral stocks were stored at −80°C. Virion-associated reverse transcriptase (RT) activity was measured as previously described (20).
Expression and purification of recombinant SIV integrase.
Escherichia coli strain XL10-Gold ultracompetent cells were used for plasmid production. All mutations were verified by DNA sequencing (Genome Québec). Plasmids encoding the integrase proteins of SIV were expressed using Escherichia coli BL21(DE3) competent cells (Stratagene, Agilent Technologies, Mississauga, ON, Canada): BF−dcm ompT hsdS(rB− mB−) gal λ(DE3). Typically, bacterial cultures were grown in 1 liter Luria-Bertani (LB) broth supplemented with 100 μg/ml at 37°C with shaking (225 rpm) until the culture reached an optical density at 600 nm (OD600) of 0.4 to 0.6; induction of protein expression was initiated by adding isopropyl-β-d-thiogalactopyranoside (IPTG) to a concentration of 1 mM. The culture was further incubated for 4 h at 37°C with shaking (225 rpm). Cells were harvested by centrifugation at 5,000 rpm at 4°C for 10 min, and cell pellets were immediately frozen at −80°C until required. Protein purification was carried out with cold buffers, and all chromatography steps were performed at 4°C. Frozen bacterial pellets were thawed and then resuspended into lysis buffer (50 mM Tris, 2 mM dithiothreitol [DTT], pH 7.5, supplemented with Complete EDTA-free protease inhibitor cocktail tablet with 1 tablet per 50 ml of resuspension [Roche]) and lysed by sonication. The lysates were centrifuged (12,500 rpm for 5 min), and supernatants were kept on ice while the pellets were resuspended in resuspension buffer (50 mM HEPES, 1 M MgCl2, 50 μM ZnCl2, 2 mM DTT, pH 7.0, supplemented with protease inhibitor cocktail tablet), lysed, and pelleted again by centrifugation (12,500 rpm for 5 min). Supernatants from lysis and resuspension steps were incubated with nickel-nitrilotriacetic acid (Ni-NTA) agarose beads (Qiagen) for 1 h at 4°C with mild agitation. The remainder of the purification of integrase recombinant proteins was performed as previously described (18). His-tagged integrase protein was then eluted by a gradient of increasing imidazole concentrations (0 to 1 M). Fractions containing purified integrase were dialyzed overnight into storage buffer (20 mM HEPES, 1 M NaCl, 1 mM EDTA, 2 mM DTT, 10% glycerol, pH 7.5). Dialyzed purified integrase proteins were aliquoted and rapidly frozen at −80°C.
Determination of protein concentrations.
Protein concentrations were measured using the absorbance at 280 nm (A280) and an extinction coefficient of 50,420 M−1 cm−1, calculated using ProtParam (21), and confirmed by the Bradford protein assay. Bovine serum albumin (BSA) was used a standard.
PAGE.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to assess the purity and estimate the molecular weights of proteins. SDS-PAGE was performed using a 4% stacking gel (pH 6.8) and 10% resolving gel (pH 8.3) as reported by Sambrook and Russell (22). A 30% acrylamide stock solution was prepared by mixing 29% (wt/vol) acrylamide and 1% (wt/vol) N,N′-methylene-bis-acrylamide in distilled H2O. Tris buffers (1.5 M at pH 8.3 and 0.5 M at pH 6.8) were prepared in distilled water, and the pH was adjusted using HCl. The acrylamide and Tris buffer solutions were stored at 4°C. Ten percent SDS (wt/vol) in distilled water was stored at room temperature, while 10% (wt/vol) ammonium persulfate was prepared in distilled water and stored at 4°C. Electrophoresis buffer was prepared with 25 mM Tris base, 250 mM glycine, and 0.1% SDS, pH 8.3. Protein samples, diluted 1:1 (vol/vol) into 2× SDS gel loading buffer (1.5 M Tris-HCl, 4% SDS, 20% glycerol, 0.2 M DTT, bromophenol blue) along with unstained protein molecular weight marker (Thermo Fisher Scientific), were boiled for 5 min before loading on the gel. The gel was electrophoresed at 80 V in order for the samples to migrate through the stacking gel, and then the voltage was increased to 150 V as the dye entered the resolving gel. Proteins were visualized by staining using a solution containing 0.25% (wt/vol) Brilliant blue R (Sigma), 40% (vol/vol) methanol, and 7% (vol/vol) acetic acid, followed by destaining in a solution consisting of 10:9:1 (vol/vol/vol) methanol-distilled water-glacial acetic acid.
Expression and purification of SIV recombinant integrase proteins and calibration of enzyme activities.
All of the HIV-1 resistance-associated substitutions introduced into the SIVmac239 integrase coding sequence and examined in this study are the same as those identified in drug selection studies in both SIVmac239 and HIV. Plasmids encoding SIV integrase carrying the WT sequence or an E92Q, T97A, G118R, Y143R, Q148R, N155H, R263K, E92Q T97A, E92Q Y143R, G140S Q148R, or R263K H51Y substitution were generated by site-directed mutagenesis. WT and variant enzymes were expressed with a hexahistidine tag and purified simultaneously. WT or mutated SIV recombinant proteins were expressed in and purified from E. coli BL-21(DE3) cells to greater than 90% homogeneity. Protein purification was monitored by SDS-PAGE analysis. The homogeneity of all variant proteins was deduced by denaturing electrophoretic analysis with Coomassie staining (Fig. 1). There was no major difference in yields between various mutant proteins.
FIG 1.

SDS-PAGE analysis of purified recombinant SIVmac239 integrase proteins. Lane 1, molecular mass ladder; lanes 2 to 13, SIVmac239 IN proteins as labeled. About 2 to 3 μg of protein was loaded into lanes 2 to 13. After nickel affinity chromatography and dialysis, protein samples were analyzed on 10% polyacrylamide gels as outlined in Materials and Methods. The arrowhead indicates a molecular mass of ∼34 kDa, corresponding to that of the recombinant SIV protein.
To determine the optimal protein concentration of SIV recombinant enzymes in the strand transfer assay, we employed microtiter plates and increasing protein concentrations in the absence of drug. The WT enzyme showed maximal activity at 400 nM (Fig. 2); the variant enzymes also attained maximal activity at a protein concentration of 400 nM (data not shown). Therefore, subsequent experiments utilized protein concentrations of 400 nM.
FIG 2.
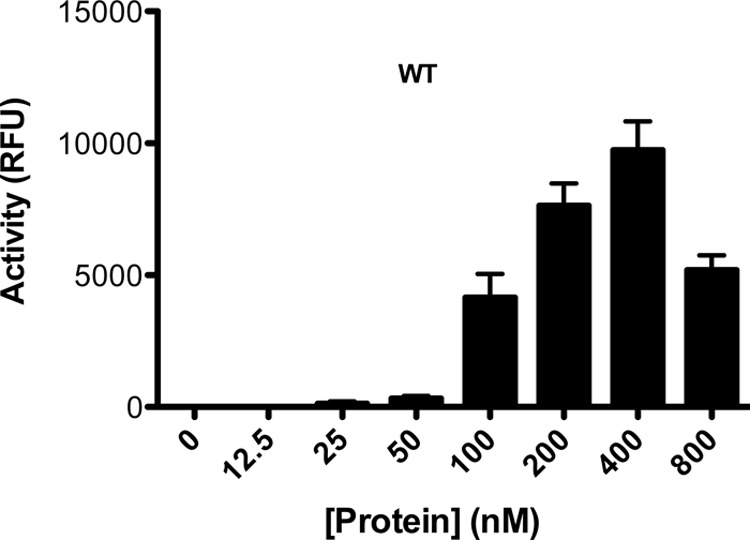
Titration for the optimal integrase concentration to be used in strand transfer assays. Various SIVmac239IN(WT) protein concentrations, ranging from 0 to 800 nM were tested. Although these experiments were performed for all of the proteins tested in this article, only results with WT protein are shown. RFU, relative fluorescence units.
DNA substrates used for in vitro assays.
All oligonucleotide substrates were purchased from Integrated DNA Technologies (IDT; Coralville, IA). To prepare functional duplexes, oligonucleotides (DNA substrates for strand transfer and 3′ processing in Table 1) were mixed in microcentrifuge tubes at a molar ratio of 1:1 of sense and antisense primers in 10 mM Tris (pH 7.8)–0.1 mM EDTA. Duplexes were annealed by heating at 95°C for 10 min and slow cooling at room temperature over a period of 4 h and were stored at −20°C until needed. “5AmMC12” refers to a reactive amino group attached to the 5′ end of the oligonucleotides by a 12-carbon linker. “BioTEG” refers to a modified 3′ end with a biotin tag attached via a triethylene glycol spacer. The BamHI and NdeI restriction sites are shown in boldface in the sequences of the Mac_NdeI and Mac_BamHI cloning primers in Table 1. All primers were designed using QuikChange Primer Design software (Agilent Technologies).
Strand transfer activity assay.
The strand transfer assay used in our laboratory has been described previously (15, 23, 24). Strand transfer assays with WT and variant enzymes were carried out using DNA-BIND 96-well plates (Corning). The preprocessed LTR oligonucleotide duplexes A/B (primers A and B in Table 1) were diluted to 80 nM in phosphate-buffered saline (PBS [pH 7.4]) (Bioshop) and were covalently linked to Costar DNA-BIND 96-well plates by incubation at 4°C overnight. The following day, plates were blocked in blocking buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 0.5% BSA) for a minimum of 2 h. Before use, the plates were washed twice with each of 1 × PBS (pH 7.4) and assay buffer consisting of 50 mM morpholinepropanesulfonic acid (MOPS [pH 6.8]), 50 μg/ml BSA, 0.15% 3-[(3-cholamidopropyl)-dimethyl-ammonio]-1-propanesulfonate (CHAPS), 50 mM NaCl, and 30 mM MnCl2. Purified SIV integrase proteins were diluted in assay buffer supplemented with 5 mM DTT, and 100 μl of purified proteins (final concentration, 400 nM) was added followed by incubation for 30 min at 37°C. This step was followed by an additional 1 h of incubation at 37°C in the presence of biotinylated target DNA duplex C/D, and then the strand transfer reaction plates were washed three times with wash buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.05% [vol/vol] Tween 20, 2 mg/ml BSA). Plates were then incubated with Eu-labeled streptavidin (PerkinElmer) diluted to 0.025 μg/ml in wash buffer in the presence of 50 μM DTPA. The plates were then rinsed with the same wash buffer, followed by the addition of Delfia enhancement solution (PerkinElmer). The enhancement solution is used to quantify europium (Eu3+) in dissociation-enhanced time resolved fluorescence (TRF). The low pH (<4) of the enhancement solution causes the ionization of conjugated Eu to free aqueous Eu3+. A FLUOStar Optima multilabel plate reader (BMG LabTech) in TRF mode was used to measure the TRF of Eu3+, where it was excited at 355 nm and emitted at 612 nm. The values of enzyme activity (V) in relative fluorescence units (RFU) and target DNA concentration (S) were fit by nonlinear regression using GraphPad Prism V 5.0 (GraphPad Software, San Diego, CA) to the following Michaelis-Menten derivative equations: V = Vmax′ × [S]/([S] + Km′) and Km′ = Vmax′ × [S]/V − [S].
In this case, the pseudo-Michaelis constant (i.e., the apparent Km, or Km′) reflects the apparent affinity of the enzyme for the target DNA substrate ([S]), and the apparent maximal activity (Vmax′) reflects the maximum activity obtainable with the same concentration of the protein in a similar assay, regardless of the substrate concentration. To determine enzyme performance, we calculated the ratio of Vmax′ to Km′.
3′-processing assay.
The determination of the 3′-processing activity of purified recombinant integrase proteins was performed as previously described (25). IN 3′-processing activity in the absence of INSTIs was measured using a TRF-based assay using a system similar to that described above for the strand transfer assay, except that unprocessed donor DNA instead of target DNA was used. First, 3′-biotinylated unprocessed LTR duplexes E/F (primers E and F in Table 1) were diluted in PBS at pH 7.4 and covalently linked at various concentrations to DNA-BIND 96-well plates under similar conditions to the strand transfer assay. Negative-control wells had only reaction buffer added without any LTR. To initiate the 3′-processing reaction, purified integrase recombinant proteins (400 nM) diluted in reaction buffer were added, and plates were incubated at 37°C for 2 h as described previously (25). After the reactions were completed, three washes with 250 μl wash buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.05% [vol/vol] Tween 20, 2 mg/ml BSA) were carried out to remove all traces of cleaved biotinylated dinucleotides and proteins. All subsequent steps of the assay were performed as described above for the strand transfer assay, except for the analysis. The maximum possible signal (3′OHmax) represented wells that contained total unprocessed LTR substrate and no protein; the observed signal (3′OHobserved) represents wells with remaining uncleaved LTR substrate. Therefore, the actual 3′-processing activity of each sample was calculated using the equation 3′OHactual = 3′OHmax − 3′OHobserved.
The data from the 3′-processing reaction were used to calculate kinetic parameters by fitting to binding saturation curves in order to obtain kinetic parameters; this was necessary since 3′ processing does not follow Michaelis-Menten kinetics (25, 26). The calculated Kd was equated to the apparent Km (Km′) in our analysis. Maximal 3′-processing activity was calculated by fitting data to binding saturation curves. Data were analyzed using GraphPad Prism 5.0, and are expressed as the mean ± standard error of the mean (SEM) on the basis of three or more independent experiments performed in triplicate.
Competitive inhibition of strand transfer activity by DTG, EVG, and RAL.
The susceptibility of SIV recombinant integrase proteins to INSTIs was assessed by conducting competitive inhibition assays in the presence of DTG, EVG, or RAL. Drug stock solutions were prepared at a concentration of 6 μM and diluted in compound dilution buffer (assay buffer plus 10% dimethyl sulfoxide [DMSO]) to concentrations between 0 and 1,000 nM. Competitive inhibition assays were performed in the presence of various target DNA concentrations (16, 32, 64, and 128 nM). Briefly, preprocessed LTR (effective LTR concentration, 80 nM) was prepared as described above and used to coat DNA-BIND plates. Purified SIV integrase proteins (effective concentration, 400 nM) in assay buffer supplemented with 5 mM DTT were added to each well and incubated for 30 min at room temperature. Twenty-five microliters of diluted INSTI was added to each well followed by 25 μl of appropriately diluted biotinylated target DNA duplex. This was followed by a 1 h of incubation at 37°C. Subsequent steps were performed as described above for the strand transfer assay. Inhibitory constants (Ki) were calculated by fitting data to a competitive inhibition algorithm using GraphPad Prism 5.0. Ki values were calculated and were transformed to fold change (FC) values by dividing by the reference Ki obtained with WT SIV protein.
Data analysis.
Unless otherwise indicated, all experiments were repeated at least 3 times. Strand transfer values in the absence of drugs were arbitrarily set at 100%. Whenever relevant, statistically significant differences between data sets for two or more integrase proteins were determined using a one-sample two-tailed t test. P values that were ≤0.05 were used to indicate statistically significant differences between different results.
RESULTS
Effect of integrase substitutions on viral infectivity.
The infectivity of mutated viruses was measured relative to the wild type (WT) using the single-cycle assay in TZM-bl cells with increasing inocula of WT or E92Q, T97A, and R263K H51Y mutant viruses in the absence of INSTIs. The addition of H51Y to R263K reduced infectivity by 63% compared to that of the WT, while the T97A substitution increased viral infectivity by 223% compared to that of the WT (Fig. 3). E92Q-mutated SIV possessed 40% diminished infectivity. This agrees with data that have been obtained for HIV-1 (27).
FIG 3.
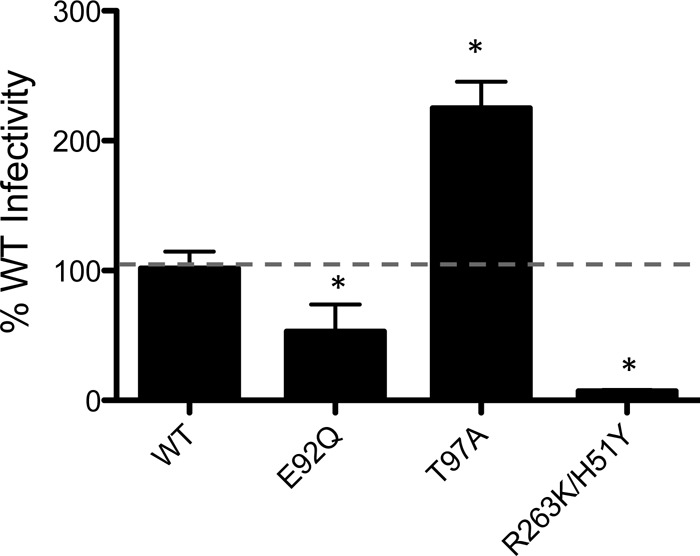
Effects of SIVmac239 integrase substitutions on viral infectivity. The infectiousness of SIVmac239IN(WT), SIVmac239IN(E92Q), SIVmac239IN(T97A), and SIVmac239IN(R263K H51Y) was assessed by quantifying luciferase activity at 48 h postinfection in TZM-bl cells. Infectivity of WT and mutant viruses is represented by means ± SEM for each of three independent TZM-bl infectivity assays normalized against WT signal, which was arbitrarily set at 100%. Asterisks indicate statistically significant differences. Statistical significance was calculated for individual pairs of data using a one sample two-tailed t test with a statistical cutoff of P ≤ 0.05.
Detection of IN substitutions in the presence of increasing pressure of INSTIs.
The dose escalation patterns of DTG, EVG, and RAL are shown in Fig. 4. INSTI concentrations were increased in a stepwise manner. In the in vitro passage experiments, each passage began in the presence of 10 nM drug. The concentration of DTG was increased to 100 nM at week 28 (Fig. 4), while the concentration of RAL was increased to 100 nM at week 26. In contrast, the concentration of EVG was increased to 50 nM at week 27, which remains lower than those of DTG and RAL (Fig. 4). Indeed, viruses treated with DTG and RAL were not able to replicate at concentrations of these drugs above 100 nM, while the concentration of EVG above which replication ceased was 50 nM (Table 2).
FIG 4.
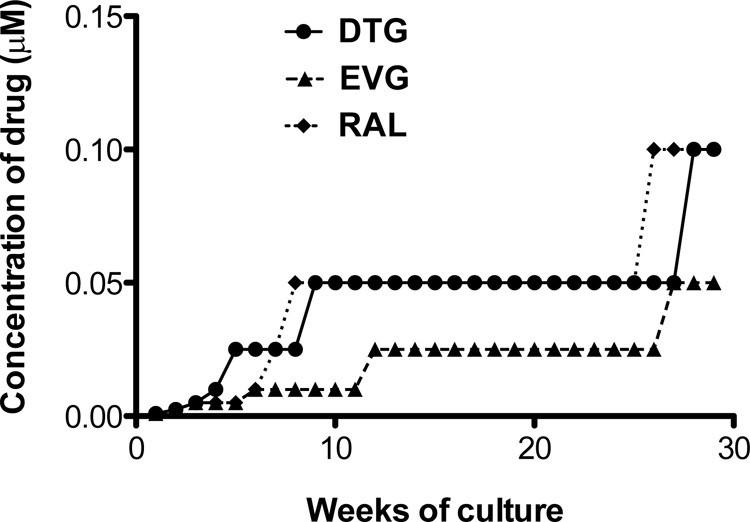
Long-term culture of infected rhesus PBMCs with increasing concentrations of DTG, EVG, or RAL. Rhesus PBMCs were infected with the SIVmac239 WT strain and passaged once weekly in the presence of INSTIs.
TABLE 2.
Emergent substitutions during serial passage in rhesus PBMCs infected with SIVmac239 in the presence of increasing concentrations of INSTIs
| Drug | Result at wk 21–29: |
|
|---|---|---|
| Concn (μM) | Acquired mutations | |
| RAL | 0.1 | None |
| EVG | 0.05 | A54A/V, E92Q, A265A/G, V277V/M |
| DTG | 0.1 | R263K, A54A/V, V277V/M |
In vitro passage experiments were performed in rhesus PBMCs under selection pressure with DTG, EVG, or RAL using WT SIVmac239 virus. Viral RNA was extracted from cell culture fluids and sequenced for any changes in the integrase coding region (Table 2). In the presence of DTG, the R263K substitution was observed at week 21. The R263K mutation has been previously characterized as a resistance substitution against DTG in HIV-1 (15, 23). Viruses harboring A54A/V, A265A/G, and V277V/M were identified at week 21 during serial passage experiments with EVG, and the E92Q substitution was detected at week 29. Viruses under RAL selective pressure did not select for any substitutions. E92Q is the most frequent substitution in HIV-1-infected individuals treated with EVG (28–30) and confers high levels of resistance against this compound (31).
Impact of IN mutations on 3′-processing activity.
To address whether the above-described mutational impact on viral replication fitness and antiviral susceptibility could be attributed to enzymatic function, we analyzed the biochemical properties of the WT and variant recombinant IN enzymes in time-resolved fluorescence cell-free 3′-processing and strand transfer assays performed in microtiter plates in the absence of INSTIs.
First, we used the 3′-processing assay to investigate whether there were differences in apparent LTR binding (Km′) and/or 3′-processing activities of the different enzymes in the presence of increasing concentrations of unprocessed LTR DNA. Integrase enzymes containing the T97A, Y143R, G140S Q148R, and R263K H51Y substitutions displayed similar Km′s to those of the WT for donor LTR DNA (Fig. 5A). In contrast, the G118R, N155H, R263K, E92Q T97A, and E92Q Y143R substitutions resulted in decreased Km′s compared to those of the WT, with the activity of the E92Q T97A mutant being the only activity that was significantly different (Fig. 5A). With regard to 3′-processing activity, all of the variants possessed activities comparable to those of the WT, with the exception of the E92Q, Y143R, and E92Q Y143R enzymes. Both the E92Q and E92Q Y143R enzymes displayed statistically higher 3′-processing activities than the WT, whereas the Y143R enzyme showed decreased activity that was not statistically different from that of the WT (Fig. 5B). The sequential addition of G140S to Q148R was innocuous with regard to 3′-processing activity (Fig. 5B).
FIG 5.

Measurement of 3′-processing activities of integrase. 3′-processing activity was measured with enzyme concentrations fixed at 400 nM and various concentrations of immobilized LTR DNA. Time-resolved fluorescence (TRF) data were calculated as described in Materials and Methods, and the data were fitted in GraphPad Prism 5.0 to binding saturation curves to obtain values of LTR DNA Km′ (A) and maximal 3′-processing activity (B). The data represent the means from at least 3 independent experiments, each performed in triplicate. Error bars represent the standard errors of the means (SEM). Asterisks indicate statistically significant differences. Statistical significance was calculated for individual pairs of data using a one-sample two-tailed t test with a statistical cutoff of P ≤ 0.05.
Effects of single and double mutations on IN strand transfer activity.
To examine strand transfer, we used preprocessed LTR DNA, thereby decoupling the strand transfer activity of integrase from its 3′-processing activity. Strand transfer experiments were performed using fixed concentrations of purified integrase proteins (400 nM) and preprocessed LTR mimic DNA (80 nM) in the presence of increasing concentrations of target DNA (0 to 300 nM): i.e., a wider range of target DNA was used for the variant enzymes that possessed higher apparent target DNA binding (Km′), therefore achieving saturating concentrations of target DNA. Calculated values for apparent strand transfer activity and target DNA Km′ are summarized in Fig. 6 and Table 3. The target DNA Km′ of the vast majority of the mutated recombinant proteins was not significantly different from that of the WT (Fig. 6; Table 3). The N155H and G140S Q148R substitutions resulted in small decreases in Km′s that were statistically significant (1.4-fold and 1.79-fold, respectively). However, the G118R substitution caused a significant decrease in Km′, ∼5.2-fold, consistent with results previously obtained in HIV (32, 33). The presence of single or double substitutions did not significantly impact strand transfer activity. The presence of N155H and G140S Q148R resulted in 1.25- and 1.5-fold decreases in strand transfer activity, respectively. The E92Q T97A and E92Q Y143R combinations of substitutions resulted in 1.24- and 1.37-fold increases in strand transfer activity, respectively.
FIG 6.
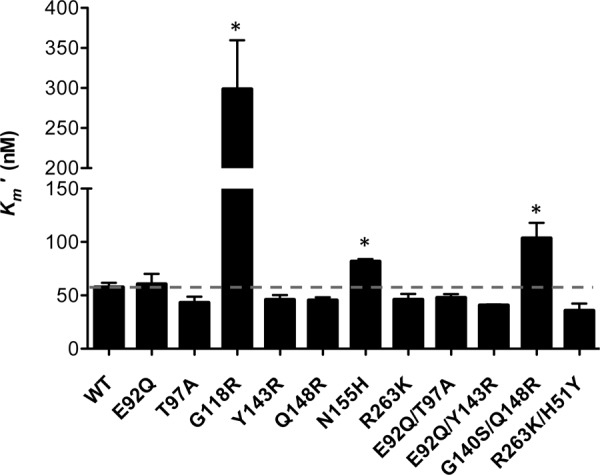
Effect of amino acid substitutions on functional binding (Km′) of target DNA by variant SIV integrase proteins. Strand transfer activities were measured using an immobilized preprocessed-LTR plate-based assay. GraphPad Prism 5.0 was used to transform TRF data using Michaelis-Menten equations to obtain target DNA Km′. Asterisks indicate statistically significant differences. Statistical significance was calculated for individual pairs of data using a one-sample two-tailed t test with a statistical cutoff of P ≤ 0.05.
TABLE 3.
Strand transfer parameters for purified recombinant SIV integrase proteins
| SIVmac239 IN protein type | Fixed [LTR] − strand transfer activitya |
|||
|---|---|---|---|---|
| Apparent maximal strand transfer activity (Vmax′ [mean ± SEM RFU/h]) | Apparent target DNA (Km′ [mean ± SEM nM]) | Vmax′/Km′ (RFU/h/nM) | FC in Vmax′/Km′ relative to WT | |
| WT | 25,751 ± 603.8 | 57.71 ± 4.085 | 446.2 | 1.0 |
| Recombinant | ||||
| E92Q | 33,247 ± 1,808 | 60.66 ± 9.523 | 548.1 | 1.2 |
| T97A | 31,062 ± 1,331 | 43.18 ± 5.693 | 719.4 | 1.6 |
| G118R | 28,266 ± 3,393 | 298.7 ± 60.85* | 94.63 | 0.2 |
| Y143R | 27,142 ± 858.5 | 45.96 ± 4.398 | 590.55 | 1.3 |
| Q148R | 31,008 ± 625.1* | 45.40 ± 2.778 | 683 | 1.5 |
| N155H | 21,494 ± 236.3* | 81.85 ± 2.195* | 262.6 | 0.6 |
| R263K | 23,851 ± 46.18 | 46.18 ± 5.280 | 516.5 | 1.2 |
| E92Q T97A | 31,916 ± 1,175* | 47.78 ± 3.355 | 667.9 | 1.5 |
| E92Q Y143R | 35,339 ± 228.4* | 40.84 ± 0.8202 | 865.3 | 1.9 |
| G140S Q148R | 16,976 ± 973.5* | 103.7 ± 14.17* | 163.7 | 0.4 |
| R263K H51Y | 29,475 ± 1,650 | 45.22 ± 6.479 | 651.8 | 1.5 |
All data represent the results from at least 3 independent experiments. Significant fold changes (FC) are indicated in boldface. *, statistically significant differences. Statistical significance was calculated for individual pairs of data using a one-sample two-tailed t test with a statistical cutoff of P ≤ 0.05.
Since in vivo enzyme activity does not differentiate between Vmax′ (apparent Vmax refers to maximal velocity [i.e., strand transfer activity]) and Km′ effects, enzyme performance calculations (Vmax′/Km′) were used to estimate the effect of substitutions in enzymes under physiological conditions, where substrates are present at limiting concentrations (24, 34). This analysis provides a more accurate analysis of single-turnover enzymes such as integrase (26). We observed that G118R, N155H, and G140S/Q148R caused 80%, 40%, and 60% reductions in Vmax′/Km′, respectively (Table 3), while the T97A and E92Q Y143R enzymes showed increased Vmax′/Km′s of ∼37% and ∼47%, respectively, compared to the WT (Table 3).
Activity of INSTIs against SIV IN enzymes in strand transfer activity assays.
We next investigated the effects of the single or double integrase substitutions on levels of resistance to DTG, EVG, and RAL. Target saturation assays were performed in the presence of these drugs (0 to 1 μM final concentration) (Fig. 7A). The data were fitted by nonlinear regression analysis to a competitive enzyme inhibition equation using GraphPad Prism 5.0 to yield an inhibitory constant, Ki, which is an indicator of how potent an inhibitor is (Table 4). In contrast to a 50% inhibitory concentration (IC50), the Ki is an intrinsic property of each enzyme-inhibitor complex and is independent of the substrate concentration (35). Ki fold change (FC) was calculated by dividing the Ki values obtained using the different enzyme variants by that of WT enzyme (Fig. 7B to D; Table 4). The results show that the WT SIV recombinant protein showed high susceptibility to all INSTIs tested, with Ki values in the nanomolar range (1 to 2.6 nM) (Table 4). The competitive inhibition model showed that most of the variant enzymes remained susceptible to DTG (Table 4). In contrast, the Q148R and E92Q Y143R proteins displayed low levels of resistance against DTG (6.4- and 2-fold, respectively), while the G140S Q148R variant enzyme showed high-level resistance (26.6-fold). Substitutions at the E92Q, T97A, Y143R, N155H, R263K, E92Q T97A, and R263K H51Y positions did not have a significant effect on resistance to DTG (Table 4). Both Q148R and G140S Q148R variant enzymes conferred cross-resistance against both RAL and EVG. The Q148R and G140S Q148R substitutions caused ∼122- and ∼254-fold resistance to EVG and ∼120- and ∼160-fold resistance to RAL. The E92Q, N155H, and E92Q Y143R recombinant proteins exhibited low levels of resistance against EVG (∼4-, 4-, and 6-fold, respectively), while the remaining variant enzymes showed susceptibility to EVG that was close to that of WT enzyme (Table 4). The Y143R recombinant protein remained susceptible to DTG and EVG but conferred moderate resistance against RAL (9.4-fold). The addition of the E92Q mutation to Y143R further decreased susceptibility to RAL (∼51-fold for the combination compared to 9.4-fold for Y143R alone). The G118R variant enzyme was associated with low resistance to all INSTIs tested (∼2- to 5.6-fold). Both E92Q and E92Q T97A conferred low levels of resistance against RAL. The R263K enzyme was susceptible to RAL and exhibited low resistance to EVG. However, the combination of H51Y and R263K slightly increased resistance to all INSTIs compared to R263K alone, although this effect was not statistically significant.
FIG 7.

Competitive inhibition of strand transfer by DTG, EVG, and RAL. Strand transfer reactions were carried out in the presence of 400 nM integrase proteins. Raw data from two independent experiments were fitted for the competitive inhibition equation using GraphPad Prism 5.0. (A) A representative plot for competitive inhibition of WT integrase in the presence of DTG is shown. (B, C, and D) The fold change susceptibility of each variant protein for DTG (B), EVG (C), and RAL (D) was calculated for each enzyme by dividing the Ki of the corresponding variant enzyme by that of the WT. Data represent the means of results from at least three independent experiments. Error bars represent standard errors of the means (SEM). Asterisks indicate statistically significant differences. Statistical significance was calculated for individual pairs of data using a one-sample two-tailed t test with a statistical cutoff of P ≤ 0.05.
TABLE 4.
Susceptibilities of purified SIV recombinant integrase proteins for DTG, EVG, and RAL
| SIVmac239 IN protein type | Result fora: |
|||||
|---|---|---|---|---|---|---|
| DTG |
EVG |
RAL |
||||
| Ki (mean ± SEM nM) | ∼FC | Ki (mean ± SEM nM) | ∼FC | Ki (mean ± SEM nM) | ∼FC | |
| WT | 1.1 ± 0.19 | 1.0 | 2.7 ± 0.29 | 1.0 | 2.1 ± 0.22 | 1.0 |
| Recombinant | ||||||
| E92Q | 1.5 ± 0.54 | 1.4 | 11.0 ± 5.0* | 4.1 | 4.7 ± 0.36* | 2.2 |
| T97A | 0.95 ± 0.10 | 0.9 | 3.3 ± 0.51 | 1.2 | 3.9 ± 1.6 | 1.8 |
| G118R | 5.9 ± 0.65* | 5.6 | 7.3 ± 2.0* | 2.7 | 5.5 ± 1.3* | 2.6 |
| Y143R | 1.8 ± 0.95 | 1.7 | 3.6 ± 1.3 | 1.3 | 19.8 ± 3.9* | 9.4 |
| Q148R | 6.7 ± 2.4* | 6.4 | 328 ± 137* | 122.7 | 252 ± 111* | 119.8 |
| N155H | 1.9 ± 0.80 | 1.8 | 11.3 ± 1.6* | 4.2 | 8.8 ± 2.3* | 4.2 |
| R263K | 1.3 ± 0.37 | 1.2 | 4.0 ± 1.8 | 1.5 | 2.0 ± 0.79 | 0.9 |
| E92Q T97A | 1.1 ± 0.56 | 1.1 | 4.3 ± 1.4 | 1.6 | 4.8 ± 2.7 | 2.3 |
| E92Q Y143R | 2.1 ± 0.73 | 2.0 | 16.5 ± 5.9* | 6.2 | 108 ± 65.5* | 51.3 |
| G140S Q148R | 27.9 ± 6.8* | 26.6 | 679 ± 354* | 253.9 | 337 ± 174* | 160.3 |
| R263K H51Y | 1.5 ± 0.90 | 1.4 | 4.6 ± 2.5 | 1.7 | 2.8 ± 0.75 | 1.3 |
All data represent the results from at least 3 independent experiments. Fold changes (FC) of >10-fold are shown in boldface. *, statistically significant differences. Statistical significance was calculated for individual pairs of data using a one-sample two-tailed t test with a statistical cutoff of P ≤ 0.05.
The results show that DTG is more potent than EVG and RAL against all single or double mutants examined in our panel. In our study, DTG showed only limited cross-resistance against RAL- and/or EVG-resistant viruses. Although DTG did show reduced potency against the most resistant SIV variants harboring the G140S Q148R substitutions (∼26.6-fold) (Table 4), it remained more effective against this combination than EVG and RAL, which showed ∼250- and 160-fold-reduced potency against this variant, respectively.
DISCUSSION
Our results add to the evidence that SIV is an important preclinical tool for testing integrase strand transfer inhibitors. Previous studies have shown that SIV and HIV share similar resistance patterns against different classes of ARVs (10, 36, 37). Additionally, tissue culture studies by our group demonstrated that SIV substitutions confer resistance against INSTIs in a similar fashion to HIV (12). Now, we performed in vitro selection experiments with DTG, EVG, and RAL using rhesus macaque PBMCs infected with SIVmac239. We also tested the effects of relevant IN HIV drug resistance-associated substitutions on SIVmac239 IN catalytic activity using purified mutated recombinant proteins.
Over a period of 21 to 29 weeks, serial passage studies with all drugs only yielded viruses that showed relatively small increases in IC50s, and drug concentrations could not be substantially increased over this period. This provides testimony to the robustness of each of the drugs RAL, EVG, and DTG (Fig. 4). Indeed, viruses treated with DTG and RAL were not able to replicate at concentrations above 100 nM, while the concentration of EVG above which replication ceased was 50 nM. However, the use of DTG selected an R263K substitution in IN that was present after 21 weeks. R263K is a nonpolymorphic mutation that also has been found in several INSTI-naive ART-experienced patients who received DTG as therapy after failing on other drugs (38). It has also been reported as a secondary mutation after failure with RAL and EVG (30, 39). Additionally, tissue culture selection experiments with DTG led to the emergence of R263K with both HIV subtype B and circulating recombinant form CRF_A/G viruses (15). The R263K mutation has also been selected in vitro under EVG pressure (30).
In our tissue culture selections, the E92Q substitution was observed in passage with EVG at week 29. E92Q is a nonpolymorphic mutation that has been selected in patients receiving either EVG (40–44) or RAL (39, 40, 45) and is associated with virological failure on EVG-based regimens (45). The A54A/V, A265A/V, and V277V/M partial substitutions were selected under INSTI pressure, but only the A265 position is conserved among HIV integrases of different subtypes. Although the influence of A54A/V, A265A/V, and V277V/M on SIV viral fitness and resistance has not yet been studied, the natural positions of these residues in HIV IN are V54 and M277. While resistance mutations were detected within 29 weeks under either DTG or EVG selection pressure, no resistance mutations emerged under pressure with RAL. A direct comparison cannot be drawn between our selection studies, which were performed using rhesus PBMCs, and those performed by others who used HIV-1 in MT-2 cell lines and showed that mutations emerged rapidly under RAL pressure (31, 46). Our selection experiments with RAL may suggest that RAL possesses a higher genetic barrier than the other INSTIs in rhesus macaques; however, a previous study revealed that SHIV-infected macaques treated with pressure from L-870812, a RAL analogue, possessed the N155H substitution after 25 days of treatment with this drug (36). We cannot exclude that further prolonged tissue culture selection experiments with RAL may have yielded resistance. Overall, our serial passage experiments and those of others (36) confirm similarities in IN resistance patterns between HIV and SIV.
Proteins were expressed at similar levels in E. coli with no signs of aggregation or insolubility (Fig. 1). Our findings are in agreement with previous reports that variant enzymes typically showed maximal activity at 400 nM (47, 48). The data suggest that there were no significant differences in the oligomerization state or stability of the various proteins that were studied here.
The panel of mutations studied included 11 different single or double mutants that confer resistance to DTG, EVG, and/or RAL. Not all possible combinations were studied, as this would have been impossible. The E92Q T97A, E92Q Y143R, and G140S Q148R combinations were chosen as they are clinically relevant and confer resistance to DTG, EVG, and/or RAL in HIV-1 (40, 49–52). We have also shown the effect of introducing single or double substitutions in the IN gene of SIVmac239 on viral infectivity and IN strand transfer and 3′-processing activity. Of all of the recombinant proteins tested, enzymes containing G118R and G140S Q148R had the greatest loss of Vmax′/Km′ relative to WT (∼80% and 60% for strand transfer, respectively), perhaps explaining why the viral infectivity of both G118R- and G140S Q148R-containing SIV is impaired (12). This is in agreement with previous reports on G118R and G140S Q148R in both HIV-1 and HIV-2 (32, 33, 53, 54). The G118R and R263K substitutions also caused decreased Km′ for donor LTR DNA, while Y143R led to seemingly lower 3′-processing activity compared to WT (i.e., an impact on integrase catalytic activity). We did not observe any significant effect of E92Q, T97A, or R263K H51Y on 3′-processing and strand transfer activities.
It is striking that DTG, EVG, and RAL had inhibitory constants of ∼1 to 2 nM against WT SIV recombinant protein: i.e., similar to that reported for HIV-1 subtype B integrase protein (32). Unsurprisingly, DTG showed significant potency against singly or doubly mutated enzymes that were examined in our panel at a level similar to that observed for WT integrase. This agrees with previous observations on the robustness of this drug for most resistance substitutions (31, 55). One exception is the G140S Q148R-containing enzyme that exhibited an ∼26-fold decrease in susceptibility against DTG compared to the WT. The G140S Q148R combination also yielded the greatest increases in resistance against both EVG (Table 4; Fig. 7C) and RAL (Table 4; Fig. 7D), consistent with previous reports in HIV (31). Q148R had a similar resistance profile to G140S Q148R with regard to all INSTIs (122.7-fold and 119.8-fold increases in Ki against EVG and RAL, respectively), except that this substitution mutant displayed increased susceptibility to DTG (6.4-fold compared 26.6-fold for G140S Q148R) (Table 4; Fig. 7B). This is consistent with reports that the preexistence of Q148 + ≥1 resistance substitutions, but not Q148 mutations alone, were statistically associated with lower success rates in the clinic with DTG than when this drug was used in first-line therapy (56). The G118R variant enzyme displayed low-level resistance against all INSTIs, also in agreement with tissue culture resistance studies using TZM-bl cells (12) and with data on HIV-1 (32, 53). The R263K substitution in integrase led to marginally higher susceptibility to RAL than the WT (0.9-fold), while the introduction of H51Y in an R263K background increased levels of resistance against RAL (Table 4), similar to what has been observed with HIV-1 (23). In contrast, the combination of H51Y and R263K in SIV conferred low-level resistance against both DTG and EVG in our study (1.4-fold and 1.7-fold, respectively) (Table 4). The E92Q variant enzyme displayed low-level resistance against EVG, consistent with results obtained for HIV-1 (31, 57), but the combination of E92Q and T97A did not alter this resistance profile, nor did E92Q T97A confer resistance against either RAL or EVG (Table 4). The HIV RAL resistance-associated substitution, Y143R, also conferred resistance to SIV against this compound but not against DTG or EVG (Table 4). The combination of E92Q and Y143R has been associated with virological failure in HIV-1-infected patients on RAL (52, 58); our data suggest that SIV replicates this finding because the SIV integrase protein harboring the E92Q Y143R combination of substitutions displayed high-level resistance against RAL (∼51-fold [Table 4]). The RAL N155H resistance substitution conferred cross-resistance to EVG but hypersusceptibility to DTG (Table 4).
Of course, biological processes such as interaction with cellular cofactors and nuclear localizations, as well as cell-based integrase catalytic activities, may influence levels of resistance to INSTIs. The HIV-based chimera stHIV-1(SCA,SVIF), in which the HIV capsid (CA) and vif regions were replaced with corresponding regions from SIV in order to be able to infect human and macaque cell lines, shows differences in levels of replication for various IN-resistant viruses between human cord blood mononuclear cells (CBMCs) versus rhesus PBMCs (14). There is no definitive evidence that integrase and CA interact directly, but CA has been shown to play a role in the nuclear import of viral DNA (59) and may therefore be important for integrase activities. Although TZM-bl cells are susceptible to HIV and SIV infections, luciferase expression is under the control of the HIV-1 LTR promoter, which may explain the lack of assay sensitivity seen in some assays.
Some caveats about using SIVmac239 as a model should be mentioned. Although sensitive to HIV nucleoside reverse transcriptase inhibitors and INSTIs, SIV has limited susceptibility to some protease inhibitors and is not as susceptible to some nonnucleoside reverse transcriptase inhibitors (12, 60, 61). Also, integrase substitutions that have been well studied in HIV, such as E92Q, T97A, G118R, G140S, Y143R, Q148R, N155H, R263K, E92Q T97A, E92Q Y143R, G140S Q148R, and R263K H51Y, also have relevance for SIVmac239 IN, but other IN positions that are associated with HIV-1 resistance to INSTIs are not present in SIVmac239 IN: i.e., L74, T124, E138, S153, E157, and G163 (12). The use of IN-SHIV or stHIV-1 may be more suitable for the study of some INSTIs, since coding sequences are conserved among each of HIV, IN-SHIV, and stHIV-1 (14, 62, 63).
Collectively, our findings suggest that SIV and HIV share similar resistance pathway profiles with regard to EVG and DTG and that SIVmac239 is a useful nonhuman primate model for studies of HIV resistance to INSTIs.
ACKNOWLEDGMENTS
This study was supported by research grants from the Canadian Institutes of Health Research (CIHR), including CanCURE, Réseau SIDA de Fonds de Recherche du Québec, and the Canadian Foundation for AIDS Research (CANFAR). S.A.H. received a doctoral studentship from the Fonds de la Recherche du Québec en Santé (FRQS). P.K.Q. received a doctoral scholarship from the CIHR.
We thank James Whitney of the Ragon Institute of MGH, MIT, and Harvard for providing proviral DNA of SIVmac239 and Yingshan Han, Melissa Wares, Jiaming Liang, Ruxandra-Ilinca Ibanescu, and Victor Kramer for valuable discussions and scientific insight. We also thank Estrella Moyal for help with manuscript preparation.
The authors declare that they have no competing interests.
REFERENCES
- 1.Delelis O, Carayon K, Saib A, Deprez E, Mouscadet JF. 2008. Integrase and integration: biochemical activities of HIV-1 integrase. Retrovirology 5:114. doi: 10.1186/1742-4690-5-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Craigie R, Bushman FD. 2012. HIV DNA integration. Cold Spring Harb Perspect Med 2:a006890. doi: 10.1101/cshperspect.a006890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kenney J, Singer R, Derby N, Aravantinou M, Abraham CJ, Menon R, Seidor S, Zhang S, Gettie A, Blanchard J, Piatak M Jr, Lifson JD, Fernandez-Romero JA, Zydowsky TM, Robbiani M. 2012. A single dose of a MIV-150/zinc acetate gel provides 24 h of protection against vaginal simian human immunodeficiency virus reverse transcriptase infection, with more limited protection rectally 8-24 h after gel use. AIDS Res Hum Retroviruses 28:1476–1484. doi: 10.1089/aid.2012.0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cranage M, Sharpe S, Herrera C, Cope A, Dennis M, Berry N, Ham C, Heeney J, Rezk N, Kashuba A, Anton P, McGowan I, Shattock R. 2008. Prevention of SIV rectal transmission and priming of T cell responses in macaques after local pre-exposure application of tenofovir gel. PLoS Med 5:e157. doi: 10.1371/journal.pmed.0050157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobard C, Sharma S, Martin A, Pau CP, Holder A, Kuklenyik Z, Lipscomb J, Hanson DL, Smith J, Novembre FJ, Garcia-Lerma JG, Heneine W. 2012. Durable protection from vaginal simian-human immunodeficiency virus infection in macaques by tenofovir gel and its relationship to drug levels in tissue. J Virol 86:718–725. doi: 10.1128/JVI.05842-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dobard C, Sharma S, Parikh UM, West R, Taylor A, Martin A, Pau CP, Hanson DL, Lipscomb J, Smith J, Novembre F, Hazuda D, Garcia-Lerma JG, Heneine W. 2014. Postexposure protection of macaques from vaginal SHIV infection by topical integrase inhibitors. Sci Transl Med 6:227ra35. doi: 10.1126/scitranslmed.3007701. [DOI] [PubMed] [Google Scholar]
- 7.Andrews CD, Yueh YL, Spreen WR, St Bernard L, Boente-Carrera M, Rodriguez K, Gettie A, Russell-Lodrigue K, Blanchard J, Ford S, Mohri H, Cheng-Mayer C, Hong Z, Ho DD, Markowitz M. 2015. A long-acting integrase inhibitor protects female macaques from repeated high-dose intravaginal SHIV challenge. Sci Transl Med 7:270ra4. doi: 10.1126/scitranslmed.3010298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andrews CD, Spreen WR, Mohri H, Moss L, Ford S, Gettie A, Russell-Lodrigue K, Bohm RP, Cheng-Mayer C, Hong Z, Markowitz M, Ho DD. 2014. Long-acting integrase inhibitor protects macaques from intrarectal simian/human immunodeficiency virus. Science 343:1151–1154. doi: 10.1126/science.1248707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Rompay KK, Singh RP, Pahar B, Sodora DL, Wingfield C, Lawson JR, Marthas ML, Bischofberger N. 2004. CD8+-cell-mediated suppression of virulent simian immunodeficiency virus during tenofovir treatment. J Virol 78:5324–5337. doi: 10.1128/JVI.78.10.5324-5337.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Rompay KK, Johnson JA, Blackwood EJ, Singh RP, Lipscomb J, Matthews TB, Marthas ML, Pedersen NC, Bischofberger N, Heneine W, North TW. 2007. Sequential emergence and clinical implications of viral mutants with K70E and K65R mutation in reverse transcriptase during prolonged tenofovir monotherapy in rhesus macaques with chronic RT-SHIV infection. Retrovirology 4:25. doi: 10.1186/1742-4690-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Lerma JG, Otten RA, Qari SH, Jackson E, Cong ME, Masciotra S, Luo W, Kim C, Adams DR, Monsour M, Lipscomb J, Johnson JA, Delinsky D, Schinazi RF, Janssen R, Folks TM, Heneine W. 2008. Prevention of rectal SHIV transmission in macaques by daily or intermittent prophylaxis with emtricitabine and tenofovir. PLoS Med 5:e28. doi: 10.1371/journal.pmed.0050028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hassounah SA, Mesplede T, Quashie PK, Oliveira M, Sandstrom PA, Wainberg MA. 2014. Effect of HIV-1 integrase resistance mutations when introduced into SIVmac239 on susceptibility to integrase strand transfer inhibitors. J Virol 88:9683–9692. doi: 10.1128/JVI.00947-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewis MG, Norelli S, Collins M, Barreca ML, Iraci N, Chirullo B, Yalley-Ogunro J, Greenhouse J, Titti F, Garaci E, Savarino A. 2010. Response of a simian immunodeficiency virus (SIVmac251) to raltegravir: a basis for a new treatment for simian AIDS and an animal model for studying lentiviral persistence during antiretroviral therapy. Retrovirology 7:21. doi: 10.1186/1742-4690-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wares M, Hassounah S, Mesplede T, Sandstrom PA, Wainberg MA. 2015. Simian-tropic HIV as a model to study drug resistance against integrase inhibitors. Antimicrob Agents Chemother 59:1942–1949. doi: 10.1128/AAC.04829-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quashie PK, Mesplede T, Han YS, Oliveira M, Singhroy DN, Fujiwara T, Underwood MR, Wainberg MA. 2012. Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. J Virol 86:2696–2705. doi: 10.1128/JVI.06591-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oliveira M, Brenner BG, Wainberg MA. 2009. Isolation of drug-resistant mutant HIV variants using tissue culture drug selection. Methods Mol Biol 485:427–433. doi: 10.1007/978-1-59745-170-3_29. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Yan Y, Zugay-Murphy J, Xu B, Cole JL, Witmer M, Felock P, Wolfe A, Hazuda D, Sardana MK, Chen Z, Kuo LC, Sardana VV. 1999. Purification, solution properties and crystallization of SIV integrase containing a continuous core and C-terminal domain. Acta Crystallogr D Biol Crystallogr 55:1906–1910. doi: 10.1107/S0907444999009610. [DOI] [PubMed] [Google Scholar]
- 18.Bar-Magen T, Donahue DA, McDonough EI, Kuhl BD, Faltenbacher VH, Xu H, Michaud V, Sloan RD, Wainberg MA. 2010. HIV-1 subtype B and C integrase enzymes exhibit differential patterns of resistance to integrase inhibitors in biochemical assays. AIDS 24:2171–2179. doi: 10.1097/QAD.0b013e32833cf265. [DOI] [PubMed] [Google Scholar]
- 19.Bar-Magen T, Sloan RD, Faltenbacher VH, Donahue DA, Kuhl BD, Oliveira M, Xu H, Wainberg MA. 2009. Comparative biochemical analysis of HIV-1 subtype B and C integrase enzymes. Retrovirology 6:103. doi: 10.1186/1742-4690-6-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu HT, Asahchop EL, Oliveira M, Quashie PK, Quan Y, Brenner BG, Wainberg MA. 2011. Compensation by the E138K mutation in HIV-1 reverse transcriptase for deficits in viral replication capacity and enzyme processivity associated with the M184I/V mutations. J Virol 85:11300–11308. doi: 10.1128/JVI.05584-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF. 1999. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 112:531–552. [DOI] [PubMed] [Google Scholar]
- 22.Sambrook J, Russell D. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 23.Mesplede T, Quashie PK, Osman N, Han Y, Singhroy DN, Lie Y, Petropoulos CJ, Huang W, Wainberg MA. 2013. Viral fitness cost prevents HIV-1 from evading dolutegravir drug pressure. Retrovirology 10:22. doi: 10.1186/1742-4690-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quashie PK, Mesplede T, Han YS, Veres T, Osman N, Hassounah S, Sloan RD, Xu HT, Wainberg MA. 2013. Biochemical analysis of the role of G118R-linked dolutegravir drug resistance substitutions in HIV-1 integrase. Antimicrob Agents Chemother 57:6223–6235. doi: 10.1128/AAC.01835-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han YS, Quashie P, Mesplede T, Xu H, Mekhssian K, Fenwick C, Wainberg MA. 2012. A high-throughput assay for HIV-1 integrase 3′-processing activity using time-resolved fluorescence. J Virol Methods 184:34–40. doi: 10.1016/j.jviromet.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Smolov M, Gottikh M, Tashlitskii V, Korolev S, Demidyuk I, Brochon JC, Mouscadet JF, Deprez E. 2006. Kinetic study of the HIV-1 DNA 3′-end processing. FEBS J 273:1137–1151. doi: 10.1111/j.1742-4658.2006.05139.x. [DOI] [PubMed] [Google Scholar]
- 27.Anstett K, Mesplede T, Oliveira M, Cutillas V, Wainberg MA. 2015. Dolutegravir resistance mutation R263K cannot coexist in combination with many classical integrase inhibitor resistance substitutions. J Virol 89:4681–4684. doi: 10.1128/JVI.03485-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goethals O, Clayton R, Van Ginderen M, Vereycken I, Wagemans E, Geluykens P, Dockx K, Strijbos R, Smits V, Vos A, Meersseman G, Jochmans D, Vermeire K, Schols D, Hallenberger S, Hertogs K. 2008. Resistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitors. J Virol 82:10366–10374. doi: 10.1128/JVI.00470-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimura K, Kodama E, Sakagami Y, Matsuzaki Y, Watanabe W, Yamataka K, Watanabe Y, Ohata Y, Doi S, Sato M, Kano M, Ikeda S, Matsuoka M. 2008. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J Virol 82:764–774. doi: 10.1128/JVI.01534-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Margot NA, Hluhanich RM, Jones GS, Andreatta KN, Tsiang M, McColl DJ, White KL, Miller MD. 2012. In vitro resistance selections using elvitegravir, raltegravir, and two metabolites of elvitegravir M1 and M4. Antiviral Res 93:288–296. doi: 10.1016/j.antiviral.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi M, Yoshinaga T, Seki T, Wakasa-Morimoto C, Brown KW, Ferris R, Foster SA, Hazen RJ, Miki S, Suyama-Kagitani A, Kawauchi-Miki S, Taishi T, Kawasuji T, Johns BA, Underwood MR, Garvey EP, Sato A, Fujiwara T. 2011. In vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob Agents Chemother 55:813–821. doi: 10.1128/AAC.01209-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quashie PK, Mesplede T, Han YS, Veres T, Osman N, Hassounah S, Sloan RD, Xu HT, Wainberg MA. 2014. Biochemical analysis of the role of G118R-linked dolutegravir drug resistance substitutions in HIV-1 integrase. Antimicrob Agents Chemother 58:3580. doi: 10.1128/AAC.02916-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quashie PK, Oliviera M, Veres T, Osman N, Han YS, Hassounah S, Lie Y, Huang W, Mesplede T, Wainberg MA. 2015. Differential effects of the G118R, H51Y, and E138K resistance substitutions in different subtypes of HIV integrase. J Virol 89:3163–3175. doi: 10.1128/JVI.03353-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cleland WW. 1975. Partition analysis and the concept of net rate constants as tools in enzyme kinetics. Biochemistry 14:3220–3224. doi: 10.1021/bi00685a029. [DOI] [PubMed] [Google Scholar]
- 35.Cleland WW. 1989. The kinetics of enzyme-catalyzed reactions with two or more substrates or products. I. Nomenclature and rate equations. 1963. Biochim Biophys Acta 1000:213–220. [PubMed] [Google Scholar]
- 36.Hazuda DJ, Young SD, Guare JP, Anthony NJ, Gomez RP, Wai JS, Vacca JP, Handt L, Motzel SL, Klein HJ, Dornadula G, Danovich RM, Witmer MV, Wilson KA, Tussey L, Schleif WA, Gabryelski LS, Jin L, Miller MD, Casimiro DR, Emini EA, Shiver JW. 2004. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science 305:528–532. doi: 10.1126/science.1098632. [DOI] [PubMed] [Google Scholar]
- 37.Van Rompay KK. 2010. Evaluation of antiretrovirals in animal models of HIV infection. Antiviral Res 85:159–175. doi: 10.1016/j.antiviral.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 38.Cahn P, Pozniak AL, Mingrone H, Shuldyakov A, Brites C, Andrade-Villanueva JF, Richmond G, Buendia CB, Fourie J, Ramgopal M, Hagins D, Felizarta F, Madruga J, Reuter T, Newman T, Small CB, Lombaard J, Grinsztejn B, Dorey D, Underwood M, Griffith S, Min S. 2013. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet 382:700–708. doi: 10.1016/S0140-6736(13)61221-0. [DOI] [PubMed] [Google Scholar]
- 39.Blanco JL, Varghese V, Rhee SY, Gatell JM, Shafer RW. 2011. HIV-1 integrase inhibitor resistance and its clinical implications. J Infect Dis 203:1204–1214. doi: 10.1093/infdis/jir025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hatano H, Lampiris H, Fransen S, Gupta S, Huang W, Hoh R, Martin JN, Lalezari J, Bangsberg D, Petropoulos C, Deeks SG. 2010. Evolution of integrase resistance during failure of integrase inhibitor-based antiretroviral therapy. J Acquir Immune Defic Syndr 54:389–393. doi: 10.1097/QAI.0b013e3181c42ea4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winters MA, Lloyd RM Jr, Shafer RW, Kozal MJ, Miller MD, Holodniy M. 2012. Development of elvitegravir resistance and linkage of integrase inhibitor mutations with protease and reverse transcriptase resistance mutations. PLoS One 7:e40514. doi: 10.1371/journal.pone.0040514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Wesenbeeck L, Rondelez E, Feyaerts M, Verheyen A, Van der Borght K, Smits V, Cleybergh C, De Wolf H, Van Baelen K, Stuyver LJ. 2011. Cross-resistance profile determination of two second-generation HIV-1 integrase inhibitors using a panel of recombinant viruses derived from raltegravir-treated clinical isolates. Antimicrob Agents Chemother 55:321–325. doi: 10.1128/AAC.01733-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeJesus E, Rockstroh JK, Henry K, Molina JM, Gathe J, Ramanathan S, Wei X, Yale K, Szwarcberg J, White K, Cheng AK, Kearney BP. 2012. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate versus ritonavir-boosted atazanavir plus co-formulated emtricitabine and tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet 379:2429–2438. doi: 10.1016/S0140-6736(12)60918-0. [DOI] [PubMed] [Google Scholar]
- 44.Sax PE, DeJesus E, Mills A, Zolopa A, Cohen C, Wohl D, Gallant JE, Liu HC, Zhong L, Yale K, White K, Kearney BP, Szwarcberg J, Quirk E, Cheng AK. 2012. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet 379:2439–2448. doi: 10.1016/S0140-6736(12)60917-9. [DOI] [PubMed] [Google Scholar]
- 45.Molina JM, Lamarca A, Andrade-Villanueva J, Clotet B, Clumeck N, Liu YP, Zhong L, Margot N, Cheng AK, Chuck SL. 2012. Efficacy and safety of once daily elvitegravir versus twice daily raltegravir in treatment-experienced patients with HIV-1 receiving a ritonavir-boosted protease inhibitor: randomised, double-blind, phase 3, non-inferiority study. Lancet Infect Dis 12:27–35. doi: 10.1016/S1473-3099(11)70249-3. [DOI] [PubMed] [Google Scholar]
- 46.Seki T, Suyama-Kagitani A, Kawauchi-Miki S, Miki S, Wakasa-Morimoto C, Akihisa E, Nakahara K, Kobayashi M, Underwood MR, Sato A, Fujiwara T, Yoshinaga T. 2015. Effects of raltegravir or elvitegravir resistance signature mutations on the barrier to dolutegravir resistance in vitro. Antimicrob Agents Chemother 59:2596–2606. doi: 10.1128/AAC.04844-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lutzke RA, Plasterk RH. 1998. Structure-based mutational analysis of the C-terminal DNA-binding domain of human immunodeficiency virus type 1 integrase: critical residues for protein oligomerization and DNA binding. J Virol 72:4841–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hickman AB, Palmer I, Engelman A, Craigie R, Wingfield P. 1994. Biophysical and enzymatic properties of the catalytic domain of HIV-1 integrase. J Biol Chem 269:29279–29287. [PubMed] [Google Scholar]
- 49.Yoshinaga T, Kobayashi M, Seki T, Miki S, Wakasa-Morimoto C, Suyama-Kagitani A, Kawauchi-Miki S, Taishi T, Kawasuji T, Johns BA, Underwood MR, Garvey EP, Sato A, Fujiwara T. 2015. Antiviral characteristics of GSK1265744, an HIV integrase inhibitor dosed orally or by long-acting injection. Antimicrob Agents Chemother 59:397–406. doi: 10.1128/AAC.03909-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hurt CB, Sebastian J, Hicks CB, Eron JJ. 2014. Resistance to HIV integrase strand transfer inhibitors among clinical specimens in the United States, 2009-2012. Clin Infect Dis 58:423–431. doi: 10.1093/cid/cit697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Canducci F, Ceresola ER, Boeri E, Spagnuolo V, Cossarini F, Castagna A, Lazzarin A, Clementi M. 2011. Cross-resistance profile of the novel integrase inhibitor dolutegravir (S/GSK1349572) using clonal viral variants selected in patients failing raltegravir. J Infect Dis 204:1811–1815. doi: 10.1093/infdis/jir636. [DOI] [PubMed] [Google Scholar]
- 52.Canducci F, Sampaolo M, Marinozzi MC, Boeri E, Spagnuolo V, Galli A, Castagna A, Lazzarin A, Clementi M, Gianotti N. 2009. Dynamic patterns of human immunodeficiency virus type 1 integrase gene evolution in patients failing raltegravir-based salvage therapies. AIDS 23:455–460. doi: 10.1097/QAD.0b013e328323da60. [DOI] [PubMed] [Google Scholar]
- 53.Bar-Magen T, Sloan RD, Donahue DA, Kuhl BD, Zabeida A, Xu H, Oliveira M, Hazuda DJ, Wainberg MA. 2010. Identification of novel mutations responsible for resistance to MK-2048, a second-generation HIV-1 integrase inhibitor. J Virol 84:9210–9216. doi: 10.1128/JVI.01164-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ni XJ, Delelis O, Charpentier C, Storto A, Collin G, Damond F, Descamps D, Mouscadet JF. 2011. G140S/Q148R and N155H mutations render HIV-2 Integrase resistant to raltegravir whereas Y143C does not. Retrovirology 8:68. doi: 10.1186/1742-4690-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mesplede T, Quashie PK, Zanichelli V, Wainberg MA. 2014. Integrase strand transfer inhibitors in the management of HIV-positive individuals. Ann Med 46:123–129. doi: 10.3109/07853890.2014.883169. [DOI] [PubMed] [Google Scholar]
- 56.Castagna A, Maggiolo F, Penco G, Wright D, Mills A, Grossberg R, Molina JM, Chas J, Durant J, Moreno S, Doroana M, Ait-Khaled M, Huang J, Min S, Song I, Vavro C, Nichols G, Yeo JM. 2014. Dolutegravir in antiretroviral-experienced patients with raltegravir- and/or elvitegravir-resistant HIV-1: 24-week results of the phase III VIKING-3 study. J Infect Dis 210:354–362. doi: 10.1093/infdis/jiu051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abram ME, Hluhanich RM, Goodman DD, Andreatta KN, Margot NA, Ye L, Niedziela-Majka A, Barnes TL, Novikov N, Chen X, Svarovskaia ES, McColl DJ, White KL, Miller MD. 2013. Impact of primary elvitegravir resistance-associated mutations in HIV-1 integrase on drug susceptibility and viral replication fitness. Antimicrob Agents Chemother 57:2654–2663. doi: 10.1128/AAC.02568-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reigadas S, Anies G, Masquelier B, Calmels C, Stuyver LJ, Parissi V, Fleury H, Andreola ML. 2010. The HIV-1 integrase mutations Y143C/R are an alternative pathway for resistance to raltegravir and impact the enzyme functions. PLoS One 5:e10311. doi: 10.1371/journal.pone.0010311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Le Sage V, Mouland AJ, Valiente-Echeverria F. 2014. Roles of HIV-1 capsid in viral replication and immune evasion. Virus Res 193:116–129. doi: 10.1016/j.virusres.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 60.Cherry E, Slater M, Salomon H, Rud E, Wainberg MA. 1997. Mutations at codon 184 in simian immunodeficiency virus reverse transcriptase confer resistance to the (−) enantiomer of 2′,3′-dideoxy-3′-thiacytidine. Antimicrob Agents Chemother 41:2763–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Witvrouw M, Pannecouque C, Switzer WM, Folks TM, De Clercq E, Heneine W. 2004. Susceptibility of HIV-2, SIV and SHIV to various anti-HIV-1 compounds: implications for treatment and postexposure prophylaxis. Antivir Ther 9:57–65. [PubMed] [Google Scholar]
- 62.Akiyama H, Ishimatsu M, Miura T, Hayami M, Ido E. 2008. Construction and infection of a new simian/human immunodeficiency chimeric virus (SHIV) containing the integrase gene of the human immunodeficiency virus type 1 genome and analysis of its adaptation to monkey cells. Microbes Infect 10:531–539. doi: 10.1016/j.micinf.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 63.Hatziioannou T, Princiotta M, Piatak M Jr, Yuan F, Zhang F, Lifson JD, Bieniasz PD. 2006. Generation of simian-tropic HIV-1 by restriction factor evasion. Science 314:95. doi: 10.1126/science.1130994. [DOI] [PubMed] [Google Scholar]