

ABSTRACT
Following natural human or experimental murine infections and in cell culture, coxsackievirus B (CVB) RNA can persist for weeks in the absence of a cytopathic effect, yet viral RNA remains detectable. Our earlier studies demonstrated that this persistence produced viral RNA with up to 49 nucleotide deletions at the genomic 5′ terminus which partially degraded the cloverleaf (or domain I), an RNA structure required for efficient viral replication. A cis-acting replication element (CRE) in the 2C protein-coding region [CRE(2C)] templates the addition of two uridine residues to the virus genome-encoded RNA replication primer VPg prior to positive-strand synthesis. Because our previous work also demonstrated that the genomes of CVB with a 5′-terminal deletion (CVB-TD) have VPg covalently linked, even though they rarely terminate in the canonical UU donated by CRE(2C)-mediated uridylylation of VPg, we hypothesized that a functional (uridylylating) CRE(2C) would be unnecessary for CVB-TD replication. Using the same 16 mutations in the CVB3 CRE(2C) structure that were considered lethal for this virus by others, we demonstrate here both in infected cell cultures and in mice that wild-type (wt) and CVB3-TD strains carrying these mutations with a nonuridylylating CRE(2C) are viable. While the wt genome with the mutated CRE(2C) displays suppressed replication levels similar to those observed in a CVB3-TD strain, mutation of the CRE(2C) function in a CVB3-TD strain does not further decrease replication. Finally, we show that replication of the parental CVB3 strain containing the mutated CRE(2C) drives the de novo generation of genomic deletions at the 5′ terminus.
IMPORTANCE In this report, we demonstrate that while CVB can replicate without a uridylylating CRE(2C), the replication rate suffers significantly. Further, deletions at the 5′ terminus of the genome are generated in this virus population, with this virus population supplanting the wild-type population. This demonstrates that VPg can prime without being specifically uridylylated and that this priming is error prone, resulting in the loss of sequence information from the 5′ terminus. These findings have significance when considering the replication of human enteroviruses, and we believe that these data are unattainable in a cell-free system due to the poor replication of these CRE-deficient viruses.
INTRODUCTION
Group B coxsackieviruses (CVBs; serotypes 1 to 6) are nonenveloped, single-stranded, positive-sense RNA viruses classified as human enteroviruses (HEV), species B, within the viral family Picornaviridae (1). CVBs cause or are etiologically associated with a variety of human diseases, including aseptic meningitis, myocarditis, pancreatitis, and type I diabetes (2–6). The small, single-stranded, positive-sense HEV RNA genome encodes 11 proteins from a single open reading frame (ORF). Upon successful viral infection of the host cell, the HEV genome is translated and then functions as a template for the synthesis of a negative-sense RNA strand, which serves, in turn, as the template for the replication of the positive-strand genomic RNA (1, 7, 8).
Four cis-acting replication elements (CREs) which modulate replication and translation have been identified in the HEV genome; these are at the 5′ and 3′ termini, within the ORF of the genomic positive strand, and at the 3′ end of the negative RNA strand (a replicative intermediate) (9, 10). The 5′ and 3′ ends of the genomic RNA are nontranslated regions (NTRs), and the 3′ NTR terminates in a poly(A) tail. A cloverleaf secondary structure termed domain I (Fig. 1A) is present at the 5′ terminus of the HEV genome (11, 12) and is believed to be the site of formation of the replication complex, which includes both viral and host cell proteins required for negative-strand synthesis (12–17). Binding of the viral protein 3CD to stem-loop d of domain I and the host cell protein poly(rC)-binding protein 2 (PCBP2) to stem-loop b of domain I [or the poly(C) tract in the spacer region between domain I and domain II of the internal ribosome entry site (IRES)] (Fig. 1C) has been proposed to be required for negative-strand synthesis (18–22). It has been demonstrated using a cell-free system that deletion of 4 nucleotides from stem-loop d of poliovirus RNA led to the loss of both negative-strand synthesis and the production of viral proteins (17), perhaps due to decreased binding or a loss of binding of 3CD. Further studies demonstrated that, with domain I left intact, deletion of portions or all of the IRES from poliovirus RNA did not affect the synthesis of either negative- or positive-strand genomic RNA in a cell-free system (23). These findings indicate that domain I but not the IRES of the 5′ NTR is crucial for the efficient replication of viral RNA.
FIG 1.

Domain I and CRE(2C) of CVB3 and the location of protein binding to these structures. (A) Domain I of CVB3, located at the 5′ terminus of the genome (the gray nucleotides indicate the sequence deleted in CVB3-TD50). (B) CRE(2C) of CVB3 located in the 2C protein-coding region from nucleotides 4365 to 4425. The two red adenines at the apex of the loop represent the experimentally determined template for the uridylylation of VPg. (C) During replication of the CVB3 genome, the viral protein 3CD and host-cell protein poly(rC) binding protein 2 (PCBP2) bind in the region of domain I and viral proteins 3CD and 3Dpol bind to CRE(2C) to mediate the uridylylation of VPg.
Our previous work identified a mechanism of CVB persistence in which domain I can largely be lost during replication in experimentally inoculated cell cultures and mice (24, 25), as well as within heart tissue of naturally infected humans (26); more recent evidence suggests that HEVs evolve populations with 5′-terminal deletions (TDs) in cardiomyopathic human heart tissues at the terminal stage of disease (27) as well as within the pancreatic tissue of experimentally inoculated mice (28). The discovery of naturally occurring enteroviral 5′-TDs modified our understanding of enteroviral biology by demonstrating not only that enteroviruses without an intact domain I are viable but also that populations of CVB with a 5′-terminal deletion (CVB-TD) replicate with a severe deficiency in positive-strand RNA replication. This was readily apparent in the reduction of positive- to negative-strand viral RNA ratios to near unity in CVB-TD-infected cells as well as by the demonstration of efficient encapsidation of the replication-incompetent negative strand (25). The inefficient replication observed with CVB-TD (24, 25) confirmed and significantly extended the well-established role of domain I as a contributor to efficient viral replication and positive-strand RNA synthesis (10, 12, 29). With much of domain I being lost in viral RNA populations with a 5′-TD (Fig. 1A), it is not surprising that CVB-TD strains replicate to low titers not detectable by standard cell culture assays using the cytopathic effect (CPE) and require enzymatic amplification for their detection. Further work by Sharma and colleagues demonstrated that CVB-TD genomic RNA is stable and replication competent when up to 49 nucleotides are deleted from the 5′ terminus, although it was stable and replication competent at a minimal level in a cell-free reporter system (22), supporting and extending our previous findings. Curiously, while we observed that CVB-TD genomes rarely terminate in the canonical UU derived from the uridylylated VPg primer during positive-strand synthesis (24–26), VPg was still linked to the 5′ end of the genome with a 5′-TD (25).
In addition to the 3′ and 5′ NTR CREs, picornaviruses also possess small hairpin structures within the genome that are necessary for efficient RNA replication and that template the uridylylation of the viral protein primer VPg (30–32); VPg is subsequently used as the RNA replication primer. These hairpin structures are located at different sites within various picornaviral genomes (9, 33–39), with that of the HEV B species being located in the 2C coding region [CRE(2C)] (Fig. 1B) (31). It has been firmly demonstrated that these CREs are location independent (34, 38, 39); for example, poliovirus remains replication competent when CRE(2C) is moved from the 2C protein-coding region of the ORF to the 5′ NTR (40). Despite diverse locations of picornaviral CREs, the location within a species is highly conserved (9). To determine the function and structural requirements of the HEV CRE(2C), both cell culture and cell-free systems have been used to analyze the effects of mutations introduced into the CRE(2C) region. These studies defined the site of the template nucleotides for the addition of uridine residues to VPg (in the loop of the predicted RNA structure [30, 41]) and defined the degree to which uridylylation and replication are disrupted by mutations in the loop or stem of the structure (17, 31, 40, 42–44). Studies of replication in cell culture not only demonstrated a loss of CPE upon transfection of some poliovirus CRE(2C) mutants but also demonstrated that some mutants were capable of a dominant-negative effect (45); replication of wild-type (wt) poliovirus within the same cell was inhibited by the presence of these single-site mutations. This effect is not complete, however: in the cell-free system, perhaps because of the noncompartmentalized nature of the replication, low-level replication is observed in the mutated CRE(2C) genomes, as evidenced by the presence of replicative-form-intermediate production, and unlike in cell culture assays, CRE(2C) mutations could be rescued in trans (46). In addition, the passage of some initially noncytopathic mutants (following transfection of T7-transcribed RNA) resulted in CPE, demonstrating that although the CRE(2C) mutations had a severely debilitating effect on virus replication, the replication of the viruses that had occurred was sufficient to permit reversion to restore the wt sequence (34). Analysis of CRE(2C) mutations in cell-free systems allows quantitative assays of replication based on the incorporation of radioactive nucleotides and demonstration of the effects upon the generation of uridylylated VPg (31, 42, 44, 46, 47). The greatest effect upon uridylylation was noted in mutations of the A5 and A6 nucleotides of the loop (Fig. 1B, red nucleotides), but considerable defects in uridylylation and RNA replication in the cell-free assays were observed when mutations were present in the stem or loop region (31, 46, 47). When 16 silent mutations were introduced into the CVB3 CRE(2C), creating the CRE(2C)-DM mutant (31), the virus was deemed unable to replicate on the basis of the failure to detect both uridylylated VPg and positive-strand synthesis and an inability to detect virus-expressed luciferase activity above the background levels 10 h after transfection of cell cultures.
Our findings (25, 26) that CVB3-TD strains replicate poorly compared to the wt and that VPg is attached to CVB3 genomic termini, despite a general lack of the 5′ UU consensus sequence, suggested the possibility that VPg is nonspecifically nucleotidylated instead of specifically uridylylated, in order to function as the primer for viral RNA replication. We therefore hypothesized that a functional (uridylylating) CRE(2C) would not be required for the replication of viruses with a TD. Using the same 16 mutations described for the CRE(2C)-DM mutant (31), we generated the wt CVB3 genome with these mutations (here termed CVB3-CKO for CVB3 with a CRE knockout [CKO]) to serve as a replication control for studies of CVB3-TD replication; we also generated the CKO mutations in the CVB3 infectious cDNA clone with a 49-nucleotide 5′-terminal deletion (CVB3-TD50-CKO). Indeed, a nonfunctional CRE(2C) was not necessary for CVB3-TD50 replication, but contrary to expectations, we also observed that CVB3-CKO was viable. In this report, we demonstrate that in cell culture or in mice, while this specific disruption of CRE(2C) in CVB3 is detrimental to viral replication, it is not lethal after all. Further, we demonstrate that when the native structure of CRE(2C) is altered in this way in a genome with a deletion at a location other than the terminus, the virus population rapidly evolves to become one containing 5′-terminal genomic deletions.
MATERIALS AND METHODS
Cells and viruses.
Coxsackievirus B3, strain 28 (48), was used as the wt strain in this work. HeLa cell monolayer cultures were maintained in Dulbecco's modified Eagle medium (DMEM; high glucose; GE Life Sciences, Logan, UT) containing 10% newborn calf serum and 50 μg/ml gentamicin (Gibco, Life Technologies, Grand Island, NY). Viral stocks were prepared in HeLa cells by electroporation of 2 × 106 cells at 100 V and 1,980 μF (Cell-Porator; Gibco, Life Technologies) with 12 μg of viral RNA (equivalent to 2.96 × 1012 genomes) transcribed from infectious cDNA clones of either wt CVB3, CVB3 in which CRE(2C) was mutationally disrupted with 16 mutations [CVB3-CKO, as described below in “Mutational disruption of CRE(2C) using overlap extension PCR”], CVB3 with a 49-nucleotide 5′ terminal deletion (CVB3-TD50) (25), and CVB3-TD50 in which the CRE(2C) was mutationally disrupted (CVB3-TD50-CKO). Cells were subsequently seeded into 100-mm dishes. After 8 days of incubation, with replacement of the medium at day 4, cultures were examined for CPE using an Olympus CKX41 inverted microscope with a DP12 camera; complete CPE was observed with the wt at 2 days postelectroporation. After freezing and thawing three times, the lysates were cleared of cell debris by centrifugation at 30,000 rpm for 30 min at 8°C (SW28.1 rotor; Beckman, Brea, CA) and then prepared as described previously (25) with RNase A treatment and pelleting through a 30% sucrose cushion to purify the virions. This preparation was termed pass 1, as previously described (25).
Mutational disruption of CRE(2C) using overlap extension PCR.
The mutations in the CVB3 CRE(2C) were generated using overlap extension mutagenesis (49). Two overlapping fragments of CVB3 cDNA were generated from the infectious cDNA clone of CVB3/28 (48) by PCR using Deep Vent DNA polymerase (New England BioLabs, Ipswich, MA) and the primer pair ID13 and CREKORev or CREKO and XBA (Table 1) with 4 mM MgSO4 and 200 μM deoxynucleoside triphosphates (dNTPs). Cycling was carried out as noted in Table 2 with modified cycle denaturation and annealing times of 30 s each. The two amplimers were gel purified, combined in Thermopol reaction buffer [20 mM Tris-HCl, 10 mM (NH4)2SO4, 10 mM KCl, 4 mM MgSO4, 0.1% Triton X-100, pH 8.8; New England BioLabs] with 200 μM dNTPs, and repeatedly denatured and annealed 10 times (1 min at 94°C, 50°C for 1 min), followed by extension with Deep Vent polymerase (2,000 U/ml) at 72°C for 2 min. The mutated overlap extension product was subsequently directly amplified using the ID13 and XBA primers (Tables 1 and 2) and Deep Vent polymerase with the modified denaturation and annealing times described above. The amplified product of this reaction was digested with BssH2 and XbaI and then ligated with the BssH2-XbaI restriction fragment of pCVB3 or pCVB3-TD50 (25) to produce pCVB3-CKO and pCVB3-TD50-CKO, respectively.
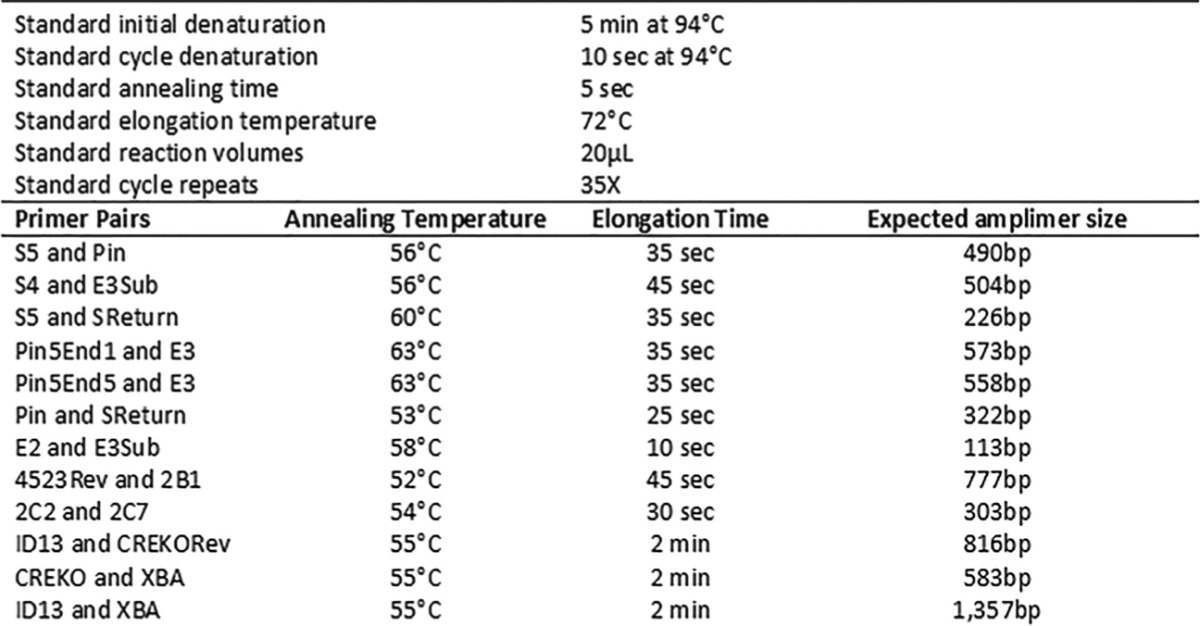
TABLE 1.
Names, annealing sites, and sequences of primers used in CKO studies
| Primer name | Region of CVB3 strain 28 genome | Strand | Nucleotide sequence (5′ to 3′)b |
|---|---|---|---|
| Sa | 1–20 | + | TTAAAACAGCCTGTGGGTTG |
| S4a | 45–74 | + | CGCTAGCACTCTGGTATCACGGTACCTTTG |
| S5 | 86–113 | + | TATACCCCCTCCCCCAACTGTAACTTAG |
| SReturn | 291–312 | − | TACACTGGGGTAGTGCTGAGCG |
| E2a | 450–464 | + | TCCGGCCCCTGAATG |
| E3a | 537–563 | − | ACACGGACACCCAAAGTAGTCGGTTCC |
| E3Sub | 535–549 | − | AGTAGTCGGTTCCGC |
| PinE3 | 537–563 | − | GGAATTCATCGATACGCGACACGGACACCCAAAGTAGTCGGTTCC |
| ID13 | 3601–3620 | + | TGGATTTTCCGAACCAGGTG |
| CREKORev | 4352–4417 | − | CAAGCAAACCGGTTCTATCCTACACTTACTTTTAAATTGAATATAATTGCTCATCTTCTTCTCAAG |
| CREKO | 4375–4434 | + | TATTCAATTTAAAAGTAAGTGTAGGATAGAACCGGTTTGCTTGCTCCTGCACGGGAGCCC |
| XBA | 4932–4958 | − | GCATGTCTAGAGAGTATCTGACCTGTG |
| 2B1 | 3746–3764 | + | GGAGTGAAGGACTATGTGG |
| 4523Rev | 4504–4523 | − | GGTCTGGCGGTAGTGAGTAC |
| 2C2 | 4450–4469 | − | CAATTAAGTTTGTTGCCACC |
| 2C7 | 4166–4186 | + | GAAAAACACGAATTCCTGAAC |
| Pin | NAc | NA | GGAATTCATCGATACGCG |
| Pin5End1 | 1–10 | + | GGAATTCATCGATACGCGTTAAAACAG |
| Pin5End5 | 15–24 | + | GGAATTCATCGATACGCGGGGTTGATCC |
Previously published in the work of Kim et al. (25).
Underlined sequences are nongenomic cDNA.
NA, not applicable.
TABLE 2.
Standard PCR cycle parameters and primer pair variations
Preparation of T7 RNA transcripts for electroporation or transfection.
The preparation of infectious cDNA copies of the CVB3 and CVB3-TD50 genomes was described previously (25). Mutational disruption of the CRE(2C) region of these clones to generate CVB3-CKO and CVB3-TD50-CKO was described above in “Mutational disruption of CRE(2C) using overlap extension PCR.” Viral RNA was transcribed from 5 μg of ClaI-linearized cloned cDNA using a T7 RiboMAX system (Promega, Madison, WI) for 1 h, followed by removal of the DNA template by digestion with RQ1 DNase (6 U). RNA was precipitated overnight at −20°C in 120 μl containing 4 mM EDTA, 80 μg of glycogen, and 2 M LiCl, pelleted, and resuspended in RNase-free water. The expected 7.4-kb size of the viral RNAs was verified on a 1.4% native agarose gel, and concentrations were determined spectrophotometrically.
Cell culture of purified virus stocks (pass 2).
CVB3-CKO replicates to very low titers and does not produce observable CPE. Consequently, inoculation of cell cultures with both CVB3-CKO and wt CVB3 virions (purified with RNase treatment and pelleting through 30% sucrose) was performed as described previously (25) using virus titers based upon the positive-strand RNA copy number in the virus preparations which were derived from reverse transcription (RT)-quantitative PCR (qPCR) analyses (see “Determination of viral RNA titer in purified virus stocks using RT-qPCR” below). Thus, for passage of purified virions in HeLa cell cultures, 3 × 105 HeLa cells (at 80 to 90% confluence) per well in 24-well plates were inoculated in duplicate with the equivalent of 3,190 positive RNA genomic strands. In addition, equivalent amounts of virus were incubated for 1 h at 37°C with a 1:400 dilution of anti-CVB3 neutralizing antibody (ATCC, Manassas, VA) prior to inoculation of cells. The plates were incubated for 3 h (with antibody; escape occurred if they were incubated overnight) or overnight (without antibody) at 37°C, and the monolayers were washed twice with phosphate-buffered saline (PBS) and then provided fresh medium and incubated with or without neutralizing antibody for 5 days. The monolayers were harvested by direct lysis with the TRIzol reagent in the wells, followed by RNA isolation per the manufacturer's protocol, isopropanol precipitation with glycogen as a carrier (50), and resuspension in nuclease-free water. Total RNA was assayed for the presence of CVB3 RNA using nested RT-PCR as described below.
Transfection of mice and detection of viral RNA in tissue homogenates using RT-PCR.
The use of mice in this study was approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee. Male A/J mice (6 to 7 weeks old; The Jackson Laboratory, Bar Harbor, ME) were inoculated intraperitoneally (i.p.) with 25 μg (equivalent to 6.2 × 1012 genomes) of CVB3 or CVB3-CKO T7 transcripts (prepared as described above in “Preparation of T7 RNA transcripts for electroporation or transfection”). Inocula (0.1 ml/mouse) were prepared in 0.4-ml aliquots containing 100 μg T7-transcribed RNA in 100 μl of nuclease-free water, 100 μl of a lipid-based in vivo transfection reagent (Altogen Biosystems, Las Vegas, NV), 20 μl transfection enhancer, and 80 μl of a 5% (wt/vol) sterile glucose solution. Hearts and spleens were collected from the mice at 5 and 8 days posttransfection (for mice inoculated with wt CVB3) or 20 days posttransfection (for mice inoculated with CVB3-CKO and mice transfected with saline as a control) and halved, one half of each organ was weighed, and then the tissue was homogenized in 500 μl TRIzol reagent (Invitrogen, Carlsbad, CA) and total RNA was extracted as described above in “Cell culture of purified virus stocks (pass 2).” To detect viral RNA, a series of four 10-fold dilutions was prepared to find the optimal RNA concentration for reverse transcription. This method was required for all RNA preparations from mouse tissue, as the required dilution varied between 100- and 1,000-fold, depending on the tissue and mouse. Dilution of RNA was required to overcome the known effects of RT inhibition in samples with low numbers of copies of RNA, in which dilution can increase the level of detection (51–53). cDNA was then transcribed from each dilution with primer E3 (Table 1) at a final concentration of 2 μM. Briefly, RNA was denatured for 5 min at 70°C and incubated for 2 min on ice. RNA was then reverse transcribed using an ImProm-II reverse transcriptase system (Promega) with 0.8 mM dNTPs, 2 mM MgCl2, and 1 μl enzyme (the numbers of units are proprietary and not provided by Promega) in 20 μl of reaction buffer for 60 min at 42°C. cDNA was ethanol precipitated and resuspended in water, and nested RT-PCR for viral RNA detection was performed as described below.
Inoculation of cell cultures with spleen homogenates.
Murine spleen was homogenized in complete tissue culture medium, frozen and thawed 3 times, and then cleared by centrifugation at 16,000 × g for 10 min. These cleared homogenates were then treated with RNase A as described above in “Cell culture of purified virus stocks (pass 2).” Duplicate HeLa cell cultures (as described above) were inoculated with 100 μl of cleared, RNase-treated splenic homogenate from CVB3-CKO-transfected mice (spleens were collected at 20 days posttransfection) or 1 μl of splenic homogenate from wt CVB3-transfected mice (spleens were collected at 8 days posttransfection; the lower volume for wt CVB3 was chosen on the basis of the greater extent of wt CVB3 replication in cell culture observed by RT-qPCR). Cell cultures were inoculated in the presence or absence of a 1:400 dilution of anti-CVB3 neutralizing antibody; preparations containing neutralizing antibody were treated as described above in “Cell culture of purified virus stocks (pass 2).” After overnight incubation (without antibody), cultures were washed 2 times with PBS and the medium was replaced in the presence or absence of neutralizing antibody. Cultures were observed daily by light microscopy for CPE. After 5 days, total RNA was extracted from each monolayer as described above in “Cell culture of purified virus stocks (pass 2).” RNA preparations were then assayed for viral RNA using nested RT-PCR (see “Nested RT-PCR for detection of viral RNA in total RNA isolated from cell culture lysates and mouse tissues” below).
Detection of positive-strand viral RNA in purified pass 1 virus preparations.
Total RNA was isolated from purified virus preparations (treated with RNase and pelleted through 30% sucrose) using the TRIzol reagent as described above in “Cell culture of purified virus stocks (pass 2),” and cDNA was prepared (as described above) using an antisense primer, Pin E3 (Table 1), designed to anneal to a region of the 5' NTR where deletions do not occur and with a 5' non-CVB3-specific oligonucleotide tag, Pin (54). Half of each cDNA reaction mixture was amplified using the primer S5 and the tag primer Pin (54) (Table 1) in GoTaq Green master mix (Promega). The reactions were cycled as noted in Table 2 for the different primer pairs, followed by electrophoresis on 2% agarose gels for analysis.
Enzymatic amplification of the CRE(2C) sequence from plasmids and T7 transcripts.
The CRE(2C) region of plasmids carrying wt CVB3 and CVB3-CKO and the cDNA of T7 transcripts of cloned cDNA were amplified using nested primers in 20-μl reaction volumes containing GoTaq Green master mix (Promega). The primers for the first enzymatic amplification were 4523Rev and 2B1 (Tables 1 and 2) at a final concentration of 0.5 μM. After ethanol precipitation, a second round of PCR was carried out with the ethanol-precipitated amplimers using primers 2C2 and 2C7 (Tables 1 and 2). The PCR products were electrophoresed on 2% agarose gels, and bands of the appropriate size (Table 2) were excised and purified using the a Zymoclean gel DNA recovery kit (Zymo Research, Irvine, CA) per the manufacturer's protocol. The recovered amplimers were sequenced with primer 2C7 (Table 1) to verify that the sequence was that of either wt CVB3 or the CRE(2C) mutant CVB3-CKO, as expected, prior to electroporation and virus preparation.
Nested RT-PCR for detection of viral RNA in total RNA isolated from cell culture lysates and mouse tissues.
To test for the presence of viral RNA in cell culture or mouse tissue, nested RT-PCR was performed to detect the 5′ NTR sequence in the CVB3 genome corresponding to nucleotides 45 to 564. Initial enzymatic amplification of all cDNA samples from viral RNA preparations (except for initial viral RNA detection in sucrose-purified preparations, described above in “Detection of positive-strand viral RNA in purified pass 1 virus preparations”) was carried out using primers S4 and E3Sub (Tables 1 and 2) in GoTaq Green master mix (Promega). A second round of PCR was carried out with primers S5 and SReturn and the ethanol-precipitated product of the PCR amplification with primers S4 and E3Sub (Tables 1 and 2). Final PCR products were analyzed on gels as described above in “Detection of positive-strand viral RNA in purified pass 1 virus preparations.”
Determination of viral RNA titer in purified virus stocks using RT-qPCR.
Because the CVB3-CKO, CVB3-TD50, and CVB3-TD50-CKO strains do not produce detectable CPE upon infection of tissue culture monolayers, the viral RNA copy number was quantitated using RT-qPCR by a method similar to that described previously (25). cDNA was transcribed using ImProm-II reverse transcriptase (Promega) and primer E3 (Table 1) as described above in “Transfection of mice and detection of viral RNA in tissue homogenates using RT-PCR.” RT-qPCR was performed using an Opticon 2 DNA engine (MJ Research, Waltham, MA) by combining the cDNA reaction mixtures with primers E2 and E3Sub (Table 1) in Maxima SYBR green-fluorescein qPCR master mix (2×; Thermo Scientific, Waltham, MA). qPCR cycling was carried out as indicated in Table 2 for the different primer pairs with a modified annealing time of 10 s. Standard curves were generated with 10-fold serial dilutions of a 500-bp amplimer of CVB3 cDNA generated with primers S and E3 (Table 1); the amplimer was present at a known concentration previously calculated using spectrophotometry. With knowledge of the cDNA copy number generated from a reverse transcriptase reaction mixture containing a specific amount of RNA, the efficiency of the cDNA transcription (number of molecules of cDNA/number of molecules of RNA) was calculated. Statistical analysis was carried out using one-way analysis of variance with the Bonferroni correction.
RT-PCR to detect 5′-terminal deletions in purified virus stocks.
cDNA from purified virion preparations was prepared using ImProm-II reverse transcriptase (Promega) and primer E3 (Table 1) as described above in “Transfection of mice and detection of viral RNA in tissue homogenates using RT-PCR.” All reverse transcriptase reaction mixtures were assembled with an equal RNA copy number for comparison of the presence of 5′-terminal sequences. Copy numbers either were calculated using the cDNA copy number determined by RT-qPCR (for the virus preparations) or were calculated on the basis of the RNA concentration from spectrophotometric analysis (for the T7-transcribed RNA controls). Tagged primers (54) and detection in the purified pass 1 virus preparations, as described above, of positive-strand viral RNA corresponding to regions of the genomic 5′ terminus were used to detect 5′-terminal deletions. Initial enzymatic amplification was carried out using primers Pin5End1 and E3 (Tables 1 and 2) and 400 copies of E3 cDNA or primers Pin5End5 and E3 (Tables 1 and 2) and 1,400 copies of E3 cDNA (at the experimentally determined limit of detection for the respective primer pairs) in GoTaq Green master mix (Promega) with a modified annealing time of 10 s. PCR products were ethanol precipitated. The second round of enzymatic amplification was performed with the ethanol-precipitated product of the first reaction in GoTaq Green master mix with primers Pin and SReturn (Tables 1 and 2). The PCR products were analyzed on agarose gels as described above in “Detection of positive-strand viral RNA in purified pass 1 virus preparations.”
RESULTS
CVB3-CKO replicates in HeLa cells.
The HEV CRE(2C) is required for efficient viral replication (31, 44, 46, 47), and it is established that various mutational disruptions of CRE(2C) in either poliovirus or CVB3 are lethal, on the basis of the absence of radiographic evidence for positive-strand (genomic) RNA replication, VPg uridylylation (31, 44, 47), or luciferase activity after 10 h of incubation in cell culture (31). Our previous findings demonstrated that a CVB-TD genome rarely terminates in the canonical UU donated by uridylylated VPg upon replication yet still has VPg linked to the genomic RNA (25). Therefore, we hypothesized that CVB-TD would not require a functional (that is, a uridylylating) CRE(2C) in order to successfully replicate. To test this, we induced the previously characterized 16 mutations [described as CVB3-CRE(2C)-DM (31)] in both wt CVB3 (initially intended for use as a negative control) and CVB3-TD50 infectious cDNA clones; the sequences of the CRE(2C) regions in the DNA clones and in the T7 transcripts from the clones (after cDNA synthesis) were verified prior to use, as described in Materials and Methods.
HeLa cells were electroporated with equivalent copy numbers of full-length wt CVB3, CVB3-CKO, CVB3-TD50, or CVB3-TD50-CKO T7-transcribed RNA and then cultured for 8 days. As expected from other work (31), no CPE was observed in the monolayers electroporated with CVB3-CKO (Fig. 2A, CVB3-CKO) compared to the electroporation control cells (Fig. 2A, TCC [tissue culture control]), nor was CPE evident in cultures transfected with any of the other viral RNAs (data not shown), except for cultures transfected with wt CVB3 RNA, which completely lysed the cultures by 48 h postelectroporation (Fig. 2A, wt CVB3). The observed absence of CPE in cells electroporated with CVB3-CKO was consistent with the findings of studies indicating that a mutated CRE(2C) suppresses or ablates viral replication (31, 42, 46, 47).
FIG 2.
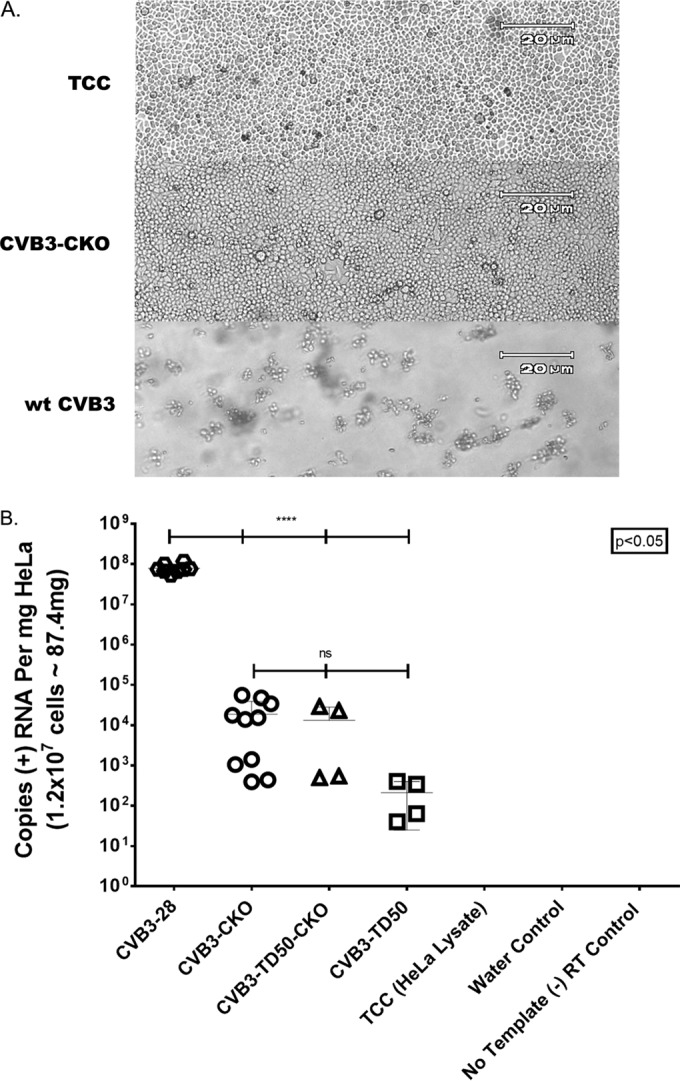
Virus without a functional CRE(2C) can be recovered in the absence of observable CPE following electroporation of HeLa cells with T7 RNA transcripts from infectious cloned genomes. (A) CVB3-CKO does not produce observable CPE at 8 days postelectroporation with 12 μg of RNA, whereas the tissue culture control (TCC) electroporated with saline only does produce observable CPE. wt CVB3 RNA induced complete CPE by 48 h postelectroporation. Magnifications, ×100. (B) Quantitation of viral RNA in RNase-treated and sucrose-purified mutant virus preparations (CVB3-CKO, CVB3-TD50, or CVB3-TD50-CKO). Loss of CRE(2C) function, deletion of 49 nucleotides from the 5′ terminus in domain I, or both, does not lead to a lethal virus phenotype but deleteriously affects replication compared to that of wt CVB3. A loss of CRE(2C) in CVB3-CKO lowers the extent of replication to a level similar to that of CVB3-TD50, but a loss of CRE(2C) in CVB3-TD50 (CVB3-TD50-CKO) does not further impact replication.
However, because our previous work demonstrated that CVB3-TD strains also showed no apparent CPE in cell cultures yet could be shown to be replicating using RT-PCR to detect viral RNA (24, 25), we also assayed all electroporated cultures for the presence of encapsidated CVB3 RNA. Viral RNA was extracted from concentrated, RNase-treated virion preparations for the detection of viral RNA in pass 1 preparations. To determine whether and to what extent CVB3-CKO and CVB3-TD50-CKO were replicating (compared to the replication of wild-type CVB3 or CVB3-TD50), viral RNA was extracted from aliquots of the RNase-treated and pelleted preparations and RT-qPCR was performed (Fig. 2B). These results (based on those from five replicate plates and 10 quantitative data points [CVB3-CKO and wt CVB3] or two replicate plates and 4 quantitative data points [CVB3-TD50 and CVB3-TD50-CKO]) indicate that CVB3-CKO is indeed replication competent, as the RNA was derived from pelleted viral particles. CVB3-CKO replicated to a level approximately 5 log units lower than that for wt CVB3 (Fig. 2B; compare CVB3-28 [wt] to CVB3-CKO), indicating that ablation of the CRE(2C) uridylylation function severely handicaps replication of the parental virus. Interestingly, these results also show that there is no significant difference in the extents of replication of CVB3-CKO and CVB3-TD50 and that the mutational disruption of CRE(2C) in a CVB3-TD strain does not significantly alter its replication (Fig. 2B; compare the results for CVB3-CKO to those for CVB3-TD50-CKO and CVB3-TD50). In summary, the ablation of the CRE(2C) function is not lethal either in wt CVB3 or in a CVB3-TD strain, but it does lower the level of replication to the level of replication for the CVB3-TD populations. Important to note is that the CVB3-TD50-CKO strain did not replicate at a lower level than either CVB3-CKO or CVB3-TD50, indicating that the functional loss of the two CREs [domain I and CRE(2C)] is not additive.
Replication of CVB3-CKO is accompanied by the generation of CVB3-CKO genomes with 5′-terminal deletions.
Because the replication of CVB3-CKO and CVB3-TD50 is similarly poor (i.e., the apparent absence of CPE and replication at a level on the order of 5 log units lower than that of wt CVB3) and the mutational disruption of CRE(2C) does not affect the overall replication of the CVB3-TD50-CKO strain more than it affects the overall replication of CVB3-TD50, we hypothesized that alterations at either of these sites were likely to result in the same type of positive-strand initiation and, consequently, that 5′-terminal genomic deletions could be occurring during the replication of CVB3-CKO in cell culture. We therefore used RT-PCR to test whether 5′-terminal deletions had arisen in the CVB3-CKO population. Using equivalent numbers of positive-sense viral RNA strands for wt CVB3 and CVB3-CKO, cDNA was transcribed using primer E3 (Table 1), followed by PCR using primers designed to detect sequences close to the 5′ terminus (Fig. 3A to C). Absence of bands in the amplifications of CVB3-CKO correlating to the 5’ terminal 20 nucleotides (Fig. 3D, arrows, lanes 2 and 3) and the presence of bands in those of wild-type CVB3 RNA (Fig. 3C, lanes 5 and 6) and the controls (Fig. 3C, lanes 8 and 9 and lanes 11 and 12) indicated that the CVB3-CKO RNA had lost 5′-terminal sequence. Water (no cDNA), no-template RT-PCR, and tissue culture controls were all negative (Fig. 3E, lanes 2 to 4 for no-cDNA PCR controls, lanes 5 to 7 for no-template RT-PCR controls, and lanes 8 to 10 for tissue culture controls). However, bands were observed by PCR of cDNA amplified from both CVB3-CKO and wt CVB3 with primers whose sequences correspond to a sequence in the 5′ NTR of the genome (Fig. 3C) greater than 50 nucleotides downstream of the 5′ terminus (Fig. 3D, lanes 4 [CVB3-CKO] and 7 [wt CVB3]), demonstrating that virus was indeed present in the CVB3-CKO preparations. Thus, the majority population of CVB3-CKO RNA lacks all or most of nucleotides 1 to 24 of the wt CVB3 genome, similar to what was observed in persisting CVBs in human and murine infections (24, 25, 28).
FIG 3.

Loss of a functional CRE(2C) leads to evolution of 5′-terminal deletions in CVB3-CKO after 8 days of replication in HeLa cells. (A) Schematic of how amplification with Pin primers works. (B and C) Relative position in the CVB3 genome of tagged primers used to amplify the 5′-terminal sequence (B) or to amplify the sequence in a region of the 5′ NTR where deletions do not occur (C). (D) Agarose gel analysis of E3-primed cDNA amplified with primers Pin5End1 and E3 or primers Pin5End5 and E3 and then subsequently amplified with primers Pin and SReturn (lanes 2 and 3, CVB3-CKO purified virus preparation; lanes 5 and 6, CVB3-CKO T7 transcripts; lanes 8 and 9, wt CVB3 preparation; lanes 11 and 12, wt CVB3 T7 transcripts). To detect virus using a region of the 5′ NTR where deletions do not occur, cDNA was made with primer PinE3 and amplified with primers S5 and Pin (lane 4, CVB3-CKO preparation; lane 7, CVB3-CKO T7 transcripts; lane 10, wt CVB3 preparations; lane 13, wt CVB3 T7 transcripts). The cDNA of 400 (Pin5End1) or 1,400 (Pin5End5) viral RNA or T7-transcribed RNA (controls) copies was used in each PCR. Arrows, the loss of sequence from the 5′ terminus of CVB3-CKO (lanes 2 and 3) compared to the T7 or wt virus controls (lanes 5 and 6, 8 and 9, and 11 and 12). However, CVB3-CKO could be detected by RT-PCR priming in a region of the 5′ NTR where deletions do not occur (compare lane 4 to lanes 7, 10, and 13). (E) Controls (lanes 2 to 4, no-cDNA PCR controls; lanes 5 to 7, no-template RT-PCR controls; lanes 8 to 10, tissue culture controls) were negative. Lanes 1 and 4 in panel D and lanes 1 and 11 in panel E contain molecular size markers.
CVB3-CKO virions can be sequentially passaged in cell culture, and productive infection is neutralized by anti-CVB3 neutralizing serum.
Previous work demonstrated that CVB3-TD strains encapsidate RNA and can be passaged in cell culture and neutralized by anti-CVB3 neutralizing antibody (25). Because we had generated stocks of virions that had been partially purified using RNase A to degrade nonencapsidated RNA followed by ultracentrifugal pelleting through a 30% sucrose cushion (Fig. 2B), we had evidence that viral RNA was encapsidated and RNA detection was not due to contamination with free viral RNA. This is in contrast to the previous observation that the mutational disruption of CRE(2C) in HEVs is lethal, on the basis of the absence of CPE in cell culture, the absence of single-stranded RNA in autoradiographic assays, and the absence of luciferase activity after only 10 h of incubation in transfected cells (31, 44, 47). As a further test that CVB3-CKO produced infectious virions, we used RNase-treated and virus preparations collected by ultracentrifugation to inoculate fresh cell cultures (termed pass 2) in the presence or absence of anti-CVB3 neutralizing antibody. HeLa cells in 24-well plates were inoculated with CVB3-CKO (equivalent to 4,123 positive RNA strands, determined on the basis of RT-qPCR quantitation of cDNA from virus preparations) with or without anti-CVB3 neutralizing antibody (Fig. 4A to F and H) as described earlier (25). After 5 days of incubation, cytopathic effects were present only in cultures inoculated with wt CVB3 in the absence of neutralizing antibody (Fig. 4; compare panels A to E to panel F), demonstrating that the amount of antibody used was sufficient to suppress a productive cytopathic infection.
FIG 4.

CVB3-CKO replicates without observable CPE and can be passaged from purified HeLa cell lysates and neutralized by anti-CVB3 neutralizing antibody. (A to F) HeLa cell monolayers at 5 days postinoculation with wt CVB3 (C, F) or CVB3-CKO (B, E) with anti-CVB3 neutralizing antibody (+ Ab) (A to C) or without antibody (− Ab) (D to E). Monolayers without virus inoculation (A, D) were also cultured. CVB3-CKO does not produce CPE after infection. (B) Neutralizing antibody prevents CPE with wt CVB3 (C) at 5 days postinoculation, whereas CPE occurs without neutralizing antibody (F). Magnifications, ×100. (G) Nested primers used to amplify the sequence in the 5′ NTR of CVB3 to detect viral RNA after inoculation of cell cultures. Primers are specific for a region of the 5′ NTR where deletions do not occur. (H) Total RNA was extracted from HeLa cell monolayers at 5 days postinoculation and assayed for the presence of viral RNA by nested PCR and gel electrophoresis. wt CVB3 was detected (lanes 8 and 9), as was noncytopathic CVB3-CKO (arrows, lanes 4 and 5). The absence of bands when neutralizing antibody was present in the cell cultures (lanes 2 and 3, CVB3-CKO; lanes 6 and 7, wt CVB3) demonstrates that the antibody effectively suppressed productive infection. Lane 1, molecular size markers. (I) All controls (lanes 2 to 4, no-cDNA PCR controls; lanes 5 to 7, no-RNA RT-PCR controls; lanes 8 to 10, tissue culture controls) were negative. Lanes 1 and 10, molecular size markers. (J) Additional virus preparations were tested to verify the observations made from panel H. CVB3-CKO can be passaged in cell culture (lane 3), whereas the T7 RNA transcript control cannot (lane 1), and CVB3-CKO can be neutralized with anti-CVB3 neutralizing antibody (lane 2). Lane 4, molecular size markers.
Successful neutralizing antibody suppression of infection should be verifiable by an inability to detect viral RNA. To test whether CVB3 RNA was present in the cultures with or without neutralizing antibody, nested RT-PCR for the detection of positive-strand viral RNA was performed on total RNA prepared from cell cultures in two separate experiments (Fig. 4G). Viral RNA was detected in wells inoculated with CVB3-CKO and wt CVB3 in the absence of neutralizing antibody (compare Fig. 4H, lanes 4 and 5 [arrows], and J, lane 3 [CVB3-CKO], with H, lanes 8 and 9 [wt CVB3]) but not in wells containing antibody (compare Fig. 4H, lanes 2 and 3, and J, lane 2 [CVB3-CKO], with H, lanes 6 and 7 [wt CVB3]). The water (no-cDNA), no-template RT-PCR, and tissue culture controls were negative (Fig. 4I, lanes 2 to 4 for the no-cDNA PCR controls, lanes 5 to 7 for the no-template RT-PCR controls, and lanes 8 to 10 for the tissue culture controls). These results demonstrate that electroporation with CVB3-CKO T7-transcribed RNA produces infectious CVB3 particles that can be isolated and transferred to fresh cell cultures.
Inoculation of CVB3 and CVB3-CKO RNA into mice generates virus.
Our observations that CVB3-CKO produced infectious virus and can be passaged in cell culture led us to ask if this was reproducible in vivo. A/J mice were chosen for use in this experiment, as long-term CVB3 persistence has been demonstrated in this strain of mouse and this strain has regularly been used in our studies of CVB3-TD replication (25, 28, 55). Mice were inoculated i.p. with lipid-conjugated CVB3-CKO or wt CVB3 T7-transcribed RNA and killed 8 days (wt CVB3) or 20 days (CVB3-CKO) postinoculation. Spleen and heart RNA preparations were assayed for the presence of viral RNA by RT-PCR. It was necessary to perform serial dilutions of RNA extracted at day 20 posttransfection from tje tissues of mice that had been transfected with CVB3-CKO in order to detect the low-level replication of this mutant virus against the high background level of tissue RNA. Viral RNA was detectable only at higher dilutions (Fig. 5A; compare lanes 4 [10-fold dilution], 5 [100-fold dilution], and 7 [10,000-fold dilution] to lane 6 [arrow, 1,000-fold dilution], though the required dilution varied slightly among the different samples. Both CVB3-CKO RNA and wt CVB3 RNA were detectable in spleen (Fig. 5B, lanes 7 and 8, respectively) and heart (Fig. 5C, lanes 6 and 7, respectively), demonstrating the infectivity of T7 transcripts of CVB3 genomes in mice.
FIG 5.

T7 RNA transcripts from CVB3 and CVB3-CKO clones can be transfected into mice, and virus replicates posttransfection. Nested RT-PCR could detect viral RNA using total RNA from the hearts and spleens of mice transfected with wt CVB3 or CVB3-CKO T7 RNA transcripts. (A) A representative gel demonstrating the necessary 10-fold serial dilutions of total extracted tissue RNA (in this case, from heart) to detect the low level of replication of CVB3-CKO in mouse tissue at 20 days posttransfection (lanes 1 and 8, molecular size marker; lane 2, no-RNA RT-PCR control; lane 3, untransfected mouse RNA; lane 4, 10-fold dilution of RNA from a CVB3-CKO-transfected mouse; lane 5, 100-fold dilution; lane 6, 1,000-fold dilution; lane 7, 10,000-fold dilution). CVB3-CKO RNA was detectable only at a 1,000-fold dilution of RNA (arrow, lane 6). (B) Detection of viral RNA in the spleens of transfected mice (lanes 1 and 9, molecular size marker; lane 2, no-cDNA PCR control; lane 3, no-RNA RT-PCR control; lane 4, HeLa cell RNA control; lane 5, 1,000 copies of CVB3 cDNA from T7 transcripts; lane 6, untransfected mouse RNA; lane 7, 1,000-fold dilution of RNA obtained from a CVB3-CKO-transfected mouse at day 20 posttransfection; lane 8, 100,000-fold dilution of RNA obtained from a wt CVB3-transfected mouse at day 8 posttransfection). (C) Detection of viral RNA in the hearts of transfected mice (lanes 1 and 8, molecular size marker; lane 2, no-cDNA PCR control; lane 3, no-RNA RT-PCR control; lane 4, untransfected mouse RNA; lane 5, 1,000 copies of CVB3 cDNA from T7 transcripts; lane 6, 100-fold dilution of RNA obtained from a CVB3-CKO-transfected mouse at day 20 posttransfection; lane 7, 10,000-fold dilution of RNA obtained from a wt CVB3-transfected mouse at day 4 posttransfection). (D to K) RNase-treated homogenates of spleens were passaged onto HeLa cells at approximately 90% confluence and incubated overnight with (D to G) or without (H to K) neutralizing antibodies (D and H, no homogenate; E and I, control mouse homogenate; F and J, homogenate obtained from CVB3-CKO-transfected mice at day 20 posttransfection; G and K, homogenate obtained from CVB3-transfected mice at day 8 posttransfection). Magnifications, ×100. CPE was observed only in cultures inoculated with homogenates from mice transfected with wt CVB3 RNA (K). (L) Following 5 days of incubation, total RNA was tested by nested RT-PCR and analyzed on 2% agarose gels (lanes 1 and 14, molecular size marker; lanes 2 and 3, CVB3-CKO plus neutralizing antibody; lanes 4 and 5, CVB3-CKO minus neutralizing antibody; lanes 6 and 7, wt CVB3 with neutralizing antibody; lanes 8 and 9, wt CVB3 without neutralizing antibody; lane 10, no-cDNA control; lane 11, no-RNA RT control; lane 12, uninfected control with neutralizing antibody; lane 13, uninfected control without neutralizing antibody), demonstrating that CVB3-CKO (arrows, lanes 4 and 5) and wt CVB3 (arrows, lanes 8 and 9) were detected in tissue homogenates following transfection of mice. The absence of amplimers in lanes 2 and 3 (CVB3-CKO) and lanes 6 and 7 (wt CVB3) demonstrates that virus is neutralized by anti-CVB3 neutralizing antibody.
To test whether the CVB3-CKO RNA detected in mice at day 20 posttransfection was encapsidated and infectious, as would be expected of a true viral infection, cleared and RNase-treated spleen homogenates were inoculated onto cell cultures either with or without prior incubation with anti-CVB3 neutralizing antibody. Spleen homogenates from mice inoculated with wt CVB3 RNA completely lysed the monolayers by 2 days postexposure, but for all other spleen homogenates, no CPE was evident by 5 days postexposure (Fig. 5D to J). No CPE was observed in cells exposed to spleen homogenates from mice inoculated with wt CVB3 transcripts if wells contained neutralizing antibody. No CPE was observed in control wells, or in wells inoculated with spleen homogenates from mice inoculated with CVB3-CKO (Fig. 5; compare panels D to J with panel K). Lysis by wt CVB3 RNA demonstrated that inoculation of conjugated CVB3 RNA generated infectious virus in the tissues of transfected mice. To assay for the presence of viral RNA in HeLa cell cultures that did not show CPE (wells inoculated with spleen homogenates from mice inoculated with CVB3-CKO and control wells), total RNA was prepared from HeLa cell monolayers and assayed using nested RT-PCR for the detection of positive-strand RNA. In wells of HeLa cell cultures inoculated with tissue homogenates from mice transfected with either CVB3-CKO or wt CVB3 RNA, viral RNA was detected at 5 days postinoculation (Fig. 5L, arrows, lanes 4 and 5 [CVB3-CKO] and lanes 8 and 9 [wt CVB3]). However, when neutralizing antibody was present, no viral RNA was detected (Fig. 5L, lanes 2 and 3 [CVB3-CKO] and lanes 6 and 7 [wt CVB3]). These results demonstrate that encapsidated, infectious virus was produced in the tissues of mice following inoculation of wt CVB3 RNA as well as CVB3-CKO RNA. Taken together, these results confirm that CVB3 is replication competent in the absence of a functional (uridylylating) CRE(2C).
DISCUSSION
Our hypothesis—that CVB3-TD strains should be able to replicate without a uridylylating CRE(2C)—is firmly supported by the results presented here. Previously, a range of 5′-terminal genomic deletions ranging from 7 to 49 nucleotides in CVB-TD populations which arose from both experimental and naturally occurring infections was characterized (24–26). In these CVB3-TD genomes, while VPg remained attached to the 5′ terminus, only 1 (of 5) of the naturally occurring deletions that were characterized terminated in the UU that would normally be donated by uridylylated VPg at the start of replication (25). If CRE(2C)-uridylylated VPg were used in CVB3-TD positive-strand initiation, CVB3-TD genomes would be expected to have a proportion of 5′-terminal uridine residues higher than the proportion that was observed. The results presented here demonstrate that mutational disruption of the CRE(2C) structure using the 16 mutations previously shown to prevent uridylylation and inhibit replication (31) does not prevent replication of CVB3-TD, as shown by an insignificant impact on the replication of viruses with a TD when assayed by RT-qPCR (Fig. 2B). We conclude that a functional CRE(2C) [defined as CRE(2C) that is able to uridylylate VPg] is not necessary for CVB3-TD replication.
However, a most unexpected result from this work came from an intended control: the mutationally disrupted CRE(2C) structure in the wt CVB3 genome (CVB3-CKO). We fully expected to show with this construct, as others had previously (31, 42, 46, 47), that a uridylylating CRE(2C) is necessary for CVB replication and that mutational disruption of the element would result in lost replication capacity. We were surprised, therefore, to observe that the strain termed CVB3-CKO, containing 16 silent mutations which ablate VPg uridylylation, was replication competent and able to be passaged in cell culture after isolation of viral particles. Our ability to isolate virus and, subsequently, viral RNA after RNase treatment and sucrose purification demonstrated that de novo replication was occurring following transfection of CVB3-CKO RNA, as only newly synthesized RNA is encapsidated (1). The additional finding that CVB3-CKO replicated as virus in mice following inoculation of RNA transcripts of the CVB3-CKO cDNA clone confirmed the in vivo findings derived from the cell culture system. van Ooij and colleagues (31) reported that the same 16 mutations introduced into CVB3 were lethal, as demonstrated by a lack of observable CPE in cell cultures, a lack of detectable luciferase activity 10 h posttransfection in BGM cells (when the P1 region was replaced with a luciferase gene), and an undetectable level of single-stranded RNA production in cell-free assays (31). The observed disparity of the effect of mutated CRE(2C) on CVB3 viability can best be explained by the type and sensitivity of the assays used to assess the viability of these viruses and the ability of these viruses to replicate at detectable levels over an extended period of time (days) in culture.
Yet another unexpected finding occurred while performing this work: the replication of the wt CVB3 genome with the mutationally disrupted CRE(2C) structure is associated with the development of a CVB3-CKO population possessing 5′-terminal deletions. As in earlier studies (25), the CVB3-CKO-TD population was detectable in the complete absence of the wt (or parental) initiating genome population and thus does not require the presence of the wt genome to replicate successfully. Terminal deletions may occur by a lack of specific initiation of replication caused by two potential mechanisms. In the first mechanism, a lack of specific VPg uridylylation on a functional CRE(2C) could lead to VPg nucleotidylation at the site of RNA replication initiation. The absence of the 2 uridine residues linked to VPg during CRE(2C)-mediated uridylylation would prevent annealing to the 2 adenine residues at the 3′ terminus of the viral negative RNA strand. In the second potential mechanism, the three-dimensional interaction with VPg in the absence of the native CRE(2C) stem-loop structure perhaps prevents the formation of the native initiation complex that would confer specificity to the 3′ end of the negative strand (56, 57). Our previous work showed that CVB3-TD populations did not arise detectably even after repeated passages of CVB3 in HeLa cells but occurred in passages of virus in nondividing primary cells (24). However, as the replication of CVB3-CKO generates 5′-terminal deletions (described above), a role for CRE(2C) in the correct initiation of positive-strand RNA can be strongly inferred, even in continuously dividing HeLa cells. From the observations that deletion of the 5′ end of the genome or a mutation in CRE(2C) results in similar levels of viral RNA replication and the presence of both lesions in a single genome does not have an additive effect on the extent of replication, it is clear that both of these sites are required for the process of efficient (and correct) positive-strand initiation.
At this juncture, we have shown that CVB3, a typical enterovirus, can replicate at a low efficiency (compared to that of wt CVB3) without two of the four known cis-acting replication elements (9, 10): namely, domain I in the 5′ NTR and CRE(2C). Thus, replication in the absence of both domain I and CRE(2C) has not been seriously considered to date due to the experimentally verified importance of these structures for HEV replication (14, 31, 34, 47). That said, it has been demonstrated that a loss of another HEV cis-acting replication element, the 3′ NTR, does not prevent replication either (58, 59). In addition, Goodfellow and colleagues (2000) demonstrated that after introducing two and five mutations into the poliovirus CRE(2C), a return to a wt lytic phenotype occurred after serial blind passage, despite the fact that no CPE was observed upon initial transfection (34). The ability of these viruses to revert demonstrates that replication occurred in the absence of a wt CRE(2C) structure and lends support to our findings that mutational disruption of CRE(2C) is not lethal.
The poor extents of replication of the CVB3-TD strains as well as the CVB3-CKO and CVB3-TD50-CKO strains led us to investigate the use of the transfection of viral genomic RNA into mice as an alternative approach to generating virus. We used only one transfection reagent, a lipid-based in vivo transfection reagent (Altogen Biosystems), but from the results obtained here (Fig. 5), it would appear that this approach has potential for studying diverse in vivo aspects of the biology of these inefficiently replicating virus strains, as well as the host response to infection by such viruses, without having to laboriously produce virus stocks from cell culture. To the best of our knowledge, this represents the first report of the generation of infectious virus by i.p. lipid-based transfection of animals with a picornaviral RNA, though others have previously described methods to inoculate mice with viral DNA (60, 61) or RNA (62). However, the method described in this report may be more efficient than the methods previously reported, as it uses a reagent designed for in vivo RNA transfection.
ACKNOWLEDGMENTS
This work was supported in part by grants from the National Institutes of Health (to N.M.C.) and the Juvenile Diabetes Research Foundation (to N.M.C. and S.T.). S.S. was supported by a University of Nebraska Medical Center College of Medicine graduate fellowship.
Those providing funding had no role in the experimental design, data gathering, data analysis, or submission of the work for publication.
REFERENCES
- 1.Ehrenfeld E, Domingo E, Roos RP (ed). 2010. The picornaviruses. ASM Press, Washington, DC. [Google Scholar]
- 2.Drescher KM, Kono K, Bopegamage S, Carson SD, Tracy S. 2004. Coxsackievirus B3 infection and type 1 diabetes development in NOD mice: insulitis determines susceptibility of pancreatic islets to virus infection. Virology 329:381–394. doi: 10.1016/j.virol.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 3.Tracy S, Drescher KM, Chapman NM. 2011. Enteroviruses and type 1 diabetes. Diabetes Metab Res Rev 27:820–823. doi: 10.1002/dmrr.1255. [DOI] [PubMed] [Google Scholar]
- 4.Chapman NM, Kim KS. 2008. Persistent coxsackievirus infection: enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr Top Microbiol Immunol 323:275–292. [DOI] [PubMed] [Google Scholar]
- 5.Modlin JF, Rotbart HA. 1997. Group B coxsackie disease in children. Curr Top Microbiol Immunol 223:53–80. [DOI] [PubMed] [Google Scholar]
- 6.Pallansch MA, Oberste MS, Whitton JL. 2013. Enteroviruses: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses, 6th ed, vol 1 Wolters Kluwer, Philadelphia, PA. [Google Scholar]
- 7.Ogram SA, Flanegan JB. 2011. Non-template functions of viral RNA in picornavirus replication. Curr Opin Virol 1:339–346. doi: 10.1016/j.coviro.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daijogo S, Semler BL. 2011. Mechanistic intersections between picornavirus translation and RNA replication. Adv Virus Res 80:1–24. doi: 10.1016/B978-0-12-385987-7.00001-4. [DOI] [PubMed] [Google Scholar]
- 9.Cordey S, Gerlach D, Junier T, Zdobnov EM, Kaiser L, Tapparel C. 2008. The cis-acting replication elements define human enterovirus and rhinovirus species. RNA 14:1568–1578. doi: 10.1261/rna.1031408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Wimmer E, Paul AV. 2009. cis-Acting RNA elements in human and animal plus-strand RNA viruses. Biochim Biophys Acta 1789:495–517. doi: 10.1016/j.bbagrm.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rivera VM, Welsh JD, Maizel JV Jr. 1988. Comparative sequence analysis of the 5′ noncoding region of the enteroviruses and rhinoviruses. Virology 165:42–50. doi: 10.1016/0042-6822(88)90656-3. [DOI] [PubMed] [Google Scholar]
- 12.Sean P, Semler BL. 2008. Coxsackievirus B RNA replication: lessons from poliovirus. Curr Top Microbiol Immunol 323:89–121. [DOI] [PubMed] [Google Scholar]
- 13.Andino R, Rieckhof GE, Achacoso PL, Baltimore D. 1993. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J 12:3587–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andino R, Rieckhof GE, Baltimore D. 1990. A functional ribonucleoprotein complex forms around the 5′ end of poliovirus RNA. Cell 63:369–380. doi: 10.1016/0092-8674(90)90170-J. [DOI] [PubMed] [Google Scholar]
- 15.Parsley TB, Towner JS, Blyn LB, Ehrenfeld E, Semler BL. 1997. Poly (rC) binding protein 2 forms a ternary complex with the 5′-terminal sequences of poliovirus RNA and the viral 3CD proteinase. RNA 3:1124–1134. [PMC free article] [PubMed] [Google Scholar]
- 16.Xiang W, Harris KS, Alexander L, Wimmer E. 1995. Interaction between the 5′-terminal cloverleaf and 3AB/3CDpro of poliovirus is essential for RNA replication. J Virol 69:3658–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barton DJ, O'Donnell BJ, Flanegan JB. 2001. 5′ cloverleaf in poliovirus RNA is a cis-acting replication element required for negative-strand synthesis. EMBO J 20:1439–1448. doi: 10.1093/emboj/20.6.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zell R, Ihle Y, Seitz S, Gundel U, Wutzler P, Gorlach M. 2008. Poly(rC)-binding protein 2 interacts with the oligo(rC) tract of coxsackievirus B3. Biochem Biophys Res Commun 366:917–921. doi: 10.1016/j.bbrc.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 19.Gamarnik AV, Andino R. 1998. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev 12:2293–2304. doi: 10.1101/gad.12.15.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gamarnik AV, Andino R. 2000. Interactions of viral protein 3CD and poly(rC) binding protein with the 5′ untranslated region of the poliovirus genome. J Virol 74:2219–2226. doi: 10.1128/JVI.74.5.2219-2226.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris KS, Xiang W, Alexander L, Lane WS, Paul AV, Wimmer E. 1994. Interaction of poliovirus polypeptide 3CDpro with the 5′ and 3′ termini of the poliovirus genome. Identification of viral and cellular cofactors needed for efficient binding. J Biol Chem 269:27004–27014. [PubMed] [Google Scholar]
- 22.Sharma N, Ogram SA, Morasco BJ, Spear A, Chapman NM, Flanegan JB. 2009. Functional role of the 5′ terminal cloverleaf in coxsackievirus RNA replication. Virology 393:238–249. doi: 10.1016/j.virol.2009.07.039. [DOI] [PubMed] [Google Scholar]
- 23.Murray KE, Steil BP, Roberts AW, Barton DJ. 2004. Replication of poliovirus RNA with complete internal ribosome entry site deletions. J Virol 78:1393–1402. doi: 10.1128/JVI.78.3.1393-1402.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim K-S, Chapman NM, Tracy S. 2008. Replication of coxsackievirus B3 in primary cell cultures generates novel viral genome deletions. J Virol 82:2033–2037. doi: 10.1128/JVI.01774-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim K-S, Tracy S, Tapprich W, Bailey J, Lee C-K, Kim K, Barry WH, Chapman NM. 2005. 5′-terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J Virol 79:7024–7041. doi: 10.1128/JVI.79.11.7024-7041.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chapman NM, Kim K-S, Drescher KM, Oka K, Tracy S. 2008. 5′ terminal deletions in the genome of a coxsackievirus B2 strain occurred naturally in human heart. Virology 375:480–491. doi: 10.1016/j.virol.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leveque N, Renois F, Talmud D, Nguyen Y, Lesaffre F, Boulagnon C, Bruneval P, Fornes P, Andreoletti L. 2012. Quantitative genomic and antigenomic enterovirus RNA detection in explanted heart tissue samples from patients with end-stage idiopathic dilated cardiomyopathy. J Clin Microbiol 50:3378–3380. doi: 10.1128/JCM.01612-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tracy S, Smithee S, Alhazmi A, Chapman N. 2015. Coxsackievirus can persist in murine pancreas by deletion of 5′ terminal genomic sequences. J Med Virol 87:240–247. doi: 10.1002/jmv.24039. [DOI] [PubMed] [Google Scholar]
- 29.Sharma N, O'Donnell BJ, Flanegan JB. 2005. 3′-terminal sequence in poliovirus negative-strand templates is the primary cis-acting element required for VPgpUpU-primed positive-strand initiation. J Virol 79:3565–3577. doi: 10.1128/JVI.79.6.3565-3577.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paul AV, Rieder E, Kim DW, van Boom JH, Wimmer E. 2000. Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J Virol 74:10359–10370. doi: 10.1128/JVI.74.22.10359-10370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Ooij MJ, Vogt DA, Paul A, Castro C, Kuijpers J, van Kuppeveld FJ, Cameron CE, Wimmer E, Andino R, Melchers WJ. 2006. Structural and functional characterization of the coxsackievirus B3 CRE(2C): role of CRE(2C) in negative- and positive-strand RNA synthesis. J Gen Virol 87:103–113. doi: 10.1099/vir.0.81297-0. [DOI] [PubMed] [Google Scholar]
- 32.Yang Y, Rijnbrand R, McKnight KL, Wimmer E, Paul A, Martin A, Lemon SM. 2002. Sequence requirements for viral RNA replication and VPg uridylylation directed by the internal cis-acting replication element (cre) of human rhinovirus type 14. J Virol 76:7485–7494. doi: 10.1128/JVI.76.15.7485-7494.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerber K, Wimmer E, Paul AV. 2001. Biochemical and genetic studies of the initiation of human rhinovirus 2 RNA replication: identification of a cis-replicating element in the coding sequence of 2A(pro). J Virol 75:10979–10990. doi: 10.1128/JVI.75.22.10979-10990.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodfellow I, Chaudhry Y, Richardson A, Meredith J, Almond JW, Barclay W, Evans DJ. 2000. Identification of a cis-acting replication element within the poliovirus coding region. J Virol 74:4590–4600. doi: 10.1128/JVI.74.10.4590-4600.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKnight KL, Lemon SM. 1998. The rhinovirus type 14 genome contains an internally located RNA structure that is required for viral replication. RNA 4:1569–1584. doi: 10.1017/S1355838298981006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lobert P-E, Escriou N, Ruelle J, Michiels T. 1999. A coding RNA sequence acts as a replication signal in cardioviruses. Proc Natl Acad Sci U S A 96:11560–11565. doi: 10.1073/pnas.96.20.11560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKnight KL. 2003. The human rhinovirus internal cis-acting replication element (cre) exhibits disparate properties among serotypes. Arch Virol 148:2397–2418. doi: 10.1007/s00705-003-0177-7. [DOI] [PubMed] [Google Scholar]
- 38.Mason PW, Bezborodova SV, Henry TM. 2002. Identification and characterization of a cis-acting replication element (cre) adjacent to the internal ribosome entry site of foot-and-mouth disease virus. J Virol 76:9686–9694. doi: 10.1128/JVI.76.19.9686-9694.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Y, Yi M, Evans DJ, Simmonds P, Lemon SM. 2008. Identification of a conserved RNA replication element (cre) within the 3Dpol-coding sequence of hepatoviruses. J Virol 82:10118–10128. doi: 10.1128/JVI.00787-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin J, Paul AV, Wimmer E, Rieder E. 2003. Functional dissection of a poliovirus cis-acting replication element [PV-cre(2C)]: analysis of single- and dual-cre viral genomes and proteins that bind specifically to PV-cre RNA. J Virol 77:5152–5166. doi: 10.1128/JVI.77.9.5152-5166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paul AV, Peters J, Mugavero J, Yin J, van Boom JH, Wimmer E. 2003. Biochemical and genetic studies of the VPg uridylylation reaction catalyzed by the RNA polymerase of poliovirus. J Virol 77:891–904. doi: 10.1128/JVI.77.2.891-904.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goodfellow IG, Kerrigan D, Evans DJ. 2003. Structure and function analysis of the poliovirus cis-acting replication element (CRE). RNA 9:124–137. doi: 10.1261/rna.2950603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rieder E, Paul AV, Kim DW, van Boom JH, Wimmer E. 2000. Genetic and biochemical studies of poliovirus cis-acting replication element cre in relation to VPg uridylylation. J Virol 74:10371–10380. doi: 10.1128/JVI.74.22.10371-10380.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morasco BJ, Sharma N, Parilla J, Flanegan JB. 2003. Poliovirus cre(2C)-dependent synthesis of VPgpUpU is required for positive- but not negative-strand RNA synthesis. J Virol 77:5136–5144. doi: 10.1128/JVI.77.9.5136-5144.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crowder S, Kirkegaard K. 2005. trans-Dominant inhibition of RNA viral replication can slow growth of drug-resistant viruses. Nat Genet 37:701–709. doi: 10.1038/ng1583. [DOI] [PubMed] [Google Scholar]
- 46.Goodfellow IG, Polacek C, Andino R, Evans DJ. 2003. The poliovirus 2C cis-acting replication element-mediated uridylylation of VPg is not required for synthesis of negative-sense genomes. J Gen Virol 84:2359–2363. doi: 10.1099/vir.0.19132-0. [DOI] [PubMed] [Google Scholar]
- 47.Murray KE, Barton DJ. 2003. Poliovirus CRE-dependent VPg uridylylation is required for positive-strand RNA synthesis but not for negative-strand RNA synthesis. J Virol 77:4739–4750. doi: 10.1128/JVI.77.8.4739-4750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tracy S, Drescher KM, Chapman NM, Kim KS, Carson SD, Pirruccello S, Lane PH, Romero JR, Leser JS. 2002. Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J Virol 76:12097–12111. doi: 10.1128/JVI.76.23.12097-12111.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 50.Tracy S. 1981. Improved rapid methodology for the isolation of nucleic acids from agarose gels. Prep Biochem 11:251–268. doi: 10.1080/00327488108061767. [DOI] [PubMed] [Google Scholar]
- 51.Gregory JB, Litaker RW, Noble RT. 2006. Rapid one-step quantitative reverse transcriptase PCR assay with competitive internal positive control for detection of enteroviruses in environmental samples. Appl Environ Microbiol 72:3960–3967. doi: 10.1128/AEM.02291-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shulman LM, Hindiyeh M, Muhsen K, Cohen D, Mendelson E, Sofer D. 2012. Evaluation of four different systems for extraction of RNA from stool suspensions using MS-2 coliphage as an exogenous control for RT-PCR inhibition. PLoS One 7:e39455. doi: 10.1371/journal.pone.0039455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levesque-Sergerie JP, Duquette M, Thibault C, Delbecchi L, Bissonnette N. 2007. Detection limits of several commercial reverse transcriptase enzymes: impact on the low- and high-abundance transcript levels assessed by quantitative RT-PCR. BMC Mol Biol 8:93. doi: 10.1186/1471-2199-8-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lanford RE, Sureau C, Jacob JR, White R, Fuerst TR. 1994. Demonstration of in vitro infection of chimpanzee hepatocytes with hepatitis C virus using strand-specific RT/PCR. Virology 202:606–614. doi: 10.1006/viro.1994.1381. [DOI] [PubMed] [Google Scholar]
- 55.Rabausch-Starz I, Schwaiger A, Grunewald K, Muller-Hermelink HK, Neu N. 1994. Persistence of virus and viral genome in myocardium after coxsackievirus B3-induced murine myocarditis. Clin Exp Immunol 96:69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kirkegaard K, Semler B. 2010. Genome replication II: the process, p 127–140. In Ehrenfeld E, Domingo E, Roos RP (ed), The picornaviruses. ASM Press, Washington, DC. [Google Scholar]
- 57.Rozovics J, Semler BL. 2010. Genome replication I: the players, p 107–125. In Ehrenfeld E, Domingo E, Roos RP (ed), The picornaviruses. ASM Press, Washington, DC. [Google Scholar]
- 58.Brown DM, Cornell CT, Tran GP, Nguyen JH, Semler BL. 2005. An authentic 3′ noncoding region is necessary for efficient poliovirus replication. J Virol 79:11962–11973. doi: 10.1128/JVI.79.18.11962-11973.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Todd S, Towner JS, Brown DM, Semler BL. 1997. Replication-competent picornaviruses with complete genomic RNA 3′ noncoding region deletions. J Virol 71:8868–8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Israel MA, Chan HW, Hourihan SL, Rowe WP, Martin MA. 1979. Biological activity of polyoma viral DNA in mice and hamsters. J Virol 29:990–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dubensky TW, Campbell BA, Villarreal LP. 1984. Direct transfection of viral and plasmid DNA into the liver or spleen of mice. Proc Natl Acad Sci U S A 81:7529–7533. doi: 10.1073/pnas.81.23.7529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hunziker IP, Cornell CT, Whitton JL. 2007. Deletions within the 5′UTR of coxsackievirus B3: consequences for virus translation and replication. Virology 360:120–128. doi: 10.1016/j.virol.2006.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]