

ABSTRACT
Despite encoding multiple viral proteins that modulate the retinoblastoma (Rb) protein in a manner classically defined as inactivation, human cytomegalovirus (HCMV) requires the presence of the Rb protein to replicate efficiently. In uninfected cells, Rb controls numerous pathways that the virus also commandeers during infection. These include cell cycle progression, senescence, mitochondrial biogenesis, apoptosis, and glutaminolysis. We investigated whether a potential inability of HCMV to regulate these Rb-controlled pathways in the absence of the Rb protein was the reason for reduced viral productive replication in Rb knockdown cells. We found that HCMV was equally able to modulate these pathways in the parental Rb-expressing and Rb-depleted cells. Our results suggest that Rb may be required to enhance a specific viral process during HCMV productive replication.
IMPORTANCE The retinoblastoma (Rb) tumor suppressor is well established as a repressor of E2F-dependent transcription. Rb hyperphosphorylation, degradation, and binding by viral oncoproteins are also codified. Recent reports indicate Rb can be monophosphorylated, repress the transcription of antiviral genes in association with adenovirus E1A, modulate cellular responses to polycomb-mediated epigenetic methylations in human papillomavirus type 16 E7 expressing cells, and increase the efficiency of human cytomegalovirus (HCMV) productive replication. Since Rb function also now extends to regulation of mitochondrial function (apoptosis, metabolism), it is clear that our current understanding of this protein is insufficient to explain its roles in virus-infected cells and tumors. Work here reinforces this concept, showing the known roles of Rb are insufficient to explain its positive impact on HCMV replication. Therefore, HCMV, along with other viral systems, provide valuable tools to probe functions of Rb that might be modulated with therapeutics for cancers with viral or nonviral etiologies.
INTRODUCTION
Retinoblastoma (Rb) protein function is modified by multiple viruses (1–3). Through transcriptional repression of the E2F-responsive genes required for DNA replication, hypophosphorylated (active) Rb impedes cell cycle transit through G1 and into S phase (4). Rb can also induce the formation of heterochromatin at E2F responsive genes, leading to permanent transcriptional silencing and replicative senescence (5, 6), providing a tumor suppressive function. As the role of Rb as a mediator of senescence and restrictor of cell cycle progression has long been acknowledged, the prevailing model in the field of DNA virology has associated viral targeting of Rb with maintaining a cell cycle state conducive to viral replication (7). Specifically, it was proposed that viruses alter the function of Rb to provide an S-phase-like environment where the enzymes and small molecule precursors necessary for DNA synthesis would be readily available for viral DNA replication. Indeed, the ability of the E7 protein of the high-risk human papillomavirus strain 16 to bind Rb is necessary for viral DNA replication (8).
However, we recently reported that transient and stable Rb knockdown reduces the efficiency of human cytomegalovirus (HCMV) DNA synthesis and productive replication (9). This result was unexpected as HCMV encodes at least four viral proteins reported to modify several biological functions of Rb (2). Therefore, the relationship between viruses and Rb appears more complicated than the current paradigm allows.
In recent years Rb has been shown to affect many facets of mitochondrial function in addition to its critical role in controlling the cell cycle. These include mitochondrial biogenesis, apoptosis, and the utilization of glutamine for the tricarboxylic acid (TCA) cycle and the production of glutathione. In the absence of Rb, cells have lower ratios of mitochondrial to cellular DNA, and this has been ascribed to defects in mitochondrial biogenesis (10, 11). Rb regulates apoptosis directly at the mitochondria by binding to Bax (12, 13). Interestingly, it is a phosphorylated form of Rb that interacts with Bax, and loss of this form can trigger apoptosis (12). Rb also impacts apoptosis indirectly in the nucleus by repressing the transcription of E2F-responsive proapoptotic genes such as Apaf1 and caspases (14). In the absence of Rb, proapoptotic proteins can accumulate, making cells more sensitive to stress-induced apoptosis. Rb also controls metabolic reactions that impinge upon the ability of mitochondria to generate ATP under conditions of stress (15, 16). Rb loss can decrease cell energy expenditure (17), and direct glutamine catabolism toward the production of glutathione and therefore away from anaplerotic supplementation of the TCA pathway (10, 18).
Provocatively, viruses, including HCMV, also modulate all of these cellular operations regulated by Rb. We reasoned that the dependence of efficient HCMV replication on the presence of Rb might be related to the control this protein exerts over these cellular processes. Therefore, we tested whether the inability of HCMV to arrest the cell cycle, invoke senescence, prevent apoptosis, alter mitochondrial abundance and morphology, or balance metabolic pathways in the absence of Rb could potentially explain the replication defect observed in the absence of this crucial tumor suppressor. We found HCMV fully capable of wild-type level manipulation of these cellular pathways in the absence of Rb. Our work points to the strong potential for a direct effect of Rb on a viral process critical for efficient HCMV replication and perhaps for the success of other viral infections as well.
MATERIALS AND METHODS
Cells, plasmids, and viruses.
Primary normal human dermal fibroblasts (NHDFs; Clonetics) transduced with retroviruses expressing scrambled shRNA (19), shRNA against Rb (Rb2), or p107 (107.2, formerly Rb-sh2 and 107-sh2, respectively [20]) were derived and cultured as previously described (9, 21). The Lonza nucleofection system (VPI-1002; Lonza) was used according to the manufacturer's instructions to transiently express HCMV UL97 (pCGN-HA-UL97) (22) and a green fluorescent protein (GFP)-lamin A fusion protein (pEGFPhLA-WT) (23). Then, 1 μg of each plasmid was transfected per 5 × 105 cells, and the cells were plated on glass coverslips in complete medium for 24 h before fixation and immunofluorescence staining, as previously described (21). Cells were infected as previously described (9) with wild-type AD169 or an AD169 derivative expressing IE2 fused to GFP (AD169 IE2-GFP) (24) when indicated. Virus titers were measured by standard plaque assay on nontransduced NHDFs.
Inhibitors, chemicals, and antibodies.
Z-VAD-FMK (20 μM; Calbiochem) dissolved in dimethyl sulfoxide (DMSO) was added at the time of infection and replenished every 48 h. Mitochondria were stained by adding MitoTracker Red CMXRos (250 nM in DMSO [Life Technologies]) for 30 min prior to fixation and immunofluorescence staining. Dimethyl 2-oxoglutarate (α-ketoglutarate, 7 mM; catalog no. 349631 [Sigma]), pyruvate (Pyr; 4 mM; catalog no. S8636 [Sigma]), oxaloacetic acid (OAA; 4 mM; catalog no. O7753 [Sigma]) or N-acetyl-l-cysteine (NAC; 5 mM; catalog no. A9165 [Sigma]) dissolved in water were added at the time of infection and replenished every 48 h as previously described (25). Nocodazole (100 ng/ml [VWR]) and phosphonoacetic acid (250 μg/ml; Sigma) were added at 12 h postinfection (hpi) and maintained on cells until harvesting at 48 hpi, as previously described (26). Commercially available primary antibodies used in these experiments are: anti-Rb (4H1 [Cell Signaling] and 554136 [BD Pharmingen]), anti-p107 (C-18; Santa Cruz), anti-E2F1 (3742; Cell Signaling), anti-tubulin (DM 1A; Sigma), anti-p53 (OP43; Calbiochem), anti-Daxx (M-112; Santa Cruz), and anti-HA (11867423001; Roche). Monoclonal antibodies against pp71 (2H10-9) and IE1 (1B12) have been described previously (27). IR dye 680- and 800-conjugated secondary antibodies (LiCor) were used for Western blot analyses, and Alexa 488- and 594-conjugated secondary antibodies (A-11017 and A-11007; Life Technologies) were used for immunofluorescence.
Immunoblots and flow cytometry.
Equal cell numbers were lysed and analyzed by SDS-PAGE and immunoblotting for indicated antibodies as previously described (9). Flow cytometry was performed as previously described (9, 26, 28).
SA-β-Gal staining.
Infected or mock-infected cells were rinsed in the cell culture plate with Dulbecco phosphate-buffered saline (DPBS; catalog no. 14190-144 [Life Technologies]) and fixed with 4% formaldehyde in DPBS at 4°C overnight. Cells were then rinsed several times with DPBS before being stained with a X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) staining solution (5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 2 mM MgCl2, 150 mM NaCl, and 1 mg of X-Gal/ml in 40 mM citric acid-sodium phosphate buffer [pH 6.0]) overnight at 4°C (29). Cells were washed once in DPBS, and nuclei were stained with 0.2 mg of Hoechst/ml in DPBS. Cells were visualized and counted by light and fluorescence microscopy on a Nikon Eclipse TE2000-S microscope. At least 70 cells were counted for each condition, and HCMV infection experiments were repeated three times. The percentages of positive cells are represented as the number of cells in a field of view with blue X-Gal staining divided by the number of Hoechst-stained nuclei.
Trypan blue exclusion viability assay.
Serum-starved scrambled or Rb knockdown cells infected at a multiplicity of infection (MOI) of 1 were harvested by trypsinization at 72 h postinfection, and cells were diluted 1:1 in a 0.2% solution of trypan blue in PBS (T8154; Sigma). At least 80 cells were counted by using a hemocytometer and blue staining identified nonviable cells. Viability is represented as the percentage of viable cells over the total number of cells counted.
Quantitative real-time PCR.
mRNA was isolated from equal numbers of cells using total RNA minikit (IB47323; IBI). cDNA was generated using SuperScript III first-strand synthesis SuperMix (catalog no. 11752; Invitrogen) for quantitative reverse transcription-PCR (qRT-PCR). Transcripts were measured for ASCT1 (forward [Fwd], 5′-CCC GTT TGC ATC ATC TCC AG-3′; reverse [Rev], 5′-TCT GGC AAA AGA CGG GGT TC-3′), ASCT2 (Fwd, 5′-TGG ACT GGC TAG TCG ACC G-3′; Rev, 5′-GCT TGG AAT GTC ACC TGG AG-3′), GLS1 (Fwd, 5′-GAC ATG GAA CAG CGG GAC TA-3′; Rev, 5′-GAG GTG TGT ACT GGA CTT GGT-3′), and GCL (Fwd, 5′-TAC AGT TGA GGC CAA CAT GC-3′; Rev, 5′-GCT TGG AAT GTC ACC TGG AG-3′) and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcripts (30). Total DNA was extracted from equal numbers of cells using a genomic DNA minikit (IB47202; IBI). Mitochondrial DNA was measured with 16S primers (Fwd, 5′-CCG CAA GGG AAA GAT GAA AAA T-3′; Rev, 5′-TCG TCT GGT TTC GGG GGT CT-3′) and normalized to cellular DNA amplified with the GAPDH primers. cDNA or total DNA were quantitated using iTaq Universal SYBR green Supermix (172-5124; Bio-Rad) on an Applied Biosystems 7900HT instrument.
RESULTS
Cells with reduced levels of Rb produce fewer infectious particles compared to cells expressing a scrambled control shRNA at multiple times postinfection.
We previously reported that HCMV produces less progeny virions at 96 h postinfection in cells with reduced levels of Rb (9). It was not clear whether the reduction in infectious virions was due to an overall decrease in the ability to produce virions or whether there was a delay in the accumulation of infectious virions. To differentiate between the two possibilities, we performed a growth curve assay measuring the production of infectious virions by scrambled control or Rb knockdown cells at multiple times postinfection. Although the viral inoculum contained comparable levels of virus (0 days postinfection), the Rb knockdown cells consistently produced fewer infectious virions than the scrambled control cells (Fig. 1). This suggests that the Rb knockdown cells have a reduced capacity to replicate HCMV virions and the decreased levels of Rb do not simply delay viral replication.
FIG 1.
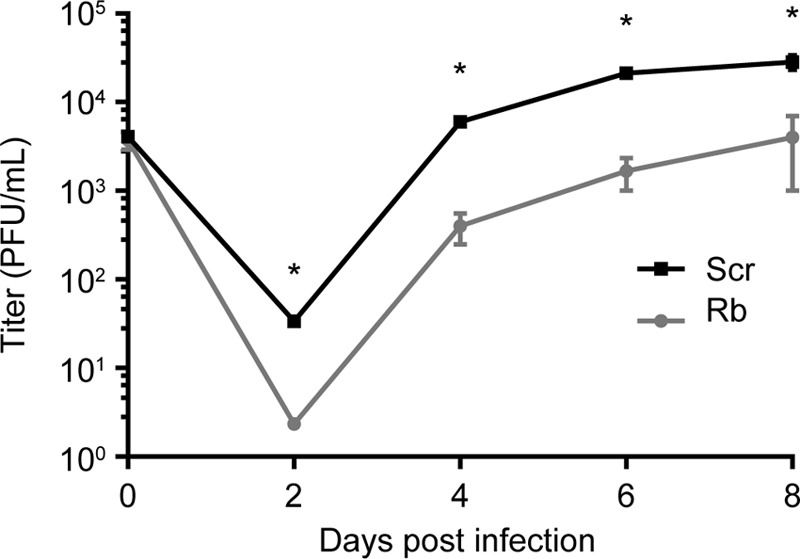
Cells with reduced levels of Rb accumulate fewer replicated virions than scrambled control cells at multiple times postinfection. Viral replication after infection with HCMV at an MOI of 1 in scrambled control (Scr) or Rb knockdown (Rb) cells was determined by plaque assay of the viral inoculum at the time of infection (day 0) and of cell-free and cell-associated virus at the indicated days postinfection. Error bars represent the standard errors from three biological replicates. Student t test was used to determine the statistical difference between Scr and Rb knockdown cells at each time point (*, P < 0.05).
HCMV arrests cell cycle progression in Rb knockdown cells.
Rb can restrict cell cycle progression through G1 and into the S phase. HCMV infection also arrests cells with a G1 DNA content (31–33). Importantly, HCMV replicates efficiently only in G1 and not in subsequent phases of the cell cycle (34). We therefore sought to determine whether HCMV replicated less efficiently in Rb knockdown cells because it could no longer arrest cell cycle progression in G1. To gauge cell cycle arrest during mock or HCMV infections, we used propidium iodide (PI) staining and flow cytometry to monitor the ability of cells released from serum starvation induced G1 synchronization to accumulate with S- or G2-phase DNA content. Rb knockdown cells synchronize in G1 by serum starvation with similar efficiency as the control cells (Fig. 2A) (9). Serum-stimulated cells were treated with the microtubule depolymerizing agent nocodazole to prevent passage through mitosis and conversion of a cell with a G2 DNA content into two cells each with a G1 DNA content. Furthermore, phosphonoacetic acid (PAA) was added to inhibit the viral DNA polymerase, a necessary supplement as accumulating viral DNA stains with PI and thus undermines the ability of the technique to define cell cycle status. G1 serum starved cells expressing either an shRNA targeting Rb or a scrambled control shRNA were equally able to enter S phase and accumulate in G2 phase upon serum stimulation and mock infection (Fig. 2B). The Rb knockdown cells displayed an apparent increase in the percentage of cells in S phase after serum stimulation (Fig. 2E.). Cells lacking Rb are reported to spend longer in S phase (35), possibly explaining this observation. Furthermore, both control and Rb knockdown cells were equally able to prevent serum-induced cell cycle progression after HCMV infection (Fig. 2C). We conclude that HCMV efficiently blocks cell cycle progression in the absence of Rb (Fig. 2D, E, and F), and thus an inability to prevent entry into the S phase is not the reason why HCMV replicates inefficiently in Rb-depleted cells.
FIG 2.

Rb is not required to maintain a cell cycle block during HCMV infection. Total cellular DNA content was measured by flow cytometry and propidium iodide (PI) staining of scrambled control (Scr) or Rb knockdown cells after serum starvation for 48 h (A), serum starvation followed by addition of serum containing media for 48 h (B), or serum starvation followed by concurrent addition of serum containing media and infection with AD169 IE2-GFP virus at an MOI of 1 before analysis at 48 h postinfection (hpi) (C). (B and C) Nocodazole and phosphonoacetic acid (PAA) were added to both serum-stimulated infected cells and mock-infected cells at 12 h after the addition of the serum and maintained until 48 h after the addition of serum-containing medium. Representative histograms from each condition from three biological replicates are shown. (D to F) The average percentages of scrambled control (Scr) or Rb knockdown cells present ± the standard deviations in each phase of the cell cycle after serum starvation (D), serum stimulation and treatment with nocodazole and PAA (E), or infection with nocodazole and PAA treatment (F) are shown. A Student t test determined there was no statistical difference in the accumulation of cells in each phase of the cell cycle between the scrambled control and Rb knockdown cells under any condition (P > 0.35).
HCMV induces senescence in Rb knockdown cells.
In addition to temporarily restricting cell cycle progression in G1, Rb can mediate a permanent withdrawal from the cell cycle into a G0 state also termed senescence (36). Senescence is induced in response to cellular stress (37), and Rb is required to establish senescence-associated heterochromatin (SAHC) at E2F-responsive promoters, rendering them permanently silenced and thus inhibiting cell cycle progression (5). HCMV infection also induces a senescence-like phenotype, and senescence induction prior to infection enhances HCMV replication (38). We therefore sought to determine whether HCMV replicated less efficiently in Rb knockdown cells because it could no longer induce senescence. To gauge senescence induction, we stained cells for senescence-associated β-galactosidase (SA-β-Gal) activity at neutral pH (29). SA-β-Gal activity was clearly evident (Fig. 3A) in primary fibroblasts induced to senesce by high-density seeding combined with prolonged exposure to growth factors (38). The same cells transiently arrested in G1 by serum starvation showed no SA-β-Gal staining (Fig. 3B). HCMV infection was equally able to induce SA-β-Gal staining in both control and Rb knockdown cells (Fig. 3C, D, and E). We conclude that HCMV efficiently induces senescence in the absence of Rb, and thus an inability to induce senescence is not the reason why HCMV replicates inefficiently in Rb-depleted cells.
FIG 3.
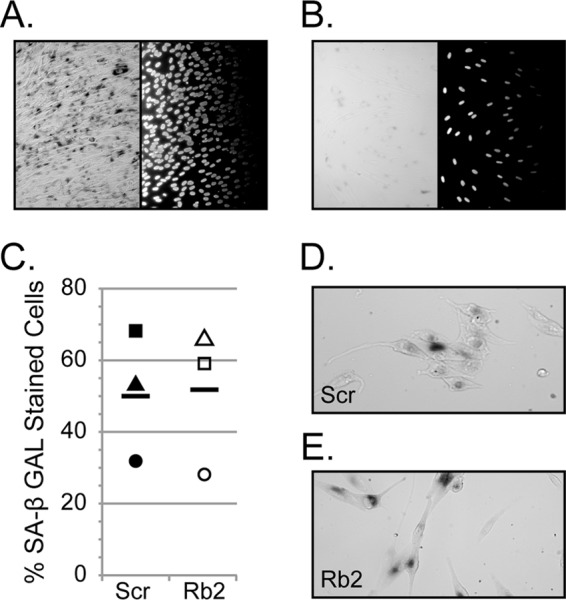
HCMV is able to efficiently induce a senescence-like phenotype in infected cells that have reduced levels of Rb. (A and B) Passage 6 fibroblasts were induced to undergo senescence by plating at a high density and maintenance in complete media for 48 h (A) or plated at a subconfluent density and serum-starved for 48 h before fixation and staining for senescence-associated β-galactosidase (SA-β-Gal, left) and Hoechst to visualize nuclei (right) (B). (C) HCMV-infected scrambled control cells were serum starved and infected at an MOI of 1, and at 72 h postinfection the cells were stained for SA-β-Gal and the percentages of positive cells were calculated. The horizontal bar represents the mean from three biological replicates, and the individual symbols represent the values from three biological replicates. A paired Student t test was used to evaluate statistical significance (not significant, P > 0.9). (D and E) Representative images of the SA-β-Gal staining of scrambled control (Scr) (D) or Rb knockdown (Rb2) (E) cells used to generate the results for panel C are shown.
HCMV replicates efficiently in p107 knockdown cells.
We considered one additional manner in which the absence of Rb could skew cell cycle status and thereby impair HCMV replication. We previously showed (9) and confirm here (Fig. 4A) that the S-phase-specific p107 protein, an Rb family member encoded by an E2F-responsive gene, accumulates upon Rb knockdown (39). Whether or how p107 impacts HCMV infection is not known. However, because an S-phase-specific gene can have a deleterious impact on HCMV infection when overexpressed (40), we sought to determine whether p107 restricts HCMV infection. Although HCMV replication was impaired in Rb knockdown cells (Fig. 4B), the virus was able to productively replicate equally in control and p107 knockdown cells (Fig. 4C). We conclude that p107 does not restrict HCMV infection and thus p107 accumulation is not the reason why HCMV replicates inefficiently in Rb-depleted cells.
FIG 4.

Rb knockdown deregulates p107 expression, but p107 is not a restrictive factor for HCMV replication. (A) Protein lysates from equal numbers of cycling primary fibroblasts stably expressing a scrambled shRNA (Scr), Rb targeting shRNA (Rb2), or p107 targeting shRNA (107.2) were analyzed with the indicated antibodies by Western blotting. (B and C) Serum-starved scrambled control (Scr) and Rb knockdown cells (B) or scrambled control (Scr) and p107 knockdown (107.2) cells (C) were infected at an MOI of 1. Combined cell-free and cell-associated virus was collected 4 days postinfection, and the virus titers were determined by standard plaque assay. The data are mean titers ± the standard deviations from six biological replicates. A Student t test was used to determine the statistical difference between Scr and Rb knockdown or 107.2 cells (*, P < 0.05; not significant [n.s.], P > 0.44).
HCMV induces mitochondrial biogenesis in Rb knockdown cells.
Rb controls the expression of genes required for mitochondrial biogenesis (41), and in a cell-type-specific manner, can regulate the number of mitochondria within a cell, assayed by the relative amount of mitochondrial genomes compared to the cellular genome. For example, mouse embryonic fibroblasts contain fewer mitochondria in the absence of Rb (10), whereas adipocytes contain more (17). HCMV infection increases mitochondrial biogenesis, which supports efficient viral replication (42). We therefore sought to determine whether HCMV replicated less efficiently in Rb knockdown cells because it could no longer induce mitochondrial biogenesis. HCMV infection was equally able to induce mitochondrial biogenesis in both control and Rb knockdown cells assayed by determining the DNA ratio of the mitochondrial genome encoded 16S rRNA gene compared to the nuclear genome encoded glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene (Fig. 5A). Furthermore, HCMV infection was equally able to induce previously observed morphological changes in mitochondria (43) in both control and Rb knockdown cells (Fig. 5B, C, and D). We conclude that HCMV efficiently induces mitochondrial biogenesis and modifies mitochondrial morphology in the absence of Rb, and thus an inability to modify mitochondrial numbers or morphology is not the reason why HCMV replicates inefficiently in Rb depleted cells.
FIG 5.
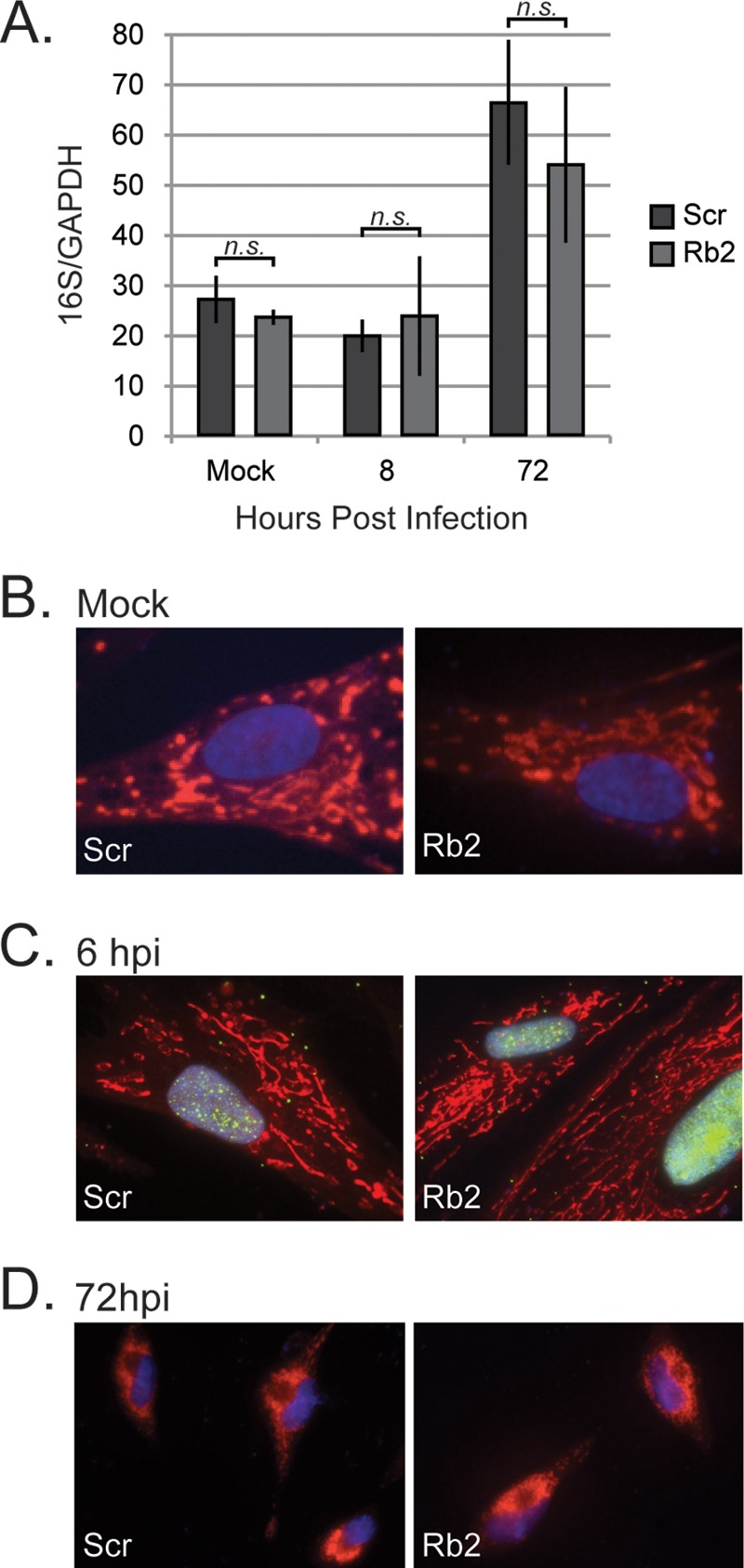
HCMV-infected cells induce mitochondrial biogenesis and mitochondrial morphological changes in the absence of Rb. (A) Serum-starved scramble control (Scr) or Rb knockdown cells (Rb) were mock infected or infected at an MOI of 1, and the ratio of mitochondrial DNA (16S) to cellular genomic DNA (GAPHD) was measured by quantitative PCR at 8 or 72 h postinfection. Bars represent the means ± the standard deviations of three biological replicates. Statistical significance was determined by using a Student t test. There was a statistically significant increase in the relative accumulation of mitochondrial DNA at 72 h compared to mock-infected cells in both the scrambled and Rb knockdown cells (P < 0.05); however, there was no statistical significance between the scrambled control and Rb knockdown cells at any time point (n.s., P > 0.3) (B to D) Representative images of serum-starved scrambled control (Scr) or Rb knockdown (Rb2) cells either mock infected (B) or infected at an MOI of 1 for 5 h (C) or 72 h (D) before staining with MitoTracker Red (red), pp71 (green), and Hoechst (blue). The green channel was omitted from the 72 hpi to aid in the visualization of the mitochondria.
A pan-caspase inhibitor does not improve HCMV productive replication in Rb knockdown cells.
Rb loss leads to the induction of E2F1 (39) (Fig. 4A). Although E2F1 is required for efficient HCMV replication (44), high-level expression combined with genotoxic stress can induce apoptosis (45). Apoptosis can reduce the ability of HCMV to replicate in the absence of viral genes that block this cell death process (46–48). We therefore sought to determine whether HCMV replicated less efficiently in Rb knockdown cells because it could no longer effectively inhibit apoptosis. HCMV infection in Rb knockdown cells was not improved by the addition of the pan caspase inhibitor Z-VAD-FMK (Fig. 6A). Likewise, the proapoptotic p53 protein, which is induced subsequent to Rb loss (49) and upon HCMV infection (50), accumulated to similar levels in control and Rb-depleted cells (Fig. 6B). In addition to apoptosis, other forms of cell death such as necroptosis can impair cytomegalovirus infections (51, 52). We found that identical percentages of cells were viable 72 h after HCMV infection of either control or Rb knockdown cells (Fig. 6C), a time when infected cells are under significant virus-induced stress (53). We conclude that HCMV efficiently avoids cell death pathways in the absence of Rb, and thus an inability to maintain cell viability is not the reason why HCMV replicates inefficiently in Rb depleted cells.
FIG 6.

The decrease in virus production is not due to infected Rb knockdown cells undergoing apoptosis. (A) Serum-starved scrambled control cells (Scr) or Rb knockdown cells (Rb2) were infected at an MOI of 1 and treated with DMSO or pan-caspase inhibitor Z-VAD-FMK at the time of infection and replenished at 48 h postinfection. Combined cell-free and cell-associated virus was collected 4 days postinfection, and the titers of virus were determined by standard plaque assay. The data are mean titers ± the standard deviations from six biological replicates. The statistical significance was determined by a permutation test (*, P < 0.02; n.s., P > 0.45). (B) Protein lysates from equal numbers of serum-starved scrambled control cells (Scr) or Rb knockdown cells (Rb2) mock infected or infected at an MOI of 1 for 6 or 72 h were analyzed with the indicated antibodies by Western blotting. (C) Serum-starved scrambled control cells (Scr) or Rb knockdown cells (Rb2) were infected at an MOI of 1, and 72 h later the cell viability was determined by trypan blue exclusion. At least 80 cells were counted per replicate, and the bars represent the mean percentages of live cells ± the standard deviations of three biological replicates. Statistical significance was assayed by using the Student t test (n.s., P > 0.17).
HCMV modulates glutaminolysis equivalently in control and Rb knockdown cells.
Rb depletion leads to oxidative stress, which is counteracted through the conversion of glutamine to glutathione, a reducing agent (10, 18). HCMV uses glutamine for anaplerotic restoration of TCA cycle intermediates that are depleted because glucose is shunted away from the TCA cycle and toward fatty acid biosynthesis in HCMV-infected cells (Fig. 7A) (25, 54). In Rb-depleted cells infected with HCMV, competing processes (glutathione production and anaplerosis) would vie for the limiting amounts of glutamine. We therefore sought to determine whether HCMV replicated less efficiently in Rb knockdown cells because it could no longer effectively regulate glutamine metabolism. The mRNA levels of two genes encoding key glutamine transporters (ASCT1 and ASCT2) were not dramatically different in HCMV-infected control and Rb knockdown cells, although the Rb knockdown cells did show statistically higher levels of ASCT2 transcripts than did control cells (Fig. 7B). The mRNA levels of glutaminase (GLS1), the enzyme that converts glutamine to glutamate plus ammonia, were also unchanged between HCMV-infected control and Rb knockdown cells (Fig. 7C). Likewise, the mRNA levels of glutamine-cysteine ligase (GCL), the enzyme that converts glutamate to glutathione, were also unchanged between HCMV-infected control and Rb knockdown cells (Fig. 7D). Thus, at the mRNA level, the glutamine metabolism enzymes are not perceptibly different between HCMV infections of control or Rb depleted cells.
FIG 7.
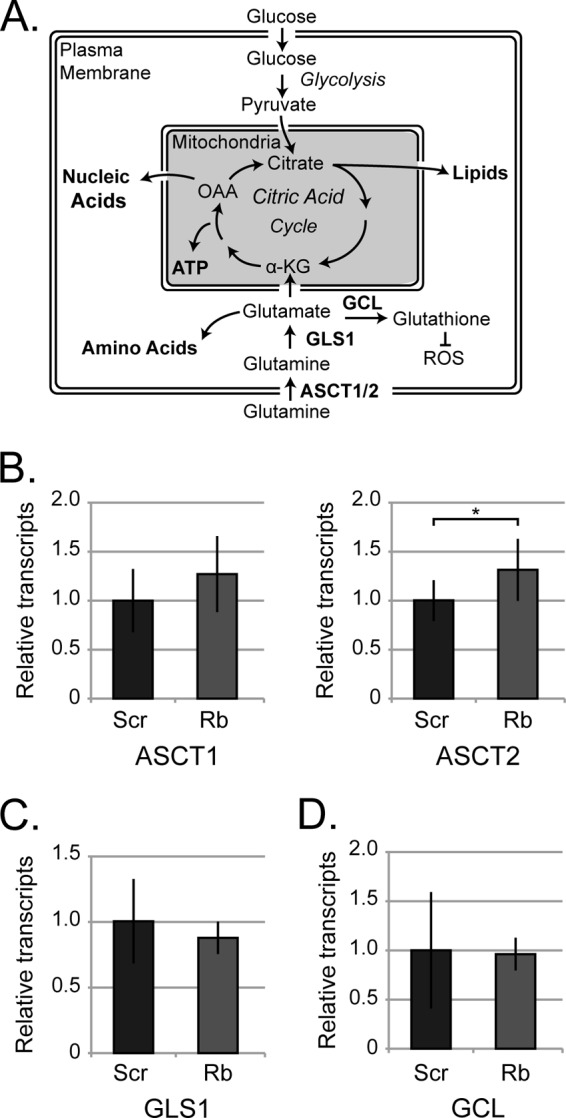
Rb knockdown cells efficiently express key glutaminolysis enzymes required to support viral replication. (A) An oversimplified illustration of pathways utilized for nucleic acids, amino acids, and lipid synthesis to illustrate the ability of citric acid cycle intermediates to facilitate macromolecule synthesis as well as oxidative phosphorylation. Citric acid cycle intermediates citrate, α-ketoglutarate (α-KG), and oxaloacetic acid (OAA) can be used as carbon sources for the synthesis of nucleic acids, amino acids, and lipids. To meet the energetic and physical requirements of HCMV replication, cells will increase glutamine uptake to replenish citric acid cycle intermediates that become depleted when glucose undergoes aerobic glycolysis and the pyruvate does not enter the citric acid cycle. The enzymes regulated by Rb that are involved in glutaminolysis and glutathione synthesis are ASCT2 (but not ASCT1), glutaminase (GLS1), and glutamine-cysteine ligase (GCL). (B) Transcript levels of glutamine transporters ASCT1, ASCT2 (C) glutaminase (GLS1), or (D) glutamate cysteine ligase (GCL) from HCMV-infected scrambled control (Scr) or Rb knockdown cells (Rb2) at 72 h postinfection were analyzed by qRT-PCR and normalized to GAPDH. (B to D) Bars represent the mean ± the standard deviation of 4 biological replicates. Prior to normalization, a paired Student t test was used to determine statistical significance (*, P < 0.05; n.s., P > 0.2).
Our transcript analysis cannot determine whether glutaminolysis enzyme levels or activity are different between HCMV-infected control and Rb knockdown cells. Therefore, we added cell permeable small molecules downstream of glutamine to Rb-depleted cells to determine whether such supplementation could enhance HCMV productive replication. Neither the TCA intermediates pyruvate (PYR) or oxaloacetic acid (OAA), nor the strong antioxidant N-acetyl cysteine (NAC) substantially improved HCMV productive replication in Rb knockdown cells (Fig. 8A). However, addition of the TCA intermediate alpha-ketoglutarate (αKG) showed a reproducible 4-fold induction of HCMV productive replication in Rb knockdown cells (Fig. 8B). However, αKG supplementation also bolstered HCMV productive replication in control cells (Fig. 8B), indicating that while αKG is limiting for HCMV infection, this condition is not specific for Rb knockdown cells. We conclude that HCMV efficiently modulates glutamine metabolism in the absence of Rb, and thus a deficiency in this phase of intermediary metabolism is not the reason why HCMV replicates inefficiently in Rb depleted cells.
FIG 8.
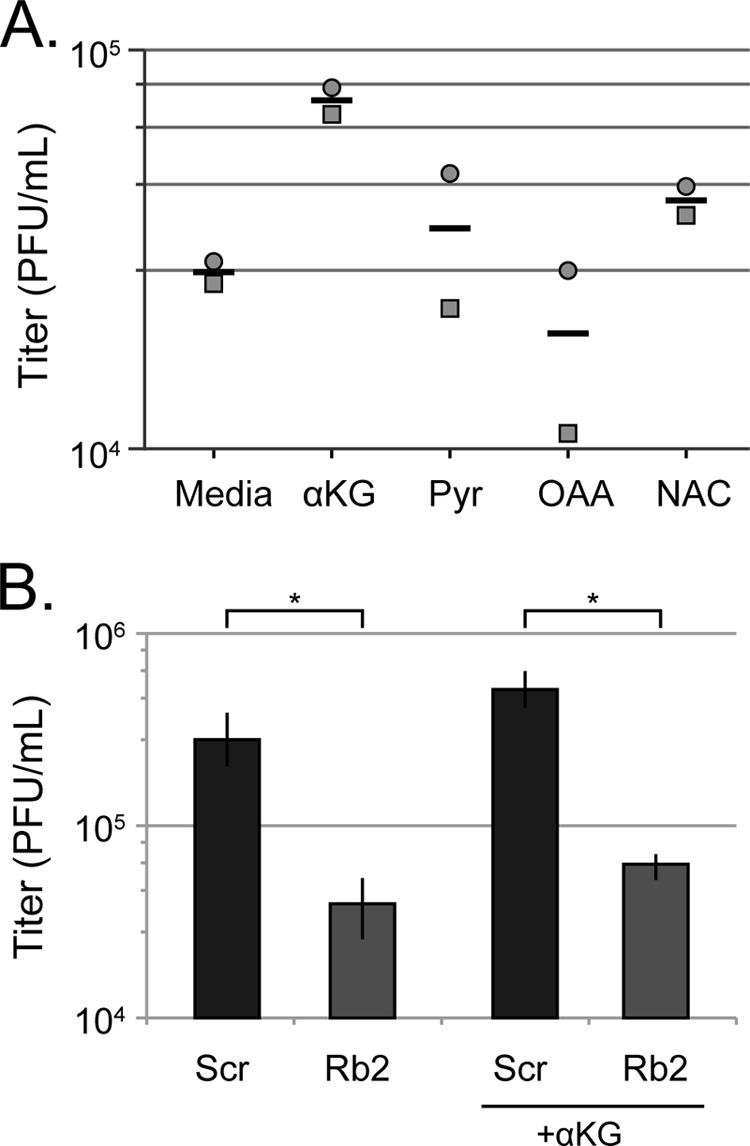
Rb knockdown cells produce sufficient citric acid cycle intermediates and antioxidants to support efficient HCMV replication (A) Serum-starved Rb knockdown cells were infected at an MOI of 1, and the medium was supplemented with α-ketoglutarate (α-KG), pyruvate (Pyr), oxaloacetic acid (OAA), or the antioxidant N-acetyl cysteine (NAC) at the time of infection and replenished at 48 h postinfection. Combined cell-free and cell-associated virus was collected 4 days postinfection, and the titers of virus were determined by standard plaque assay. The symbols represent individual experiments and horizontal bars are the mean from two experiments. (B) Serum-starved scrambled control (Scr) and Rb knockdown cells were infected at an MOI of 1 and left in normal medium or supplemented with α-KG, as in panel A. Combined cell-free and cell-associated virus was collected at 4 days postinfection, and the titers of virus were determined by standard plaque assay. The data are mean titers ± the standard deviations from six biological replicates. Statistical significance was determined by using the Student t test (*, P < 0.001). A Sen-Adichie test for parallelism determined there was no significant difference between the α-KG mediated increase in virus titers in the scrambled control and Rb knockdown cells (P > 0.3).
HCMV proteins that inactivate Rb do not require Rb to function.
Having exhausted the known cellular pathways controlled by Rb, we turned to viral proteins that may require this tumor suppressor to exert their positive effects on HCMV replication. We focused on the only two HCMV proteins demonstrated to regulate Rb during HCMV infection, pp71 and UL97. pp71 is a tegument protein that binds to and degrades the hypophosphorylated form of Rb during HCMV infection (55–57). pp71 also degrades Daxx in a reaction required for efficient viral immediate-early gene expression (21). Daxx and Rb can both localize to promyelocytic leukemia nuclear bodies (PML-NBs) (58), and thus pp71 could potentially require the presence of Rb to help it target Daxx at PML-NBs. We therefore sought to determine whether HCMV infection promoted Daxx degradation less efficiently in Rb knockdown cells. HCMV infection was equally able to degrade Daxx in both control and Rb knockdown cells (Fig. 9A).
FIG 9.
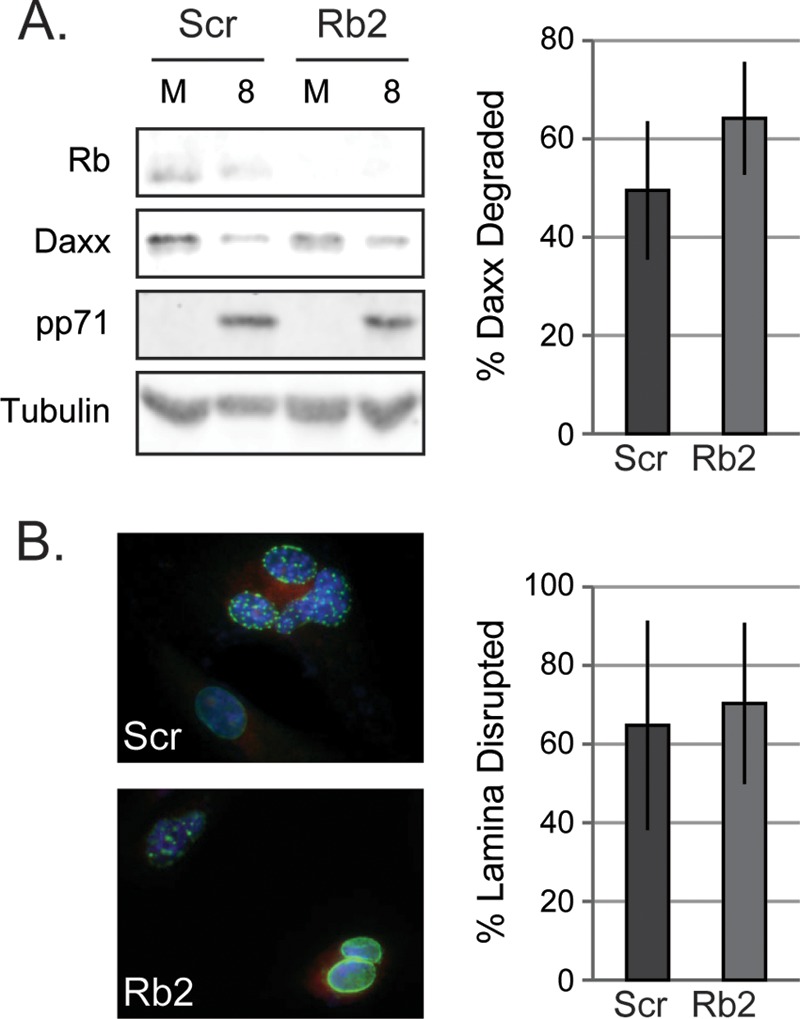
Viral proteins that interact with Rb are still functional in the absence of Rb. (A) Protein lysates from equal numbers of serum-starved scrambled control (Scr) or Rb knockdown cells (Rb2) were mock infected or infected at an MOI of 1 and harvested at 8 h postinfection and were then analyzed with the indicated antibodies by Western blotting. The percentages of Daxx degraded (Daxx/tubulin in mock-infected cells minus Daxx/tubulin at 8 hpi normalized to Daxx/tubulin in the mock-infected cells) from three biological replicates are shown. The bars represent the means ± the standard deviations from three biological replicates analyzed by quantitative Western blot analysis. The statistical significance was determined by using the Student t test (n.s., P > 0.2). (B) Representative images from scrambled control (Scr) or Rb knockdown cells (Rb2) transfected with plasmids expressing HA-UL97 and GFP-lamin A showing intact or disrupted nuclear lamina are shown. The percentage of GFP lamina disrupted in scrambled control (Scr) or Rb knockdown cells (Rb2) from three biological replicates were analyzed by indirect immunofluorescence. The bars represent the means ± the standard deviations from three biological replicates. Statistical significance was determined by using a Student t test (n.s., P > 0.5).
UL97 is a v-Cdk that phosphorylates and inactivates Rb (55, 59, 60). UL97 also phosphorylates lamin proteins to induce partial lamina breakdown at the inner nuclear envelope (61) that theoretically promotes nuclear capsid egress and thus efficient HCMV productive replication. Rb can also interact with lamin proteins (62, 63), and thus UL97 could potentially require the presence of Rb to help it target lamin proteins for phosphorylation. We therefore sought to determine whether UL97 could disrupt the nuclear lamina in Rb knockdown cells. Transfected UL97 was equally able to displace an ectopically expressed GFP-lamin A fusion protein from the nuclear lamina in both control and Rb knockdown cells (Fig. 9B). We conclude that the known HCMV Rb modulating proteins pp71 and UL97 efficiently perform their respective tasks that promote HCMV productive replication in the absence of Rb, and thus deficiencies in their function is not the reason why HCMV replicates inefficiently in Rb depleted cells. In total our work points toward a novel, likely viral process directly impacted by Rb that is required for efficient HCMV replication.
DISCUSSION
Despite encoding at least four viral proteins reported to target aspects of Rb function, HCMV replicates less efficiently in cells with reduced levels of the cellular tumor suppressor protein. There is significant overlap between the cellular pathways that Rb regulates and those that are modified during HCMV infection, providing ample opportunities for Rb to contribute in a positive way to efficient HCMV replication. However, we show here that HCMV remains able to modulate these overlapping pathways of cell cycle (Fig. 2), cell death (Fig. 6), and mitochondrial biology (Fig. 5) and chemistry (Fig. 7) in Rb-depleted cells. How Rb promotes efficient HCMV replication remains to be discovered.
Aberrantly modified cellular pathways do not seem responsible for the impaired replication of HCMV in the absence of Rb. However, our targeted approach analyzed pathways known to be regulated by Rb. Thus, it remains possible that HCMV exploits a yet to be determined function of Rb for efficient replication. Unbiased, global, hypothesis-generating experiments comparing epigenomes, transcriptomes, proteomes, and metabolomes between HCMV-infected control and Rb-depleted cells could provide clues as to how Rb supports efficient HCMV replication. The defect in HCMV replication when Rb is reduced could be used as a tractable assay to discover new functions for Rb that might be important for its tumor suppressor function.
An alternative hypothesis is that Rb controls a critical viral process. The two HCMV proteins known to modulate Rb during infection (pp71 and UL97) efficiently perform their other essential tasks (Daxx degradation and lamina disruption, respectively) when Rb is reduced (Fig. 9). A third viral protein, IE1, interacts with p107 in transfection and infection assays (64). However, since p107 is dispensable for efficient HCMV replication (Fig. 3), it seems unlikely that this interaction contributes to the replication defect observed in the absence of Rb. The fourth viral protein reported to interact with Rb in vitro is IE2 (65, 66), a protein that arrests cell cycle progression (33, 67), stimulates cellular E2F-responsive transcription (65, 68), and can both positively and negatively regulate viral transcription (65, 69, 70). HCMV remained able to arrest cell cycle progression (Fig. 2), and E2F-responsive genes are expressed (Fig. 4) in Rb-depleted cells, so deficiencies in these activities of IE2 do not appear to cause the drop in replication observed in Rb knockdown cells. Whether or not the multiple effects of IE2 on viral transcription are altered in Rb-depleted cells remains to be directly explored, although we previously detected little to no effect of Rb knockdown on HCMV immediate-early or early protein accumulation (9), so IE2 seems capable of regulating viral transcription when the levels of Rb are reduced.
In addition to the four proteins implicated in Rb regulation listed above, HCMV encodes 14 other proteins with canonical Rb-binding LxCxE motifs (9) that conceivably could require Rb for their functions. UL77 and UL93, two of the LxCxE containing proteins, are essential for viral replication, while several more (US26, UL20, UL29, and UL69) enhance viral replication (71). Determining whether any of these proteins use their LxCxE motifs to interact with Rb, support efficient viral replication, or both could help us to understand the role of Rb during viral replication.
It is possible that the function of Rb required for efficient HCMV replication combines both cellular and viral pathways. For example, recent evidence indicates that adenovirus E1A, an Rb-binding LxCxE-containing protein, redirects Rb and its associated chromatin remodeling complexes to cellular genes that encode antiviral proteins (72). This correlates with silencing of their transcription and presumably, enhanced adenoviral replication. HCMV could invoke a similar strategy, utilizing a viral Rb-binding protein to direct Rb to viral or cellular genes to modulate their transcription.
Rb accumulates in phosphorylated forms during HCMV infection (31, 55, 60). Classically, phosphorylated Rb is considered inactive (73); thus, it was surprising that HCMV replication was inefficient in the absence of a posttranslationally modified form of a protein already considered to be devoid of activity. Our results seem to indicate an unknown role for the phosphorylated form of Rb may exist. Although the pathway controlled by Rb is aberrant in most human tumors, the Rb gene itself is mutated or lost infrequently (74). Any activity of the phosphorylated form of Rb important for maintenance of the transformed phenotype could be exploited for cancer therapies. HCMV infection of Rb knockdown cells may prove a useful tool in identifying functions of phosphorylated Rb, increasing our understanding of this critical tumor suppressor and potentially identifying targets for novel cancer therapeutics.
ACKNOWLEDGMENTS
We thank Phil Balandyk for expert technical assistance, Paul Lambert and Hiroyuki Sakai for shRNA retroviral plasmids, Nathan Sherer for sharing equipment, and Norman Drinkwater for help with statistical analysis.
This study was supported by National Institutes of Health grant AI080675 to R.F.K. H.R.V. was supported by training grant T32 CA009135.
REFERENCES
- 1.Bellacchio E, Paggi MG. 2013. Understanding the targeting of the RB family proteins by viral oncoproteins to defeat their oncogenic machinery. J Cell Physiol 228:285–291. doi: 10.1002/jcp.24137. [DOI] [PubMed] [Google Scholar]
- 2.Hume AJ, Kalejta RF. 2009. Regulation of the retinoblastoma proteins by the human herpesviruses. Cell Div 4:1. doi: 10.1186/1747-1028-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Munger K, Jones DL. 2015. Human papillomavirus carcinogenesis: an identity crisis in the retinoblastoma tumor suppressor pathway. J Virol 89:4708–4711. doi: 10.1128/JVI.03486-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weinberg RA. 1995. The retinoblastoma protein and cell cycle control. Cell 81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 5.Narita M, N u nez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. 2003. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113:703–716. doi: 10.1016/S0092-8674(03)00401-X. [DOI] [PubMed] [Google Scholar]
- 6.Talluri S, Isaac CE, Ahmad M, Henley SA, Francis SM, Martens AL, Bremner R, Dick FA. 2010. A G1 checkpoint mediated by the retinoblastoma protein that is dispensable in terminal differentiation but essential for senescence. Mol Cell Biol 30:948–960. doi: 10.1128/MCB.01168-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Helt A-M, Galloway DA. 2003. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 24:159–169. doi: 10.1093/carcin/24.2.159. [DOI] [PubMed] [Google Scholar]
- 8.Collins AS, Nakahara T, Do A, Lambert PF. 2005. Interactions with pocket proteins contribute to the role of human papillomavirus type 16 E7 in the papillomavirus life cycle. J Virol 79:14769–14780. doi: 10.1128/JVI.79.23.14769-14780.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.VanDeusen HR, Kalejta RF. 2015. The retinoblastoma tumor suppressor promotes efficient human cytomegalovirus lytic replication. J Virol 89:5012–5021. doi: 10.1128/JVI.00175-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG, Dean DC, Clem BF. 2014. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 33:556–566. doi: 10.1038/onc.2012.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sankaran VG, Orkin SH, Walkley CR. 2008. Rb intrinsically promotes erythropoiesis by coupling cell cycle exit with mitochondrial biogenesis. Genes Dev 22:463–475. doi: 10.1101/gad.1627208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antonucci LA, Egger JV, Krucher NA. 2014. Phosphorylation of the retinoblastoma protein (Rb) on serine-807 is required for association with Bax. Cell Cycle Georget Tex 13:3611–3617. doi: 10.4161/15384101.2014.964093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilgendorf KI, Leshchiner ES, Nedelcu S, Maynard MA, Calo E, Ianari A, Walensky LD, Lees JA. 2013. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev 27:1003–1015. doi: 10.1101/gad.211326.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moroni MC, Hickman ES, Lazzerini Denchi E, Caprara G, Colli E, Cecconi F, Müller H, Helin K. 2001. Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol 3:552–558. doi: 10.1038/35078527. [DOI] [PubMed] [Google Scholar]
- 15.Clem BF, Chesney J. 2012. Molecular pathways: regulation of metabolism by RB. Clin Cancer Res Off J Am Assoc Cancer Res 18:6096–6100. doi: 10.1158/1078-0432.CCR-11-3164. [DOI] [PubMed] [Google Scholar]
- 16.Fajas L. 2013. Re-thinking cell cycle regulators: the cross-talk with metabolism. Front Oncol 3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dali-Youcef N, Mataki C, Coste A, Messaddeq N, Giroud S, Blanc S, Koehl C, Champy M-F, Chambon P, Fajas L, Metzger D, Schoonjans K, Auwerx J. 2007. Adipose tissue-specific inactivation of the retinoblastoma protein protects against diabesity because of increased energy expenditure. Proc Natl Acad Sci U S A 104:10703–10708. doi: 10.1073/pnas.0611568104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nicolay BN, Gameiro PA, Tschöp K, Korenjak M, Heilmann AM, Asara JM, Stephanopoulos G, Iliopoulos O, Dyson NJ. 2013. Loss of RBF1 changes glutamine catabolism. Genes Dev 27:182–196. doi: 10.1101/gad.206227.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pasque V, Gillich A, Garrett N, Gurdon JB. 2011. Histone variant macroH2A confers resistance to nuclear reprogramming. EMBO J 30:2373–2387. doi: 10.1038/emboj.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ueno T, Sasaki K, Yoshida S, Kajitani N, Satsuka A, Nakamura H, Sakai H. 2006. Molecular mechanisms of hyperplasia induction by human papillomavirus E7. Oncogene 25:4155–4164. doi: 10.1038/sj.onc.1209445. [DOI] [PubMed] [Google Scholar]
- 21.Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80:3863–3871. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuny CV, Chinchilla K, Culbertson MR, Kalejta RF. 2010. Cyclin-dependent kinase-like function is shared by the beta- and gamma-subsets of the conserved herpesvirus protein kinases. PLoS Pathog 6:e1001092. doi: 10.1371/journal.ppat.1001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee C-P, Huang Y-H, Lin S-F, Chang Y, Chang Y-H, Takada K, Chen M-R. 2008. Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J Virol 82:11913–11926. doi: 10.1128/JVI.01100-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanchez V, Clark CL, Yen JY, Dwarakanath R, Spector DH. 2002. Viable human cytomegalovirus recombinant virus with an internal deletion of the IE2 86 gene affects late stages of viral replication. J Virol 76:2973–2989. doi: 10.1128/JVI.76.6.2973-2989.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chambers JW, Maguire TG, Alwine JC. 2010. Glutamine metabolism is essential for human cytomegalovirus infection. J Virol 84:1867–1873. doi: 10.1128/JVI.02123-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayashi ML, Blankenship C, Shenk T. 2000. Human cytomegalovirus UL69 protein is required for efficient accumulation of infected cells in the G1 phase of the cell cycle. Proc Natl Acad Sci U S A 97:2692–2696. doi: 10.1073/pnas.050587597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nowak B, Sullivan C, Sarnow P, Thomas R, Bricout F, Nicolas JC, Fleckenstein B, Levine AJ. 1984. Characterization of monoclonal antibodies and polyclonal immune sera directed against human cytomegalovirus virion proteins. Virology 132:325–338. doi: 10.1016/0042-6822(84)90039-4. [DOI] [PubMed] [Google Scholar]
- 28.Kalejta RF, Brideau AD, Banfield BW, Beavis AJ. 1999. An integral membrane green fluorescent protein marker, Us9-GFP, is quantitatively retained in cells during propidium iodide-based cell cycle analysis by flow cytometry. Exp Cell Res 248:322–328. doi: 10.1006/excr.1999.4427. [DOI] [PubMed] [Google Scholar]
- 29.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Juckem LK, Boehme KW, Feire AL, Compton T. 2008. Differential initiation of innate immune responses induced by human cytomegalovirus entry into fibroblast cells. J Immunol 180:4965–4977. doi: 10.4049/jimmunol.180.7.4965. [DOI] [PubMed] [Google Scholar]
- 31.Jault FM, Jault JM, Ruchti F, Fortunato EA, Clark C, Corbeil J, Richman DD, Spector DH. 1995. Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb, and p53, leading to cell cycle arrest. J Virol 69:6697–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalejta RF, Shenk T. 2002. Manipulation of the cell cycle by human cytomegalovirus. Front Biosci J Virtual Libr 7:295–306. doi: 10.2741/kalejta. [DOI] [PubMed] [Google Scholar]
- 33.Wiebusch L, Hagemeier C. 1999. Human cytomegalovirus 86-kilodalton IE2 protein blocks cell cycle progression in G1. J Virol 73:9274–9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salvant BS, Fortunato EA, Spector DH. 1998. Cell cycle dysregulation by human cytomegalovirus: influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J Virol 72:3729–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Classon M, Salama S, Gorka C, Mulloy R, Braun P, Harlow E. 2000. Combinatorial roles for pRB, p107, and p130 in E2F-mediated cell cycle control. Proc Natl Acad Sci U S A 97:10820–10825. doi: 10.1073/pnas.190343497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, Dickins RA, Narita M, Zhang M, Lowe SW. 2010. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 17:376–387. doi: 10.1016/j.ccr.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ben-Porath I, Weinberg RA. 2005. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol 37:961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 38.Noris E, Zannetti C, Demurtas A, Sinclair J, De Andrea M, Gariglio M, Landolfo S. 2002. Cell cycle arrest by human cytomegalovirus 86-kDa IE2 protein resembles premature senescence. J Virol 76:12135–12148. doi: 10.1128/JVI.76.23.12135-12148.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sage J, Miller AL, Pérez-Mancera PA, Wysocki JM, Jacks T. 2003. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 424:223–228. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- 40.Bogdanow B, Weisbach H, von Einem J, Straschewski S, Voigt S, Winkler M, Hagemeier C, Wiebusch L. 2013. Human cytomegalovirus tegument protein pp150 acts as a cyclin A2-CDK-dependent sensor of the host cell cycle and differentiation state. Proc Natl Acad Sci U S A 110:17510–17515. doi: 10.1073/pnas.1312235110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blanchet E, Annicotte J-S, Lagarrigue S, Aguilar V, Clapé C, Chavey C, Fritz V, Casas F, Apparailly F, Auwerx J, Fajas L. 2011. E2F transcription factor-1 regulates oxidative metabolism. Nat Cell Biol 13:1146–1152. doi: 10.1038/ncb2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaarbø M, Ager-Wick E, Osenbroch PØ, Kilander A, Skinnes R, Müller F, Eide L. 2011. Human cytomegalovirus infection increases mitochondrial biogenesis. Mitochondrion 11:935–945. doi: 10.1016/j.mito.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 43.McCormick AL, Smith VL, Chow D, Mocarski ES. 2003. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J Virol 77:631–641. doi: 10.1128/JVI.77.1.631-641.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.E X, Pickering MT, Debatis M, Castillo J, Lagadinos A, Wang S, Lu S, Kowalik TF. 2011. An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog 7:e1001342. doi: 10.1371/journal.ppat.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ginsberg D. 2002. E2F1 pathways to apoptosis. FEBS Lett 529:122–125. doi: 10.1016/S0014-5793(02)03270-2. [DOI] [PubMed] [Google Scholar]
- 46.McCormick AL, Roback L, Livingston-Rosanoff D, St Clair C. 2010. The human cytomegalovirus UL36 gene controls caspase-dependent and -independent cell death programs activated by infection of monocytes differentiating to macrophages. J Virol 84:5108–5123. doi: 10.1128/JVI.01345-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reboredo M, Greaves RF, Hahn G. 2004. Human cytomegalovirus proteins encoded by UL37 exon 1 protect infected fibroblasts against virus-induced apoptosis and are required for efficient virus replication. J Gen Virol 85:3555–3567. doi: 10.1099/vir.0.80379-0. [DOI] [PubMed] [Google Scholar]
- 48.Terhune S, Torigoi E, Moorman N, Silva M, Qian Z, Shenk T, Yu D. 2007. Human cytomegalovirus UL38 protein blocks apoptosis. J Virol 81:3109–3123. doi: 10.1128/JVI.02124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Macleod KF, Hu Y, Jacks T. 1996. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J 15:6178–6188. [PMC free article] [PubMed] [Google Scholar]
- 50.Muganda P, Mendoza O, Hernandez J, Qian Q. 1994. Human cytomegalovirus elevates levels of the cellular protein p53 in infected fibroblasts. J Virol 68:8028–8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Omoto S, Guo H, Talekar GR, Roback L, Kaiser WJ, Mocarski ES. 2015. Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J Biol Chem 290:11635–11648. doi: 10.1074/jbc.M115.646042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Upton JW, Kaiser WJ, Mocarski ES. 2010. Virus Inhibition of RIP3-dependent necrosis. Cell Host Microbe 7:302–313. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alwine JC. 2008. Modulation of host cell stress responses by human cytomegalovirus. Curr Top Microbiol Immunol 325:263–279. [DOI] [PubMed] [Google Scholar]
- 54.Munger J, Bennett BD, Parikh A, Feng X-J, McArdle J, Rabitz HA, Shenk T, Rabinowitz JD. 2008. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol 26:1179–1186. doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hume AJ, Finkel JS, Kamil JP, Coen DM, Culbertson MR, Kalejta RF. 2008. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320:797–799. doi: 10.1126/science.1152095. [DOI] [PubMed] [Google Scholar]
- 56.Kalejta RF, Bechtel JT, Shenk T. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol Cell Biol 23:1885–1895. doi: 10.1128/MCB.23.6.1885-1895.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kalejta RF, Shenk T. 2003. Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc Natl Acad Sci U S A 100:3263–3268. doi: 10.1073/pnas.0538058100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alcalay M, Tomassoni L, Colombo E, Stoldt S, Grignani F, Fagioli M, Szekely L, Helin K, Pelicci PG. 1998. The promyelocytic leukemia gene product (PML) forms stable complexes with the retinoblastoma protein. Mol Cell Biol 18:1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prichard MN, Sztul E, Daily SL, Perry AL, Frederick SL, Gill RB, Hartline CB, Streblow DN, Varnum SM, Smith RD, Kern ER. 2008. Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J Virol 82:5054–5067. doi: 10.1128/JVI.02174-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iwahori S, Hakki M, Chou S, Kalejta RF. 2015. Molecular determinants for the inactivation of the retinoblastoma tumor suppressor by the viral cyclin-dependent kinase UL97. J Biol Chem 290:19666–19680. doi: 10.1074/jbc.M115.660043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hamirally S, Kamil JP, Ndassa-Colday YM, Lin AJ, Jahng WJ, Baek M-C, Noton S, Silva LA, Simpson-Holley M, Knipe DM, Golan DE, Marto JA, Coen DM. 2009. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog 5:e1000275. doi: 10.1371/journal.ppat.1000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson BR, Nitta RT, Frock RL, Mounkes L, Barbie DA, Stewart CL, Harlow E, Kennedy BK. 2004. A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc Natl Acad Sci U S A 101:9677–9682. doi: 10.1073/pnas.0403250101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Markiewicz E, Dechat T, Foisner R, Quinlan RA, Hutchison CJ. 2002. Lamin A/C binding protein LAP2alpha is required for nuclear anchorage of retinoblastoma protein. Mol Biol Cell 13:4401–4413. doi: 10.1091/mbc.E02-07-0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Poma EE, Kowalik TF, Zhu L, Sinclair JH, Huang ES. 1996. The human cytomegalovirus IE1-72 protein interacts with the cellular p107 protein and relieves p107-mediated transcriptional repression of an E2F-responsive promoter. J Virol 70:7867–7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hagemeier C, Caswell R, Hayhurst G, Sinclair J, Kouzarides T. 1994. Functional interaction between the HCMV IE2 transactivator and the retinoblastoma protein. EMBO J 13:2897–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sommer MH, Scully AL, Spector DH. 1994. Transactivation by the human cytomegalovirus IE2 86-kilodalton protein requires a domain that binds to both the TATA box-binding protein and the retinoblastoma protein. J Virol 68:6223–6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murphy EA, Streblow DN, Nelson JA, Stinski MF. 2000. The human cytomegalovirus IE86 protein can block cell cycle progression after inducing transition into the S phase of permissive cells. J Virol 74:7108–7118. doi: 10.1128/JVI.74.15.7108-7118.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Song Y-J, Stinski MF. 2002. Effect of the human cytomegalovirus IE86 protein on expression of E2F-responsive genes: a DNA microarray analysis. Proc Natl Acad Sci U S A 99:2836–2841. doi: 10.1073/pnas.052010099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cherrington JM, Khoury EL, Mocarski ES. 1991. Human cytomegalovirus ie2 negatively regulates alpha gene expression via a short target sequence near the transcription start site. J Virol 65:887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang L, Stinski MF. 1995. Binding of cellular repressor protein or the IE2 protein to a cis-acting negative regulatory element upstream of a human cytomegalovirus early promoter. J Virol 69:7612–7621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A 100:14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferrari R, Gou D, Jawdekar G, Johnson SA, Nava M, Su T, Yousef AF, Zemke NR, Pellegrini M, Kurdistani SK, Berk AJ. 2014. Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host Microbe 16:663–676. doi: 10.1016/j.chom.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morris EJ, Dyson NJ. 2001. Retinoblastoma protein partners. Adv Cancer Res 82:1–54. doi: 10.1016/S0065-230X(01)82001-7. [DOI] [PubMed] [Google Scholar]
- 74.Sherr CJ, McCormick F. 2002. The RB and p53 pathways in cancer. Cancer Cell 2:103–112. doi: 10.1016/S1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]