

ABSTRACT
Vesicular stomatitis virus (VSV) assembly requires condensation of the viral ribonucleoprotein (RNP) core with the matrix protein (M) during budding from the plasma membrane. The RNP core comprises the negative-sense genomic RNA completely coated by the nucleocapsid protein (N) and associated by a phosphoprotein (P) with the large polymerase protein (L). To study the assembly of single viral particles, we tagged M and P with fluorescent proteins. We selected from a library of viruses with insertions in the M gene a replication-competent virus containing a fluorescent M and combined that with our previously described virus containing fluorescent P. Virus particles containing those fusions maintained the same bullet shape appearance as wild-type VSV but had a modest increase in particle length, reflecting the increased genome size. Imaging of the released particles revealed a variation in the amount of M and P assembled into the virions, consistent with a flexible packaging mechanism. We used the recombinants to further study the importance of the late domains in M, which serve to recruit the endosomal sorting complex required for transport (ESCRT) machinery during budding. Mutations in late domains resulted in the accumulation of virions that failed to pinch off from the plasma membrane. Imaging of single virions released from cells that were coinfected with M tagged with enhanced green fluorescent protein and M tagged with mCherry variants in which the late domains of one virus were inactivated by mutation showed a strong bias against the incorporation of the late-domain mutant into the released virions. In contrast, the intracellular expression and membrane association of the two variants were unaltered. These studies provide new tools for imaging particle assembly and enhance our resolution of existing models for assembly of VSV.
IMPORTANCE Assembly of vesicular stomatitis virus (VSV) particles requires the separate trafficking of the viral replication machinery, a matrix protein (M) and a glycoprotein, to the plasma membrane. The matrix protein contains a motif termed a “late domain” that engages the host endosomal sorting complex required for transport (ESCRT) machinery to facilitate the release of viral particles. Inactivation of the late domains through mutation results in the accumulation of virions arrested at the point of release. In the study described here, we developed new tools to study VSV assembly by fusing fluorescent proteins to M and to a constituent of the replication machinery, the phosphoprotein (P). We used those tools to show that the late domains of M are required for efficient incorporation into viral particles and that the particles contain a variable quantity of M and P.
INTRODUCTION
Vesicular stomatitis virus (VSV) forms particles with an ordered bullet shape. Those particles contain a lipid envelope modified by the insertion of the viral attachment and fusion glycoprotein (G). Inside the particle is a ribonucleoprotein (RNP) core that is delivered to the cytoplasm to initiate the process of infection. The RNP core comprises the negative-sense genomic RNA completely encased within a nucleocapsid protein (N) sheath. The N-RNA is associated with the large protein (L) RNA-dependent RNA polymerase via a phosphoprotein (P) cofactor, which bridges this interaction. During assembly and budding, the N-RNA:P-L complex is associated with and condensed by the viral matrix protein (M) and buds through the plasma membrane to acquire the G-containing lipid envelope. Cryo-electron microscopy (cryo-EM) reconstructions show that M is present as a discrete helix of ∼1,200 molecules per particle under the membrane. This helical M surrounds the helical N-RNA complex of ∼1,200 molecules of N encasing the genomic RNA (1). G and the polymerase complex are not visible, reflecting a lack of symmetry or poorly ordered density. Earlier electron microscopy (EM) and biochemical approaches estimated that approximately 1,826 molecules of M, 1,258 molecules of N, 466 molecules of P, and 50 molecules of L were present per virion (2). The difference in the number of molecules of M per particle from the cryo-EM reconstruction and the biochemical measurements may also reflect the presence of M inside the N-RNA helix in a poorly symmetric central cigar-like structure (1, 3).
Biochemical experiments support distinct pathways for assembly of RNP, M, and G (4). In infected cells, cotranslational insertion of G into the endoplasmic reticulum leads to its traffic to the plasma membrane, where it forms microdomains (5). G is not essential, however, as bald particles or those decorated with different envelope proteins assemble (6, 7), yet assembly is more efficient in the presence of G (7, 8), leading to a model in which internal components of the particle favor G incorporation and G microdomains promote the assembly of internal virion components (5). M is synthesized as a soluble cytoplasmic protein, of which less than 10% is transported to the plasma membrane (9), where it forms separate microdomains (10). Transport of M to the plasma membrane occurs independently of the transport of other viral components (9). At sites of virus assembly, the M and G microdomains coalesce into larger microdomains that appear to be the sites of particle release (10).
During replication, the RNA chain is encapsidated by N, and the resulting RNP complex, i.e., N-RNA:P-L, is transported to the plasma membrane. This complex undergoes condensation by M to form the nucleocapsid-matrix protein (NCM) complex (11). Although M can bind RNPs fully stripped of their M, this binding does not recapitulate the high-affinity binding observed in NCM complexes, suggesting an additional reversible step in assembly (12). While NCM complex-like structures can be assembled in vitro, these artificial complexes contain less stable interactions between M and the RNP, illustrating the importance of this additional step (13).
M comprises two domains: an N-terminal disordered region (amino acids 1 to 57) and a C-terminal globular domain whose atomic structure is known (14–16). Mutational analysis maps multiple functions to the N-terminal region, including self-association (17), binding to N (9, 18), and membrane association (19, 20). Membrane association is mediated by the association of positively charged residues found within the N-terminal 20 amino acids with the negatively charged phospholipids on the plasma membrane (20). The N terminus also contains two late-domain motifs that facilitate virus budding (21, 22). Those motifs were first identified to be position-independent motifs in the group-specific antigen protein (Gag) of retroviruses (23, 24). Such motifs recruit the cellular endosomal sorting complex required for transport (ESCRT) machinery to release enveloped virus particles (25). Disruption of late-domain motifs results in assembly of particles that remain trapped during pinching off from the plasma membrane. For VSV, electron micrographs of cells infected with particles containing late-domain mutations show that the surface of the cell is littered with particles that appear to be fully assembled (21).
The objective of the present study was to develop tools to image the assembly of single viral particles so that we might interrogate the order of events. Earlier work has allowed the visualization of M protein by appending a tetracysteine tag that permits labeling using a biarsenic dye (26, 27). While such a pulse-labeling approach permits the analysis of a subset of the M molecules in the cell, we elected to use fluorescent protein fusions to permit visualization of the entire population of M. A C-terminal fusion of monomeric red fluorescent protein to M was previously shown to be nonfunctional for assembly of viral particles (26). Consequently, we elected to use a different approach to generate a fluorescent M fusion that involved insertional mutagenesis within the gene followed by selection for variants competent for replication and assembly. We also generated replication-competent viruses containing fluorescent protein fusions to both M and P. By characterizing the properties of the released viral particles, we show that virions contain a distribution of M and P and that there is no correlation between the amount of M and P in the particle, consistent with independent incorporation. Using these tools, we tracked the fate of two distinct M proteins into single particles following coinfection of cells with a fluorescently tagged wild type and a late-domain mutant. The two variants were expressed at equal levels and displayed equal membrane association, yet the late-domain mutant was largely excluded from the released virions. This finding has implications for VSV assembly.
MATERIALS AND METHODS
Cells, viruses, and mutagenesis.
BS-C-1 (CCL-26; American Type Culture Collection, Manassas, VA) and BSR T7/5 (a kind gift from K. K. Conzelmann [28]) cells were grown at 37°C with 5% CO2. Cells were infected in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA) but maintained in DMEM supplemented with 10% fetal bovine serum (FBS; Tissue Culture Biologicals, Tulare, CA). Plaque assays were performed by overlaying infected cells with 0.25% (wt/vol) agarose–10% (vol/vol) FBS–DMEM. They were fixed in 3.75% formaldehyde in phosphate-buffered saline (PBS) and stained with 0.5% (wt/vol) crystal violet and 10% (vol/vol) ethanol in water. Recombinant VSV was generated using the infectious clone pVSV1(+) (29). Restriction sites were introduced into the backbone to facilitate cloning of M. CTCGAG (XhoI) was inserted immediately before the M start codon (ATG), and TGGCCA (MscI) was inserted after the stop codon (TGA). The late-domain mutants contained the point mutations 24-PPPY-27 to AAPA (21) and 37-PSAP-40 to AAAA (22), annotated for wild-type M. These mutations were generated through site-directed mutagenesis using the primers CTAAGAAATTAGGGATCGCAGCAGCCCCTGCTGAAGAGGACACTAGCATGG for PPPY and GCTGTACAAGGGCGGCCGCACTGCTGCTGCTGCAATTGACAAATCCTATTTTGG for PSAP (only the sequences of the positive-sense primers are reported, and point mutations are underlined). Viruses containing enhanced green fluorescent protein (eGFP)-tagged P (P-eGFP) were derived from the previously published clone that contains an N-terminal fusion to P (30).
The VSV M-coding sequence was mutagenized using a mutation generation system (F-701; Finnzymes, Vantaa, Finland) according to the instructions of the manufacturer. The transposon (Tn) was replaced with eGFP via the NotI sites, and the M-eGFP library was cloned into a VSV genomic analog comprising 3′-Leader (Le)-M-G-Trailer (Tr)-5′. Infectious particles were recovered as previously described (29).
Biochemical characterization of viruses.
VSV was purified on a 15 to 45% sucrose gradient as previously described (31), and virion composition was analyzed by low-bis sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with 10% (wt/vol) acrylamide and 0.13% (wt/vol) bisacrylamide. Protein was visualized with Coomassie brilliant blue G (70 μg ml−1, pH 1.5; Sigma, St. Louis, MO), by Western blotting with anti-M antibody 23H12 (a kind gift from D. S. Lyles [32]) and anti-mouse immunoglobulin conjugated with horseradish peroxidase (Sigma) and BioMax MR film (Kodak, Rochester, NY), or by Western blotting with anti-mouse immunoglobulin conjugated with IRDye 680RD (LI-COR, Lincoln, NE) and Odyssey Classic software (LI-COR). Radioactive protein was visualized with a storage phosphor screen (Amersham Biosciences, General Electric [GE] Healthcare, Piscataway, NJ) and a Typhoon 9400 variable-mode imager using the control software Typhoon Scanner Control (v4.0, build 4.0.0301.2100; Amersham). Quantification was performed with standard curves and ImageQuant TL software (v7; GE Healthcare), and statistical tests were performed with R (The R Foundation for Statistical Computing, Vienna, Austria; http://www.r-project.org/).
Immunofluorescence.
Cells were plated on a number 1.5 micro-cover glass (Electron Microscopy Sciences, Hatfield, PA), infected at a multiplicity of infection (MOI) of 3, fixed in 2% paraformaldehyde at 8 h postinfection (hpi), permeabilized with 0.2% Triton X-100, and blocked with 1% (wt/vol) bovine serum albumin in PBS. They were stained with 2 μM 4′,6-diamidino-2-phenylindole (DAPI), anti-M antibody 23H12 (32), anti-green fluorescent protein (anti-GFP) antibody (Abcam, Cambridge, MA), anti-mouse antibody (Molecular Probes Life Technologies, Grand Island, NY), and/or anti-rabbit antibody (Molecular Probes) and mounted on microslides (VWR, Radnor, PA) with ProLong Gold antifade reagent (Invitrogen, Eugene, OR). Samples were placed in a computer-controlled Piezo Z PZ-2000 and xy-stage MS-2000 with linear encoders (Applied Scientific Instrumentation, Eugene, OR) and imaged with a Marianas system (Intelligent Imaging Innovations [3i], Denver, CO) with a Zeiss Axio-Observer microscope (Carl Zeiss AG, Oberkochen, Germany) and SlideBook control software (v5.0; 3i). A Laser Launch light source (3i) with a 405-nm, 488-nm, 561-nm, and 640-nm laser and Semrock CF HQE long-pass filter sets (Chroma Technology Corp., Rockingham, VT) with an excitation (Ex) of 405/3 nm (filter center/bandpass width is stated) and an emission (Em) of 452/45 nm for DAPI, an Ex of 488/3 nm and an Em of 525/50 nm for eGFP, an Ex of 561/3 nm and an Em of 607/36 nm Alexa Fluor 594, and an Ex of 640/3 nm and an Em of 680 nm for Alexa Fluor 647 were used. The system is equipped with a Plan-Apo 63× oil objective (numerical aperture, 1.40) with an additional ×1.2 magnification lens for 0.212 μm/pixel (Carl Zeiss AG), spherical aberration correction (3i), a model CSU-X1 spinning disk unit (Yokogawa Electric Corporation, Tokyo, Japan), and a QuantEM:512SC electron multiplication camera (Photometrics, Tucson, AZ).
Transmission electron microscopy and analysis of particle dimensions.
Purified virus was adhered to glow-discharged Formvar and carbon-coated copper grids. Samples were stained with 2% (wt/vol) phosphotungstic acid, pH 7.5, in H2O and imaged with a Tecnai G2 Spirit BioTWIN transmission electron microscope (FEI, Hillsboro, OR) with an AMT 2K charge-coupled-device camera (Advanced Microscopy Techniques Corp., Woburn, MA). Particle dimensions were measured with ImageJ software (U.S. National Institutes of Health, Bethesda, MD; http://rsb.info.nih.gov/ij/), and statistical tests were performed with R (The R Foundation for Statistical Computing).
Quantification of virion fluorescence intensity.
Purified virus was placed between two number 1.5 micro-cover glasses, which were loaded into an Attofluor cell chamber (Invitrogen, Carlsbad, CA) and used in a modified heated-stage insert (20/20 Technology, Wilmington, NC) in a computer-controlled Piezo Z PZ-2000 and xy-stage MS-2000 with linear encoders (Applied Scientific Instrumentation). The Marianas system (3i) with a Zeiss 200M microscope (Carl Zeiss AG) and control software (SlideBook, v5.0; 3i) was used to acquire images. A Laser Launch light source (3i) used a 491-nm and 561-nm laser with Semrock CF HQE filter sets (Chroma Technology Corp.) with Ex at 491/3 nm and Em at 525/50 nm to image eGFP and Ex at 561/3 nm and Em at 620/60 nm to image mCherry. The system is equipped with a Plan-Apo 63× oil objective (numerical aperture, 1.40) with an additional ×1.5 magnification lens for 0.169 μm/pixel (Carl Zeiss AG), a model CSU-22 spinning disk unit (Yokogawa Electric Corporation, Tokyo, Japan), and a QuantEM:512SC electron multiplication camera (Photometrics, Tucson, AZ). z-stacks 7 μm tall with 0.1-μm spacing were acquired, and values were averaged to compensate for slanted samples. Single particle intensities were measured with a custom MATLAB (MathWorks, Natick, MA) script (Innovative Microscopy Applications for Biology [IMAB]) (33) and normalized to the peak of the histogram. To control for fluorophore and microscope noise, data sets were determined by imaging the same field of view twice and, for each particle, calculating the ratio of the intensity between the images. Statistical tests were performed with R (The R Foundation for Statistical Computing).
Isolation of plasma membrane.
The plasma membrane was isolated as previously described (34) with minor modifications. Briefly, BSR T7/5 cells were infected at an MOI of 20, and at 7 hpi, cells were harvested in 10 mM EDTA in PBS, resuspended in homogenization buffer (HB; 10 mM Tris, pH 7.4, 10 mM NaCl, 0.25 mM MgCl2), and subjected to 30 strokes in a Dounce chamber. Following centrifugation at 2,000 × g for 5 min at 4°C, 500 μl of the postnuclear fraction was mixed with 2.5 ml of 66% (wt/vol) sucrose in HB, transferred to an SW 41 Ti tube, and overlaid with 6 ml of 40% (wt/vol) sucrose in HB followed by 3 ml of 10% (wt/vol) sucrose in HB. Following centrifugation at 160,000 × g for 15 h at 4°C, the plasma membrane was collected from the 10 to 40% interface, diluted to 12 ml in HB, and subsequently recovered by centrifugation at 160,000 × g for 1 h at 4°C. The resulting pellet was resuspended in SDS sample buffer and analyzed by SDS-PAGE. Statistical tests were performed with R (The R Foundation for Statistical Computing).
Fitting of the M crystal structure into the M helix obtained by cryo-EM.
The fitting of the crystal structure of full-length VSV New Jersey M (PDB accession number 2W2R) (15) into the cryo-EM electron density map of VSV virions (EMDataBank accession number EMD 1663) (1), was performed with the UCSF Chimera program (v1.7; University of California, San Francisco, San Francisco, CA). Correlations were calculated with a map simulated from the M crystal structure at the resolution of the cryo-EM reconstruction, i.e., 10.6 Å.
RESULTS
A functional M-eGFP fusion identified from a screen.
To generate functional M-eGFP, we took the approach of creating a library of fusion proteins. Using transposon (Tn) mutagenesis, we first identified permissive insertion sites within M. The Tn was subsequently replaced with eGFP, and the resulting M-eGFP library was cloned into a VSV genomic analog comprising 3′-Le-M-G-Tr-5′ (Fig. 1A). The diversity of the library was verified by double digestion with EcoRI and NotI, which excises both M and Tn (Fig. 1B). Each insertion site creates a fragment with a unique size representing the distance of the Tn from the start of M. A smear of DNA visible on the agarose gel is consistent with Tn insertions at multiple locations in M (Fig. 1B, lane 4), and sequence analysis revealed the presence of secondary peaks in the chromatogram (Fig. 1C). To determine whether any M variants were functional for particle budding, the genomic analog library was transfected into cells along with the necessary trans-acting replication machinery, N, P, and L (Fig. 1D). The functional M expressed by the genomic analog permits the assembly and budding of particles that are subsequently amplified by infection of cells expressing N, P, and L at a low MOI. Sequence analysis of the resulting genomic analogs identified a single permissive site for insertion of eGFP in M after residue 37 (Fig. 1E), which is located within the disordered N terminus of the protein (15).
FIG 1.

Selection of a viable M-eGFP fusion protein. (A) cDNA of VSV M (blue) was subjected to transposon mutagenesis. The Tn (orange) was replaced with eGFP (green), and the resulting M-eGFP library was cloned into a VSV genomic analog consisting of 3′-Le-M-G-Tr-5′ (gray). (B) The diversity of the library was verified by restriction enzyme digestion. A schematic of the cDNA highlighting the position of restriction sites, including the upstream XhoI site and the downstream MscI site, is shown. The library was digested with HindIII, EcoRI, NotI, or EcoRI-NotI and compared to wild-type (wt) M (digested with EcoRI) (lanes 1 to 5, respectively). Multiple Tn insertion sites are indicated by the smear of DNA (*) smaller than wild-type M in lane 4. (C) Sequence analysis of the M-eGFP library. Primers specific for sequences upstream and downstream of the highlighted (green) XhoI and MscI sites, which flank the coding sequence, were used to sequence the cDNA, and the diversity is indicated by the secondary peaks in the chromatogram, underlined in red. (D) Selection of viable M-eGFP fusions. VSV genomic analog particles were recovered from the library by infection of cells with a T7-expressing vaccinia virus (vTF7-3) and transfection (red arrow) of the cells with the library and plasmids carrying the trans-acting viral replication machinery, N, P, and L. The particles that were produced (black arrow) were subsequently used to infect cells that were also infected with vTF7-3 and transfected with N, P, and L. The genomes of the released virions were sequenced. (E) The sequence of the viable fusion selected for further analysis is shown. The numbers listed above represent nucleotide positions, and those listed below represent the amino acid positions in the cDNA of wild-type M.
Fusion of M with eGFP attenuates virus replication in cell culture.
To determine the impact of the M fusion on virus replication, we inserted this M-eGFP fusion or an equivalent fusion, M-mCherry, into an infectious cDNA clone of VSV (29). To facilitate this, we engineered unique cloning sites to allow M exchange. We similarly engineered M-mCherry into a previously described infectious variant in which P is fused to eGFP (30). The latter recombinant, termed “VSV Dual,” was designed to permit the analysis of both M and P during assembly. Fusion of M with eGFP attenuated viral replication, as shown by single-step growth curves and a reduction in viral plaque size (Fig. 2A and B). As expected, purified particles contained M-eGFP, as evidenced by reactivity with antibodies against M and eGFP in Western blots and a corresponding alteration in the mobility (Fig. 2C and D). In addition to full-length M-eGFP, the Western blot illustrated smaller products that likely correspond to previously described alternate translational start sites within M (35) and the effect of posttranslational modifications on M (36, 37).
FIG 2.

Recombinant VSV M-eGFP and VSV M-mCherry P-eGFP (VSV Dual) replicate with slower kinetics. (A) Plaque assays of viruses fixed and stained at 48 hpi. (B) Single-step growth curves of the recombinant viruses containing the indicated fluorescent fusions of M and P. BSR T7/5 cells were infected at an MOI of 3, and the supernatant was sampled at the indicated times. VSV Control, virus in which wild-type M is flanked by the restriction sites that were used to facilitate cloning. Those insertions are inert with regard to virus replication in cell culture. (C and D) Purified viruses were analyzed on an SDS-polyacrylamide gel and stained with Coomassie blue (C) or analyzed on an SDS-polyacrylamide gel and then analyzed by Western blotting for M or eGFP (D).
Wild-type M migrates to the nucleus, where it blocks the Rae1 mRNP export pathway (32, 38). The intracellular distribution of M-eGFP appeared to be indistinguishable from that of wild-type M (Fig. 3), showing both nuclear and cytoplasmic accumulation. These data show that although insertion of eGFP into M attenuates virus replication, the functions of M for viral replication and assembly, as well as the subcellular distribution, appear to be similar to those of wild-type M.
FIG 3.

Subcellular localization of M-eGFP in infected cells. BS-C-1 cells were infected with wild-type VSV or VSV M-eGFP at an MOI of 3, fixed at 8 hpi, and stained with antibodies against eGFP (red) and M (blue). The merged image also includes DAPI (purple). Images were acquired under the same microscope settings and renormalized to the same values; i.e., identical gray value ranges were used for illustration. Bar, 10 μm.
M-eGFP alters virion protein composition but not shape.
VSV M-eGFP and VSV Dual particles remained bullet shaped, as shown by transmission EM (Fig. 4A). The M-eGFP particles were 12% longer than the wild-type particles (Fig. 4B) but had the same width as the wild-type particles (Fig. 4C), consistent with the fact that the M-eGFP genome is 7% larger (39). Analysis of purified virions by SDS-PAGE (Fig. 4D) showed similar amounts of P but a reduction in the amounts of G and L and 56% less M-eGFP per virion compared to the amounts of these proteins from wild-type VSV (Fig. 4E). On the basis of previous biochemical measurements of M (2), this predicts that there are 800 ± 30 molecules of M-eGFP per virion, on average. Using confocal fluorescence microscopy, we found that individual viral particles showed a distribution of fluorescence intensities consistent with a flexible packaging mechanism for M-eGFP (Fig. 5A). Examination of VSV Dual particles (Fig. 5B) illustrated the similar distributions of the fluorescence intensities for the incorporation of M-mCherry and P-eGFP (Fig. 5C and D). There was no correlation between the fluorescence intensities of the two proteins in a single virion, consistent with their independent incorporation into particles (Fig. 5E). We note that the M-mCherry signal displayed a distribution broader than the M-eGFP signal. Whether this reflects a distinction in the intrinsic fluorescence of the two proteins or subtle alterations in the fusion protein conformation was not determined.
FIG 4.

Dimensions and compositions of VSV M-eGFP particles. (A) Purified viruses were negatively stained and imaged on a transmission electron microscope. Bar, 100 nm. (B and C) The lengths (B) and widths (C) of 50 virions were measured, and the lengths but not the widths were significantly different (Student's t test, P = 10−14 and P = 0.3, respectively). (D) 35S-labeled viruses were analyzed on an SDS-polyacrylamide gel, and the viral bands were quantified by densitometry. (E) The intensities were normalized to the number of cysteines and methionines in each protein and then normalized to the intensity of N. SEMs (n = 3) are shown. ***, P < 10−5; **, P < 10−2; *, P < 0.05 (all P values were determined by Student's t test).
FIG 5.
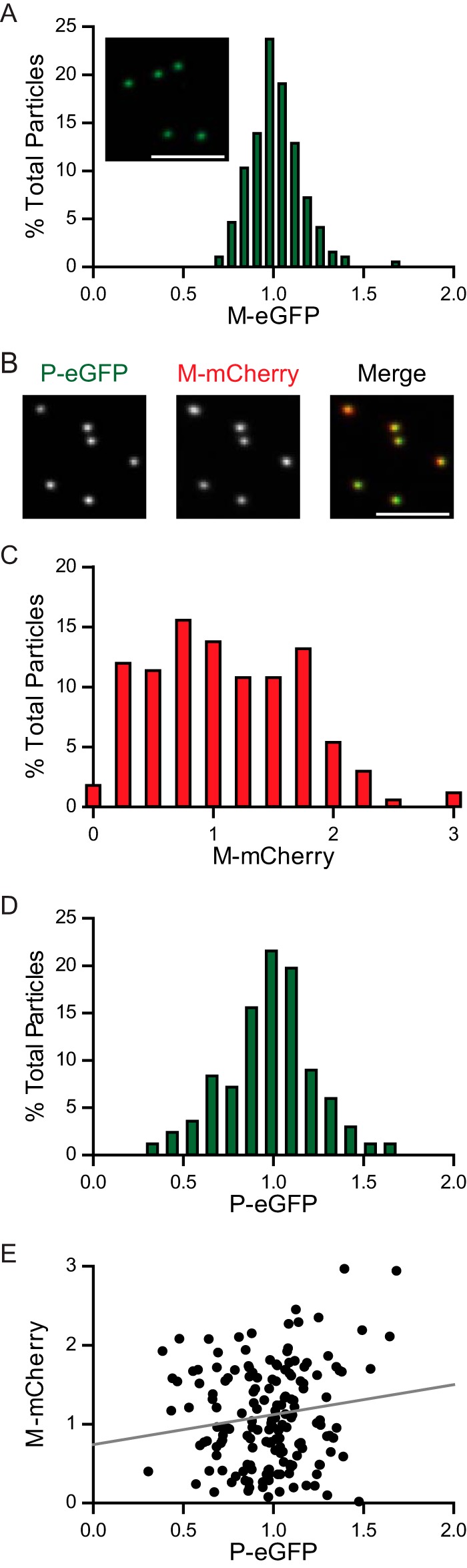
Fluorescence intensity of VSV M-eGFP and VSV Dual particles. (A) Gradient-purified VSV M-eGFP was imaged with a confocal fluorescence microscope, and the fluorescence intensity, in arbitrary units, of individual particles was plotted to generate a histogram (n = 194). (Inset) Fluorescent image of purified virions. (B to E) VSV Dual particles (n = 167) were imaged as described in the legend to panel A (B) and used to generate a histogram of M-mCherry fluorescence intensity (C), a histogram of P-eGFP fluorescence intensity (D), and a scatter plot of their fluorescence intensities (E) in arbitrary units. Histogram intensities are normalized to the peak. A control data set was used to measure the acquisition error, which was calculated by determination of the variation in the intensity of the particles between multiple acquisitions. The distributions of M-eGFP, M-mCherry, and P-eGFP in that data set were all significantly different from those in their control data sets (Kolmogorov-Smirnov test, P < 10−8, P < 10−12, and P < 10−6, respectively), and there was no correlation between M-mCherry and P-eGFP intensities (Spearman's rank correlation [ρ] = 0.054, P = 0.6). The line of best fit is shown in gray in panel E. Bars, 5 μm.
Incorporation of fluorescently distinct M proteins into single particles.
We next examined whether the fluorescent M protein could permit identification of the genetic origin of M in particles released from coinfected cells. To do this, we infected cells with both VSV M-eGFP and VSV M-mCherry (Fig. 6A). As expected, cells infected with both viruses expressed both fluorescently tagged versions of M and the released particles (Fig. 6B) contained a distribution of each M (Fig. 6C and D). The M-eGFP and M-mCherry were copackaged into particles, and their intensities were negatively correlated with each other, consistent with the random incorporation of each variant (Fig. 6E).
FIG 6.

Fluorescence intensity of particles from cells expressing M-eGFP and M-mCherry. (A) Schematic of the experimental setup. BSR T7/5 cells were coinfected with VSV M-eGFP and VSV M-mCherry at an MOI of 10 each. The virus that was produced was assayed for whether it contained equal or unequal amounts of M-eGFP and M-mCherry. (B) Fluorescent images of particles. Bar, 5 μm. (C and D) Histograms (n = 103) of the eGFP (C) and mCherry (D) intensities are shown. (E) A scatter plot demonstrates a negative correlation between eGFP and mCherry intensities (Spearman's rank correlation [ρ] = −0.31, P < 10−2). The line of best fit is shown in gray.
Functional late domains are required for efficient incorporation into particles.
Prior estimates suggested that there are 1,826 molecules of M per virion; we thus hypothesized that not all of the molecules would need a functional late domain for assembly. Incorporation of a few molecules of wild-type M at a late stage of assembly might compensate for the defect in release due to mutation of these motifs. To investigate this, we coinfected cells with M-eGFP and M-mCherry with and without functional late domains. Mutations in both late domains of M reduced its incorporation into the released virion irrespective of which fluorescent fusion was altered (Fig. 7A and B). This result suggests that although the manifest phenotype of a late-domain mutant is to restrict the pinching off stage of a fully assembled virion, the function of the late domain of VSV M is not simply restricted to this late role in particle assembly.
FIG 7.

Competition between M with and without functional late domains for incorporation into particles. The same experimental setup described in the legend to Fig. 6A was used, except that both late domains in one of the viruses in the coinfection were mutated. (A) Fluorescent images of particles that were acquired under the same microscope settings and renormalized to the same values; i.e., identical gray value ranges were used for illustration. Bar, 5 μm. (B) The particles were quantified, and the ratio of eGFP/mCherry was calculated for each particle and graphed on histograms (n = 103, 151, and 295 for coinfections with M-eGFP and M-mCherry with functional late domains, M-eGFP with a mutated late domain, and M-mCherry with a mutated late domain, respectively). The ratio values are normalized to the value at the peak of the coinfection, where both viruses had functional late domains. The results for coinfection with mutated M-eGFP are shifted to the left, and those for coinfection with mutated M-mCherry are shifted to the right (Kolmogorov-Smirnov test, P < 10−15 and P < 10−13, respectively). LD−, mutated both late domains.
To confirm that inactivation of the late domains was associated with a defect in the release of infectious particles that could be complemented, we measured the titers of the released virus particles. As expected, mutation of the late domains of VSV M-eGFP decreased the virus yield 48 ± 8-fold (n = 3). When cells were infected with the M-eGFP late-domain mutant and a wild-type M, the level of M-eGFP mutant virus budding was restored to 50% ± 40% (n = 5) of that of wild-type M-eGFP.
The reduced incorporation of the late-domain mutants in virions is not due to their reduced expression in cells since both variants of M were expressed at similar levels (Fig. 8A). Similarly, both wild-type and late-domain mutant M associated equally well with the plasma membrane in a flotation assay, suggesting that differences in membrane association do not account for the lack of the late-domain mutant M in virions (Fig. 8B). These results show that late-domain mutants are efficiently synthesized and transported to the plasma membrane but are not efficiently incorporated into virions in the presence of wild-type M.
FIG 8.

Comparison of the cellular expression and presence at the plasma membrane of M with and without functional late domains. BSR T7/5 cells were infected at an MOI of 20, and the whole-cell extract (A) or plasma membrane (B) was extracted at 7 hpi. The amounts of G and M present were determined by Western blotting. By using G as a loading control, the amount of M expressed in cells or present at the plasma membrane was quantified. SEMs (n = 3) are shown.
DISCUSSION
In this study, we report new tools to characterize the protein composition of individual VSV particles, identify sites at which heterologous proteins can be inserted into M and retain function, reveal the variable protein composition of single viral particles, and enhance the resolution of current models of particle assembly.
Virus composition.
Variations in the amounts of M and P present within single viral particles have not been previously reported. Biochemical estimates suggest that there are, on average, 466 molecules of P per virion (2). P is an oligomeric protein that forms an L2-P4 complex with L and can bind the N-RNA. The estimated number of L molecules per virion is 50, and thus, the L2-P4 complex accounts for 100 of those P molecules. It is thought that during the assembly process the polymerase is frozen—perhaps randomly—along the genome. It is therefore not surprising that the abundance of P per particle can vary. An earlier study using a virus with a fluorescent L or P suggested that a variable amount of both of these proteins is packaged into single particles (40). The fluorescent M fusions result in viruses that are attenuated with regard to their growth, which may reflect the fact that the presence of the fluorophore itself influences the packaging of M.
The cryo-EM reconstruction of VSV particles reveals an ordered helical assembly of M around the RNP (1). That helical assembly accounts for ∼1,200 molecules of M per particle. Here we found that replacement of M with M-eGFP resulted in an average of 800 copies of M per particle without altering the expected dimensions of the particle. This measurement is incompatible with all ∼1,200 M positions in the cryo-EM helix (1) being occupied by M and suggests flexibility in the requirements for the formation of the helical M assembly.
We suggest that the following possibilities might explain the apparent disparity in the number of molecules and positions of M. Both M (14–16) and GFP (41, 42) are globular proteins of similar size, and consequently, the two presumed domains of M-eGFP could each occupy a position in the helix normally adopted by wild-type M. An ability of eGFP to occupy an M position in the virion suggests flexibility in the requirements for formation of the bullet shape. The orientation(s) of M-eGFP in the helix would emphasize which intermolecular interactions are required for virion formation and which can be disrupted. There are several possible ways that M-eGFP can be organized within this structure, and those are depicted in our model (Fig. 9). Fitting of the crystal structure of M (15) into the cryo-EM electron density map (1) suggests a better fit when the N terminus is facing away from the membrane than when it is facing toward the membrane (correlation values of 0.63 versus 0.41, respectively, with a contour map value of 1.15σ, where σ is the standard deviation of the map intensities) (1). If, however, we adjust the contour map value to 1.7σ, M fits equally well when it is facing away from and toward the membrane (correlation values of 0.828 versus 0.839, respectively) (Fig. 9). The suggestion that M can adopt such alternate orientations is consistent with the detection of M filaments in the crystal space group (15). Inside virions, how M is able to simultaneously associate with other M molecules, N, and the membrane is not fully understood. Assembly of M into the helix in multiple orientations would allow a subset of M to form each of these interactions. In addition, GFP itself is able to assemble into antiparallel dimers (42). This association could facilitate the packing of M-eGFP into the helix, with some monomers facing away from the membrane and some facing toward the membrane. The ability of eGFP to replace M at some positions in the helix supports the notion that assembly of the M helix does not have rigid requirements.
FIG 9.

Orientation of M-eGFP in the virion. The crystal structure of M (15) was fit into the cryo-EM reconstruction of VSV virions in the published orientation (1) (A) or in an orientation rotated 180° from the published orientation (1) (B). With the electron density map segmented at a contour map value of 1.7σ, this generates correlation values of 0.828 and 0.839, respectively. (C) Schematics illustrate the relative orientation of N (red) and the M (blue) and eGFP (green) domains of M-eGFP in the virion M helix if M packs as described previously (1), packs as the crystal structure space group (15), or packs as GFP antiparallel dimers (42) (shown in order).
We would also suspect that the interactions between the M helix and the G-modified membrane would be affected. Although the density of the G protein was not fully apparent from cryo-EM (1), some density thought to correspond to the cytoplasmic tail of G was visible in a position that would permit interaction with 3 molecules of M. One anticipated outcome of altering the geometry of the M helix would therefore be to alter the amount of G incorporated into the virions. Consistent with this, measurements of the composition of our population of purified M-eGFP particles showed a reduction in the amount of G. This finding supports the model where G interactions facilitate assembly (7, 8, 10) but are not essential (6, 7).
We also noticed a reduction in the amount of L in the virion. We did not, however, measure the distribution of L among the particles. The modest reduction in the amount of L in the population of particles could be mediated by the presence of a physical occlusion or a more specific interaction between M or another viral component, but more definitive experiments would be required to discriminate among those possibilities.
Virus assembly.
The viruses reported here represent new tools to study assembly, and we examined how mutations in M influence particle composition. Cells coinfected with eGFP- and mCherry-tagged M produced particles that contained both tagged proteins in the virion, and the relative abundance of the two demonstrates that they were incorporated equivalently, consistent with a stochastic process for M incorporation. In contrast, analysis of particles released from cells coinfected with both the wild type and the late-domain mutant showed that the wild-type M is preferentially incorporated into particles. This result is surprising, as it has generally been assumed that the late-domain mutants of VSV stuck at the plasma membrane are fully assembled and thus contain a full complement of M. In a coinfected cell, the expectation would be that both the late-domain mutant and wild-type versions of M would incorporate equally well into those particles, resulting in equivalent amounts in the released virions. The observed biased particle content and the inability of wild-type M to fully complement the late-domain mutant in trans suggest that the M late domains not only function to recruit the ESCRT pathway, as has been previously thought, but likely also play an additional role in assembly. The possible explanations for this result have implications for our current understanding of VSV assembly.
Although we expect bullet-shaped viral particles stuck at the plasma membrane to contain a full complement of M, there is no evidence to support this. Recent work has shown that N-RNA alone can adopt the classic bullet shape of the interior of the virion (43). This raises the possibility that the particles stuck at the membrane are viral particles that lack significant M. Earlier in vitro dissociation and reassociation experiments with NCM complexes proposed that the association of the RNP with a minor virion component mediates conversion to the NCM complex (12, 13). When those data are combined with our data illustrating that the VSV bullet shape can be formed with a fraction of the M found in wild-type virions, it seems possible that the trapped particles contain only a few molecules of M which might aid the N-RNA with the formation of the bullet-shaped structure.
The bias against the late-domain mutant in the released virions could simply reflect the fact that the mutation alters the rate at which any individual M molecule is incorporated into a virion. This would bias against the incorporation of defective M in cells coinfected with the wild type. In cells infected with the late-domain mutant alone, this would result in a traffic jam effect, in which particles would appear to be otherwise stuck at the plasma membrane. One function of the late domain is to recruit the cellular ESCRT machinery and help accomplish the budding process by driving fission of the viral and cellular membranes. The simplistic model that the ESCRT machinery is recruited after assembly is completed is not compatible with our observed bias against the incorporation of the late-domain mutant M in coinfected cells. Our data are consistent with those from studies of the late domains in the Gag proteins of retroviruses that have demonstrated an additional role of these motifs in viral assembly. For Mason-Pfizer monkey virus (MPMV), both the PPPY and PSAP motifs are required for maturation of the procapsid (44), and for human immunodeficiency virus type 1 (HIV-1), assembly requires recruitment of the host factor Angiomotin and binding of this protein to Nedd4, a component of the ESCRT pathway (45). An alternative possibility that we cannot eliminate is that there is a thresholding effect, where a minimal amount of the ESCRT machinery must be recruited by M to mediate fission, which results in the observed bias in the composition of released particles.
Insertion of eGFP in M.
Our insertional mutagenesis screen identified the disordered N terminus of M to be a permissible site for insertion of eGFP. This is consistent with the structures of M (14–16) that would suggest that the most likely site for such an insertion to be tolerated is the disordered N terminus or loops between β sheets. Although we cannot rule out the possibility that all other positions are possible insertion sites, we found that other locations in the disordered N terminus were not tolerant of insertion. For example, insertions at residues 46 and 50 were also identified in the screen, but we were unable to recover viruses with insertions at those residues. While the approach used here was successful in generating an autonomously replicating virus, it is important to emphasize that VSV M-eGFP exhibited slower replication kinetics in cell culture than wild-type M, demonstrating that the insertion is not inert with regard to viral replication. An N-terminal fusion to M was found to reduce the budding activity (27), and the PSAP proximal sequences, which include residues 33 to 37, are involved in plaque size and cytopathy (46). Consequently, it seems possible that the insertion of a fluorescent protein at this position attenuates viral growth. Nevertheless, the viruses described in this study represent useful tools to track the entry of incoming viral particles (47) and the composition of released virions. Combining the visualization of the released particles with coinfection of cells also allows a higher level of resolution in genetic complementation studies.
ACKNOWLEDGMENTS
We thank Jennifer C. Waters at the Nikon Imaging Center at Harvard Medical School for helpful discussions and Ramiro H. Massol for use of and support with the use of IMAB.
This work was funded by National Institutes of Health (NIH) grant AI081842 (to S.P.J.W.) and a Postgraduate Scholarships-Doctoral Program (PGS D) award from the Natural Sciences and Engineering Research Council of Canada (NSERC) (to T.K.S.).
REFERENCES
- 1.Ge P, Tsao J, Schein S, Green TJ, Luo M, Zhou ZH. 2010. Cryo-EM model of the bullet-shaped vesicular stomatitis virus. Science 327:689–693. doi: 10.1126/science.1181766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas D, Newcomb WW, Brown JC, Wall JS, Hainfeld JF, Trus BL, Steven AC. 1985. Mass and molecular composition of vesicular stomatitis virus: a scanning transmission electron microscopy analysis. J Virol 54:598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barge A, Gaudin Y, Coulon P, Ruigrok RW. 1993. Vesicular stomatitis virus M protein may be inside the ribonucleocapsid coil. J Virol 67:7246–7253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knipe DM, Baltimore D, Lodish HF. 1977. Separate pathways of maturation of the major structural proteins of vesicular stomatitis virus. J Virol 21:1128–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown EL, Lyles DS. 2003. Organization of the vesicular stomatitis virus glycoprotein into membrane microdomains occurs independently of intracellular viral components. J Virol 77:3985–3992. doi: 10.1128/JVI.77.7.3985-3992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takada A, Robison C, Goto H, Sanchez A, Murti KG, Whitt MA, Kawaoka Y. 1997. A system for functional analysis of Ebola virus glycoprotein. Proc Natl Acad Sci U S A 94:14764–14769. doi: 10.1073/pnas.94.26.14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robison CS, Whitt MA. 2000. The membrane-proximal stem region of vesicular stomatitis virus G protein confers efficient virus assembly. J Virol 74:2239–2246. doi: 10.1128/JVI.74.5.2239-2246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitt MA, Chong L, Rose JK. 1989. Glycoprotein cytoplasmic domain sequences required for rescue of a vesicular stomatitis virus glycoprotein mutant. J Virol 63:3569–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chong LD, Rose JK. 1994. Interactions of normal and mutant vesicular stomatitis virus matrix proteins with the plasma membrane and nucleocapsids. J Virol 68:441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swinteck BD, Lyles DS. 2008. Plasma membrane microdomains containing vesicular stomatitis virus M protein are separate from microdomains containing G protein and nucleocapsids. J Virol 82:5536–5547. doi: 10.1128/JVI.02407-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newcomb WW, Brown JC. 1981. Role of the vesicular stomatitis virus matrix protein in maintaining the viral nucleocapsid in the condensed form found in native virions. J Virol 39:295–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flood EA, Lyles DS. 1999. Assembly of nucleocapsids with cytosolic and membrane-derived matrix proteins of vesicular stomatitis virus. Virology 261:295–308. doi: 10.1006/viro.1999.9856. [DOI] [PubMed] [Google Scholar]
- 13.Lyles DS, McKenzie MO. 1998. Reversible and irreversible steps in assembly and disassembly of vesicular stomatitis virus: equilibria and kinetics of dissociation of nucleocapsid-M protein complexes assembled in vivo. Biochemistry 37:439–450. doi: 10.1021/bi971812j. [DOI] [PubMed] [Google Scholar]
- 14.Gaudier M, Gaudin Y, Knossow M. 2002. Crystal structure of vesicular stomatitis virus matrix protein. EMBO J 21:2886–2892. doi: 10.1093/emboj/cdf284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graham SC, Assenberg R, Delmas O, Verma A, Gholami A, Talbi C, Owens RJ, Stuart DI, Grimes JM, Bourhy H. 2008. Rhabdovirus matrix protein structures reveal a novel mode of self-association. PLoS Pathog 4:e1000251. doi: 10.1371/journal.ppat.1000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quan B, Seo HS, Blobel G, Ren Y. 2014. Vesiculoviral matrix (M) protein occupies nucleic acid binding site at nucleoporin pair (Rae1 · Nup98). Proc Natl Acad Sci U S A 111:9127–9132. doi: 10.1073/pnas.1409076111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaudier M, Gaudin Y, Knossow M. 2001. Cleavage of vesicular stomatitis virus matrix protein prevents self-association and leads to crystallization. Virology 288:308–314. doi: 10.1006/viro.2001.1062. [DOI] [PubMed] [Google Scholar]
- 18.Capone J, Ghosh HP. 1984. Association of the nucleocapsid protein N of vesicular stomatitis virus with phospholipid vesicles containing the matrix protein M. Can J Biochem Cell Biol 62:1174–1180. doi: 10.1139/o84-151. [DOI] [PubMed] [Google Scholar]
- 19.Lenard J, Vanderoef R. 1990. Localization of the membrane-associated region of vesicular stomatitis virus M protein at the N terminus, using the hydrophobic, photoreactive probe 125I-TID. J Virol 64:3486–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye Z, Sun W, Suryanarayana K, Justice P, Robinson D, Wagner RR. 1994. Membrane-binding domains and cytopathogenesis of the matrix protein of vesicular stomatitis virus. J Virol 68:7386–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jayakar HR, Murti KG, Whitt MA. 2000. Mutations in the PPPY motif of vesicular stomatitis virus matrix protein reduce virus budding by inhibiting a late step in virion release. J Virol 74:9818–9827. doi: 10.1128/JVI.74.21.9818-9827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Obiang L, Raux H, Ouldali M, Blondel D, Gaudin Y. 2012. Phenotypes of vesicular stomatitis virus mutants with mutations in the PSAP motif of the matrix protein. J Gen Virol 93:857–865. doi: 10.1099/vir.0.039800-0. [DOI] [PubMed] [Google Scholar]
- 23.Gottlinger HG, Dorfman T, Sodroski JG, Haseltine WA. 1991. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc Natl Acad Sci U S A 88:3195–3199. doi: 10.1073/pnas.88.8.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demirov DG, Orenstein JM, Freed EO. 2002. The late domain of human immunodeficiency virus type 1 p6 promotes virus release in a cell type-dependent manner. J Virol 76:105–117. doi: 10.1128/JVI.76.1.105-117.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Votteler J, Sundquist WI. 2013. Virus budding and the ESCRT pathway. Cell Host Microbe 14:232–241. doi: 10.1016/j.chom.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das SC, Panda D, Nayak D, Pattnaik AK. 2009. Biarsenical labeling of vesicular stomatitis virus encoding tetracysteine-tagged M protein allows dynamic imaging of M protein and virus uncoating in infected cells. J Virol 83:2611–2622. doi: 10.1128/JVI.01668-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mire CE, Dube D, Delos SE, White JM, Whitt MA. 2009. Glycoprotein-dependent acidification of vesicular stomatitis virus enhances release of matrix protein. J Virol 83:12139–12150. doi: 10.1128/JVI.00955-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buchholz UJ, Finke S, Conzelmann KK. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J Virol 73:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whelan SP, Ball LA, Barr JN, Wertz GT. 1995. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc Natl Acad Sci U S A 92:8388–8392. doi: 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schott DH, Cureton DK, Whelan SP, Hunter CP. 2005. An antiviral role for the RNA interference machinery in Caenorhabditis elegans. Proc Natl Acad Sci U S A 102:18420–18424. doi: 10.1073/pnas.0507123102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, Whelan SP. 2009. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog 5:e1000394. doi: 10.1371/journal.ppat.1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lyles DS, Puddington L, McCreedy BJ Jr. 1988. Vesicular stomatitis virus M protein in the nuclei of infected cells. J Virol 62:4387–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Massol RH, Boll W, Griffin AM, Kirchhausen T. 2006. A burst of auxilin recruitment determines the onset of clathrin-coated vesicle uncoating. Proc Natl Acad Sci U S A 103:10265–10270. doi: 10.1073/pnas.0603369103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chong LD, Rose JK. 1993. Membrane association of functional vesicular stomatitis virus matrix protein in vivo. J Virol 67:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jayakar HR, Whitt MA. 2002. Identification of two additional translation products from the matrix (M) gene that contribute to vesicular stomatitis virus cytopathology. J Virol 76:8011–8018. doi: 10.1128/JVI.76.16.8011-8018.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaptur PE, McKenzie MO, Wertz GW, Lyles DS. 1995. Assembly functions of vesicular stomatitis virus matrix protein are not disrupted by mutations at major sites of phosphorylation. Virology 206:894–903. doi: 10.1006/viro.1995.1012. [DOI] [PubMed] [Google Scholar]
- 37.Harty RN, Brown ME, McGettigan JP, Wang G, Jayakar HR, Huibregtse JM, Whitt MA, Schnell MJ. 2001. Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J Virol 75:10623–10629. doi: 10.1128/JVI.75.22.10623-10629.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Faria PA, Chakraborty P, Levay A, Barber GN, Ezelle HJ, Enninga J, Arana C, van Deursen J, Fontoura BM. 2005. VSV disrupts the Rae1/mrnp41 mRNA nuclear export pathway. Mol Cell 17:93–102. doi: 10.1016/j.molcel.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 39.Schnell MJ, Buonocore L, Kretzschmar E, Johnson E, Rose JK. 1996. Foreign glycoproteins expressed from recombinant vesicular stomatitis viruses are incorporated efficiently into virus particles. Proc Natl Acad Sci U S A 93:11359–11365. doi: 10.1073/pnas.93.21.11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hodges J, Tang X, Landesman MB, Ruedas JB, Ghimire A, Gudheti MV, Perrault J, Jorgensen EM, Gerton JM, Saffarian S. 2013. Asymmetric packaging of polymerases within vesicular stomatitis virus. Biochem Biophys Res Commun 440:271–276. doi: 10.1016/j.bbrc.2013.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. 1996. Crystal structure of the Aequorea victoria green fluorescent protein. Science 273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- 42.Yang F, Moss LG, Phillips GN Jr. 1996. The molecular structure of green fluorescent protein. Nat Biotechnol 14:1246–1251. doi: 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]
- 43.Desfosses A, Ribeiro EA Jr, Schoehn G, Blondel D, Guilligay D, Jamin M, Ruigrok RW, Gutsche I. 2013. Self-organization of the vesicular stomatitis virus nucleocapsid into a bullet shape. Nat Commun 4:1429. doi: 10.1038/ncomms2435. [DOI] [PubMed] [Google Scholar]
- 44.Gottwein E, Bodem J, Muller B, Schmechel A, Zentgraf H, Krausslich HG. 2003. The Mason-Pfizer monkey virus PPPY and PSAP motifs both contribute to virus release. J Virol 77:9474–9485. doi: 10.1128/JVI.77.17.9474-9485.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mercenne G, Alam SL, Arii J, Lalonde MS, Sundquist WI. 2015. Angiomotin functions in HIV-1 assembly and budding. eLife 4:03778. doi: 10.7554/eLife.03778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Irie T, Carnero E, Okumura A, Garcia-Sastre A, Harty RN. 2007. Modifications of the PSAP region of the matrix protein lead to attenuation of vesicular stomatitis virus in vitro and in vivo. J Gen Virol 88:2559–2567. doi: 10.1099/vir.0.83096-0. [DOI] [PubMed] [Google Scholar]
- 47.Jae LT, Raaben M, Herbert AS, Kuehne AI, Wirchnianski AS, Soh TK, Stubbs SH, Janssen H, Damme M, Saftig P, Whelan SP, Dye JM, Brummelkamp TR. 2014. Virus entry. Lassa virus entry requires a trigger-induced receptor switch. Science 344:1506–1510. doi: 10.1126/science.1252480. [DOI] [PMC free article] [PubMed] [Google Scholar]