

ABSTRACT
Adult T cell leukemia/lymphoma (ATL) is an aggressive cancer of CD4/CD25+ T lymphocytes, the etiological agent of which is human T-cell lymphotropic virus type 1 (HTLV-1). ATL is highly refractory to current therapies, making the development of new treatments a high priority. Oncolytic viruses such as vesicular stomatitis virus (VSV) are being considered as anticancer agents since they readily infect transformed cells compared to normal cells, the former appearing to exhibit defective innate immune responses. Here, we have evaluated the efficacy and safety of a recombinant VSV that has been retargeted to specifically infect and replicate in transformed CD4+ cells. This was achieved by replacing the single VSV glycoprotein (G) with human immunodeficiency virus type 1 (HIV-1) gp160 to create a hybrid fusion protein, gp160G. The resultant virus, VSV-gp160G, was found to only target cells expressing CD4 and retained robust oncolytic activity against HTLV-1 actuated ATL cells. VSV-gp160G was further noted to be highly attenuated and did not replicate efficiently in or induce significant cell death of primary CD4+ T cells. Accordingly, VSV-gp160G did not elicit any evidence of neurotoxicity even in severely immunocompromised animals such as NOD/Shi-scid, IL-2Rγ-c-null (NSG) mice. Importantly, VSV-gp160G effectively exerted potent oncolytic activity in patient-derived ATL transplanted into NSG mice and facilitated a significant survival benefit. Our data indicate that VSV-gp160G exerts potent oncolytic efficacy against CD4+ malignant cells and either alone or in conjunction with established therapies may provide an effective treatment in patients displaying ATL.
IMPORTANCE Adult T cell leukemia (ATL) is a serious form of cancer with a high mortality rate. HTLV-1 infection is the etiological agent of ATL and, unfortunately, most patients succumb to the disease within a few years. Current treatment options have failed to significantly improve survival rate. In this study, we developed a recombinant strain of vesicular stomatitis virus (VSV) that specifically targets transformed CD4+ T cells through replacement of the G protein of VSV with a hybrid fusion protein, combining domains from gp160 of HIV-1 and VSV-G. This modification eliminated the normally broad tropism of VSV and restricted infection to primarily the transformed CD4+ cell population. This effect greatly reduced neurotoxic risk associated with VSV infection while still allowing VSV to effectively target ATL cells.
INTRODUCTION
Adult T cell leukemia (ATL) is a highly aggressive malignancy of activated mature CD4/CD25+ T lymphocytes (1) that has been linked etiologically to human T-cell lymphotropic virus type 1 (HTLV-1) infection. An estimated 15 to 20 million people are infected with HTLV-1, predominantly in southern Japan, the Caribbean, Central and South America, intertropical Africa, and northern Iran (2–5). Of those infected, a small percentage (6.6% for male and 2.1% for female) will develop ATL after a long latency period of anywhere between 20 and 80 years (6). ATL is generally classified into four clinical subtypes: acute, lymphoma, chronic, and smoldering (7), with the median survival of patients in the acute phase being only 6 to 9 months (8).
ATL patients suffer from a multitude of problems due to organ complications arising from infiltrating leukemic cells (9), and opportunistic infections resulting from immune suppression (10). Studies report that dendritic cells isolated from HTLV-1 carriers have impaired alpha interferon (IFN-α) production (11) and reduced capacity to mature into antigen-presenting cells (12). Natural killer cells have significantly decreased cytotoxic activity, allowing the escape of infected CD4+ T lymphocytes from immune destruction (13). In addition, several reports have demonstrated that HTLV-1-infected cells have a blunted type I IFN response, thereby inhibiting the induction of antiviral genes (14). The HTLV-1 proteins Tax and HBZ have been implicated in suppressing the IFN signaling pathway (15–18). HTLV-1 infection also induces the expression of miR-155 and miR-146a (19, 20), which are known to downregulate components of IRF3 (21) and TLR and RLR signaling, respectively (22, 23). Collectively, HTLV-1 infection disrupts multiple levels of host immunity, allowing opportunistic infections and leukemogenesis.
Mechanistically, HTLV-1's Tax protein exerts multiple functions and is likely responsible for leukemogenesis through the activation of growth regulatory pathways, as well as repression of several tumor suppressor genes (24). Tax is known to cause the constitutive activation of NF-κB (25), resulting in the expression of progrowth and prosurvival lymphokines such as interleukin-6 (IL-6), granulocyte-macrophage colony-stimulating factor, transforming growth factor β, IL-2Rα, c-fos, c-egr, and c-jun (26–32). Tax has been shown to promote T cell survival, proliferation, and override cell senescence, leading to immortalization, and ultimately, the transformation of human primary CD4+ T cells (24, 32, 33). In addition to upregulating growth and survival pathways, Tax mediates the accumulation of genetic changes, which can lead to Tax independent proliferation and escape from cytotoxic-T-lymphocyte (CTL) targeted destruction, since Tax is a preferential target of the immune response (34). Interestingly, most ATL patients are Tax negative, indicating that Tax is necessary for oncogenesis but not required for maintenance of the malignant phenotype (35).
Despite significant progress since ATL's discovery in 1977 (36), there is no effective treatment regimen for ATL. ATL is highly refractory to most treatment options and, although survival benefit has been seen using combination IFN-α and zidovudine or with allogeneic hematopoietic stem-cell transplant, ATL recurrence rates are high. There are a variety of clinical trials for ATL, both ongoing and in preparation. However, the need for new therapies is dire as most patients succumb to the disease within 5 years after diagnosis (37).
Oncolytic virotherapy is under evaluation as a therapeutic option for a variety of diseases. Oncolytic virotherapy is the use of natural or engineered viruses as a therapeutic against a variety of cancers. Most transformed cells accumulate defects in innate immunity and translational control mechanisms since this confers a growth and survival advantage. However, this same advantage leaves them highly susceptible to viral infection (38). Oncolytic virotherapy takes advantage of this differential to preferentially target cancer cells while sparing primary cells.
A promising oncolytic vector against ATL is vesicular stomatitis virus (VSV). VSV is an 11-kb negative-stranded enveloped RNA virus of the Rhabdoviridae family. Recombinant VSV has several advantages as an oncolytic vector over other oncolytic viruses. For instance, VSV is a RNA virus that remains in the cytoplasm during its life cycle; there is no known risk of its integration into the genome or transformation. VSV is genetically malleable, allowing new strains to be created with ease, and there is low seroprevalence in the general population, reducing the likelihood of a preexisting immunity that would render the virus ineffective as an oncolytic vector (39). VSV's oncoselectivity is mediated by its sensitivity to the IFN pathway. For instance, transfection of various IFN-stimulated genes (ISGs) renders normally permissive cells highly resistant to VSV replication (40), and murine models harboring a defective IFN system are highly sensitive to normally innocuous exposure to VSV (41).
As already discussed, ATL cells exhibit defects in IFN signaling and previous studies have shown VSV-HR Indiana serotype able to induce apoptosis in ex vivo ATL cells (34), making ATL a highly attractive target for the development of new recombinant VSV oncolytic strains. However, due to the immunocompromised nature of ATL patients, VSV may prove to be pathogenic, especially since the VSV G protein is highly tropic for numerous cell types (8). In the present study, we circumvented plausible problems associated with toxicity by creating a recombinant VSV strain that is specific for CD4+ T cells, an event which effectively prevented off-target infection. This was achieved by replacing the VSV-G with gp160G, a fusion hybrid glycoprotein combining human immunodeficiency virus 1 (HIV-1) gp160 with VSV-G. A similar construct was developed earlier for the purpose of creating an HIV vaccine. Here, we utilize it as a directed oncolytic vector against ATL (42, 43).
We demonstrate that VSV-gp160G is replication competent, has excellent specificity for CD4+ cells, remains attenuated against primary cell types, and retains potent oncolytic activity against transformed ATL cells. Furthermore, we investigated VSV-gp160G efficacy in vivo by treating ATL in NOD/Shi-scid, IL-2Rγ-c-null (NSG) mice. NSG mice are immunocompromised and are excellent recipients for transformed human cell lines, including ATL (44). Normally, NSG mice succumb rapidly to VSV infection regardless of the injection route. However, we found that VSV-gp160G displayed no toxicity in mice, indicating that the vector is specific for human CD4. In vivo analysis indicated that VSV-gp160G successfully exhibited oncolytic activity in ATL tumor-bearing hosts and provided a significant survival benefit by delaying metastasis and impeding tumor growth. Collectively, our data demonstrate that VSV-gp160G is a promising agent for the treatment of CD4+ T cell-related malignancies.
MATERIALS AND METHODS
Cells.
BHK-21-WI cells (generously provided by M. A. Whitt) and HeLa cells were maintained in Dulbecco modified Eagle medium (DMEM; Gibco/Invitrogen) supplemented with 10% fetal bovine serum (FBS), 5% penicillin-streptomycin. HeLa CD4+ cells were maintained in DMEM supplemented with 15% FBS, 5% penicillin streptomycin, and 200 μg of G418 sulfate (Calbiochem)/ml. The ATL cells, MT-2, MT-4, TLO-m1, and TLO-m1-luc (which were generously provided and developed by J. C. Ramos) and the B cell line, BJAB, were maintained in RPMI 1640 Medium (Gibco/Invitrogen) supplemented with 10% FBS and 5% penicillin-streptomycin.
Generation of rVSV expressing gp160G in lieu of VSV-G.
A fusion protein between HIV-gp160 and VSV-G was generated using overlap extension PCR with Pfx Super Mix (Invitrogen) and incorporated into the VSV construct (VSV-XN2) (45). To generate the HIV portion of the fusion protein, the first 750 amino acids of HIV gp160 were PCR amplified from the pNL4-3 HIV plasmid using the forward primer P1 (5′-CCGGACGCGTATGAGAGTGAAGGAGAAGTA-3′; the underlined region indicates the restriction site for MluI, and the boldfacing indicates the HIV portion) and the reverse primer P2 (5′-ATGGATACCAACTCGGGATCCGTTCACTAA-3′; the boldfacing indicates the HIV portion of the primer, and the rest is homologous to the VSV-G cytoplasmic tail region). The last 30 amino acids of VSV-G (containing the cytoplasmic tail region) were PCR amplified from pVSV-XN2 using the forward primer P3 (5′-TTAGTGAACGGATCCCGAGTTGTATCCAT-3′; the boldfacing indicates the VSV portion of the primer, and the rest is the HIV portion) and reverse primer P4 (5′-CCGGCTCGAGTTACTTTCCAAGTCGGTTC-3′; the underlined region indicates the restriction site for XhoI, and the boldfacing indicates the VSV-G portion). Next, both PCR fragments were mixed together and PCR amplified to generate the full-length fusion protein gp160G with MluI and XhoI restriction sites. The gp160G gene was then cloned into pVSV-XN2 after restriction enzyme digestion with MluI and XhoI (NEB) to remove VSV-G and create compatible ends to ligate gp160G into the VSV-XN2 cDNA plasmid using quick T4 DNA ligation (NEB).
Recovery and purification of VSV-gp160G.
VSV-gp160G was recovered using a modified version of established VSV recovery methods (46, 47). In brief, BHK-21-WI cells in six-well plates (∼70% confluent) were infected with vTF7-3 (45) at a multiplicity of infection (MOI) of 5 on day 0. After 45 min, vTF7-3-infected BHK-21-WI cells were washed with serum-free medium and transfected using Lipofectamine 2000 (Invitrogen) with 0.5 μg of pBS-N, 0.83 μg of pBS-P, 0.17 μg of pBS–L, 2 μg of pcDNA-G, and 5 μg of pVSV-gp160G in DMEM with 5% low-IgG FBS (Life Technologies). After 3 to 5 h of incubation, the cells were given fresh medium and then incubated at 37°C for 48 h. On day 1, a second six-well plate of BHK-21 cells was transfected with 2 μg of pcDNA-G. After 48 hpi, the medium from the VTF7-3-infected cells was passed through a 0.2-μm-pore-size syringe filter twice to remove the vTF7-3 vaccinia virus before being transferred to the BHK-21-G-transfected cells. If a cytopathic effect (CPE) was observed, indicating a viable VSV recovery, the medium was transferred to HeLa CD4+ cells for amplification of non-VSV-G-pseudotyped progeny. The virus was plaque purified and further amplified using HeLa CD4+ cells. Purification and concentration were achieved by pelleting the virus using ultracentrifugation with a low-density 10% OptiPrep cushion (Sigma). VSV was resuspended in phosphate-buffered saline (PBS) aliquots and kept at −80°C. Virus titers were determined by standard plaque assays using HeLa CD4+ cells in 24-well plates.
Virus infections.
Cells were seeded in 6- or 12-well plates. Adherent cells were grown to 80% confluence, while cells in suspension were immediately infected after plating with rVSV at the indicated MOI. Adherent cells were infected with rVSVs at the indicated MOI in a reduced volume of serum-free DMEM for 1 h with agitation at 15-min intervals. The cells were then washed with 1× PBS twice, and complete medium was added back to the cells. Cells in suspension were pelleted and resuspended in serum-free RPMI medium containing rVSV at the indicated MOI. After 1 h of incubation with agitation at 15-min intervals, the cells were washed twice in 1× PBS, and resuspended in complete medium.
Western blotting.
Infected cells were collected and incubated in lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate [SDS], 1× protease inhibitor cocktail [Sigma]) (48) for 30 min at 4°C with gentle agitation. Cell debris was removed by centrifugation for 10 min at 15,000 × g. Protein concentration was quantitated using Coomassie blue (Thermo Scientific), and the optical density was read at 595 nm. Equal amounts of protein were separated using SDS–10% PAGE and transferred to a polyvinylidene difluoride membrane (PVDF). Membranes were blocked with 5% milk powder in PBS–0.1% Tween 20 at room temperature and then probed with antibodies against HIV gp41 (ARRRP 2F5, 1:5,000; PBS-Tween, 0.1%), HIV gp120 (Santa Cruz, 1:5,000; PBS-Tween, 0.1%), VSV-G (Sigma, 1:10,000; 5% milk in 0.1% PBS-Tween), or β-actin (Sigma, 1:20,000; 5% milk in 0.1% PBS-Tween) overnight at 4°C with gentle rocking. The membranes were probed with horseradish peroxidase-conjugated anti-human, anti-goat, or anti-mouse antibody (Santa Cruz) diluted 1:10,000 in blocking buffer. Membranes were washed with PBS 0.1% Tween 20, and then the image was resolved using chemiluminescence (Thermo Scientific) and captured by autoradiography (Kodak Film).
Growth kinetic assays.
For adherent cells, i.e., HeLa and HeLa CD4+ cells, a total of 105 cells/well were seeded in a six-well plate. Cells were infected with either VSV-XN2 or VSV-gp160G at an MOI of 0.001 in serum-free DMEM for 1 h, with agitation every 15 min. Next, the cells were washed three times with 1× PBS, and 3 ml of complete medium was added to each well. The culture supernatants were harvested at the indicated times and kept at −80°C until virus titer was measured using a standard plaque assay with HeLa CD4+ cells. For cells in suspension (MT-2, MT-4, TLO-m1, and BJAB), 105 cells were collected and resuspended in rVSV inoculum with serum-free RPMI at an MOI of 0.001 for 1 h, with agitation every 15 min. Next, cells were washed three times in 1× PBS and seeded in six-well plate with 3 ml of complete RPMI supplemented with 10% FBS. Then, a 100-μl sample was clarified of cell debris by mild centrifugation, and the culture supernatant was collected and stored at −80°C until the virus titer was measured by a standard plaque assay using HeLa CD4+ cells. The remaining pelleted cells were resuspended in 100 μl of RPMI–10% FBS and transferred back to the well.
Flow cytometry and viability assessment.
Cells were seeded at 2 × 105/well in their respective media and infected with the indicated MOIs of either VSV-XN2 or VSV-gp160G. After incubation for the indicated times, the cells were collected, washed in PBS, and suspended in annexin V buffer (eBioscience). Annexin V (Southern Biotech) and propidium iodide (PI; BD Pharmingen) staining was performed according to the protocol supplied by the manufacturer. Cell staining was analyzed using flow cytometry (with an Aria IIu cell sorter). The cells were considered dead if they stained positive for annexin V.
VSV neutralization assays.
VSV infections were performed in the presence of neutralizing antibody against either CD4 (BioLegend clone SK3) or HIV-gp120 (NIH AIDS Reagent Program, clone 2G12). Cells were seeded in a 12-well plate and then incubated with neutralizing antibody against CD4 (1 μg) for 1 h at 37°C before VSV was added. The VSV inoculum was incubated with neutralizing antibody against gp120 (2 μg) for 1 h at 37°C before being added to the cells. At 24 h postinfection (hpi) cells were analyzed by flow cytometry using annexin V-PI staining. Additional wells were photographed once significant syncytia occurred in the control wells (at 24 hpi for MT-2 and MT-4 and at 96 hpi for TLO-m1).
Ex vivo testing on primary human lymphocytes.
Ex vivo-purified primary CD4 and CD8 T cells were obtained from the blood of a healthy donor using Rosettesep CD4 and CD8 T cell enrichment cocktails, respectively (Stemcell Technologies). Primary T lymphocytes were maintained in RPMI 1640 supplemented with 10% FBS and 30 U of rIL-2/ml. T cells were expanded using Dynabeads human T-activator CD3/CD28 (Life Technologies) immediately after isolation. The purity and activation status of CD4 and CD8 T cells were determined by using flow cytometry and antibodies against CD4 (BD Biosciences), CD8 (BioLegend, clone RPA-T8), CD3 (BD Pharmingen), and CD25 (BioLegend, clone BC96). The resting cell population was removed from T-cell receptor (TCR) stimulation 24 h prior to the experiment, and the activation status was determined at the time of plating for the viability test. Viability was determined using the fixable viability dye eFlour 660 (eBioscience) at 24 hpi with VSV-XN2 or VSV-gp160G at the indicated MOI. Activated primary CD4 T cells were also infected with wild-type VSV Indiana serotype at an MOI of 0.01 at 24 hpi and measured for viability compared to VSV-XN2 and VSV-gp160G. Prior to analysis, cells were fixed in 2% paraformaldehyde. Cells were analyzed using LSR-Fortessa-HTS.
Mouse studies.
Male and female NOD/Shi-scid, IL-2Rγ-c-null (NSG) mice were purchased from Jackson Laboratory and used to establish a breeding colony in the University of Miami (UM) animal facilities. All mice were housed under pathogen-free conditions. Female mice 6 to 8 weeks old were used for all studies.
Toxicity.
Female NSG mice (n = 7) were inoculated intranasally under isoflurane anesthesia with rVSV in 20 μl of PBS (10 μl/nostril). The mice were monitored for survival. Mice were euthanized if they displayed gross morbidity, signs of neurotoxicity, or hind limb paralysis.
Tumor studies.
Female NSG mice (n = 7) were injected with 4 × 105 TLO-m1-luc cells through intraperitoneal (i.p.) injection. After tumor inoculation, mice were injected with 2 × 106 PFU of VSV-gp160G on day 3 and 1 × 107 PFU on day 18. Mice were monitored for survival.
In vivo imaging of mice.
Tumor-bearing NSG mice were transferred to the UM in vivo imaging system (IVIS) facility for imaging before and then weekly after VSV-gp160G inoculation (Caliper/Xenogen IVIS Spectrum). During imaging, mice were injected with luciferin (Caliper Life Science; 150 mg/kg diluted in PBS), anesthetized using isoflurane, and imaged 15 min after luciferin injection with the time postinjection matching between groups. The measurement region of interest (ROI) was expressed as flux (p/s), and the dimensions of ROI were kept constant for each weekly measurement.
Histology.
VSV-gp160G-treated TLO-m1-bearing NSG mice were sacrificed through cervical dislocation under anesthesia using an i.p. injection of ketamine-xylazine. Organs were explanted and fixed in 4% paraformaldehyde overnight. Samples were embedded in paraffin, sectioned, and stained with hematoxylin-eosin.
VSV detection in organ homogenates.
VSV-treated NSG mice were sacrificed through cervical dislocation under anesthesia using an i.p. injection of ketamine-xylazine. Organs were explanted and stored at −80°C for later use. Organs were weighed and then resuspended in 1 ml of cold PBS. Organs were homogenized and passed through a 0.2-μm-pore-size mesh filter. Organ homogenates were then used in a standard plaque assay using HeLa CD4+ cells to determine virus titer.
Statistics.
The data are presented as means ± the standard deviations. The statistical significance was estimated with a Student t test or a log-ranked test. P values of ≤0.05 were considered statistically significant.
RESULTS
Oncolytic rVSV expressing gp160G in lieu of VSV-G.
Recombinant VSV with endogenous VSV-G has a wide tropism and is able to infect most mammalian cell types (49). In murine models, VSV can exhibit toxicity when administered at high doses due to the onset of neuropathy. To eliminate this potential problem and to create an oncolytic vector that targets only ATL cells, which are CD4+, we generated a VSV vector with the G protein substituted with a hybrid fusion protein containing extracellular and transmembrane domains from HIV1 gp160 and the cytoplasmic region of VSV-G. HIV-1 utilizes its glycoprotein, gp160, to gain entry into T cells through entry association with CD4 (Fig. 1A and B) (48). Due to the lack of human CD4 in the BHK-21-WI cells, VSV-gp160G was first recovered with the inclusion of a support VSV-G plasmid to VSV-G pseudotype the virions and enable infection of BHK-21-WI cells. Then, after successful recovery, VSV-gp160G was grown in HeLa CD4+ cells to amplify the progeny virions sans VSV-G.
FIG 1.

VSV-gp160G selectively infects and replicates within CD4+ cells. (A) Schematic representation of VSV-XN2 construct and VSV-gp160G construct with gp160G fusion protein replacing VSV-G. (B) Graphical representation of VSV-XN2 and VSV-gp160G virions depicting the replacement of VSV-G with an HIV gp160–VSV-G fusion. The relevant glycoproteins are VSV-G, HIV-gp160, and the HIV-gp160–VSV-G fusion protein gp160G (left, right, bottom). (C) Bright-field microscopy of HeLa and HeLa CD4+ cells infected with either VSV-XN2 or VSV-gp160G at an MOI of 0.01 at 24 hpi. (D) Immunoblot analysis results for viral glycoprotein expression of either construct in infected HeLa or HeLa CD4+ cells infected at an MOI of 0.01 at 24 hpi. (E) Growth kinetic assay results for HeLa or HeLa CD4+ cells infected with either VSV-XN2 or VSV-gp160G at an MOI of 0.001. Supernatant was analyzed by a standard plaque assay using HeLa CD4+ cells (Student t test, two tailed, equal variance; **, P < 0.001).
To determine that VSV-gp160G was successfully retargeted to CD4+ cells, HeLa and HeLa CD4+ cells were infected with either VSV-XN2 (that contain VSV-G) or VSV-gp160G at an MOI of 0.01. Each rVSV was found to have a distinct CPE that was visible under bright-field microscopy within 24 hpi. As expected, VSV-XN2 induced cell rounding (48, 50) in both HeLa and HeLa CD4+ cells. However, HeLa cells were resistant to VSV-gp160G CPE, whereas HeLa CD4+ cells were highly susceptible to infection, as indicated by the significant syncytium formation, consistent with gp120 and CD4 interaction (Fig. 1C) (51).
Infected cells were subsequently harvested for immunoblot analysis using antibodies against HIV-gp120, gp41, VSV-G, and β-actin. This analysis indicated that HIV gp120 and gp41 were only detected in HeLa CD4+ cells infected with VSV-gp160G and that HeLa cells are resistant to VSV-gp160G infection (Fig. 1D). The double band in the gp41 immunoblot indicates that gp160G is cleaved into two separate polypeptides after processing, as expected (52). Growth kinetic assays were performed to quantify the replicative abilities of VSV-XN2 compared to VSV-gp160G in both HeLa and HeLa CD4 cells. VSV-XN2 displayed robust replication kinetics in a multicycle infection (MOI = 0.001) in both HeLa and HeLa CD4+ cells, whereas VSV-gp160G was only able to replicate in HeLa CD4+ cells, with a significant reduction in its replicative abilities (Fig. 1E).
VSV-gp160G's ability to induce apoptosis in HeLa CD4+ cells was confirmed using flow cytometry with annexin V-PI staining. HeLa or HeLa CD4+ cells were infected with either VSV-XN2 or VSV-gp160G at an MOI of 0.1, and representative gating results at 24 hpi are shown (Fig. 2A). Cells and supernatant were collected 6, 12, 24, and 48 hpi to establish the kinetics of VSV-mediated apoptosis for each cell line at an MOI of 0.1. VSV-XN2 exerted oncolytic activity against both HeLa and HeLa CD4+ cells and induced robust cell death within 24 to 48 h of infection. In contrast, VSV-gp160G induced significant apoptosis only in HeLa CD4+ cells with the HeLa cells being completely resistant. Despite its attenuated growth kinetics, VSV-gp160G was able to induce comparable levels of apoptosis in HeLa CD4+ cells (Fig. 2B). To confirm that VSV-gp160G is dependent upon cellular CD4 and viral gp120 interaction, infections were performed in the presence of neutralizing antibodies against either hCD4 or HIV-gp120. VSV-XN2's ability to induce apoptosis was not affected in the presence of either antibody, whereas the ability of VSV-gp160G to induce syncytia or apoptosis was severely attenuated in the presence of the neutralizing antibodies (Fig. 2C and D). Thus, VSV-gp160G specifically infects and kills transformed CD4+ cells in vitro via gp160G.
FIG 2.
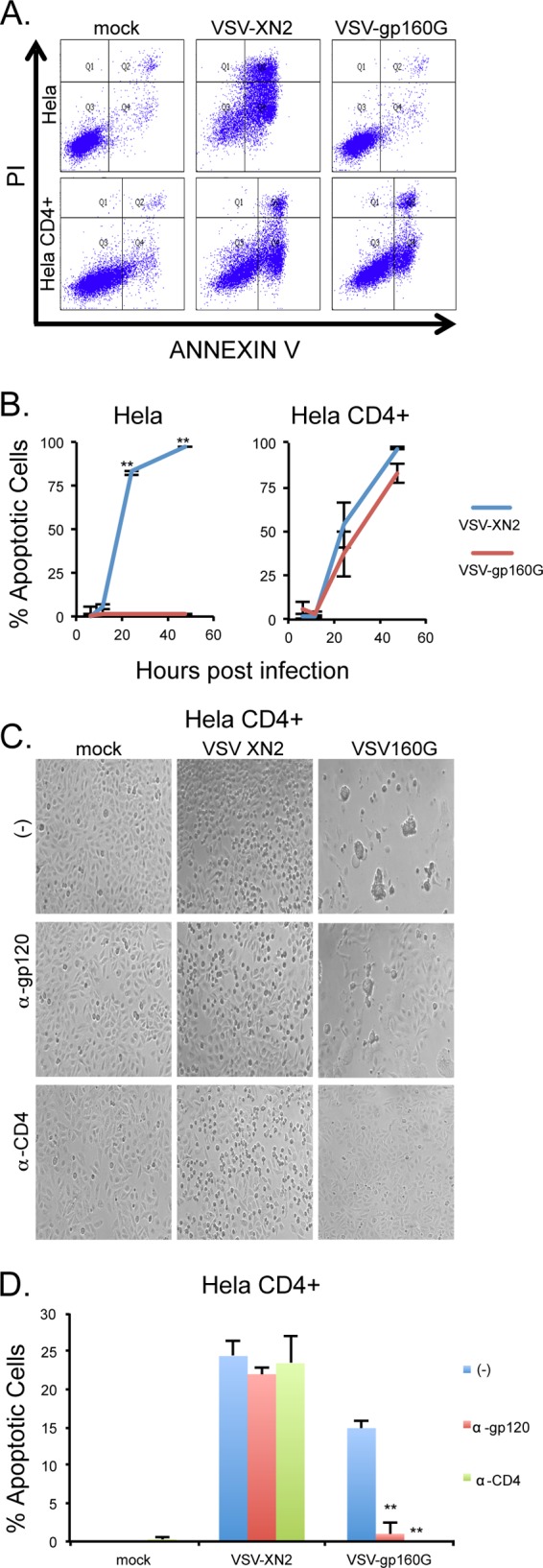
VSV-gp160G remains potently oncolytic and is dependent upon CD4 and gp120 interaction. (A and B) HeLa and HeLa CD4+ cells were infected with either VSV-XN2 or VSV-gp160G at an MOI of 0.1. Cells and supernatants were collected at 6, 12, 24, and 48 hpi. Cell death at each time point was determined using annexin V-propidium iodide (PI) staining. Representative gating results are shown. (C) Bright-field microscopy of infected HeLa CD4+ cells pretreated with neutralizing antibody against CD4 (1 μg/well, HeLa CD4+) or gp120 (2 μg/well, VSV inoculum) 1 h prior to infection at 24 hpi. (D) Cell death of VSV-infected HeLa CD4+ cells pretreated with neutralizing antibody was determined using annexin V-PI staining at an MOI of 0.01 at 24 hpi (Student t test, two tailed, equal variance; **, P < 0.001 [panel D, untreated control versus antibody treated]).
VSV-gp160G is potently oncolytic against ATL cell lines.
After confirming VSV-gp160G specificity for HeLa CD4+ cells, we determined the virus's ability to selectively infect and replicate within ATL lines. The CD4+ ATL lines MT-2, MT-4, and TLO-m1 and the CD4− Burkitt's B cell lymphoma cell line BJAB were all infected with either VSV-XN2 or VSV-gp160G at an MOI of 0.01 and analyzed for their respective CPEs at 24 hpi. Again, as expected, VSV-XN2 induced cell rounding against all cells lines, whereas VSV-gp160G induced syncytium formation only against the CD4+ ATL lines (Fig. 3A). MT-2, MT-4, TLO-m1, and BJAB cells were infected with either VSV-XN2 or VSV-gp160G at the indicated MOIs and then harvested at 24 hpi for immunoblotting to check for viral glycoprotein expression. HIV-1 glycoproteins gp120 and gp41 were only detected in ATL cell lines infected with VSV-gp160G, and BJAB cells did not express HIV-1 glycoproteins. Using α-HIVgp41 antibody, full-length gp160G and the gp41G fragments were detected in all CD4+ cells infected with VSV-gp160G (Fig. 3B). However, the gp41G subunit was not readily detected in MT-4 cells for reasons that remain unclear but may involve aberrant glycosylation events that could occur in these cells and that may affect antibody recognition. The detection of the heavy and light bands using α-VSV-G antibody in VSV-gp160G-infected cells correspond to recognition of the cytoplasmic tail of the VSV-G protein that was fused to gp160.
FIG 3.

VSV-gp160G is specific for transformed CD4+ ATL cells. (A) Bright–field microscopy of VSV-XN2- and VSV-gp160G-infected ATL (MT-2, MT-4, and TLO-m1) and BJAB cells at an MOI of 0.01 at 24 hpi. (B) Immunoblot analysis for viral glycoprotein expression in ATL and BJAB cells infected with VSV-XN2 or VSV-gp160G at MOIs of 0.01 or 0.1 at 24 hpi. (C) Growth kinetic assay results for ATL or BJAB cells infected with either VSV-XN2 or VSV-gp160G at an MOI of 0.001. Supernatant was analyzed by a standard plaque assay using HeLa CD4+ cells. (Student t test, two tailed, equal variance; *, P < 0.01; **, P < 0.001).
Next, growth kinetic assays were performed in a multicycle infection (MOI of 0.001) to analyze the replicative abilities of VSV-gp160G and VSV-XN2 in ATL cells. VSV-XN2 was able to replicate in all cell lines infected, whereas VSV-gp160G was only able to effectively replicate within the CD4+ ATL cell lines. Again, the growth kinetics of VSV-gp160G were significantly attenuated compared to VSV-XN2 (Fig. 3C). VSV-gp160G's ability to induce apoptosis was confirmed using flow cytometry and annexin V-PI staining. ATL cells were infected with either VSV-XN2 or VSV-gp160G at an MOI of 0.1, and representative gating results at 24 hpi are shown (Fig. 4A). The kinetics of VSV-mediated apoptosis in ATL cells were determined at 6, 12, 24, and 48 hpi. MT-2 cells were found to be extremely susceptible to VSV-gp160G-mediated apoptosis, even significantly more than they were to VSV-XN2. The extreme syncytia in the MT-2 cells at 48 hpi likely interfere with the flow cytometry analysis, since we expected the reading to be significantly higher considering the phenotype of the cells. MT-4 cells were highly sensitive to both constructs and TLO-m1 cells were slightly more resistant to VSV-gp160G than to VSV-XN2, with the difference being significant at 48 hpi (Fig. 4B). This analysis indicated that VSV-gp160G was able to effectively induce apoptosis in all ATL lines.
FIG 4.

VSV-gp160G is capable of inducing apoptosis in ATL cells and depends upon CD4 and gp120 interaction. (A and B) ATL cells were infected with either VSV-XN2 or VSV-gp160G at an MOI of 0.01. Cells and supernatants were collected at 6, 12, 24, and 48 hpi. Cell death at each time point was determined by using annexin V-PI staining. Representative gating results are shown. (C) Bright-field microscopy of infected ATL cells pretreated with neutralizing antibody against CD4 (1 μg/well, MT-2, MT-4, TLO-m1) or gp120 (2 μg, VSV inoculum) for 1 h prior to infection at an MOI of 0.01. MT-2 and MT-4 cells were assessed at 24 hpi, and TLO-m1 cells were assessed at 96 hpi. (D) Cell death of infected ATL cells pretreated with neutralizing antibody was determined using annexin V-PI staining at 24 hpi. MT-2, MT-4, or TLO-m1 cells were infected with VSV-XN2 at an MOI of 0.5, 0.01, or 0.05, respectively, or infected with VSV-gp160G at an MOI of 0.1, 0.01, or 0.5, respectively. (Student t test, two tailed, equal variance; *, P < 0.01; **, P < 0.001 [panel D, untreated control versus antibody treated]).
To verify that the oncolytic ability of VSV-gp160G was specific to gp120-CD4 interaction, we performed infection assays in the presence of neutralizing antibody against either HIV-gp120 or hCD4 as in Fig. 2. VSV-gp160G's ability to induce syncytia and apoptosis was severely attenuated when using either antibody, whereas VSV-XN2 was not affected (Fig. 4C and D). Our data indicate that VSV-gp160G is selectively oncolytic against CD4+ ATL cells and should not affect CD4− populations.
Analysis of VSV-gp160G in ex vivo primary T lymphocytes.
The retargeting of rVSV to CD4+ cells significantly reduces possible off target problems such as infection of neurons (53). However, while VSV is known not to effectively replicate or induce apoptosis in primary cells, the consequences of infection upon primary CD4+ T cells, using VSV-gp160G, is unclear. Previous work using a recombinant VSV vector expressing an HIV 89.6 Env–VSV-G hybrid indicates that these vectors are nonpathogenic and do not affect primary CD4 T cell blood counts in rhesus monkeys. Furthermore, the VSV vector worked as a vaccine adjuvant and helped stimulate an antiviral response against SHIV. Theoretically, VSV-gp160G could help stimulate antitumor immunity by producing tumor-associated antigens through ATL cell death (54).
In an effort to elucidate the potential pathogenicity of our vector, human primary CD4 and CD8 T lymphocytes were isolated from the peripheral blood of a healthy donor and expanded using TCR stimulation in the presence of IL-2. The purity of the isolated T cells was confirmed using flow cytometry and antibodies to CD4, CD8, and CD3 (Fig. 5A). It is known that activated T cells are more permissive to wild-type VSV (Indiana serotype) infection and cell death than resting primary T cells. T cell activation leads to the activation of ERK, JNK, and AKT pathways, which promote cell cycle transition from G0 to G1 phase. The activation of these pathways is crucial for VSV replication (55). However, recombinant VSV clones are attenuated compared to the wild-type VSV used to infect primary CD4 T cells in previous studies (34, 56), and the effect of VSV-gp160G on activated primary T lymphocytes needs to be determined.
FIG 5.
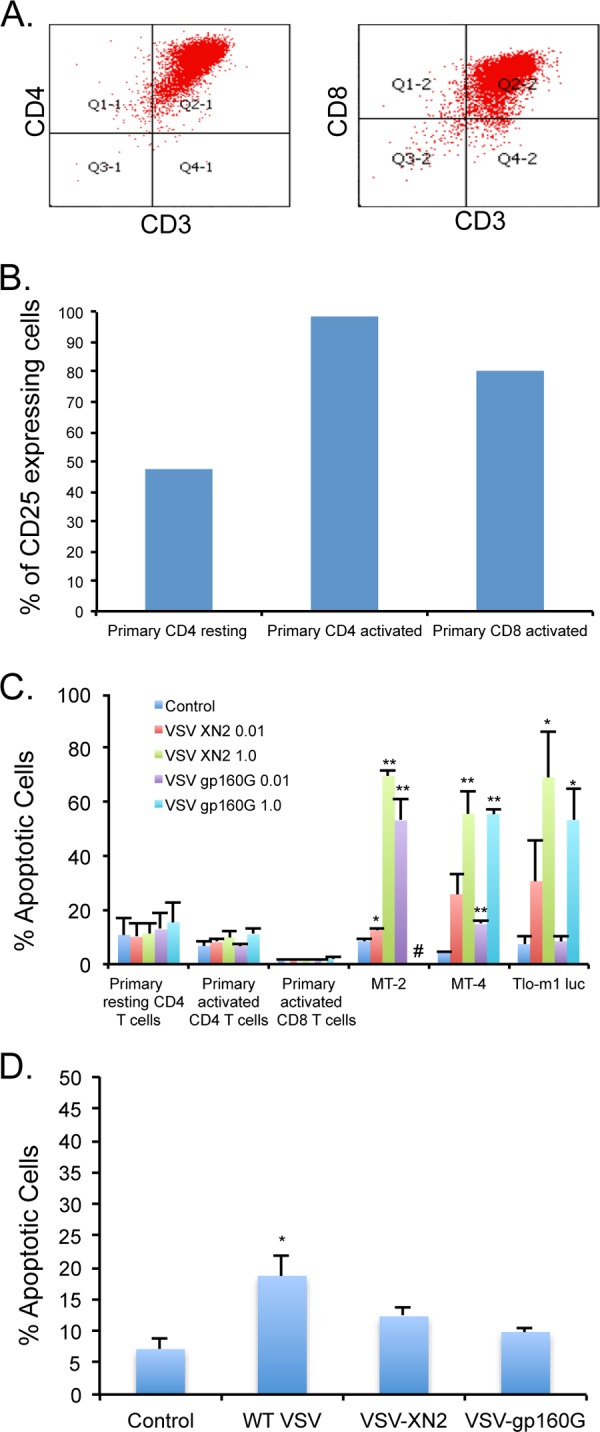
Primary lymphocytes are highly resistant to VSV-gp160G infection and apoptosis. (A) Primary CD4 and CD8 T cells were isolated from whole blood of a healthy donor. The populations were determined to be over 98% pure CD4 T cells and over 94% pure CD8 T cells after isolation. (B) Expression of CD25 activation marker was assessed at the time of VSV infection to determine the T cell activation status. (C) Primary CD4 and CD8 T cells and ATL cell lines were infected with VSV-XN2 or VSV-gp160G at MOIs of 0.01 and 1.0, and cell death was determined using fixable viability dye at 24 hpi. #, Cases where extreme syncytia in the MT-2 cells infected with VSV-gp160G at an MOI of 1.0 interfered with the flow cytometry assay. (D) Primary activated CD4 T cells were infected with wild-type VSV (Indiana serotype), recombinant VSV-XN2, or VSV-gp160G at an MOI of 0.01, and cell death was determined using fixable viability dye at 24 hpi. (Student t test, two tailed, equal variance: *, P < 0.01; **, P < 0.001 [panels C and D, infected versus uninfected control]).
Primary CD4 and CD8 T cells and ATL (MT-2, MT-4, and TLO-m1-luc) cells were infected with VSV-XN2 or VSV-gp160G at MOIs of 0.01 and 1.0. Cell death was measured at 24 hpi using flow cytometry with fixable viability dye. Prior to infection, the activation status of the primary cells was determined through CD25 expression. The resting CD4 T cell population was removed from TCR stimulation prior to plating and allowed to revert to a resting state. The cells were considered resting if <50% of the population expressed CD25 and were considered activated if >90% of the population expressed CD25 (Fig. 5B). As expected, none of the primary cell populations were significantly affected by either VSV-XN2 or VSV-gp160G exposure at either MOI. The ATL lines were all significantly affected by either VSV construct at an MOI of 1.0 (Fig. 5C). We also compared the cytotoxicity of wild-type VSV Indiana serotype to recombinant VSV-XN2 and VSV-gp160G in primary activated CD4 T cells. We found that wild-type VSV induces significant levels of apoptosis at an MOI of 0.01 by 24 hpi (Fig. 5D). These results indicate that primary CD4 T cells are not adversely affected by the administration of VSV-gp160G and that ATL cells are far more susceptible to infection and apoptosis.
VSV-gp160G efficacy against ATL in NSG mice.
To evaluate the safety of VSV-gp160G in immunocompromised hosts, we performed preliminary toxicity assays using NSG (NOD/Shi-scid, IL-2Rγ-c-null) mice. NSG mice are severely immunocompromised, lacking mature T cells, B cells, and functional NK cells. They are also deficient in cytokine signaling. All of which makes them both highly susceptible to VSV infection and excellent recipients for engraftment of human cells. NSG animals (n = 7) were inoculated intranasally with either 3 × 105 PFU of VSV-gp160G or with 1 × 103 or 3 × 105 PFU of VSV-XN2. We observed that NSG mice inoculated with VSV-XN2 at the higher dose succumbed rapidly to neurotoxicity, while the lower dose resulted in ca. 60% survival. In contrast, mice inoculated with VSV-gp160G did not experience any symptoms, and there were no mortalities (Fig. 6A).
FIG 6.

VSV-gp160G is nonpathogenic within immunodeficient NSG mice and mediates tumor burden relief in ATL bearing hosts. (A) Survival curve of NSG mice (n = 7) inoculated with various doses of VSV-gp160G, VSV-XN2, or heat-inactivated VSV-XN2. VSV vectors were prepared in PBS and delivered intranasally to anesthetized NSG mice. (B) Survival curve of NSG (n = 7) mice were inoculated with 4 × 105 TLO-m1-luc i.p. on day 0 and treated with two injections of VSV-gp160G with 2 × 106 PFU on day 3 and 1 × 107 PFU on day18 (*, P = 0.039, log-rank test). (C) Control or VSV-gp160G-treated mice from panel B were anesthetized and injected with a luciferin substrate i.p., and the uciferase activity was detected on days 3, 7, 15, and 22 using IVIS. The average flux (p/s) emitted is an indicator of tumor burden and was quantified using Living Image software (*, P = 0.014; Student t test two tailed, equal variance). (D) Representative images acquired on day 30 of the experiment when statistical significance was achieved. (E) Numbers of NSG mice inoculated with TLO-m1-luc that developed luciferase activity in areas other than the primary injection site, indicative of significant metastasis. (F) The luciferase activity detected in the metastatic lesions for each group was quantified using Living Image software with control mice (n = 7) and VSV-gp160G-treated mice (n = 3).
After determining that VSV-gp160G did not result in neurotoxicity in immunocompromised hosts, we performed further in vivo analysis to evaluate whether VSV-gp160G is able to relieve ATL tumor burden. To accomplish this, we used TLO-m1-luc, a patient-derived ATL line that expresses the luciferase gene. The advantage of TLO-m1-luc is that it allows in vivo monitoring of tumor growth using an IVIS. Measurement of luciferase activity can be used to assess relative tumor size and its dissemination throughout the host. Through this technology, we were able to quantitatively track tumor progression throughout the disease course and detect when metastatic lesions developed.
In our model, female NSG mice (n = 7) were injected with 4 × 105 of TLO-m1-luc i.p. on day 0. TLO-m1-luc inoculated mice received two different VSV-gp160G i.p. injections of 2 × 106 PFU (day 3) and 1 × 107 PFU (day 18), while the control group received PBS injections. Throughout the experiment, mice were imaged weekly using IVIS to track tumor progression and formation of metastasis. We observed that mice treated with VSV-gp160G survived for longer than the PBS control-treated mice (P = 0.039) (Fig. 6B). The luciferase activity measured at the injection site (flux [p/s]) increased at a much lower rate in the VSV-gp160G-treated mice and was significantly different at 30 days postinoculation (P = 0.014) (Fig. 6C and D). Mice treated with VSV-gp160G also displayed a reduced tendency to develop metastatic lesions, with fewer than 50% of the VSV-gp160G-treated mice showing detectable metastasis after 22 days. In contrast, 100% of PBS-treated mice had detectable metastasis by day 15 (Fig. 6E and F). Together, these results show that VSV-gp160G is able to mediate tumor burden relief in an in vivo model.
To determine the biodistribution of both VSV-XN2 and VSV-gp160G in a naive NSG mouse model, 106 PFU of either virus were injected into the peritoneal cavity of individual mice on day 0, and then the mice (n = 4) were euthanized on days 1, 7, 14, and 21 postinfection. The presence of VSV in brain, lung, liver, and kidney was determined by plaque assay. Residual VSV-gp160G was only detectable in the kidney 1 day after inoculation. In contrast, VSV-XN2 grew to a considerable titer in the brain and kidneys of the mouse sacrificed on day 14, and the mouse exhibited symptoms consistent with VSV-mediated neurotoxicity (Fig. 7A).
FIG 7.
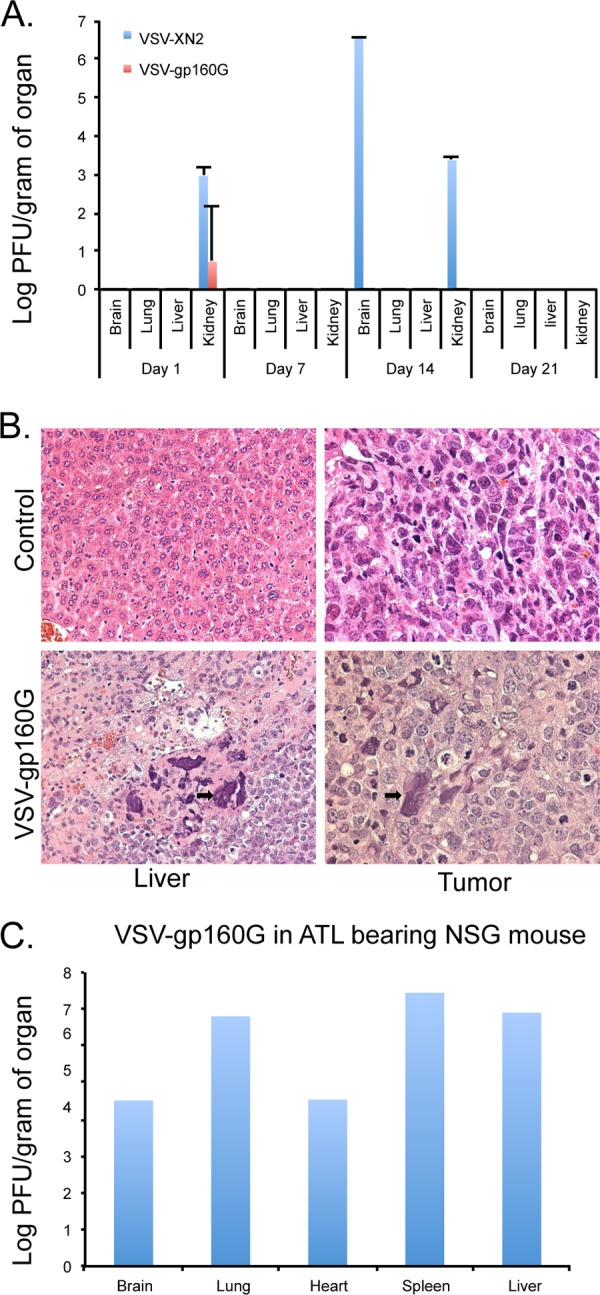
Biodistribution of VSV-gp160G in NSG mice. (A) Naive NSG mice (n = 4) were injected with 106 PFU of VSV-XN2 or VSV-gp160G i.p. and, at the indicated time points, a mouse was sacrificed, and tissue homogenates were analyzed for the presence of VSV virions using standard plaque assay with HeLa CD4+ cells. (B) Histology hematoxylin-eosin-stained samples taken from a TLO-m1 bearing NSG mouse either mock infected or treated with 106 PFU of VSV-gp160G. Syncytia were detected at VSV-gp160G ATL sites (black arrows) 76 days after TLO-m1-luc inoculation. (C) VSV-gp160G in organ homogenates of a TLO-m1-luc-bearing mouse (from panel B) treated with VSV-gp160G.
To examine the nature of VSV-gp160G-mediated survival benefit a TLO-m1 luc bearing mouse treated with VSV-gp160G was sacrificed 76 days postinoculation, and its organs were taken for histological analysis. Syncytium formation inside the tumor lesions in both metastatic sites and the primary tumor were detected, indicating that the virus is successfully replicating and causing tumor destruction (Fig. 7B). In addition, organ sections from the mouse were homogenized, and the presence of VSV-gp160G was detected by standard plaque assay (Fig. 7C). Our data thus indicate that VSV-gp160G does vacate the host if ATL is not present but is able to continually target ATL cells in vivo and mediate tumor burden relief.
DISCUSSION
Oncolytic viruses offer an intelligent targeted therapy to be used in the fight against cancer. The present study demonstrates that VSV can be retargeted to ATL cells while remaining replication competent and potently oncolytic. This was achieved by substituting the VSV-G for an HIV-1 gp160–VSV-G hybrid fusion protein to restrict the virus's tropism to CD4+ cells. HIV-gp160 is translated as a precursor and cleaved at the C5 domain by the convertases, kexin and furin, to yield external viral gp120 and transmembrane gp41. Cleavage of gp160 is highly conserved among HIV strains at the Arg-508-Gln-Lys-Arg-511 site. This tryptic cleavage site in gp160 has been shown to be essential for the fusion process and infectivity since site-directed mutagenesis at this site abolishes gp160's ability to induce fusion (52, 57). Accordingly, our data indicated that the gp160 portion expressed by VSV-gp160G was efficiently cleaved in the cell lines examined, as indicated by the two bands in the gp120 and gp41 immunoblots.
We observed that the VSV-gp160G construct induced syncytia in CD4+ transformed cells. Syncytium formation is a method evolved by several viruses to mediate cell-to-cell spread without having to bud from the host cell. By fusing membranes together, creating giant multinucleated cells, viral particles are able to spread without neutralizing antibody significantly affecting viral spread within the tumor mass (58). In our VSV neutralization assay we observed that either gp120 or CD4 antibody was extremely effective at neutralizing VSV-gp160G in the HeLa CD4 cells. However, in the ATL cell lines, we observed that although gp120 and CD4 antibody significantly attenuated the virus, it was not as effective as in the HeLa CD4 cells. The most likely explanation for this discrepancy is how the gp160G fusion glycoprotein mediates viral spread. The formation of syncytia does not require virions to bud off the cell and reattach but instead a multimolecular structure known as a virological synapse forms to transfer virions between cells (59). This drastically reduces the distance viral particles must travel to infect neighboring cells and may account for the reduction in the ability of the neutralizing antibody (NAb) to block viral infection within the ATL cells in suspension. The HeLa CD4 cells are plated onto a flat surface, so there is less range of movement, and thus the virus will have to travel further to reach other cells and is more likely to encounter NAbs. Meanwhile, ATL cells tend to form aggregate masses, and this may prevent NAbs from associating with their targets, allowing the virions to spread within these cellular masses without encountering NAbs.
The formation of syncytia in immunocompetent hosts may greatly increase the efficacy of a fusogenic rVSV versus a nonfusogenic rVSV since the efficacy of NAbs will be reduced in neutralizing viral spread. Deep within a tumor mass, viral spread is limited for a variety of factors, including intratumoral pressure gradients, limited extravasation from blood vessels, and interference from the extracellular matrix (ECM) (60). The formation of syncytia may be able to circumvent some of these barriers, and it is possible that that the fusogenic properties of VSV-gp160G could aid in viral spread within the tumor mass since our histology analysis reveals syncytium formation deep within the tumor and since others have reported an increase in viral spread when incorporating fusogenic membrane glycoproteins (FMGs) into their viral constructs (61–63).
In addition, cancer cell death through syncytium formation can lead to necrosis formation in vivo, which is a highly efficient immune activator (64). During syncytium formation large numbers of tumor-derived, exosome-like vesicles are produced. These vesicles can carry tumor-associated antigens, are efficiently picked up by immature dendritic cells, and can be presented to T cells to generate systemic antitumor immunity (65). The bystander killing effect of FMGs on oncolytic viruses has been reported as being 10 times higher than the effect of expressed suicide genes such as cytosine deaminase or thymidine kinase (63, 66). This may enhance the antitumor effects of VSV within a tumor mass since the cell death induced by viral gene expression can act synergistically with the syncytium formation to mediate a significant oncolytic effect. This effect is supported through the comparable levels of apoptosis between the constructs despite VSV-gp160G having significantly reduced growth kinetics (62).
Uncontrolled syncytium formation is a potential safety concern (67). However, in VSV, expression of FMGs depends directly on viral RNA replication. This effectively limits the fusogenic potential in normal cells since VSV-gp160G was still attenuated in normal cells. In addition, no syncytia were observed in normal tissue from histology and were limited to the ATL metastatic foci in organ tissue. Previous studies also report no significant damage to normal tissue using a fusogenic oncolytic vector in both a VSV and herpes simplex virus (HSV) model (61, 62).
VSV-induced mortality of NSG mice involves neurotoxicity. During infection, wild-type VSV can travel to the central nervous system (CNS) via the olfactory bulb. VSV then spreads trans-synaptically using both anterograde and retrograde transport and through the cerebrospinal fluid (68). The lethal dose of VSV is significantly lower via intranasal inoculation compared to i.p. injection since the former provides direct access to the CNS. VSV was rarely detected and only in trace amounts in the kidney organ homogenates directly after i.p. injection, most likely as a result of residual VSV from the injection and not a product of robust viral replication. It was not until the mouse exhibited signs of neurotoxicity that high titers of VSV-XN2 could be detected in the brain, although it still was not present in most other organs, except for the kidney. The presence of VSV-XN2 in the kidney was somewhat surprising considering that it was primary tissue and VSV-XN2 should have minimal expression within primary cells. A possible explanation is that the proximity of the organ to the injection site or an accidental puncture to the organ resulted in the kidney being exposed to a higher titer of VSV-XN2 and the virus was therefore able to overwhelm the natural innate antiviral defenses of the kidney epithelial cells. Many transformed kidney epithelial cells are excellent producers of infectious VSV, and if a viable infection is established then, theoretically, VSV would be able to grow within the kidney. In addition, since these animals are severely immunodeficient, there is no adaptive immunity to effectively neutralize the virus or CTL response to kill infected cells. Regardless, VSV-gp160G was barely detectable immediately after inoculation and then quickly dropped to undetectable levels in all organs. The initial reading of VSV-gp160G in the kidney was likely due to residual VSV-gp160G being present and not indicative of replication. This indicates that VSV-gp160G was unable to replicate within a non-tumor-bearing host and was rapidly cleared from the host.
Ex vivo analysis indicated that normal human CD4+ or CD8+ T cells were not destroyed by VSV-gp160G, suggesting that VSV-gp160G should be a safe therapeutic option when used in human patients. VSV-XN2 is able to infect many types of cells, including T cells, although there has been no indication that T-cell repertoires are targeted or significantly destroyed by VSV in experimental conditions. In a similar study to ours, the more virulent VSV-HR (Indiana serotype) was able to induce marginal apoptosis in primary activated T cells but not in resting T cells (34, 55). Here, we demonstrate that wild-type VSV Indiana serotype was able to induce significant levels of apoptosis in activated CD4 T cells, whereas recombinant VSV-XN2 and our VSV-gp160G did not affect the viability of activated primary CD4 T cells. The recombinant VSV-XN2 is more attenuated compared to the wild-type VSV strain. The reasons for this are not entirely clear, but it can probably be attributed to spontaneous mutations generated during the reverse genetics process or other sequence differences (49, 56).
The ability of VSV to effectively replicate and destroy transformed cells and not normal cells is likely due to defective antiviral innate immune mechanisms being prevalent in the latter population (41, 69). VSV has repeatedly been shown to be highly sensitive to the actions of the IFN pathway, and the IFN deficiencies in ATL cells render them highly susceptible to VSV-mediated oncolysis (34). Type I IFN defects are a common occurrence in ATL cells, and it has been demonstrated that other viruses also take advantage of this differential (70). Thus, VSV-gp160G may provide a safe, effective means for the eradication of transformed CD4+ ATL cells.
We observed that VSV-gp160G could be detected for extended periods (76 days) within an ATL bearing host but was unable to fully clear the tumor from the system. We believe that the lack of complete tumor clearance while VSV-gp160G is present within the host indicates that the ATL tumor mass is so dense within this model that it prevented the efficient spread of the virus throughout the mass and VSV-gp160G was unable to access the CD4+ ATL cells. Considering that ATL is a blood cancer, this observation is likely to be an artifact of our chosen model, and in human patients this scenario is not likely to be encountered. In addition, tumor masses retrieved from ATL-bearing mice remained highly sensitive to VSV-gp160G and when extracted from the mouse and placed in suspension were readily infected with VSV-gp160G, which indicates that the tumor did not become resistant to the virus but rather was inaccessible to the virus.
The issue of safety that comes from using an HIV-1 fusogenic glycoprotein to target CD4+ T cells is a valid concern, but we believe that the properties of VSV will override the cytopathic potential of HIV-1 gp160. VSV-gp160G possesses far slower growth kinetics than its parental VSV-XN2, and our ex vivo studies indicate that VSV-gp160G is still highly attenuated against primary cell types, including CD4+ T cells. In addition, the use of a similar VSV construct was previously reported in studies where it was used as an HIV vaccine and was able to generate a robust humoral immune response while not adversely affecting CD4 T cell counts (43, 71). This is a strong indicator that our construct will be cleared from the host, since neutralizing antibody is highly effective against VSV and the spread outside of tumor masses should be highly inhibited (72). It is unlikely that VSV-gp160G will behave similarly to HIV in any way besides tropism.
VSV-gp160G is a prototype virus that specifically targets CD4+ ATL cells. By removing the glycoprotein and restricting the virus to a CD4+ tropism, we greatly reduced the possibility of neurotoxic side effects or off-target infection. We observed that the virus's growth kinetics have been somewhat reduced, but it is still potently oncolytic against ATL cells. Logistically, this could be problematic for clinical use since the yield of VSV-gp160G is significantly lower than that of VSV-XN2. There are several ways to optimize virus production. First, we could investigate additional cell types for producing VSV-gp160G. It is known that BHK cells produce a far greater amount of infectious VSV per cell than HeLa cells, and so a logical first step would be to generate additional cell lines that could more efficiently produce VSV (73). An additional method would be to evolve the virus through serial passaging. VSV has a high mutation rate, averaging about 1 × 10−4 to 4 × 10−4 per base site, and typically exists as a mixture of genetic variants called quasispecies. Through natural selection, genetic mutants will compete during serial passaging until a superior arises that will overgrow and displace the other mutants. This technique was performed previously using a VSV-expressing chimeric Sindbis virus glycoprotein, which included a single-chain antibody directed to the human Her2/neu receptor. The evolved VSV had a 10,000-fold improvement in viral yield while maintaining its specificity for Her2/neu-expressing cells (74). A similar protocol for our viruses is worth investigating in the future if clinically relevant concentrations of VSV-gp160G are needed.
Our results demonstrate that VSV-gp160G delivers a very real therapeutic benefit to immunocompromised ATL bearing NSG mice and may be a promising new therapeutic option. In addition, VSV-gp160G is still contains an open transgene site for the insertion of an additional gene to generate more potent strains of VSV-gp160G. The incorporation of a suicide gene such cytosine deaminase is already being considered and developed. We believe that CD has potential to greatly increase VSV oncolytic potential without compromising safety or the ability of the virus to replicate since we have already incorporated CD into an earlier construct with wild-type VSV-G (75). Alternatively, the expression of matrix metalloproteinase 9 was used in an oncolytic HSV-1 vector in a glioblastoma model, and there was a significant increase in viral vector distribution (76). The use of a similar MMP could enhance the spread of VSV-gp160G by degrading the ECM and thus allowing improved distribution of the virus throughout the tumor mass. Also, there are a variety of treatment options that can be used alongside VSV-gp160G to improve its therapeutic effect. A promising option is radiation treatment, as numerous studies have shown oncolytic therapy is often synergistic with radiotherapy (77–82).
There is increasing appreciation for the use of oncolytic vectors as an immunotherapy. It was originally thought that the immune system was antagonistic to the outcome of virotherapy due to premature clearance of the oncolytic virus but recently studies have demonstrated the immunostimulatory properties of viral oncolysis to stimulate antitumor immunity, which will aid in tumor destruction and potentially prevent relapse (reviewed in reference 83). Tumor cell death from VSV-gp160G should be highly immunogenic due to the necrotic form of cell death induced by the fusogenic properties of gp160G, as discussed above. However, VSV-gp160G is highly specific for human CD4, and generating a syngeneic immunocompetent ATL tumor was beyond the scope of this study, as we were restricted to immunodeficient xenograft models. We believe that an active immune system will be beneficial to the overall survival of an ATL-bearing host. The fusogenic properties of VSV-gp160G enables the virus to spread without neutralizing antibodies significantly affecting intratumoral spread while inducing a known highly immunogenic form of cell death through syncytium formation. In addition, during tumorigenesis ATL cells generate massive amounts of mutations that form a wide variety of tumor-associated antigens and should provide plenty of targets for the CTL response if the immunotolerance can be broken (84, 85). There is a humanized mouse ATL model that will be considered in future studies that provides an attractive target for immunocompetent testing of our construct in which the mice develop adaptive CTL responses (86).
The use of oncolytic viruses allows a new treatment option in the fight against cancer that is highly customizable and is able to target the tumor in a way that can act complementary to existing treatments, greatly improving the efficacy of either treatment alone. We believe that VSV-gp160G is an important platform from which we can develop new treatment regiments that can help alleviate the burden of ATL and extend the life span of patients.
ACKNOWLEDGMENTS
These studies were supported by National Institutes of Health (NIH) grant 5P01CA128115. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We thank Toni Yeasky for technical assistance with animal work, Lan Toomey for creating TLO-m1-luc, and D. Watkins for supplying several fluorescent antibodies and reagents.
The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program, Divisions of AIDS, NIAID, NIH: HIV-1 gp120 MAb (2G12), catalog no. 1476; HIV-1 gp41 (2F5), catalog no. 1475; and HeLa CD4+ (1022), catalog no. 1109, from Hermann Katinger (antibodies) and Bruce Chesebro (HeLa CD4+ cells).
REFERENCES
- 1.Tsukasaki K, Tobinai K. 2013. Biology and treatment of HTLV-1 associated T-cell lymphomas. Best Pract Res Clin Haematol 26:3–14. doi: 10.1016/j.beha.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Abbaszadegan MR, Gholamin M, Tabatabaee A, Farid R, Houshmand M, Abbaszadegan M. 2003. Prevalence of human T-lymphotropic virus type 1 among blood donors from Mashhad, Iran. J Clin Microbiol 41:2593–2595. doi: 10.1128/JCM.41.6.2593-2595.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Proietti FA, Carneiro-Proietti AB, Catalan-Soares BC, Murphy EL. 2005. Global epidemiology of HTLV-I infection and associated diseases. Oncogene 24:6058–6068. doi: 10.1038/sj.onc.1208968. [DOI] [PubMed] [Google Scholar]
- 4.Paun L, Ispas O, Del Mistro A, Chieco-Bianchi L. 1994. HTLV-I in Romania. Eur J Haematol 52:117–118. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan JE, Khabbaz RF. 1993. The epidemiology of human T-lymphotropic virus types I and II. Rev Med Virol 3:137–148. doi: 10.1002/rmv.1980030304. [DOI] [Google Scholar]
- 6.Arisawa K, Soda M, Endo S, Kurokawa K, Katamine S, Shimokawa I, Koba T, Takahashi T, Saito H, Doi H, Shirahama S. 2000. Evaluation of adult T-cell leukemia/lymphoma incidence and its impact on non-Hodgkin lymphoma incidence in southwestern Japan. Int J Cancer 85:319–324. doi: 10.1002/(SICI)1097-0215(20000201)85:3<319::AID-IJC4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 7.Shimoyama M. 1991. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma: a report from the Lymphoma Study Group (1984-87). Br J Haematol 79:428–437. [DOI] [PubMed] [Google Scholar]
- 8.Mahieux R, Gessain A. 2007. Adult T-cell leukemia/lymphoma and HTLV-1. Curr Hematol Malignancy Rep 2:257–264. doi: 10.1007/s11899-007-0035-x. [DOI] [PubMed] [Google Scholar]
- 9.Bazarbachi A, Ghez D, Lepelletier Y, Nasr R, de The H, El-Sabban ME, Hermine O. 2004. New therapeutic approaches for adult T-cell leukaemia. Lancet Oncol 5:664–672. doi: 10.1016/S1470-2045(04)01608-0. [DOI] [PubMed] [Google Scholar]
- 10.White JD, Zaknoen SL, Kasten-Sportes C, Top LE, Navarro-Roman L, Nelson DL, Waldmann TA. 1995. Infectious complications and immunodeficiency in patients with human T-cell lymphotropic virus I-associated adult T-cell leukemia/lymphoma. Cancer 75:1598–1607. doi: 10.1002/1097-0142(19950401)75:7<1598::AID-CNCR2820750708>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 11.Hishizawa M, Imada K, Kitawaki T, Ueda M, Kadowaki N, Uchiyama T. 2004. Depletion and impaired interferon-alpha-producing capacity of blood plasmacytoid dendritic cells in human T-cell leukaemia virus type I-infected individuals. Br J Haematol 125:568–575. doi: 10.1111/j.1365-2141.2004.04956.x. [DOI] [PubMed] [Google Scholar]
- 12.Al-Dahoodi ZM, Takemoto S, Kataoka S. 2003. Dysfunction of dendritic and T cells as the cause of immune suppression in HTLV-I infected individuals. J Clin Exp Hematopathol 43:43–49. doi: 10.3960/jslrt.43.43. [DOI] [Google Scholar]
- 13.Banerjee P, Feuer G, Barker E. 2007. Human T-cell leukemia virus type 1 (HTLV-1) p12I down-modulates ICAM-1 and -2 and reduces adherence of natural killer cells, thereby protecting HTLV-1-infected primary CD4+ T cells from autologous natural killer cell-mediated cytotoxicity despite the reduction of major histocompatibility complex class I molecules on infected cells. J Virol 81:9707–9717. doi: 10.1128/JVI.00887-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng X, Ratner L. 2008. Human T-cell leukemia virus type 1 blunts signaling by interferon alpha. Virology 374:210–216. doi: 10.1016/j.virol.2007.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuoka M, Jeang KT. 2011. Human T-cell leukemia virus type 1 (HTLV-1) and leukemic transformation: viral infectivity, Tax, HBZ and therapy. Oncogene 30:1379–1389. doi: 10.1038/onc.2010.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Currer R, Van Duyne R, Jaworski E, Guendel I, Sampey G, Das R, Narayanan A, Kashanchi F. 2012. HTLV tax: a fascinating multifunctional coregulator of viral and cellular pathways. Front Microbiol 3:406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basbous J, Arpin C, Gaudray G, Piechaczyk M, Devaux C, Mesnard JM. 2003. The HBZ factor of human T-cell leukemia virus type I dimerizes with transcription factors JunB and c-Jun and modulates their transcriptional activity. J Biol Chem 278:43620–43627. doi: 10.1074/jbc.M307275200. [DOI] [PubMed] [Google Scholar]
- 18.Isono O, Ohshima T, Saeki Y, Matsumoto J, Hijikata M, Tanaka K, Shimotohno K. 2008. Human T-cell leukemia virus type 1 HBZ protein bypasses the targeting function of ubiquitination. J Biol Chem 283:34273–34282. doi: 10.1074/jbc.M802527200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bellon M, Lepelletier Y, Hermine O, Nicot C. 2009. Deregulation of microRNA involved in hematopoiesis and the immune response in HTLV-I adult T-cell leukemia. Blood 113:4914–4917. doi: 10.1182/blood-2008-11-189845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pichler K, Schneider G, Grassmann R. 2008. MicroRNA miR-146a and further oncogenesis-related cellular microRNAs are dysregulated in HTLV-1-transformed T lymphocytes. Retrovirology 5:100. doi: 10.1186/1742-4690-5-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, Fabbri M, Alder H, Liu CG, Calin GA, Croce CM. 2007. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol 179:5082–5089. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- 22.Hou J, Wang P, Lin L, Liu X, Ma F, An H, Wang Z, Cao X. 2009. MicroRNA-146a feedback inhibits RIG-I-dependent type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol 183:2150–2158. doi: 10.4049/jimmunol.0900707. [DOI] [PubMed] [Google Scholar]
- 23.Taganov KD, Boldin MP, Chang KJ, Baltimore D. 2006. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A 103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshida M. 2001. Multiple viral strategies of HTLV-1 for dysregulation of cell growth control. Annu Rev Immunol 19:475–496. doi: 10.1146/annurev.immunol.19.1.475. [DOI] [PubMed] [Google Scholar]
- 25.Yoshida M. 1993. HTLV-1 Tax: regulation of gene expression and disease. Trends Microbiol 1:131–135. doi: 10.1016/0966-842X(93)90127-D. [DOI] [PubMed] [Google Scholar]
- 26.Inoue J, Seiki M, Taniguchi T, Tsuru S, Yoshida M. 1986. Induction of interleukin 2 receptor gene expression by p40x encoded by human T-cell leukemia virus type 1. EMBO J 5:2883–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muraoka O, Kaisho T, Tanabe M, Hirano T. 1993. Transcriptional activation of the interleukin-6 gene by HTLV-1 p40tax through an NF-κB-like binding site. Immunol Lett 37:159–165. doi: 10.1016/0165-2478(93)90026-X. [DOI] [PubMed] [Google Scholar]
- 28.Miyatake S, Seiki M, Yoshida M, Arai KI. 1988. T-cell activation signals and human T-cell leukemia-virus type I-encoded P40x protein activate the mouse granulocyte-macrophage colony-stimulating factor gene through a common DNA element. Mol Cell Biol 8:5581–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim SJ, Kehrl JH, Burton J, Tendler CL, Jeang KT, Danielpour D, Thevenin C, Kim KY, Sporn MB, Roberts AB. 1990. Transactivation of the transforming growth factor beta 1 (TGF-β1) gene by human T lymphotropic virus type 1 tax: a potential mechanism for the increased production of TGF-beta 1 in adult T cell leukemia. J Exp Med 172:121–129. doi: 10.1084/jem.172.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujii M, Tsuchiya H, Chuhjo T, Minamino T, Miyamoto KI, Seiki M. 1994. Serum response factor has functional roles both in indirect binding to the Carg Box and in the transcriptional activation function of human T-cell leukemia-virus type-I Tax. J Virol 68:7275–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujii M, Sassone-Corsi P, Verma IM. 1988. c-fos promoter trans-activation by the tax1 protein of human T-cell leukemia virus type I Proc Natl Acad Sci U S A 85:8526–8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grassmann R, Berchtold S, Radant I, Alt M, Fleckenstein B, Sodroski JG, Haseltine WA, Ramstedt U. 1992. Role of human T-cell leukemia virus type 1 X region proteins in immortalization of primary human lymphocytes in culture. J Virol 66:4570–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franchini G. 1995. Molecular mechanisms of human T-cell leukemia/lymphotropic virus type I infection. Blood 86:3619–3639. [PubMed] [Google Scholar]
- 34.Cesaire R, Oliere S, Sharif-Askari E, Loignon M, Lezin A, Olindo S, Panelatti G, Kazanji M, Aloyz R, Panasci L, Bell JC, Hiscott J. 2006. Oncolytic activity of vesicular stomatitis virus in primary adult T-cell leukemia. Oncogene 25:349–358. [DOI] [PubMed] [Google Scholar]
- 35.Cheng H, Ren T, Sun SC. 2012. New insight into the oncogenic mechanism of the retroviral oncoprotein Tax. Protein Cell 3:581–589. doi: 10.1007/s13238-012-2047-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uchiyama T, Yodoi J, Sagawa K, Takatsuki K, Uchino H. 1977. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood 50:481–492. [PubMed] [Google Scholar]
- 37.Tsukasaki K, Tobinai K. 2012. Clinical trials and treatment of ATL. Leuk Res Treatment 2012:101754. doi: 10.1155/2012/101754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Russell SJ, Peng KW, Bell JC. 2012. Oncolytic virotherapy. Nat Biotechnol 30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barber GN. 2004. Vesicular stomatitis virus as an oncolytic vector. Viral Immunol 17:516–527. doi: 10.1089/vim.2004.17.516. [DOI] [PubMed] [Google Scholar]
- 40.Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. 2012. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci U S A 109:4239–4244. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Obuchi M, Fernandez M, Barber GN. 2003. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J Virol 77:8843–8856. doi: 10.1128/JVI.77.16.8843-8856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Owens RJ, Rose JK. 1993. Cytoplasmic domain requirement for incorporation of a foreign envelope protein into vesicular stomatitis-virus. J Virol 67:360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson JE, Schnell MJ, Buonocore L, Rose JK. 1997. Specific targeting to CD4+ cells of recombinant vesicular stomatitis viruses encoding human immunodeficiency virus envelope proteins. J Virol 71:5060–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K, Heike T, Nakahata T. 2002. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 100:3175–3182. doi: 10.1182/blood-2001-12-0207. [DOI] [PubMed] [Google Scholar]
- 45.Lawson ND, Stillman EA, Whitt MA, Rose JK. 1995. Recombinant vesicular stomatitis viruses from DNA. Proc Natl Acad Sci U S A 92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whitt MA. 2010. Generation of VSV pseudotypes using recombinant ΔG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J Virol Methods 169:365–374. doi: 10.1016/j.jviromet.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diallo JS, Vaha-Koskela M, Le Boeuf F, Bell J. 2012. Propagation, purification, and in vivo testing of oncolytic vesicular stomatitis virus strains. Methods Mol Biol 797:127–140. doi: 10.1007/978-1-61779-340-0_10. [DOI] [PubMed] [Google Scholar]
- 48.Foley HD, Otero M, Orenstein JM, Pomerantz RJ, Schnell MJ. 2002. Rhabdovirus-based vectors with human immunodeficiency virus type 1 (HIV-1) envelopes display HIV-1-like tropism and target human dendritic cells. J Virol 76:19–31. doi: 10.1128/JVI.76.1.19-31.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hastie E, Grdzelishvili VZ. 2012. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J Gen Virol 93:2529–2545. doi: 10.1099/vir.0.046672-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kopecky SA, Lyles DS. 2003. The cell-rounding activity of the vesicular stomatitis virus matrix protein is due to the induction of cell death. J Virol 77:5524–5528. doi: 10.1128/JVI.77.9.5524-5528.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sakamoto T, Ushijima H, Okitsu S, Suzuki E, Sakai K, Morikawa S, Muller WEG. 2003. Establishment of an HIV cell-cell fusion assay by using two genetically modified HeLa cell lines and reporter gene. J Virol Methods 114:159–166. doi: 10.1016/j.jviromet.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 52.Moulard M, Decroly E. 2000. Maturation of HIV envelope glycoprotein precursors by cellular endoproteases. Biochim Biophys Acta 1469:121–132. doi: 10.1016/S0304-4157(00)00014-9. [DOI] [PubMed] [Google Scholar]
- 53.Trottier MD Jr, Palian BM, Reiss CS. 2005. VSV replication in neurons is inhibited by type I IFN at multiple stages of infection. Virology 333:215–225. doi: 10.1016/j.virol.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 54.Rose NF, Marx PA, Luckay A, Nixon DF, Moretto WJ, Donahoe SM, Montefiori D, Roberts A, Buonocore L, Rose JK. 2001. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell 106:539–549. doi: 10.1016/S0092-8674(01)00482-2. [DOI] [PubMed] [Google Scholar]
- 55.Oliere S, Arguello M, Mesplede T, Tumilasci V, Nakhaei P, Stojdl D, Sonenberg N, Bell J, Hiscott J. 2008. Vesicular stomatitis virus oncolysis of T lymphocytes requires cell cycle entry and translation initiation. J Virol 82:5735–5749. doi: 10.1128/JVI.02601-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Majid AM, Barber GN. 2006. Recombinant vesicular stomatitis virus (VSV) and other strategies in HCV vaccine designs and immunotherapy, p 423–450. In Tan SL. (ed), Hepatitis C viruses: genomes and molecular biology. Horizon Bioscience, Norfolk, United Kingdom. [PubMed] [Google Scholar]
- 57.Bour S, Geleziunas R, Wainberg MA. 1995. The human immunodeficiency virus type 1 (HIV-1) CD4 receptor and its central role in promotion of HIV-1 infection. Microbiol Rev 59:63–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Young DF, Randall RE, Hoyle JA, Souberbielle BE. 1990. Clearance of a persistent paramyxovirus infection is mediated by cellular immune responses but not by serum-neutralizing antibody. J Virol 64:5403–5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sattentau QJ. 2010. Cell-to-cell spread of retroviruses. Viruses 2:1306–1321. doi: 10.3390/v2061306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heise CC, Williams A, Olesch J, Kirn DH. 1999. Efficacy of a replication-competent adenovirus (ONYX-015) following intratumoral injection: intratumoral spread and distribution effects. Cancer Gene Ther 6:499–504. doi: 10.1038/sj.cgt.7700071. [DOI] [PubMed] [Google Scholar]
- 61.Fu X, Zhang X. 2002. Potent systemic antitumor activity from an oncolytic herpes simplex virus of syncytial phenotype. Cancer Res 62:2306–2312. [PubMed] [Google Scholar]
- 62.Ebert O, Shinozaki K, Kournioti C, Park MS, Garcia-Sastre A, Woo SL. 2004. Syncytium induction enhances the oncolytic potential of vesicular stomatitis virus in virotherapy for cancer. Cancer Res 64:3265–3270. doi: 10.1158/0008-5472.CAN-03-3753. [DOI] [PubMed] [Google Scholar]
- 63.Higuchi H, Bronk SF, Bateman A, Harrington K, Vile RG, Gores GJ. 2000. Viral fusogenic membrane glycoprotein expression causes syncytium formation with bioenergetic cell death: implications for gene therapy. Cancer Res 60:6396–6402. [PubMed] [Google Scholar]
- 64.Melcher A, Todryk S, Hardwick N, Ford M, Jacobson M, Vile RG. 1998. Tumor immunogenicity is determined by the mechanism of cell death via induction of heat shock protein expression. Nat Med 4:581–587. doi: 10.1038/nm0598-581. [DOI] [PubMed] [Google Scholar]
- 65.Bateman AR, Harrington KJ, Kottke T, Ahmed A, Melcher AA, Gough MJ, Linardakis E, Riddle D, Dietz A, Lohse CM, Strome S, Peterson T, Simari R, Vile RG. 2002. Viral fusogenic membrane glycoproteins kill solid tumor cells by nonapoptotic mechanisms that promote cross presentation of tumor antigens by dendritic cells. Cancer Res 62:6566–6578. [PubMed] [Google Scholar]
- 66.Diaz RM, Bateman A, Emiliusen L, Fielding A, Trono D, Russell SJ, Vile RG. 2000. A lentiviral vector expressing a fusogenic glycoprotein for cancer gene therapy. Gene Ther 7:1656–1663. doi: 10.1038/sj.gt.3301277. [DOI] [PubMed] [Google Scholar]
- 67.Fu X, Tao L, Jin A, Vile R, Brenner MK, Zhang X. 2003. Expression of a fusogenic membrane glycoprotein by an oncolytic herpes simplex virus potentiates the viral antitumor effect. Mol Ther 7:748–754. doi: 10.1016/S1525-0016(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 68.Detje CN, Meyer T, Schmidt H, Kreuz D, Rose JK, Bechmann I, Prinz M, Kalinke U. 2009. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J Immunol 182:2297–2304. doi: 10.4049/jimmunol.0800596. [DOI] [PubMed] [Google Scholar]
- 69.Balachandran S, Barber GN. 2000. Vesicular stomatitis virus (VSV) therapy of tumors. IUBMB Life 50:135–138. doi: 10.1080/713803696. [DOI] [PubMed] [Google Scholar]
- 70.MP MC, Fernandez Landes SA, Huey K, Lairmore D, Niewiesk MS. 2014. Success of measles virotherapy in ATL depends on type I interferon secretion and responsiveness. Virus Res 189:206–213. doi: 10.1016/j.virusres.2014.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rose NF, Roberts A, Buonocore L, Rose JK. 2000. Glycoprotein exchange vectors based on vesicular stomatitis virus allow effective boosting and generation of neutralizing antibodies to a primary isolate of human immunodeficiency virus type 1. J Virol 74:10903–10910. doi: 10.1128/JVI.74.23.10903-10910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gobet R, Cerny A, Ruedi E, Hengartner H, Zinkernagel RM. 1988. The role of antibodies in natural and acquired resistance of mice to vesicular stomatitis virus. Exp Cell Biol 56:175–180. [DOI] [PubMed] [Google Scholar]
- 73.Barber GN. 2005. VSV-tumor selective replication and protein translation. Oncogene 24:7710–7719. doi: 10.1038/sj.onc.1209042. [DOI] [PubMed] [Google Scholar]
- 74.Gao Y, Whitaker-Dowling P, Watkins SC, Griffin JA, Bergman I. 2006. Rapid adaptation of a recombinant vesicular stomatitis virus to a targeted cell line. J Virol 80:8603–8612. doi: 10.1128/JVI.00142-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Porosnicu M, Mian A, Barber GN. 2003. The oncolytic effect of recombinant vesicular stomatitis virus is enhanced by expression of the fusion cytosine deaminase/uracil phosphoribosyltransferase suicide gene. Cancer Res 63:8366–8376. [PubMed] [Google Scholar]
- 76.Hong CS, Fellows W, Niranjan A, Alber S, Watkins S, Cohen JB, Glorioso JC, Grandi P. 2010. Ectopic matrix metalloproteinase-9 expression in human brain tumor cells enhances oncolytic HSV vector infection. Gene Ther 17:1200–1205. doi: 10.1038/gt.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dilley J, Reddy S, Ko D, Nguyen N, Rojas G, Working P, Yu DC. 2005. Oncolytic adenovirus CG7870 in combination with radiation demonstrates synergistic enhancements of antitumor efficacy without loss of specificity. Cancer Gene Ther 12:715–722. doi: 10.1038/sj.cgt.7700835. [DOI] [PubMed] [Google Scholar]
- 78.Touchefeu Y, Vassaux G, Harrington KJ. 2011. Oncolytic viruses in radiation oncology. Radiother Oncol 99:262–270. doi: 10.1016/j.radonc.2011.05.078. [DOI] [PubMed] [Google Scholar]
- 79.Kim W, Seong J, Oh HJ, Koom WS, Choi KJ, Yun CO. 2011. A novel combination treatment of armed oncolytic adenovirus expressing IL-12 and GM-CSF with radiotherapy in murine hepatocarcinoma. J Radiat Res 52:646–654. doi: 10.1269/jrr.10185. [DOI] [PubMed] [Google Scholar]
- 80.Rogulski KR, Wing MS, Paielli DL, Gilbert JD, Kim JH, Freytag SO. 2000. Double suicide gene therapy augments the antitumor activity of a replication-competent lytic adenovirus through enhanced cytotoxicity and radiosensitization. Hum Gene Ther 11:67–76. doi: 10.1089/10430340050016166. [DOI] [PubMed] [Google Scholar]
- 81.Liu C, Sarkaria JN, Petell CA, Paraskevakou G, Zollman PJ, Schroeder M, Carlson B, Decker PA, Wu W, James CD, Russell SJ, Galanis E. 2007. Combination of measles virus virotherapy and radiation therapy has synergistic activity in the treatment of glioblastoma multiforme. Clin Cancer Res 13:7155–7165. doi: 10.1158/1078-0432.CCR-07-1306. [DOI] [PubMed] [Google Scholar]
- 82.Ganesh S, Gonzalez-Edick M, Gibbons D, Ge Y, VanRoey M, Robinson M, Jooss K. 2009. Combination therapy with radiation or cisplatin enhances the potency of Ad5/35 chimeric oncolytic adenovirus in a preclinical model of head and neck cancer. Cancer Gene Ther 16:383–392. doi: 10.1038/cgt.2008.90. [DOI] [PubMed] [Google Scholar]
- 83.Pol J, Bloy N, Obrist F, Eggermont A, Galon J, Cremer I, Erbs P, Limacher JM, Preville X, Zitvogel L, Kroemer G, Galluzzi L. 2014. Trial watch:: oncolytic viruses for cancer therapy. Oncoimmunology 3:e28694. doi: 10.4161/onci.28694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kawahara M, Hori T, Matsubara Y, Okawa K, Uchiyama T. 2006. Identification of HLA class I-restricted tumor-associated antigens in adult T cell leukemia cells by mass spectrometric analysis. Exp Hematol 34:1496–1504. doi: 10.1016/j.exphem.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 85.Matsuoka M. 2005. Human T-cell leukemia virus type I (HTLV-I) infection and the onset of adult T-cell leukemia (ATL). Retrovirology 2:27. doi: 10.1186/1742-4690-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tezuka K, Tei M, Ueno T, Xun R, Iha H, Fujisawa J-I. 2014. Inhibition of ATL development in humanized mouse model by AZT/IFN treatment. Retrovirology 11:P43. doi: 10.1186/1742-4690-11-S1-P43. [DOI] [Google Scholar]