

ABSTRACT
Anti-hepatitis B virus (HBV) drugs are currently limited to nucleos(t)ide analogs (NAs) and interferons. A challenge of drug development is the identification of small molecules that suppress HBV infection from new chemical sources. Here, from a fungus-derived secondary metabolite library, we identify a structurally novel tricyclic polyketide, named vanitaracin A, which specifically inhibits HBV infection. Vanitaracin A inhibited the viral entry process with a submicromolar 50% inhibitory concentration (IC50) (IC50 = 0.61 ± 0.23 μM), without evident cytotoxicity (50% cytotoxic concentration of >256 μM; selectivity index value of >419) in primary human hepatocytes. Vanitaracin A did not affect the HBV replication process. This compound was found to directly interact with the HBV entry receptor sodium taurocholate cotransporting polypeptide (NTCP) and impaired its bile acid transport activity. Consistent with this NTCP targeting, antiviral activity of vanitaracin A was observed with hepatitis D virus (HDV) but not hepatitis C virus. Importantly, vanitaracin A inhibited infection by all HBV genotypes tested (genotypes A to D) and clinically relevant NA-resistant HBV isolate. Thus, we identified a fungal metabolite, vanitaracin A, which was a potent, well-tolerated, and broadly active inhibitor of HBV and HDV entry. This compound, or its related analogs, could be part of an antiviral strategy for preventing reinfection with HBV, including clinically relevant nucleos(t)ide analog-resistant virus.
IMPORTANCE For achieving better treatment and prevention of hepatitis B virus (HBV) infection, anti-HBV agents targeting a new molecule are in great demand. Although sodium taurocholate cotransporting polypeptide (NTCP) has recently been reported to be an essential host factor for HBV entry, there is a limited number of reports that identify new compounds targeting NTCP and inhibiting HBV entry. Here, from an uncharacterized chemical library, we isolated a structurally new compound, named vanitaracin A, which inhibited the process of entry of HBV and hepatitis D virus (HDV). This compound was suggested to directly interact with NTCP and inhibit its transporter activity. Importantly, vanitaracin A inhibited the entry of all HBV genotypes examined and of a clinically relevant nucleos(t)ide analog-resistant HBV isolate.
INTRODUCTION
Chronic hepatitis B virus (HBV) infection, constituting a public health problem, with an estimated 240 million carriers worldwide (1), elevates the risk of development of liver cirrhosis and hepatocellular carcinoma (2). Antiviral agents against HBV include nucleos(t)ide analogs (NAs) and interferons (IFNs), which can achieve significant reductions in HBV loads (3). Although IFN-α and its pegylated form (peg-IFN-α) modulate host immune responses to HBV infection or directly inhibit HBV replication in hepatocytes, these regimens show low tolerability because of serious adverse effects (3, 4). NAs, including lamivudine (LMV), adefovir, entecavir (ETV), tenofovir, and telbivudine, inhibit reverse transcription to suppress HBV replication, but long-term treatment with some of these NAs often results in selection for a significant number of drug-resistant viruses, which decreases treatment efficacy; i.e., the introduction of two substitutions, L180M and M204V, in the polymerase region leads to resistance to LMV, and an additional mutation of either T184, S202, or M250 with the L180M/M204V mutations confers further ETV resistance (5). More notably, it is difficult for the above-mentioned anti-HBV drugs to completely eliminate HBV from infected cells. Future antiviral strategies include multidrug treatment with an existing drug and a new anti-HBV agent. Consequently, there is a high demand for development of alternative drugs that specifically inhibit HBV infection via a different mode of action (2).
Sodium taurocholate cotransporting polypeptide (NTCP), a hepatic membrane transporter for bile acid uptake (6–8), was recently reported to be an HBV entry receptor (9). The pre-S1 region of the HBV large surface protein (LHBs) interacts with NTCP, which is known to be essential for HBV infection, in mediating the viral entry process (9, 10). This finding disclosed one of the most essential requirements for host cells to support HBV infection, and the HBV entry step has thus emerged as an attractive target for the development of HBV-specific inhibitors. An advantage of targeting this step is that interference with viral entry can efficiently block virus replication before the formation of covalently closed circular DNA (cccDNA), which is a persistent viral reservoir that is generally difficult to eliminate via NA treatment (11–13). Previously, we established a highly HBV-susceptible cell line, HepG2-hNTCP-C4, which overexpresses the human NTCP gene, and proved that it is useful for drug screening (14). In the present study, we took advantage of this cell line and identified a new anti-HBV compound.
Natural products, possessing a wide range of structural and bioactive diversity with drug-like structures, have served as a rich source of drugs for treatment of a variety of diseases (15, 16): 34% of drugs approved in the last 30 years consist of compounds from natural sources or optimized from natural products, and 47% of anti-infectives are derived from natural products (16). Here, we used our in-house chemical library derived from fungal strains, which comprises 214 compounds. We identified a new tricyclic polyketide, named “viral and NTCP inhibitor from Talaromyces-acin A” (vanitaracin A), which strongly inhibited infection by HBV and hepatitis D virus (HDV). Vanitaracin A interacted with NTCP and inhibited its bile acid uptake activity. Importantly, vanitaracin A showed pangenotypic anti-HBV activity and inhibited infection by a clinically relevant ETV-resistant HBV in primary human hepatocyte cultures. These findings indicate that vanitaracin A or its related compounds represent strong candidates for leading a new class of anti-HBV agents.
MATERIALS AND METHODS
Cell culture.
HepG2-hNTCP-C4 cells, Hep38.7-Tet cells, Huh-7.5.1 cells (kindly provided by Francis Chisari at The Scripps Research Institute), and primary human hepatocytes (PhoenixBio Co., Ltd.) were cultured as described previously (14, 17, 18).
HBV preparation and infection.
HBV used in this study was derived mainly from Hep38.7-Tet cells (genotype D), prepared as described previously (17). For the experiments in Fig. 7B, HBV (genotype A, C, or C carrying the L180M/S202G/M204V mutations) was prepared from HepG2 cells transfected with the corresponding expression plasmid, as described previously (19). For the experiments in Fig. 7A, HBV derived from serum of an infected patient was used (genotypes A, B, and C). HBV was infected at 12,000 genome equivalents (GEq)/cell (see Fig. 1C to H and 4A), 1,000 GEq/cell (see Fig. 2 and 7B), 100 GEq/cell (see Fig. 7A), and 2,700 and 900 GEq/cell (see Fig. 1I) in the presence of 4% polyethylene glycol 8000 (PEG 8000) (except for Fig. 2E) for 16 h, as described previously (20).
FIG 7.

Vanitaracin A shows pangenotype anti-HBV activity. (A) Primary human hepatocytes were pretreated with or without the indicated compounds (100 nM pre-S1 peptide or 16 μM vanitaracin A) for 5 h and then inoculated with blood-borne HBV (genotypes A, B, and C) in the presence or absence of the indicated compounds for 16 h. After washing out free HBV and compounds, cells were cultured in the absence of compounds for an additional 12 days, and HBV infection was evaluated by measuring levels of HBs antigens in the culture supernatant. (B) Primary human hepatocytes were pretreated with or without the indicated compounds (100 nM pre-S1 peptide or 16 μM vanitaracin A) for 5 h and then inoculated with HBVs (genotypes A, C, and C carrying the L180M/S202G/M204V mutations), which were prepared from cell culture as described in the legend of Fig. 1A and Materials and Methods. HBV infection was evaluated by measuring levels of HBs antigens in the culture supernatant.
FIG 1.

Vanitaracin A inhibits HBV infection. (A) Schematic representation of the schedule for chemical screening for HepG2-hNTCP-C4-based HBV infection. HepG2-hNTCP-C4 cells were pretreated with individual compounds for 5 h and then inoculated with HBV in the presence of the compounds for 16 h. After washing out free HBV and compounds, cells were cultured with medium in the absence of compounds for an additional 12 days, and HBV infection was evaluated by measuring HBs antigen levels in the culture supernatant. Black and white bars show periods of treatment and nontreatment, respectively. (B) Chemical structure of vanitaracin A. (C to H) HepG2-hNTCP-C4 cells were treated with or without 100 nM pre-S1 peptide or 45 μM vanitaracin A according to the protocol described above for panel A, and HBs (D) and HBe (E) antigens in the culture supernatant and cccDNA (F) and HBc protein (G and H) in cells were detected by CLIA, real-time PCR, and immunofluorescence analyses. Cell viability was also measured by an MTT assay (C). HBc-positive cells were counted and are shown as relative numbers of HBV-positive cells in panel H. OD450, optical density at 450 nm; S/CO, signal to cut off. (I) HBV infection assays were performed with HBV at 2,700 and 900 GEq/cell inoculum, by monitoring the HBs antigen level. The data show the means of data from three independent experiments. SDs are also shown as error bars. Statistical significance was determined by using Student's t test (*, P < 0.05; **, P < 0.01).
FIG 4.
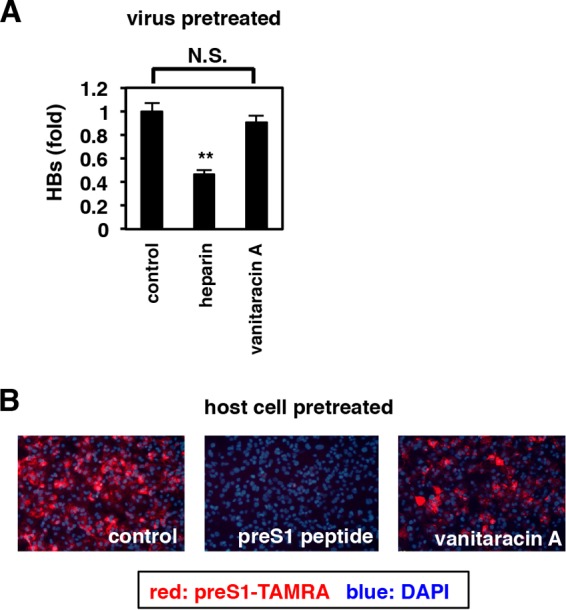
Vanitaracin A targets host cells to inhibit HBV attachment. (A) HBV particles were pretreated with or without the indicated compounds (50 U/ml heparin and 45 μM vanitaracin A) for 30 min, and the compounds were then removed in two rounds of ultrafiltration. These compound-pretreated HBVs were used for HBV infection assays in the absence of compounds. N.S., not significant. (B) HepG2-hNTCP-C4 cells were pretreated with or without the indicated compounds (100 nM pre-S1 peptide and 45 μM vanitaracin A) for 5 h and then washed extensively. These compound-pretreated cells were used for a pre-S1 binding assay (Fig. 3C) in the absence of compounds.
FIG 2.
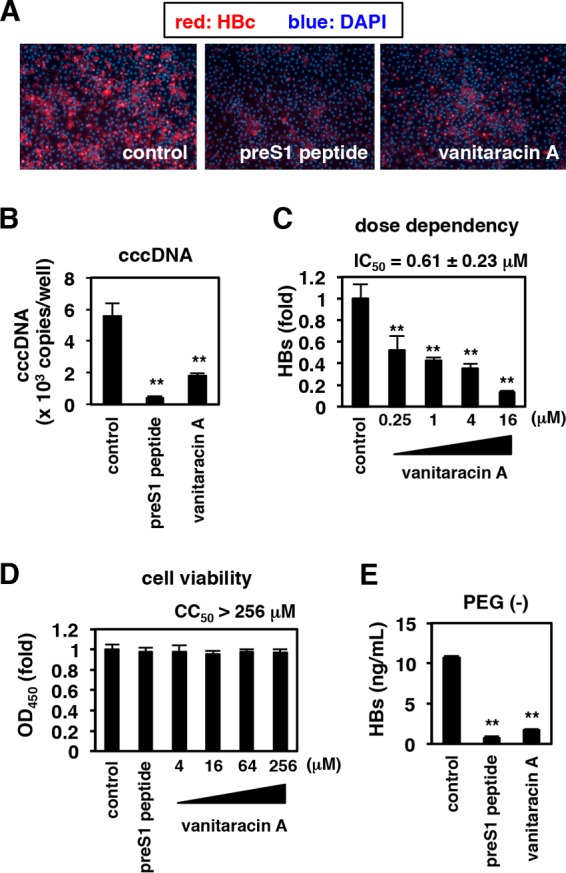
Anti-HBV effect of vanitaracin A on primary human hepatocytes. Primary human hepatocytes were treated with or without vanitaracin A (16 μM [A, B, and E]; 0.25, 1, 4, and 16 μM [C]; and 4, 16, 64, and 256 μM [D]) or pre-S1 peptide as a positive control (A, D, and E), as shown in Fig. 1A. (A to C) HBV infection was evaluated by measuring HBc protein (A) and cccDNA (B) levels in cells and HBs antigen levels in the culture supernatant (C). (D) Cell viability was also quantified by an MTT assay. (E) HBV infection was performed in the absence of PEG 8000.
Natural-product library.
Natural products were extracted from culture broths of fungal strains isolated from seaweeds, mosses, and other plants, as described previously (21, 22), and an in-house natural-product library consisting of 214 isolated compounds was prepared.
Real-time PCR.
Real-time PCR for quantification of HBV cccDNA was performed by using 5′-CGTCTGTGCCTTCTTCTCATCTGC-3′ and 5′-GCACAGCTTGGACGCTTGAA-3′ as a primer set and 5′-CTGTAGGCATAAATTGGT-MGB-3′ as a probe (14).
Detection of HBs and HBe antigens.
HBe antigen was detected by a chemiluminescence immunoassay (CLIA) (LSI Medience). The level of HBs was quantified by an enzyme-linked immunosorbent assay (ELISA), essentially as described previously (20), or by a CLIA (LSI Medience).
Indirect-immunofluorescence analysis.
Immunofluorescence analysis was conducted by using an anti-HBc antibody (catalog number B0586; Dako) at a dilution of 1:1000, essentially as described previously (20).
MTT assay.
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) cell viability assays were performed with XTT cell proliferation kit II (Roche), as described previously (17).
HBV replication assay.
After 3 days in a postseeding culture with tetracycline, Hep38.7-Tet cells were treated with compounds without tetracycline for 9 days. Nucleocapsid-associated HBV DNAs in the cells were recovered and quantified by real-time PCR, as described previously (20).
Southern blot analysis.
Isolation of nucleocapsid-associated DNA and Southern blot analysis to detect HBV DNAs were performed as previously described (20).
HBV attachment assay.
HepG2-hNTCP-C4 and HepG2 cells pretreated with compounds at 4°C for 2 h were exposed to HBV at 6,000 GEq/cell in the presence of the compound at 4°C for 30 min. After washing out free HBV, HBV DNA on the cell surface was recovered with a QIAamp minikit (Qiagen) and quantified by real-time PCR. To show NTCP-dependent hepatic attachment, amounts of HBV DNA on the surface of HepG2-hNTCP-C4 cells, subtracted by that on the surface of HepG2 cells, are shown.
Pre-S1 binding assay.
To examine binding between a pre-S1 peptide and host cells, we treated HepG2-hNTCP-C4 cells with 40 nM 6-carboxytetramethylrhodamine (TAMRA)-labeled pre-S1 peptide (pre-S1–TAMRA) for 30 min and then washed out free peptide, fixed the cells with paraformaldehyde, and finally stained the samples with 4′,6-diamidino-2-phenylindole (DAPI), as described previously (19).
Pretreatment of HBV particles or host cells with compounds.
To evaluate the effect of compounds on HBV, virions were pretreated with or without compounds for 30 min at 37°C before the compounds were removed by two rounds of ultrafiltration (AmiconUltra; Millipore). Theoretically, the ultrafiltration process leads to a 1/900 dilution of the compounds. The ultrafiltration step itself slightly reduced (to ∼70%) the infectivity of HBV, but the infectivity value was high enough to be used to evaluate the pretreatment effect on HBV.
To examine the effect of the compound on host cells, HepG2-hNTCP-C4 cells were pretreated with compounds for 2 h at 37°C and then washed out three times. These compound-pretreated cells were subjected to a pre-S1 binding assay.
Synthesis of recombinant NTCP protein.
The synthesized DNA fragment (operon) encoding the NTCP gene with the tobacco etch virus (TEV) protease site and a C-terminal His tag was digested with NdeI and XhoI and ligated into multicloning-site-modified plasmid pIVEX2.3d (5′Prime) for cell-free protein synthesis. Cell-free protein synthesis was performed by using 1 ml RTS500 Escherichia coli HY (5′Prime) with 10 μg plasmid and a 0.4% final concentration of digitonin for solubilization of expressed NTCP. After incubation at 37°C for 16 h, the reaction solution was collected and centrifuged at 21,500 × g at 4°C for 10 min. The supernatant and 5 ml wash buffer (50 mM Tris-HCl [pH 8.0], 0.5 M NaCl, 0.1% n-dodecyl-β-d-maltopyranoside (DDM), 15% glycerol, 20 mM imidazole) were mixed and agitated at 4°C with 2 ml Ni-nitrilotriacetic acid (NTA) for 1 h. Ni-NTA was washed with 50 ml wash buffer, and protein was eluted with a solution containing 50 mM Tris-Cl (pH 8.0), 0.5 M NaCl, 0.5 M imidazole, and 0.05% DDM. The fractions containing NTCP were pooled and dialyzed against a solution containing 50 mM Tris (pH 8.0), 0.5 M NaCl, and 0.1% DDM.
Surface plasmon resonance.
Surface plasmon resonance was performed with Biacore 3000 (Biacore, Herts, UK). Vanitaracin A (6.25 to 100 µM) as analyte was injected across the recombinant NTCP and BSA immobilized on a CM5 chip in HEPES (N-2-hydroxyethylpiperazine-NN-2-ethanesulfonic acid) saline (pH 7.4) supplemented with 150 mM NaCl. An analyte-free buffer was incubated for 120 s. The data were analyzed by BIA evaluation (GE Healthcare).
NTCP transporter assay.
HepG2-hNTCP-C4 cells pretreated with compounds for 30 min were incubated with [3H]taurocholic acid (TCA) in the presence of compounds at 37°C for 15 min to allow substrate uptake into the cells. After removal of free [3H]taurocholic acid, cells were lysed to measure intracellular radioactivity (17).
HCV infection assay.
Hepatitis C virus (HCV) envelope-mediated viral entry was evaluated with the HCV pseudoparticle (HCVpp) system, which was kindly provided by Francois-Loic Cosset at the Universite de Lyon (23). Huh-7.5.1 cells preincubated with compounds for 1 h were inoculated with HCVpp in the presence of compounds for 4 h. After washing out virus and compounds, cells were incubated for an additional 72 h and lysed to measure luciferase activity (22, 23).
HDV infection assay.
HDV was recovered from culture supernatants of Huh-7 cells transfected with pSVLD3 (kindly provided by John Taylor at the Fox Chase Cancer Center) and pT7HB2.7 (24, 25). HepG2-hNTCP-C4 cells were incubated with HDV at 15 GEq/cell in 5% PEG 8000 for 16 h, followed by washing out free virus and culturing of the cells for six additional days, according to a protocol described previously (26). Intracellular HDV RNA was quantified by real-time reverse transcription-PCR (RT-PCR) using 5′-GGACCCCTTCAGCGAACA-3′ and 5′-CCTAGCATCTCCTCCTATCGCTAT-3′ as a primer set and 5′-AGGCGCTTCGAGCGGTAGGAGTAAGA-3′ as a probe (26).
Statistics.
We basically repeated the experiments, except for those using primary human hepatocytes, three times, and the means of data from three independent experiments as well as standard deviations (SDs) are shown. Statistical significance was determined by using Student's t test.
RESULTS
Vanitaracin A inhibits HBV infection.
We screened for natural products that inhibited HBV infection using HepG2-hNTCP-C4 cells, an HBV-susceptible cell line expressing the human NTCP gene in HepG2 cells (14). In this study, we screened an in-house natural-product library that comprises 214 fungus-derived second metabolites (21, 22). As shown in Fig. 1A, HepG2-hNTCP-C4 cells were pretreated with individual compounds for 5 h and then inoculated with HBV in the presence of compounds for 16 h. After washing out free HBV and compounds, cells were cultured for an additional 12 days in the absence of compounds. HBV infection and cell viability were evaluated by measuring HBs antigen in the culture supernatant and MTT activity in cells, respectively. In this screening, one compound that has not been chemically reported previously (Fig. 1B) demonstrated the strongest anti-HBV activity; we named this novel compound vanitaracin A (“viral and NTCP inhibitor from Talaromyces-acin A”) based on subsequent observations.
Treatment with vanitaracin A, as shown in Fig. 1A, dramatically decreased the levels of HBe as well as HBs antigens in the culture supernatant after HBV infection, without significant cytotoxicity, as is the case with a known HBV entry inhibitor, pre-S1 peptide (9, 27) (Fig. 1C to E). Additionally, cccDNA (Fig. 1F) and HBc protein (Fig. 1G and H) levels in cells were consistently decreased upon vanitaracin A treatment. A similar anti-HBV effect was observed with inoculation of a smaller amount of HBV in the infection experiment (Fig. 1I). These data showed that vanitaracin A inhibited HBV infection in HepG2-hNTCP-C4 cells.
Anti-HBV effect of vanitaracin A in primary human hepatocytes.
To examine the effect of vanitaracin A under more physiologically relevant conditions, we used primary human hepatocytes to evaluate HBV infection. Treatment with vanitaracin A decreased HBc and cccDNA levels in cultures of primary human hepatocytes inoculated with HBV (Fig. 2A and B). Vanitaracin A showed a dose-dependent reduction in the level of secreted HBsAg (Fig. 2C); the 50% inhibitory concentration (IC50) was calculated to be 0.61 ± 0.23 μM. This compound was not cytotoxic to primary human hepatocyte cultures, with a 50% cytotoxic concentration (CC50) of >256 μM (Fig. 2D). The selectivity index (CC50/IC50 ratio) of vanitaracin A was calculated to be >419. In the above-described experiments (Fig. 1 and 2A to D), HBV was incubated with cells in the presence of PEG 8000 to facilitate infection efficiency. As shown in Fig. 2E, the anti-HBV effect of vanitaracin A was also seen when the infection assay was conducted in the absence of PEG 8000, under more physiologically relevant conditions (Fig. 2E).
Vanitaracin A blocks processes required for viral attachment to host cells.
We investigated which step in the HBV life cycle was blocked by vanitaracin A. The HBV life cycle comprises multiple steps, including the early phase (attachment/entry, trafficking to the nucleus, and cccDNA formation) and the late replication phase (transcription, encapsidation, reverse transcription, envelopment, and release) (17). We examined the effect of vanitaracin A on the HBV replication process using Hep38.7-Tet cells, which support HBV replication under depletion of tetracycline but not viral entry (17). HBV replication was induced by the depletion of tetracycline in the presence or absence of compounds for 9 days, and nucleocapsid-associated HBV DNA was detected by Southern blotting. Entecavir, a nucleoside analog used as a positive control, drastically decreased the HBV DNA level, while vanitaracin A had little effect (Fig. 3A). Next, we assessed HBV attachment to the host cell surface by incubating HepG2-hNTCP-C4 cells with HBV at 4°C, which allows viral attachment but not internalization. Cells preincubated with the compounds were exposed to HBV in the presence or absence of compounds for 30 min at 4°C, and free virus was washed out to detect HBV DNA attached to the cell surface. Vanitaracin A significantly reduced HBV attachment to the cell surface (Fig. 3B). It is known that the pre-S1 region of the LHBs envelope protein is critically involved in the HBV entry process through binding to an entry receptor, NTCP (9, 27–29). Therefore, we next used a fluorescence-labeled pre-S1 peptide as a probe to assess the effect of compounds on the attachment of pre-S1 to target cells. Treatment with vanitaracin A or nonlabeled pre-S1 peptide dramatically blocked the attachment of fluorescence-labeled pre-S1 to cells (Fig. 3C). Taken together, these findings indicated that vanitaracin A at least blocked the pre-S1-mediated attachment of HBV to host cells, although it may also affect another step(s) in the HBV life cycle.
FIG 3.
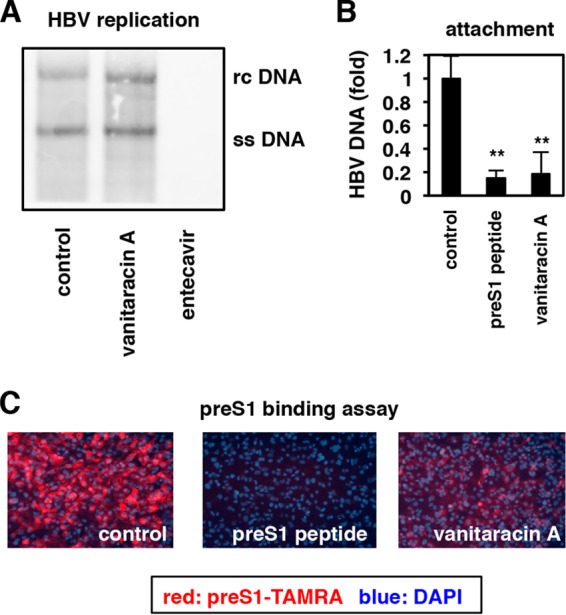
Vanitaracin A blocks HBV attachment to host cells. (A) HBV replication was induced in Hep38.7-Tet cells by depletion of tetracycline in the presence or absence of the indicated compounds (45 μM vanitaracin A or 100 nM entecavir as a positive control) for 9 days. Nucleocapsid-associated HBV DNA was detected by Southern blot analysis. rc DNA, relaxed circular DNA; ss DNA, single-stranded DNA. (B) HepG2-hNTCP-C4 and HepG2 cells were pretreated with or without the indicated compounds (100 nM pre-S1 peptide or 45 μM vanitaracin A) at 4°C for 2 h and then exposed to HBV at 4°C for 30 min in the presence or absence of compounds to allow HBV attachment to cells. After washing out free HBV, cell surface HBV DNA was extracted and quantified by real-time PCR. The data show the levels of HBV DNA in HepG2-hNTCP-C4 cells, which were subtracted by the level of HBV DNA in HepG2 cells, as a background level. (C) HepG2-hNTCP-C4 cells pretreated with or without the indicated compounds (1 μM pre-S1 peptide and 45 μM vanitaracin A) were exposed to 40 nM TAMRA-conjugated pre-S1 peptide (pre-S1–TAMRA) at 37°C for 30 min. Pre-S1–TAMRA attached to the cells was visualized as described in Materials and Methods.
Vanitaracin A targets host cells and blocks HBV attachment.
We next examined whether vanitaracin A targeted HBV particles or host cells. First, we measured the infectivity of compound-pretreated HBV particles using HepG2-hNTCP-C4 cells. HBV particles were pretreated with or without the compounds for 30 min at 37°C before they were removed by ultrafiltration. As shown in Fig. 4A, infectivity of HBV was significantly reduced upon pretreatment of the HBV inoculum with heparin, which targets HBV particles (30, 31) and was used as a positive control, but not with vanitaracin A (Fig. 4A). We next examined the susceptibility of compound-pretreated host cells to pre-S1 binding. HepG2-hNTCP-C4 cells were pretreated with compounds for 2 h and then washed out extensively to eliminate the compounds. These compound-pretreated cells were then used for a pre-S1 binding assay in the absence of compounds. As shown in Fig. 4B, the attachment of the fluorescence-labeled pre-S1 peptide was reduced in cells pretreated with either vanitaracin A or a nonlabeled pre-S1 peptide, a host-targeting HBV entry inhibitor, as a positive control (Fig. 4B). From this analysis, it was concluded that vanitaracin A targeted host cells, not HBV particles, to inhibit HBV attachment.
Vanitaracin A interacts with NTCP.
Thus, vanitaracin A inhibited pre-S1–cell binding by targeting host cells, which prompted us to investigate the interaction between NTCP and vanitaracin A. We immobilized recombinant His-tagged NTCP or bovine serum albumin (BSA), as a negative control, on sensor chips to detect interactions with vanitaracin A by surface plasmon resonance. As shown in Fig. 5A, injection of vanitaracin A across recombinant NTCP produced signals in a dose-dependent manner, and these signals were dissociated following the removal of vanitaracin A at 120 s (Fig. 5A, left). These vanitaracin A-dependent signals were not evident with the BSA-immobilized chip (Fig. 5A, right). Some of the drugs known to bind NTCP, including pre-S1 peptide, cyclosporine, irbesartan, ezetimibe, and ritonavir, reportedly inhibit the transporter activity of NTCP as well as block viral entry (32–37). We then determined whether vanitaracin A could affect NTCP-dependent bile acid uptake. The uptake of bile acids by HepG2-hNTCP-C4 cells was measured by incubating these cells with [3H]taurocholic acid in either a sodium-free or a sodium-containing buffer in the presence or absence of compounds. As shown in Fig. 5B, bile acid uptake was remarkably observed in sodium-containing buffer, which indicated NTCP transporter activity (Fig. 5B, lane 2). As previously reported, pre-S1 peptide greatly decreased NTCP transporter activity (Fig. 5B, lane 4). Treatment with vanitaracin A also reduced the sodium-dependent uptake of taurocholic acid in a dose-dependent manner (Fig. 5B, lanes 6, 8, 10, and 12). Thus, our study clearly revealed that vanitaracin A targeted NTCP, a receptor for HBV entry.
FIG 5.

Interaction of vanitaracin A with NTCP. (A) Vanitaracin A was used as an analyte, injected into a sensor chip bearing immobilized recombinant NTCP (left) or BSA (right), and analyzed with a BIAcore 3000 instrument. Real-time binding isotherms for increasing concentrations of vanitaracin A (6.25, 12.5, 25, 50, and 100 μM) are shown. The analyte was removed at 120 s. RU, resonance units. (B) NTCP transporter activity was measured by using [3H]taurocholic acid as a substrate in HepG2-hNTCP-C4 cells in the presence or absence of the indicated compounds (200 nM pre-S1 peptide and 2, 4, 8, or 16 μM vanitaracin A) in either sodium-free or sodium-containing buffer, as described in Materials and Methods.
Vanitaracin A inhibits infection by HDV but not by HCV.
We further examined the effect of vanitaracin A on other hepatitis viruses, HCV and HDV. HDV entry into host cells depends on NTCP, but HCV entry does not (9, 11, 38). HCV entry, evaluated by using the HCV pseudoparticle (HCVpp) system (22, 23), was drastically reduced by treatment with a known HCV entry inhibitor, bafilomycin A (39), but vanitaracin A had no effect (Fig. 6A). In contrast, HDV infection of HepG2-hNTCP-C4 cells was drastically reduced in the presence of vanitaracin A in a dose-dependent manner (Fig. 6B), which was consistent with the usage of NTCP for HDV entry. Vanitaracin A also inhibited HDV infection in the absence of PEG 8000, which is frequently used to enhance the infectivity of HBV/HDV (Fig. 6C).
FIG 6.

Vanitaracin A inhibits infection by HDV but not infection by HCV. (A) HCVpp assay. Huh-7.5.1 cells were preincubated with or without compounds (10 nM bafilomycin A1 or 5, 15, and 45 μM vanitaracin A) for 1 h and then inoculated with HCVpp in the presence or absence of compounds for 4 h. After washing out virus and compounds, cells were incubated for an additional 72 h and were recovered for quantification of luciferase activity that was driven by HCVpp infection. (B and C) HDV infection assay. HepG2-hNTCP-C4 cells were treated with HDV in the presence (B) or absence (C) of 5% PEG 8000 and treated with compounds (100 nM pre-S1 peptide or 6.25, 25, and 50 μM vanitaracin A) for 16 h. After washing out virus and compounds, the cells were further cultured for 6 days, and the amount of HDV RNA in the cells was quantified by real-time RT-PCR analysis.
Pangenotypic and robust anti-HBV effect on entecavir-resistant HBV.
All of the above-described results were obtained by using an HBV genotype D strain that was prepared from cell cultures. To further examine the relevance of the effect of vanitaracin A, we used blood-borne HBVs of different genotypes. As shown in Fig. 7A, the anti-HBV activity of vanitaracin A was conserved for all HBV genotypes examined (genotypes A, B, and C), which were derived from HBV-infected patients (Fig. 7A). Importantly, vanitaracin A was also found to be effective against infection by a clinically relevant entecavir-resistant HBV strain [C(L180M/S202G/M204V)] to a similar extent as the corresponding wild-type genotype C strain (Fig. 7B).
Thus, in this study, we identified a novel anti-HBV polyketide, vanitaracin A, and revealed its mode of action, which involves NTCP.
DISCUSSION
In this study, we identified a newly isolated tricyclic polyketide, named vanitaracin A, with strong activity in inhibiting HBV infection. The isolation of this compound from fungal metabolites and its chemical identification have been reported (40). A number of small molecules derived from natural sources have been reported to be inhibitors of vial infections, including those of human immunodeficiency virus, HCV, herpesviruses, and influenza virus (41–45). However, the poor understanding of the mode of action in most of these cases has hampered the development of antiviral drugs based on these findings, suggesting that mechanistic analysis, especially the identification of the target molecule, is a very important issue for drug development. In this study, we demonstrated that vanitaracin A interacted with NTCP to inhibit HBV attachment to host hepatocytes. As the compound's effect was pre-S1 and NTCP dependent, vanitaracin A also inhibited infection by HDV, which requires the same envelope-receptor involvement (9). This is the first report that identifies a new small molecule with anti-HBV properties from a natural chemical source and further describes its molecular target by using an NTCP-mediated HBV infection system.
As HBV entry is essential for the initiation, spread, and maintenance of HBV infection (46), this process represents an attractive target for the development of antiviral agents (11, 46–48). HBV entry inhibitors should be useful for inhibiting HBV reinfection after liver transplantation, for preventing vertical transmission, and for postexposure prophylaxis (49). These agents are also expected to be effective in treating chronic hepatitis B by cotreatment with other anti-HBV agents such as NAs (46). Myrcludex-B, a pre-S1 peptide that strongly inhibits HBV infection and the NTCP transporter, is currently under clinical development in a phase II study (46). Detailed adverse effects of this NTCP transporter inhibitor have not been defined. Although there are no available reports showing detailed side effects, myrcludex-B is well tolerated so far (46). These evidences support the notion that NTCP can serve as a potential target for the development of specific HBV inhibitors. Myrcludex-B is a lipopeptide and is therefore difficult to administer orally. To date, we and another group have reported that cyclosporine inhibits HBV infection by interrupting the interaction between LHBs and NTCP (17, 35). As the immunosuppressive activity of cyclosporine was dispensable for anti-HBV activity, we showed that nonimmunosuppressive derivatives, including alisporivir, which is under clinical development for treatment of chronic hepatitis C, significantly inhibited HBV infection, suggesting that these compounds are potential anti-HBV candidates (17). In addition, three FDA-approved agents, irbesartan, ezetimibe, and ritonavir, which are already known to inhibit NTCP transporter activity, reduced LHBs-dependent viral infection (32-34, 36). Although already known NTCP inhibitors have been reported to inhibit viral infection, there is no report identifying novel compounds that target NTCP and inhibit HBV infection. (−)-Epigallocatechin-3-gallate (EGCG) and Ro41-5253 decreased cell surface NTCP expression and reduced HBV infection (19, 50). Vanitaracin A was shown to have a greater potential to inhibit HBV infection (IC50 of <1 μM) than these compounds (Table 1). To obtain further insight into the potential therapeutic application of vanitaracin A, we showed that this compound was highly tolerated in primary cultures of human hepatocytes without toxicity at concentrations of up to 256 μM. Moreover, the antiviral effect was conserved for HBV genotypes A to D and a clinically relevant entecavir-resistant HBV strain.
TABLE 1.
Antiviral activities and cytotoxicities of compounds reported so far
| Small molecule | Mol wt | Antiviral activity (IC50 [μM]) | Cytotoxicity (CC50 [μM])a | Experimental system (virus/cells) | Reference |
|---|---|---|---|---|---|
| Vanitaracin A | 447 | 0.61 ± 0.23 | >256 | HBV/primary human hepatocytes | |
| Cyclosporine | 1,203 | 1.17 ± 0.22 | >10 | HBV/HepaRG | 37 |
| 0.8–4 | NA | HBV/HepaRG, HuH7-hNTCP, HepG2-hNTCP | 35 | ||
| 1.25 | NA | HBV/HepG2-hNTCP-C4 | 14 | ||
| Ezetimibe | 409 | 1–40 | >100 | HBV/HepaRG | 34 |
| 6.25–12.5 | >50 | HDV/Huh7-hNTCP | 32 | ||
| Irbesartan | 429 | 6.25–12.5 | >50 | HDV/Huh7-hNTCP | 32 |
| 3–10 | >3,000 | HBV/HepG2.N9 | 36 | ||
| 35 | NA | HBV/HepG2-NTCP | 33 | ||
| Ritonavir | 721 | 6.25–12.5 | >50 | HDV/Huh7-hNTCP | 32 |
| EGCG | 458 | 10–20 | 100–200 | HBV/HuS-E2 | 50 |
| Ro41-5253 | 485 | 5–10 | >20 | HBV/HepaRG | 19 |
NA, not applicable.
In addition, vanitaracin A was also effective against HDV infection, as in the case of anti-HBV entry inhibitors reported so far (32, 35, 51). Coinfection of HDV with HBV occurs in ∼15 million people worldwide, and HDV infection causes more severe viral hepatitis than does hepatitis B (52–54). However, there is no specific anti-HDV treatment currently clinically available, and thus, a new treatment option for HDV infection is needed. HDV lacks the ability to synthesize its own envelope protein but utilizes HBV envelope proteins to assemble and produce infectious particles in HBV- and HDV-coinfected cells. As HBV and HDV share the same envelope, they are likely to follow a common pathway to enter host cells that involves NTCP as an entry receptor. Our analysis supports that both viruses use the same entry mechanism and suggests that vanitaracin A targets this common mechanism for HBV and HDV entry, which is an advantage for blocking infection by both HBV and HDV. These findings strongly encourage further analysis of vanitaracin derivatives for developing a novel class of antiviral agents against both HBV and HDV.
ACKNOWLEDGMENTS
Huh-7.5.1 cells were kindly provided by Francis Chisari at The Scripps Research Institute. Plasmids for the HCVpp system and pSVLD3 were kind gifts from Francois-Loic Cosset at the University of Lyon and John Taylor at the Fox Chase Cancer Center, respectively.
This study was partly supported by grants-in-aid from the Ministry of Health, Labor, and Welfare, Japan; the Ministry of Education, Culture, Sports, Science, and Technology, Japan; Japan Society for the Promotion of Science KAKENHI grants 26460565 and 26102747; and the Research Program on Hepatitis from the Japan Agency for Medical Research and Development, AMED.
REFERENCES
- 1.Ott JJ, Stevens GA, Groeger J, Wiersma ST. 2012. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 30:2212–2219. doi: 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- 2.Zeisel MB, Lucifora J, Mason WS, Sureau C, Beck J, Levrero M, Kann M, Knolle PA, Benkirane M, Durantel D, Michel ML, Autran B, Cosset FL, Strick-Marchand H, Trepo C, Kao JH, Carrat F, Lacombe K, Schinazi RF, Barre-Sinoussi F, Delfraissy JF, Zoulim F. 2015. Towards an HBV cure: state-of-the-art and unresolved questions-report of the ANRS workshop on HBV cure. Gut 64:1314–1326. doi: 10.1136/gutjnl-2014-308943. [DOI] [PubMed] [Google Scholar]
- 3.Pawlotsky JM, Dusheiko G, Hatzakis A, Lau D, Lau G, Liang TJ, Locarnini S, Martin P, Richman DD, Zoulim F. 2008. Virologic monitoring of hepatitis B virus therapy in clinical trials and practice: recommendations for a standardized approach. Gastroenterology 134:405–415. doi: 10.1053/j.gastro.2007.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belloni L, Allweiss L, Guerrieri F, Pediconi N, Volz T, Pollicino T, Petersen J, Raimondo G, Dandri M, Levrero M. 2012. IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest 122:529–537. doi: 10.1172/JCI58847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tenney D, Levine S, Rose R, Walsh A, Weinheimer S, Discotto L, Plym M, Pokornowski K, Yu C, Angus P. 2004. Clinical emergence of entecavir-resistant hepatitis B virus requires additional substitutions in virus already resistant to lamivudine. Antimicrob Agents Chemother 48:3498–3507. doi: 10.1128/AAC.48.9.3498-3507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anwer MS, Stieger B. 2014. Sodium-dependent bile salt transporters of the SLC10A transporter family: more than solute transporters. Pflugers Arch 466:77–89. doi: 10.1007/s00424-013-1367-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klaassen CD, Aleksunes LM. 2010. Xenobiotic, bile acid, and cholesterol transporters: function and regulation. Pharmacol Rev 62:1–96. doi: 10.1124/pr.109.002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stieger B. 2011. The role of the sodium-taurocholate cotransporting polypeptide (NTCP) and of the bile salt export pump (BSEP) in physiology and pathophysiology of bile formation. Handb Exp Pharmacol 2011:205–259. doi: 10.1007/978-3-642-14541-4_5. [DOI] [PubMed] [Google Scholar]
- 9.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, Falth M, Stindt J, Koniger C, Nassal M, Kubitz R, Sultmann H, Urban S. 2014. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 146:1070–1083. doi: 10.1053/j.gastro.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 11.Baumert TF, Meredith L, Ni Y, Felmlee DJ, McKeating JA, Urban S. 2014. Entry of hepatitis B and C viruses—recent progress and future impact. Curr Opin Virol 4:58–65. doi: 10.1016/j.coviro.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Levrero M, Pollicino T, Petersen J, Belloni L, Raimondo G, Dandri M. 2009. Control of cccDNA function in hepatitis B virus infection. J Hepatol 51:581–592. doi: 10.1016/j.jhep.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 13.Tong S, Li J. 2014. Identification of NTCP as an HBV receptor: the beginning of the end or the end of the beginning? Gastroenterology 146:902–905. doi: 10.1053/j.gastro.2014.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwamoto M, Watashi K, Tsukuda S, Aly HH, Fukasawa M, Fujimoto A, Suzuki R, Aizaki H, Ito T, Koiwai O, Kusuhara H, Wakita T. 2014. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem Biophys Res Commun 443:808–813. doi: 10.1016/j.bbrc.2013.12.052. [DOI] [PubMed] [Google Scholar]
- 15.Cragg GM, Newman DJ. 2013. Natural products: a continuing source of novel drug leads. Biochim Biophys Acta 1830:3670–3695. doi: 10.1016/j.bbagen.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newman DJ, Cragg GM. 2012. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod 75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogura N, Watashi K, Noguchi T, Wakita T. 2014. Formation of covalently closed circular DNA in Hep38.7-Tet cells, a tetracycline inducible hepatitis B virus expression cell line. Biochem Biophys Res Commun 452:315–321. doi: 10.1016/j.bbrc.2014.08.029. [DOI] [PubMed] [Google Scholar]
- 18.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsukuda S, Watashi K, Iwamoto M, Suzuki R, Aizaki H, Okada M, Sugiyama M, Kojima S, Tanaka Y, Mizokami M, Li J, Tong S, Wakita T. 2015. Dysregulation of retinoic acid receptor diminishes hepatocyte permissiveness to hepatitis B virus infection through modulation of sodium taurocholate cotransporting polypeptide (NTCP) expression. J Biol Chem 290:5673–5684. doi: 10.1074/jbc.M114.602540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watashi K, Liang G, Iwamoto M, Marusawa H, Uchida N, Daito T, Kitamura K, Muramatsu M, Ohashi H, Kiyohara T. 2013. Interleukin-1 and tumor necrosis factor-α trigger restriction of hepatitis B virus infection via a cytidine deaminase activation-induced cytidine deaminase (AID). J Biol Chem 288:31715–31727. doi: 10.1074/jbc.M113.501122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myobatake Y, Takemoto K, Kamisuki S, Inoue N, Takasaki A, Takeuchi T, Mizushina Y, Sugawara F. 2014. Cytotoxic alkylated hydroquinone, phenol, and cyclohexenone derivatives from Aspergillus violaceofuscus Gasperini. J Nat Prod 77:1236–1240. doi: 10.1021/np401017g. [DOI] [PubMed] [Google Scholar]
- 22.Nakajima S, Watashi K, Kamisuki S, Tsukuda S, Takemoto K, Matsuda M, Suzuki R, Aizaki H, Sugawara F, Wakita T. 2013. Specific inhibition of hepatitis C virus entry into host hepatocytes by fungi-derived sulochrin and its derivatives. Biochem Biophys Res Commun 440:515–520. doi: 10.1016/j.bbrc.2013.09.100. [DOI] [PubMed] [Google Scholar]
- 23.Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med 197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuo MY, Chao M, Taylor J. 1989. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: role of delta antigen. J Virol 63:1945–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sureau C, Guerra B, Lee H. 1994. The middle hepatitis B virus envelope protein is not necessary for infectivity of hepatitis delta virus. J Virol 68:4063–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gudima S, Meier A, Dunbrack R, Taylor J, Bruss V. 2007. Two potentially important elements of the hepatitis B virus large envelope protein are dispensable for the infectivity of hepatitis delta virus. J Virol 81:4343–4347. doi: 10.1128/JVI.02478-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gripon P, Cannie I, Urban S. 2005. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J Virol 79:1613–1622. doi: 10.1128/JVI.79.3.1613-1622.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glebe D, Urban S, Knoop EV, Cag N, Krass P, Grun S, Bulavaite A, Sasnauskas K, Gerlich WH. 2005. Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and tupaia hepatocytes. Gastroenterology 129:234–245. doi: 10.1053/j.gastro.2005.03.090. [DOI] [PubMed] [Google Scholar]
- 29.Le Seyec J, Chouteau P, Cannie I, Guguen-Guillouzo C, Gripon P. 1999. Infection process of the hepatitis B virus depends on the presence of a defined sequence in the pre-S1 domain. J Virol 73:2052–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leistner CM, Gruen-Bernhard S, Glebe D. 2008. Role of glycosaminoglycans for binding and infection of hepatitis B virus. Cell Microbiol 10:122–133. [DOI] [PubMed] [Google Scholar]
- 31.Schulze A, Gripon P, Urban S. 2007. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 46:1759–1768. doi: 10.1002/hep.21896. [DOI] [PubMed] [Google Scholar]
- 32.Blanchet M, Sureau C, Labonte P. 2014. Use of FDA approved therapeutics with hNTCP metabolic inhibitory properties to impair the HDV lifecycle. Antiviral Res 106:111–115. doi: 10.1016/j.antiviral.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 33.Ko C, Park WJ, Park S, Kim S, Windisch MP, Ryu WS. The FDA approved drug irbesartan inhibits HBV-infection in HepG2 cells stably expressing sodium taurocholate co-transporting polypeptide. Antivir Ther doi: 10.3851/IMP2965. [DOI] [PubMed] [Google Scholar]
- 34.Lucifora J, Esser K, Protzer U. 2013. Ezetimibe blocks hepatitis B virus infection after virus uptake into hepatocytes. Antiviral Res 97:195–197. doi: 10.1016/j.antiviral.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 35.Nkongolo S, Ni Y, Lempp FA, Kaufman C, Lindner T, Esser-Nobis K, Lohmann V, Mier W, Mehrle S, Urban S. 2014. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J Hepatol 60:723–731. doi: 10.1016/j.jhep.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 36.Wang XJ, Hu W, Zhang TY, Mao YY, Liu NN, Wang SQ. 2015. Irbesartan, an FDA approved drug for hypertension and diabetic nephropathy, is a potent inhibitor for hepatitis B virus entry by disturbing Na(+)-dependent taurocholate cotransporting polypeptide activity. Antiviral Res 120:140–146. doi: 10.1016/j.antiviral.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 37.Watashi K, Sluder A, Daito T, Matsunaga S, Ryo A, Nagamori S, Iwamoto M, Nakajima S, Tsukuda S, Borroto-Esoda K, Sugiyama M, Tanaka Y, Kanai Y, Kusuhara H, Mizokami M, Wakita T. 2014. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter, sodium taurocholate cotransporting polypeptide (NTCP). Hepatology 59:1726–1737. doi: 10.1002/hep.26982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor JM. 2013. Virus entry mediated by hepatitis B virus envelope proteins. World J Gastroenterol 19:6730–6734. doi: 10.3748/wjg.v19.i40.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, McKeating JA, Rice CM. 2006. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J Virol 80:1734–1741. doi: 10.1128/JVI.80.4.1734-1741.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsunaga H, Kamisuki S, Kaneko M, Yamaguchi Y, Takeuchi T, Watashi K, Sugawara F. 2015. Isolation and structure of vanitaracin A, a novel anti-hepatitis B virus compound from Talaromyces sp. Bioorg Med Chem Lett 25:4325–4328. doi: 10.1016/j.bmcl.2015.07.067. [DOI] [PubMed] [Google Scholar]
- 41.Calland N, Dubuisson J, Rouille Y, Seron K. 2012. Hepatitis C virus and natural compounds: a new antiviral approach? Viruses 4:2197–2217. doi: 10.3390/v4102197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang Y, Ng TB, Wang CR, Zhang D, Cheng ZH, Liu ZK, Qiao WT, Geng YQ, Li N, Liu F. 2010. Inhibitors from natural products to HIV-1 reverse transcriptase, protease and integrase. Mini Rev Med Chem 10:1331–1344. doi: 10.2174/138955710793564133. [DOI] [PubMed] [Google Scholar]
- 43.Son M, Lee M, Sung GH, Lee T, Shin YS, Cho H, Lieberman PM, Kang H. 2013. Bioactive activities of natural products against herpesvirus infection. J Microbiol 51:545–551. doi: 10.1007/s12275-013-3450-9. [DOI] [PubMed] [Google Scholar]
- 44.Sun F, Huang R. 2014. The medicinal potential of natural products for the development of anti-influenza agents. Curr Drug Targets 15:175–183. doi: 10.2174/13894501113146660203. [DOI] [PubMed] [Google Scholar]
- 45.Zhou X, Liu J, Yang B, Lin X, Yang XW, Liu Y. 2013. Marine natural products with anti-HIV activities in the last decade. Curr Med Chem 20:953–973. doi: 10.2174/092986713805219118. [DOI] [PubMed] [Google Scholar]
- 46.Urban S, Bartenschlager R, Kubitz R, Zoulim F. 2014. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology 147:48–64. doi: 10.1053/j.gastro.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 47.Fofana I, Jilg N, Chung RT, Baumert TF. 2014. Entry inhibitors and future treatment of hepatitis C. Antiviral Res 104:136–142. doi: 10.1016/j.antiviral.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Lindenbach BD, Rice CM. 2013. The ins and outs of hepatitis C virus entry and assembly. Nat Rev Microbiol 11:688–700. doi: 10.1038/nrmicro3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petersen J, Dandri M, Mier W, Lutgehetmann M, Volz T, von Weizsacker F, Haberkorn U, Fischer L, Pollok JM, Erbes B, Seitz S, Urban S. 2008. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat Biotechnol 26:335–341. doi: 10.1038/nbt1389. [DOI] [PubMed] [Google Scholar]
- 50.Huang HC, Tao MH, Hung TM, Chen JC, Lin ZJ, Huang C. 2014. (−)-Epigallocatechin-3-gallate inhibits entry of hepatitis B virus into hepatocytes. Antiviral Res 111:100–111. doi: 10.1016/j.antiviral.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 51.Barrera A, Guerra B, Notvall L, Lanford RE. 2005. Mapping of the hepatitis B virus pre-S1 domain involved in receptor recognition. J Virol 79:9786–9798. doi: 10.1128/JVI.79.15.9786-9798.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ciancio A, Rizzetto M. 2014. Chronic hepatitis D at a standstill: where do we go from here? Nat Rev Gastroenterol Hepatol 11:68–71. doi: 10.1038/nrgastro.2013.164. [DOI] [PubMed] [Google Scholar]
- 53.Taylor JM. 2012. Virology of hepatitis D virus. Semin Liver Dis 32:195–200. doi: 10.1055/s-0032-1323623. [DOI] [PubMed] [Google Scholar]
- 54.Wedemeyer H, Manns MP. 2010. Epidemiology, pathogenesis and management of hepatitis D: update and challenges ahead. Nat Rev Gastroenterol Hepatol 7:31–40. doi: 10.1038/nrgastro.2009.205. [DOI] [PubMed] [Google Scholar]