

ABSTRACT
The white sucker Catostomus commersonii is a freshwater teleost often utilized as a resident sentinel. Here, we sequenced the full genome of a hepatitis B-like virus that infects white suckers from the Great Lakes Region of the United States. Dideoxy sequencing confirmed that the white sucker hepatitis B virus (WSHBV) has a circular genome (3,542 bp) with the prototypical codon organization of hepadnaviruses. Electron microscopy demonstrated that complete virions of approximately 40 nm were present in the plasma of infected fish. Compared to avi- and orthohepadnaviruses, sequence conservation of the core, polymerase, and surface proteins was low and ranged from 16 to 27% at the amino acid level. An X protein homologue common to the orthohepadnaviruses was not present. The WSHBV genome included an atypical, presumptively noncoding region absent in previously described hepadnaviruses. Phylogenetic analyses confirmed WSHBV as distinct from previously documented hepadnaviruses. The level of divergence in protein sequences between WSHBV and other hepadnaviruses and the identification of an HBV-like sequence in an African cichlid provide evidence that a novel genus of the family Hepadnaviridae may need to be established that includes these hepatitis B-like viruses in fishes. Viral transcription was observed in 9.5% (16 of 169) of white suckers evaluated. The prevalence of hepatic tumors in these fish was 4.9%, and only 2.4% of fish were positive for both virus and hepatic tumors. These results are not sufficient to draw inferences regarding the association of WSHBV and carcinogenesis in white sucker.
IMPORTANCE We report the first full-length genome of a hepadnavirus from fishes. Phylogenetic analysis of this genome indicates divergence from genomes of previously described hepadnaviruses from mammalian and avian hosts and supports the creation of a novel genus. The discovery of this novel virus may better our understanding of the evolutionary history of hepatitis B-like viruses of other hosts. In fishes, knowledge of this virus may provide insight regarding possible risk factors associated with hepatic neoplasia in the white sucker. This may also offer another model system for mechanistic research.
INTRODUCTION
The white sucker Catostomus commersonii is a freshwater teleost that is endemic to river systems in the midwestern and northeastern United States. The widespread distribution and life history of white suckers has made them a target species in numerous contaminant-monitoring and effects studies (1–3). The prevalence of tumors in white sucker is currently used as an indicator of exposure to environmental contaminants and is also a criterion used in the assessment and listing or delisting of areas of concern (AOCs) throughout the Great Lakes region. Fish tumors or other deformities, specifically in white sucker or brown bullhead, are listed as one of the “beneficial use” impairments at Great Lakes AOCs (4). In 2010, the Great Lakes Restoration Initiative specifically targeted certain priorities, one of which was the evaluation and monitoring of progress in AOCs (5). One component of this program was the assessment of wild populations present at potentially impacted sites. A suite of biomarkers ranging from the molecular to the organismal level was developed for multiple fish species (6). During the development of a hepatic transcriptome for white sucker, we identified the presence of a novel hepatitis B-like virus.
Hepatitis B virus (HBV) is an enveloped, reverse-transcribing DNA virus of the family Hepadnaviridae. Hepadnaviruses are small (∼42 nm), spherical viruses characterized by a compact, circular, partially double-stranded DNA genome approximately 3.2 kb in length. The genome utilizes all three forward frames to carry transcripts of three to four overlapping, canonical open reading frames (ORFs): the pre-C/C ORF, the pre-S/S ORF, the polymerase (Pol) ORF, and the X ORF. These reading frames encode the nucleocapsid protein (core antigen), envelope proteins (surface antigen), polymerase/reverse transcriptase polyprotein, and regulatory transactivating protein, respectively. The life cycle of HBV can be broadly divided into three stages that begin with infectious virions containing circular, partially double-stranded but not covalently closed DNA (relaxed circular DNA [rcDNA]). These virions attach to carbohydrate side chains of hepatocyte-associated heparin sulfate proteoglycans on the host cell membrane that initiate a multistep entry process (7). Once within the host cell, rcDNA is converted into covalently closed circular DNA (cccDNA). This serves as a template for the transcription of pregenomic RNA (pgRNA) and mRNA (8, 9).
The family Hepadnaviridae is comprised of two recognized genera, Orthohepadnavirus (type species, Hepatitis B virus [HBV]) and Avihepadnavirus (type species, Duck hepatitis B virus [DHBV]). The orthohepadnaviruses infect humans and other mammals, including the Old World great apes (10–13), while the avihepadnaviruses infect avian species (14–19). In general, these viruses have narrow host ranges, and infection leads to variable presentation and outcomes. These viruses exhibit a tropism for hepatocytes and are typically associated with acute and chronic liver diseases, including fibrosis, cirrhosis, cholangiocarcinoma (CC) (20), and hepatocellular carcinoma (HCC) (21, 22). It is estimated that 350 million people are chronically infected with HBV (23). Consequently, HCC is the fifth most frequent human cancer (24). In birds, liver pathology is less commonly observed with hepadnavirus infections (25). Viral replication is noncytopathic, and tissue pathology is typically associated with the immune response to viral antigens. Current evidence suggests that birds are the ancestral host of the mammalian orthohepadnaviruses (26). To date, hepadnaviruses have not been identified in fishes. Here, we report the complete genome of a novel hepadnavirus isolated from the white sucker and present molecular and morphological evidence to support its assignment as the type species of a new genus in the family Hepadnaviridae. We also examine the prevalence of viral transcription in white sucker collected from five rivers of the Great Lakes region and the association of virus with hepatic neoplasia.
MATERIALS AND METHODS
Sampling.
We collected liver tissue from wild-caught, white sucker (n = 169) inhabiting five rivers in the Great Lakes region (Fig. 1; see also Table S1 in the supplemental material). These samples were preserved in RNAlater (Life Technologies, Grand Island, NY) for a transcriptome assembly project (PRJNA282680) and quantitative gene expression analyses. A liver sample from a white sucker collected from Michael Brook near Carmel, NY, was included for this purpose as well. Tissue was not collected for DNA applications from these fish. In an attempt to collect samples suitable for DNA analysis, we collected white sucker (n = 20) from the Root River (Racine, WI) in the spring of 2014. Liver tissue was excised and stored in 90% ethanol for subsequent DNA extraction. All fish were euthanized with a lethal dose of tricane methanesulfonate (MS-222, Finquel; Argent Laboratories, Redmond, WA) according to approved Animal and Care Safety protocols (U.S. Geological Survey, Leetown Science Center, Kearneysville, WV). Prior to necropsy, blood was collected using heparinized syringes, transferred to heparinized Vacutainers, and stored on wet ice until centrifuged. Plasma samples were stored at −80°C until analyzed. Additional liver tissue was preserved in Z-fix (Anatech Ltd., Battle Creek, MI) from all individuals for subsequent histopathological assessment.
FIG 1.

Map of white sucker sample locations. These include the St. Louis River, Green Bay and Lower Fox River, Milwaukee Estuary, Detroit River and Maumee River areas of concern, and the Root River.
High-throughput sequencing.
Total RNA was extracted using an E.Z.N.A. Total RNA kit (Omega Biotek, Norcross, GA) from nine individuals, pooled, and enriched for non-rRNA using a Ribo-Zero rRNA removal kit (Epicentre, Maidson, WI). An RNA sequencing library was prepared, and paired-end (2 by 100 bp) ultradeep sequencing was carried out at the Institute for Genome Sciences (Baltimore, MD) on an Illumina HiSeq2000.
Read pairs were quality screened and adapter trimmed prior to de novo assembly into contigs with CLC Genomics Workbench, version 7. As part of routine efforts to screen for contaminating sequences, blastx searches identified contigs with sequence similarity to hepadnaviruses, which we then culled from our white sucker transcriptome assembly, which is to be described elsewhere.
Full-genome resequencing and mapping.
The initial indication that a hepadnavirus was present in our pooled liver sample was based on blastx results of de novo-assembled RNA transcripts (Fig. 2). To avoid the inclusion of pgRNA or viral mRNA in the genome model, we isolated DNA from an ethanol-preserved liver. Resequencing was conducted on a single, PCR-positive fish collected from the Root River (white sucker hepatitis B virus [WSHBV] RR173). The DNA was extracted using a DNeasy kit (Qiagen, Valencia, CA) as per the manufacturer's protocols. Primers were designed to resequence the complete genome and confirm a circular architecture. We used Primer3, version 2.3.4 (27), to design these primers. Default parameters were modified to amplify 280- to 797-bp products (see Table S2 in the supplemental material). Sequence overlap ranged from 19 to 461 bp, with a mean overlap of 189 bp (see Fig. S1). PCR conditions are noted in Table S2 in the supplemental material. All PCR amplicons were purified using a QIAquick purification kit (Qiagen, Valencia, CA). Dideoxy sequencing was conducted using BigDye Terminator, version 3.1, chemistry and an ABI 3100 genetic analyzer (Applied Biosystems, Forster City, CA). The resulting sequences were assembled into a single circular sequence using Geneious (version R7; Biomatters) (Fig. 3).
FIG 2.

Graphic summary of blastx results identify genomic organization and signature conserved domains consistent with those of hepadnaviruses. NTP, nucleoside triphosphate; RF, reading frame.
FIG 3.

Genome organization of the WSHBV. The complete genome consists of 3,542 nucleotides of double-stranded DNA that encode three partially or completely overlapping ORFs (RFs +1, +2, and +3). Open reading frames encoding the core, polymerase, and surface proteins are indicated in black. Conserved domains are indicated in gray.
Per-base coverage of the genome model in Illumina RNA reads was assessed by mapping the reads with bowtie2 (28) using the “end-to-end” and “very-sensitive” quick switches and specifying a maximum fragment length of 700 bp. Mapping quality and mismatches/polymorphisms were investigated with the alignment summary metrics package of Picard (Broad Institute [http://broadinstitute.github.io/picard/]) and with the Tablet alignment viewer (29). In addition, we searched for polyadenylated RNA sequences consistent with functional polyadenylation sites. To do this, we remapped Illumina reads from the liver cDNA library using bowtie2 and targeting genomic windows from 100 positions 5′ to 100 positions 3′ of each candidate site. The bowtie2 mapping mode was local rather than end to end to allow A homopolymers to remain unmapped while allowing templated mRNA upstream of the homopolymer to be mapped. The remaining bowtie2 parameters were the default values. A total of 16,384 unique fragments (read pairs or singletons) mapped to these seven (partly overlapping) regions.
In addition to resequencing, we assessed the presence of WSHBV DNA in all fish collected from the Root River via endpoint PCR (primer set and conditions are given in Table S2 in the supplemental material). This was also conducted to confirm that the viral sequence was not endogenous. Template DNA from both liver and plasma was used for this screening. DNA was extracted from plasma using a DNeasy kit (Qiagen, Valencia, CA) as per the manufacturer's protocols.
Electron microscopy.
Plasma samples from fish testing positive by PCR were diluted in TNE buffer (50 mm Tris-HCl, 100 mm NaCl, 0.1 mm EDTA, pH 7.4) to a volume of 5 ml and pelleted by ultracentrifugation through a 200-μl cushion of 25% (wt/wt) sucrose in TNE buffer (150,000 × g for 1 h at 10°C) (Beckman SW50.1 rotor). The pellet was resuspended in 0.5 ml of TNE buffer and layered onto a gradient composed of 1.5 ml of 30% (wt/wt) CsCl, 1.5 ml of 35% (wt/wt) CsCl, and 1.5 ml of 40% (wt/wt) CsCl in TNE buffer. After centrifugation to equilibrium for 16 h (115,000 × g at 10°C) (Beckman SW 50.1 rotor), 250-μl fractions were collected from the gradient, and the densities determined using a refractometer. Fractions with a density at or near 1.22 g/ml were pooled and diluted with TNE buffer to a volume of 4.5 ml, and the virus was pelleted through a 200-μl cushion of 25% (wt/wt) sucrose (150,000 × g for 2 h at 10°C) (Beckman SW50.1 rotor). The pellet of gradient-purified virus was dried and diluted in 50 μl of distilled water. Samples of the virus were adsorbed onto 400-mesh Formvar/silicone monoxide-coated copper grids (Electron Microscopy Sciences) for 3 min and negatively stained either with 1% phosphotungstic acid (PTA), pH 6.5, or with 0.5% or 1% uranyl acetate (UA) for 1 min. Grids were sent to the electron microscopy facility at the University of Montana, where they were examined using a Hitachi H-7100 transmission electron microscope (Hitachi High Technologies America, Inc.) and photographed digitally.
Survey of hepadnavirus prevalence in white sucker.
In order to assess the general prevalence and geographic distribution of WSHBV in wild-caught white sucker, we targeted WSHBV core protein RNA using a custom Nanostring CodeSet designed for a separate, ongoing study (6). The hybridization CodeSet targeted the RNA sequence in the coding region of the core protein. The hybridization code set was designed by Nanostring Technologies, and nCounter analysis was conducted at the University of Pittsburgh, Genomics Research Core. This hybridization analysis method specifically targets RNA and not DNA. Liver samples (mass, 15 to 25 mg) collected from fish inhabiting waters in the Great Lakes region and preserved in RNAlater were homogenized in TRK lysis buffer (OmegaBiotek, Norcross, GA) using 5-mm stainless steel balls in a TissueLyzer (Qiagen, Valencia, CA) at 30 Hz for 8 min. Lysate was centrifuged at 13,000 × g for 10 min. The clarified lysate was then frozen at −80°C for subsequent nCounter analysis.
In order to normalize the transcript count data and conservatively filter false positives, we utilized the internal negative-control (background) count data output from the analysis. We set the background value to subtract from target counts equal to the mean plus three standard deviations of all negative controls from all sample counts. Data were then normalized to the tissue mass.
Relationship between WSHBV and liver tumor prevalence.
Hepatocellular carcinoma is associated with HBV infection in humans. Hepatic tumors are not uncommon in white sucker inhabiting the Great Lakes regions, and prevalence of this pathology is used as an indication of contaminant exposure. Liver tissue preserved in Z-fix (Anatech Ltd., Battle Creek, MI) was therefore processed for histopathological observation via graded alcohols, paraffin infiltration, and embedding. Tissues were sectioned at 5 μm and stained with hematoxylin and eosin. Hepatic tumors included HCC, hepatic adenoma (HA), cholangioma (CO), and cholangiocarcinoma (CC). Tumor and virus data were converted to a binary data set, and Jaccard binary dichotomy coefficients were determined to evaluate dissimilarity.
Sequence comparisons and phylogenetic analysis.
Once a complete genome of the WSHBV was constructed, we performed additional blastn and blastx searches within the NCBI nucleotide collection (nonredundant nucleotide [nr/nt]). Although hepadnaviruses from fish have not been reported previously, we suspected that related sequences may exist in public nucleotide data. We executed tblastn queries in the transcriptome shotgun assembly (restricted to bony fishes; NCBI taxid:7898) database using translated gene products from all three predicted ORFs. Additionally, given that hepadnaviruses are integrating viruses and that relic genomes (endogenous viral elements [EVEs]) have been identified in avian genomes, we executed tblastx queries against the reference genomic sequences (restricted to bony fishes; taxid:7897) to further assess the possibility of endogenization events of hepadnaviruses in fishes.
Phylogenetic relationships of the white sucker hepatitis B virus (WSHBV) genome and predicted gene products (core, polymerase, and surface) were compared to those of members of the genera Avihepadnavirus and Orthohepadnavirus. We used nucleotide and protein sequences from the duck HBV (DHBV), Ross's goose HBV (RGHBV), sheldgoose HBV (ShHBV), heron HBV (HHBV), parrot HBV (PHBV), snow goose HBV (SGHBV), horseshoe bat HBV (HBHBV), bat HBV (BtHBV), roundleaf bat HBV (RBHBV), tent-making bat HBV (TBHBV), ground squirrel HBV (GSHBV), woodchuck HBV (WHBV), and human HBV (HuHBV). Accession numbers of included HBV protein or nucleotide sequences are listed in Tables 1 and 2.
TABLE 1.
Size and biochemical properties of predicted hepadnavirus core, polymerase, and surface proteins
| Virus | Polymerase protein |
Surface protein |
Core protein |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GenBank accession no. | Size (aa)a | pI | Theoretical mass (kDa) | GenBank accession no. | Size (aa) | pI | Theoretical mass (kDa) | GenBank accession no. | Size (aa) | pI | Theoretical mass (kDa) | |
| WSHBV | AKT95195 | 789 | 9.76 | 87.3 | AKT95194.2 | 346 | 9.51 | 39.6 | AKT95193 | 213 | 9.68 | 24.3 |
| PHBV | YP_004956864 | 795 | 9.77 | 90.3 | YP_004956865 | 375 | 9.31 | 41.4 | YP_004956862 | 305 | 9.55 | 34.8 |
| HHBV | NP_040998 | 788 | 9.93 | 90.0 | NP_040999 | 335 | 8.84 | 37.2 | NP_040997 | 305 | 9.69 | 34.9 |
| ShHBV | YP_024974 | 796 | 9.91 | 90.6 | YP_024975 | 338 | 8.88 | 37.8 | YP_024972 | 305 | 9.71 | 35.2 |
| SGHBV | YP_031695 | 787 | 10.08 | 90.0 | YP_031696 | 329 | 9.18 | 36.6 | YP_031693 | 305 | 9.49 | 34.9 |
| RGHBV | YP_024968 | 785 | 9.91 | 89.4 | YP_024969 | 325 | 8.90 | 36.4 | YP_024967 | 305 | 9.39 | 35.0 |
| DHBV | NP_039822 | 788 | 9.92 | 89.3 | NP_039824 | 330 | 8.35 | 37.0 | ADP55743 | 262 | 9.85 | 30.2 |
| HBHBV | YP_009045995 | 902 | 10.01 | 100.6 | YP_009045996 | 224 | 8.41 | 25.4 | YP_009045998 | 217 | 9.57 | 24.9 |
| RBHBV | YP_009045991 | 899 | 10 | 99.9 | YP_009045992 | 224 | 8.41 | 25.4 | YP_009045994 | 217 | 9.61 | 25.0 |
| BtHBV | YP_007677999 | 853 | 9.91 | 95.2 | YP_007678000 | 399 | 7.91 | 44.4 | YP_007678002 | 217 | 9.58 | 24.9 |
| TBHBV | YP_009045999 | 827 | 9.78 | 93.2 | YP_009046000 | 223 | 8.27 | 25.0 | YP_009046002 | 188 | 9.35 | 21.7 |
| GSHBV | NP_040994 | 881 | 9.57 | 100.0 | NP_040995 | 282 | 6.85 | 31.8 | NP_040993 | 217 | 9.34 | 25.2 |
| WHBV | NP_671813 | 884 | 9.71 | 99.4 | NP_671814 | 431 | 8.51 | 48.9 | NP_671816 | 188 | 10.12 | 21.7 |
| HuHBV | NP_647604 | 843 | 9.83 | 94.6 | YP_355333 | 400 | 8.21 | 43.7 | YP_355335 | 212 | 9.49 | 24.3 |
Number of amino acids (aa).
TABLE 2.
Percent nucleotide identity compared to WSHBV of hepadnaviruses partitioned by open reading frame
| Virus (GenBank accession no.) | % Identity at the indicated level |
|||||
|---|---|---|---|---|---|---|
| Polymerase protein |
Surface protein |
Core protein |
||||
| Nucleotide | Amino acid | Nucleotide | Amino acid | Nucleotide | Amino acid | |
| PHBV (NC_016561) | 40.54 | 29.82 | 40.44 | 22.25 | 33.89 | 22.26 |
| HHBV (NC_001486) | 40.30 | 30.06 | 42.22 | 23.51 | 33.67 | 21.92 |
| RGHBV (NC_005888) | 40.94 | 30.17 | 40.99 | 25.82 | 35.79 | 21.92 |
| SGHBV (NC_005950) | 41.97 | 29.21 | 40.55 | 24.05 | 35.12 | 21.92 |
| ShHBV (NC_005890) | 40.97 | 29.45 | 40.28 | 24.53 | 35.34 | 21.23 |
| DHBV (NC_001344) | 41.59 | 28.97 | 41.24 | 24.86 | 36.72 | 24.50 |
| TBHBV (NC_024445) | 35.55 | 23.79 | 25.00 | 27.07 | 27.16 | 16.82 |
| HuHBV (NC_003977) | 35.63 | 22.85 | 36.25 | 20.00 | 28.05 | 16.31 |
| GSHBV (NC_001484) | 36.12 | 25.52 | 39.69 | 15.33 | 31.17 | 19.31 |
| WHBV (NC_004107) | 37.28 | 26.13 | 36.38 | 18.82 | 28.69 | 20.69 |
| BtHBV (NC_020881) | 36.99 | 25.25 | 33.69 | 18.33 | 29.58 | 21.46 |
| RBHBV (NC_024443) | 36.66 | 25.00 | 44.30 | 16.21 | 29.32 | 16.74 |
| HBHBV (NC_024444) | 36.76 | 24.73 | 43.87 | 15.46 | 29.71 | 17.17 |
Following Cui and Holmes (30), we aligned the protein sequences using default settings in MUSCLE (31). We then determined the appropriate amino acid substitution model with ProtTest, version 2.4 (32). Maximum-likelihood phylogenetic analyses were executed in PhyML (33) using the LG (Le and Gascuel; core), WAG (Whelan and Goldman; polymerase), and JTT (Jones, Taylor, and Thornton; surface) models of amino acid substitution with 1,000 bootstrap replicates to evaluate the phylogenetic relationships of mammalian, avian, and white sucker HBVs. Nucleotide sequences for these ORFs were aligned in MUSCLE as well to determine nucleotide identity. Complete genomes were rearranged such that the first nucleotide corresponded to the start codon of the core ORF. We then aligned the sequences using MUSCLE and determined the appropriate substitution model with MEGA, version 6.0 (34). Phylogenetic relationships were determined with PhyML using the general time-reversible (GTR) nucleotide substitution model.
Nucleotide sequence accession number.
The complete genome sequence of WSHBV RR173 was deposited in the GenBank under accession number KR229754.
RESULTS
Sequencing the viral genome and ORF organization.
De novo assembly of ribosome-depleted RNA isolated from white sucker included a linear contig of 3,519 bp with blastx similarity to hepadnaviruses. Our finished genome, 2014 WSHBV RR173 (GenBank accession number KR229754), was 3,542 bp. Dideoxy sequencing confirmed a circular architecture of the genome (Fig. 3). Remapping Illumina reads to the complete genome included 131,080 reads. Genome coverage was high (average coverage, 3,184×; maximum coverage, 7,215×) with a mismatch frequency of 0.2% (Fig. 4). The genome size of WSHBV was larger than the sizes of other previously described hepadnaviruses genomes (3,542 versus 3,377 bp from HBHBV) and included an atypical, presumably noncoding, region comprised of 679 bp (nucleotides [nt] 2538 to 3214). We confirmed the presence of this region as authentic viral sequence using primer sets anchored in the polymerase and core ORFs or in one of these ORFs and the noncoding region (see Fig. S2 and Table S2 in the supplemental material). This presumably noncoding region terminated with a noncanonical polyadenylation signal (TATAAA; nt 3211 to 3216). Three additional polyadenylation signals were identified at nt 596 to 601, 1954 to 1959, and 3148 to 4158. Traditional polyadenylation signals were located at nt 2544 to 2549, 3201 to 3206, and 3425 to 3430. Local remapping of the short reads identified the noncanonical hexanucleotide polyadenylation signal (TATAAA; nt 3211 to 3216) that terminated the presumptively noncoding region as the single polyadenylation site. The GC content of the complete genome was 42% while the noncoding region had a considerably lower GC content of 34%.
FIG 4.
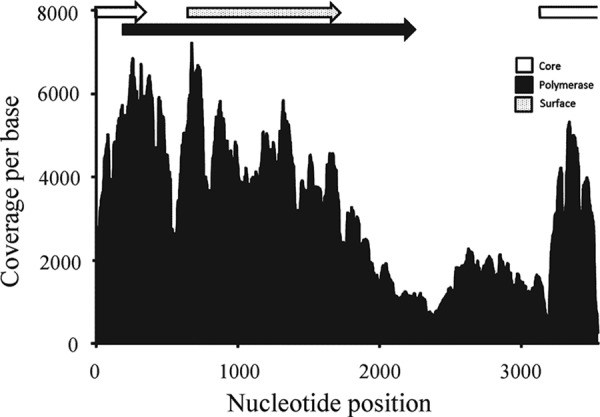
Genome coverage per base of the WSHBV in the pooled liver sample utilized for high-throughput sequencing of RNA transcripts (RNA-seq) analysis (131,080 reads). Locations of the open reading frames that encode the core, polymerase, and surface proteins are indicated at the top of the figure.
The genome organization was similar to that of other hepadnaviruses. In silico translation identified three partially or completely overlapping reading frames (RF; +1, +2, and +3). These corresponded to the core protein, polymerase protein, and surface protein of prototypical hepadnaviruses. A hepatitis B X protein homologue common to orthohepadnaviruses was not present. The numbering of base pairs in the hepadnavirus genome is typically assigned based on a conserved cleavage site for EcoRI. This restriction site is absent from the WSHBV genome. We therefore manually aligned the WSHBV to the genome of the DHBV (GenBank accession number NC_001344), such that the start codons of the polymerase protein (nt 176) corresponded.
Open reading frame 1 (RF +1; nt 3216 to 3542, 1 to 315) encodes the core polyprotein of 213 amino acids with a theoretical average molecular mass of 24,255.67 Da (Table 1). The size of this ORF was in the range of that of other hepadnaviruses but was more similar (in size) to the ORF sizes of the orthohepadnaviruses. We identified the hepatitis core antigen conserved domain within this ORF (nt 46 to 204) (Fig. 3; see also Table S3 in the supplemental material). Pairwise alignment with avian and mammalian hepadnaviruses revealed 16 to 25% amino acid identity (Table 2; see also Fig. S3). Based on amino acid identity, the WSHBV was more similar to avihepadnaviruses. The isoelectric point of this polyprotein was 9.68, which is within the range reported for avian and mammalian hepadnaviruses (pI 9.34 to 10.12). The core ORF of HHBV contains a hydrophobic N-terminal extension of 29 amino acids that forms a signal peptide (35). We identified a similar, but shorter (21 amino acids), hydrophobic N-terminal extension in the WSHBV RR173 genome.
Open reading frame 2 (RF +2; nt 170 to 2536) encodes the viral polymerase protein of 789 amino acids and is similar in size to other hepadnavirus P proteins (785 to 902 amino acids) (Table 2). This ORF contains four conserved domains that include viral DNA polymerases and reverse transcriptase (Fig. 3; see also Table S3 in the supplemental material). Pairwise alignment with avian and mammalian hepadnaviruses indicated 23 to 30% amino acid identity (Table 2; see also Fig. S4). The terminal region of this ORF associated with RNase H activity had the greatest GC content in the genome (65%). This ORF partially overlaps ORF +1 (core protein) and completely overlaps ORF +3 (surface protein).
Open reading frame 3 of WSHBV (RF +3; nt 684 to 1724) was homologous to the large surface protein of hepadnaviruses. We identified a conserved domain for major surface antigen from hepadnavirus (vMSA) in this ORF (nt 1281 to 1487) (Fig. 3; see also Table S3 in the supplemental material). An atypical conserved domain for amelogenin was also identified (nt 834 to 1094). Pairwise alignment with avian and mammalian hepadnaviruses indicated 15 to 27% amino acid identity (Table 2; see also Fig. S5). The EcoRI site typically present in this ORF was not present, as noted above.
Identification of additional fish hepadnavirus sequence.
The best blastn hit for the complete genome of the WSHBV was to the large S gene of HHBV (isolate Kyoto-LS-2006; GenBank accession number AB809504). The E value of this alignment was 8e−27 with 74% identity; however, the query coverage was only 6%. A blastx search identified crane HBV polymerase (GenBank accession number CAD29588) as the best match. The E value was 7e−104 with 35% identity and 58% query coverage. The most convincing evidence that we had identified a genome of a novel hepadnavirus was the graphic summary in which conserved domains of HBV were identified in three reading frames and in the appropriate genomic organization (Fig. 2).
A tblastn query in the transcriptome shotgun assembly (restricted to bony fishes; NCBI taxid:7898) database using the P protein identified a 2,186-bp transcript from a Lake Tanganyika African cichlid (GenBank accession number JL559376; Ophthalmotilapia ventralis) liver library. The E value for this was 4e−49 with 32% identity and 66% query coverage. When this contig was used in a blastx query, the most significant alignment produced was to stork HBV (E value, 4e−53; 34% identity; 67% query coverage).
A tblastx search performed using the complete genome of WSHBV against all available reference genomic sequences of bony fishes identified the best matches as northern pike (GenBank accession numbers NC_025975.1 and NW_011545020.1); however, both had low query coverage (14% and 9%) and E value (0.30) scores. This cursory paleovirological exploration did not identify evidence of endogenized hepadnaviruses in fish genomes.
Phylogenetic analysis and amino acid conservation.
Phylogenetic analyses of predicted WSHBV proteins with respect to those of previously described hepadnaviruses depict well-supported monophyletic groups comprised of the orthohepadnaviruses (mammals) and avihepadnaviruses (birds) which do not include the WSHBV (Fig. 5). Analysis which included the putative hepadnavirus polymerase protein from African cichlid depicts poorly resolved paraphyletic groups between fish species that are distinct from the known viruses of mammalian and bird hosts (Fig. 5A). A similar tree topology was identified for the nucleotide analysis of the complete genomes (Fig. 6). Multiple alignments of the core, polymerase, and surface proteins with a subset of mammalian and avian hepadnaviruses identified amino acid conservation within critical conserved domains (see Fig. S3, S4, and S5 in the supplemental material).
FIG 5.

Unrooted phylogram illustrating the phylogenetic relationships of mammalian, avian, and fish hepadnavirus proteins: polymerase protein (A), surface protein (B), and core protein (C). Results are from maximum-likelihood phylogenetic analysis using PhyML (1,000 replicates). Analysis of the polymerase protein includes that from another fish (African cichlid hepatitis B virus [ACHBV]). Asterisks denote bootstrap support of 100% (***), >90% (**), or >70% (*).
FIG 6.

Unrooted phylogram denoting the phylogenetic relationships of mammalian, avian, and fish hepadnavirus genomes. Results are from maximum-likelihood phylogenetic analysis using PhyML (1,000 replicates). Asterisks denote bootstrap support of 100% (***), >90%(**), or >70% (*).
Electron microscopy.
Examination of the grids prepared using plasma from fish testing positive by PCR showed two types of typical hepadnavirus-like particles. These included complete particles of approximately 40 nm in diameter that were morphologically similar to the Dane particles of hepatitis B virus (Fig. 7). These larger particles were greatly outnumbered by smaller particles of approximately 20 to 25 nm, thought to be composed of virus surface antigen.
FIG 7.

Transmission electron micrographs showing both complete virions of approximately 40 nm in diameter and smaller particles assumed to be composed of self-assembled virus surface proteins. The two types of particles from gradient-purified white sucker serum have a strong resemblance to those of other hepatitis B-like viruses from mammals and birds. Virus was stained using 1% phosphotungstic acid (A) and 0.5% uranyl acetate (B). Scale bar, 100 nm. Images were processed using Adobe Photoshop and included the adjustment of brightness and contrast and application of the unsharp mask filter.
Survey of hepadnavirus prevalence in white sucker.
Core protein RNA was identified in fish sampled from four of five sites in the Great Lakes region (Fig. 8). The highest prevalence was observed in Milwaukee, WI (Milwaukee River), where expression of the core protein gene was noted in 40% of sampled fish (n = 20). At three other sites, Duluth, MN (St. Louis River; n = 86), Maumee, OH (Swan Creek; n = 37), and Green Bay, WI (Fox River; n = 16), core protein RNA was reported in 3 to 7% of sampled fish. In Detroit, MI (Detroit River; n = 10), no core protein RNA was detected.
FIG 8.
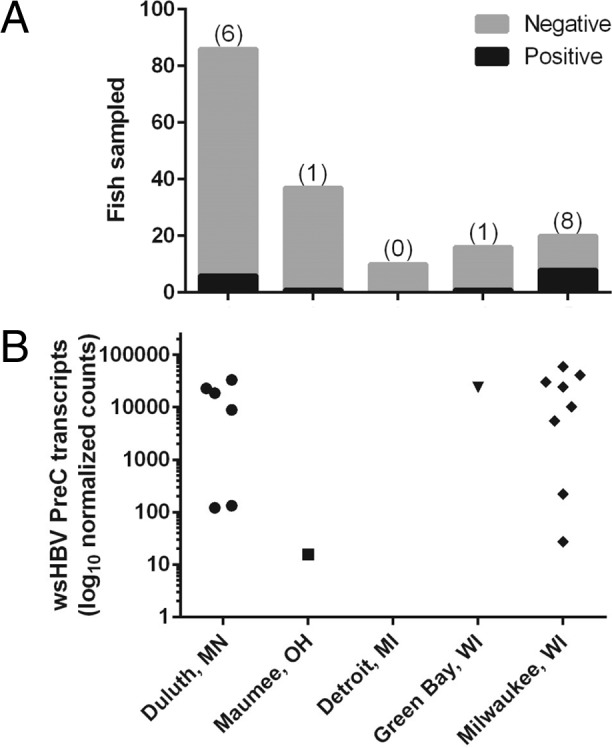
Prevalence of core protein RNA transcription in liver tissue of white sucker collected in the Great Lakes (United States) region. (A) Absolute number of fish with detectable transcripts. (B) Normalized copy number of transcripts.
Assessment of the presence of WSHBV DNA in white sucker from the Root River (Racine, WI) via endpoint PCR identified a prevalence of 20% among sampled fish (n = 20). The prevalence of virus was not statistically different from that observed at the geographically proximate Milwaukee River site (Fisher's exact test, P = 0.3). We detected viral DNA in the plasma samples from the same four Root River individuals that were positive by PCR (data not shown). Absence of amplicons from numerous individuals confirmed that this PCR primer set does not amplify an endogenous viral relic.
Association of WSHBV replication and liver neoplasms.
We compared the prevalence of intrahepatic tumors (HCC, CO, CC, and HA) with the prevalence of viral transcription in white suckers sampled throughout the Great Lakes region (n = 169). The multisite composite prevalence of virus (9.5%) and tumors (4.9%) was low. The Jaccard similarity coefficient between virus prevalence and tumor prevalence was low (0.16). Fish were identified that were virus positive/tumor negative (7.1%), tumor positive/virus negative (3.0%), and positive for both (2.4%) (Table 3). Only one HCC was identified and that fish was virus positive. Four individuals were diagnosed with CC, two of which were also virus positive.
TABLE 3.
Prevalence of viral transcription and tumors in white suckers from each AOC collection site
| Site | Sample size (no. of fish) | No. of fish positive for: |
||
|---|---|---|---|---|
| Virus only | Tumor only | Virus and tumor | ||
| St. Louis River | 86 | 5 | 2 | 1 |
| Maumee | 37 | 1 | 0 | 0 |
| Detroit River | 10 | 0 | 0 | 0 |
| Fox River | 16 | 0 | 0 | 1 |
| Milwaukee River | 20 | 6 | 3 | 2 |
DISCUSSION
Here, we identify the first complete genome of a hepadnavirus associated with fish. The confirmation of a circular genome, observation of complete virions in fish plasma, and presence of the virus in some, but not all, fish or locations provide strong evidence that the WSHBV sequence reported here is that of an infectious agent and not that of an endogenous virus. Additionally, we have identified a putative hepadnavirus polymerase sequence associated with an African cichlid hepatic transcriptome shotgun assembly that provides further evidence that there are other hepadnaviruses that infect fishes. Differences in predicted proteins between WSHBV and hepadnaviruses from mammals and birds are sufficient to support proposing the creation of a novel genus containing WSHBV as the type species. Here, we refrain from suggesting a specific genus name prior to acceptance by the International Committee on Taxonomy of Viruses. Due to the identification of only one viral transcript for the African cichlid hepadnavirus, additional information will be needed before its taxonomic status is clear.
The evolutionary origin of hepadnaviruses remains controversial. Until recently, records of endogenization in the genomes of extant avian, rodent, and primate hosts were lacking. This forced insights into the evolutionary history of members of the family Hepadnaviridae to be based solely on sequence analysis of extant HBVs. Using this approach, the last common ancestor of Orthohepadnavirus and Avihepadnavirus was dated to 30,000 years (36) or 125,000 (37) years ago. Paleovirological analyses have found HBV endogenous viral elements (EVEs) to be widely distributed in birds (38), including the genome of the zebra finch, a species not documented as an extant host of HBV (39). Phylogenetic analyses of endogenous zebra finch HBV-derived EVEs suggest that the common ancestor of viruses in the family Hepadnaviridae infected birds and that the mammalian hepadnaviruses emerged following a bird-mammal host switch (26). Suh et al. (26) suggested that the ancestor of the current members of the family Hepadnaviridae likely infected an amniote ancestor living >324 million years ago, which is significantly earlier than previous estimates. The complete WSHBV genome is 165 bp larger than the longest reported hepadnavirus genome (roundleaf bat), lacks the X protein (or X-like protein), and contains a large, presumptively noncoding region where the X protein is located in orthohepadnaviruses. Duck hepatitis B virus contains what has been described as a vestigial X open reading frame (40). It has been suggested that the common ancestor of Hepadnaviridae did not encode a fourth ORF (X or X-like protein) (41), which is the case for the WSHBV. It is possible that the noncoding region of WSHBV is a precursor to the X open reading frame and that the noncoding region of the WSHBV genome provided noncoding nucleotides as a substrate for overprinting and the emergence of an X protein. Conversely, it is possible that this region devolved from an ancestral X gene that was not necessary in fish hosts. We suggest that the origin of this virus family may predate amniotes. We did not find evidence of endogenization of WSHBV-like viruses in publicly available fish genomes, but few such genomes are presently available for paleovirological investigation.
The prevalence of WSHBV was variable between sampling sites, as assessed via WSHBV core protein mRNA transcription. Virus-positive individuals were identified from four of five sites representing three of the Great Lakes: Superior, Michigan, and Erie. Geographically, fish positive for replicating virus spanned a distance of approximately 1,164 nautical km. In humans, HBVs are represented by eight different genotypes with a cosmopolitan distribution (42). In bats, evidence of different virus strains separated by 430 km has been reported (43). Therefore, the geographic distribution of our sampled virus may include multiple genotypes. We have evidence that nucleotide 3413 of the Root River WSHBV differs from that from the pool of hepatic RNA used for the initial virus discovery. Here, we should emphasize that the prevalence of WSHBV is likely higher at these sites than indicated by our estimates, given that endpoint PCR is less sensitive than quantitative PCR (qPCR) (44) and that measuring viral RNA interjects a virus life cycle bias. Development of a nonlethal, rapid qPCR for future sampling of wild fish is a priority for subsequent assessment of the prevalence of WSHBV as well as investigations into genetic diversity and spatial distribution. These data could facilitate the reconstruction of the evolutionary history of the virus and provide preliminary knowledge of the potential of the virus to be transmitted between localities.
Early investigation of viral replication and antiviral drugs for HBV has relied heavily on animal models, particularly the duck and woodchuck. The discovery of WSHBV and the putative HBV in Ophthalmotilapia ventralis may provide an opportunity for the development of other model systems. This could assist further investigation of hepadnavirus replication, evolution, drug testing, and liver pathogenesis. A hepatitis E virus isolated from the cutthroat trout has provided similar opportunities (45). Fish represent the largest, most diverse group of vertebrates, with over 30,000 extant species. Although tumors have been identified in more than 300 species of fish (46), there is no evidence to support oncovirus induction of hepatic neoplasia in these species. Viruses of the family Herpesviridae and Retroviridae are associated with skin neoplasia in a number of fish species (47, 48). The tropism of HBV for hepatocytes and known association with acute and chronic liver diseases, many of which have historically been attributed to contaminants in previous studies of white sucker, warrant the investigation of the association of HBV with liver pathology. We identified a small percentage (2.4%; 4 of 169) of fish that were both virus and tumor positive. One of these virus-positive fish was diagnosed with a hepatocellular carcinoma. Two additional virus-positive fish were diagnosed with cholangiocarcinoma. This data set, however, is not sufficient to infer relationships between virus and carcinogenesis. Initiation of neoplasia by orthohepadnaviruses often requires decades of chronic infection in humans; thus, the relationship between viral infection and neoplasia is often not straightforward. Of note, the WSHBV described here lacks the X protein associated with the induction of neoplasia (49). To date, attempts have not been made to isolate and propagate this virus via cell culture. Considerable future research is necessary to investigate a mechanistic relationship of WSHBV with liver pathology, including necrosis, inflammation, and preneoplastic and neoplastic changes, as well as advance our understanding of the molecular biology and immunobiology of hepadnaviruses. Identification of this fish host and novel hepadnavirus may offer an additional research model system.
Supplementary Material
ACKNOWLEDGMENTS
We thank Patricia Mazik, Ryan Braham, and Rachel Harrison for assistance with fish collections and processing. We also thank Jim Driver at the University of Montana for operation of the electron microscope.
The research was funded through Great Lakes Restoration Initiative grants to the U.S. Fish and Wildlife Service and the Minnesota Pollution Control Board and by the U.S. Geological Survey's Environmental Health (Contaminant Biology), Ecosystems (Fisheries), and Cooperative Units programs.
Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01278-15.
REFERENCES
- 1.Blazer VS, Hoffman J, Walsh HL, Braham RP, Hahn C, Collins P, Jorgenson Z, Ledder T. 2014. Health of white sucker within the St. Louis River area of concern associated with habitat usage as assessed using stable isotopes. Ecotoxicology 23:236–251. doi: 10.1007/s10646-013-1167-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bowron LK, Munkittrick KR, McMaster ME, Tetreault G, Hewitt LM. 2009. Responses of white sucker (Catostomus commersoni) to 20 years of process and waste treatment changes at a bleached kraft pulp mill, and to mill shutdown. Aquat Toxicol 95:117–132. doi: 10.1016/j.aquatox.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 3.Munkittrick KR, Dixon DG. 1989. Use of white sucker (Catostomus commersoni) populations to assess the health of aquatic ecosystems exposed to low-level contaminant stress. Can J Fish Aquat Sci 46:1455–1462. doi: 10.1139/f89-185. [DOI] [Google Scholar]
- 4.Rafferty SD, Blazer VS, Pinkney AE, Grazio JL, Obert EC, Boughton L. 2009. A historical perspective on the “fish tumors or other deformities” beneficial use impairment at Great Lakes areas of concern. J Great Lakes Res 35:496–506. doi: 10.1016/j.jglr.2009.07.005. [DOI] [Google Scholar]
- 5.U.S. Environmental Protection Agency and the Great Lakes Restoration Initiative Action Plan Panel. 2010. Great Lakes restoration initiative action plan, FY2010–FY2014. U.S. Environmental Protection Agency, Washington, DC. [Google Scholar]
- 6.Blazer VS, Mazik PM, Iwanowicz LR, Braham R, Hahn C, Walsh HL, Sperry A. 2014. Monitoring of wild fish health at selected site in the Great Lakes basin—methods and preliminary results. Open-file report 2014-1027. U.S. Geological Survey, Reston, VA. [Google Scholar]
- 7.Schulze A, Gripon P, Urban S. 2007. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 46:1759–1768. doi: 10.1002/hep.21896. [DOI] [PubMed] [Google Scholar]
- 8.Sai L-T, Yao Y-Y, Guan Y-Y, Shao L-H, Ma R-P, Ma L-Z. 31 October 2014. Hepatitis B virus infection and replication in a new cell culture system established by fusing HepG2 cells with primary human hepatocytes. J Microbiol Immunol Infect doi: 10.1016/j.jmii.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 9.Beck J, Nassal M. 2007. Hepatitis B virus replication. World J Gastroenterol 13:48–64. doi: 10.3748/wjg.v13.i1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marion PL, Oshiro LS, Regnery DC, Scullard GH, Robinson WS. 1980. A virus in Beechey ground squirrels that is related to hepatitis B virus of humans. Proc Natl Acad Sci U S A 77:2941–2945. doi: 10.1073/pnas.77.5.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robertson BH, Margolis HS. 2002. Primate hepatitis B viruses—genetic diversity, geography and evolution. Rev Med Virol 12:133–141. doi: 10.1002/rmv.348. [DOI] [PubMed] [Google Scholar]
- 12.Lanford RE, Chavez D, Brasky KM, Burns RB, Rico-Hesse R. 1998. Isolation of a hepadnavirus from the woolly monkey, a New World primate. Proc Natl Acad Sci 95:5757–5761. doi: 10.1073/pnas.95.10.5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Summers J, Smolec JM, Snyder R. 1978. A virus similar to human hepatitis B virus associated with hepatitis and hepatoma in woodchucks. Proc Natl Acad Sci U S A 75:4533–4537. doi: 10.1073/pnas.75.9.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mason WS, Seal G, Summers J. 1980. Virus of Pekin ducks with structural and biological relatedness to human hepatitis B virus. J Virol 36:829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piasecki T, Kurenbach B, Chrzastek K, Bednarek K, Kraberger S, Martin DP, Varsani A. 2012. Molecular characterisation of an avihepadnavirus isolated from Psittacula krameri (ring-necked parrot). Arch Virol 157:585–590. doi: 10.1007/s00705-011-1197-3. [DOI] [PubMed] [Google Scholar]
- 16.Guo H, Mason WS, Aldrich CE, Saputelli JR, Miller DS, Jilbert AR, Newbold JE. 2005. Identification and characterization of avihepadnaviruses isolated from exotic anseriformes maintained in captivity. J Virol 79:2729–2742. doi: 10.1128/JVI.79.5.2729-2742.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prassolov A, Hohenberg H, Kalinina T, Schneider C, Cova L, Krone O, Frölich K, Will H, Sirma H. 2003. New hepatitis B virus of cranes that has an unexpected broad host range. J Virol 77:1964–1976. doi: 10.1128/JVI.77.3.1964-1976.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pult I, Netter HJ, Bruns M, Prassolov A, Sirma H, Hohenberg H, Chang SF, Frölich K, Krone O, Kaleta EF, Will H. 2001. Identification and analysis of a new hepadnavirus in white storks. Virology 289:114–128. doi: 10.1006/viro.2001.1115. [DOI] [PubMed] [Google Scholar]
- 19.Chang SF, Netter HJ, Bruns M, Schneider R, Frölich K, Will H. 1999. A new avian hepadnavirus infecting snow geese (Anser caerulescens) produces a significant fraction of virions containing single-stranded DNA. Virology 262:39–54. doi: 10.1006/viro.1999.9844. [DOI] [PubMed] [Google Scholar]
- 20.Li M, Li J, Li P, Li H, Su T, Zhu R, Gong J. 2012. Hepatitis B virus infection increases the risk of cholangiocarcinoma: a meta-analysis and systematic review. J Gastroenterol Hepatol 27:1561–1568. doi: 10.1111/j.1440-1746.2012.07207.x. [DOI] [PubMed] [Google Scholar]
- 21.Di Bisceglie AM. 2009. Hepatitis B and hepatocellular carcinoma. Hepatology 49:S56–S60. doi: 10.1002/hep.22962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou H, Wang H, Zhou D, Wang H, Wang Q, Zou S, Tu Q, Wu M, Hu H. 2010. Hepatitis B virus-associated intrahepatic cholangiocarcinoma and hepatocellular carcinoma may hold common disease process for carcinogenesis. Eur J Cancer 46:1056–1061. doi: 10.1016/j.ejca.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 23.World Health Organization. 2002. Hepatitis B. World Health Organization, Geneva, Switzerland: http://www.who.int/csr/disease/hepatitis/HepatitisB_whocdscsrlyo2002_2.pdf?ua=1. [Google Scholar]
- 24.Souza BF, Drexler JF, de Lima RS, Rosário MDO, Netto EM. 2014. Theories about evolutionary origins of human hepatitis B virus in primates and humans. Braz J Infect Dis 18:535–543. doi: 10.1016/j.bjid.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Funk A, Mhamdi M, Will H, Sirma H. 2007. Avian hepatitis B viruses: molecular and cellular biology, phylogenesis, and host tropism. World J Gastroenterol 13:91–103. doi: 10.3748/wjg.v13.i1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suh A, Brosius J, Schmitz J, Kriegs JO. 2013. The genome of a Mesozoic paleovirus reveals the evolution of hepatitis B viruses. Nat Commun 4:1791. doi: 10.1038/ncomms2798. [DOI] [PubMed] [Google Scholar]
- 27.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. 2012. Primer3—new capabilities and interfaces. Nucleic Acids Res 40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F, Marshall D. 2010. Tablet—next generation sequence assembly visualization. Bioinformatics 26:401–402. doi: 10.1093/bioinformatics/btp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui J, Holmes EC. 2012. Endogenous hepadnaviruses in the genome of the budgerigar (Melopsittacus undulatus) and the evolution of avian hepadnaviruses. J Virol 86:7688–7691. doi: 10.1128/JVI.00769-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edgar R. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acid Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abascal F, Zardoya R, Posada D. 2005. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 33.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 34.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bruss V. 2004. Envelopment of the hepatitis B virus nucleocapsid. Virus Res 106:199–209. doi: 10.1016/j.virusres.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 36.Orito E, Mizokami M, Ina Y, Moriyama EN, Kameshima N, Yamamoto M, Gojobori T. 1989. Host-independent evolution and a genetic classification of the hepadnavirus family based on nucleotide sequences. Proc Natl Acad Sci U S A 86:7059–7062. doi: 10.1073/pnas.86.18.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Hemert FJ, van de Klundert MA, Lukashov VV, Kootstra NA, Berkhout B, Zaaijer HL. 2011. Protein X of hepatitis B virus: origin and structure similarity with the central domain of DNA glycosylase. PLoS One 6:e23392. doi: 10.1371/journal.pone.0023392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui J, Zhao W, Huang Z, Jarvis ED, Gilbert MT, Walker PJ, Holmes EC, Zhang G. 2014. Low frequency of paleoviral infiltration across the avian phylogeny. Genome Biol 15:539. doi: 10.1186/s13059-014-0539-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gilbert C, Feschotte C. 2010. Genomic fossils calibrate the long-term evolution of hepadnaviruses. PLoS Biol 8:e1000495. doi: 10.1371/journal.pbio.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin B, Anderson DA. 2000. A vestigial X open reading frame in duck hepatitis B virus. Intervirology 43:185–190. doi: 10.1159/000025037. [DOI] [PubMed] [Google Scholar]
- 41.Suh A, Weber CC, Kehlmaier C, Braun EL, Green RE, Fritz U, Ray DA, Ellegren H. 2014. Early Mesozoic coexistence of amniotes and Hepadnaviridae. PLoS Genet 10:e1004559. doi: 10.1371/journal.pgen.1004559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saeed U, Waheed Y, Ashraf M. 2014. Hepatitis B and hepatitis C viruses: a review of viral genomes, viral induced host immune responses, genotypic distributions and worldwide epidemiology. Asian Pac J Trop Dis 4:88–96. doi: 10.1016/S2222-1808(14)60322-4. [DOI] [Google Scholar]
- 43.He B, Zhang F, Xia L, Hu T, Chen G, Qiu W, Fan Q, Feng Y, Guo H, Tu C. 2015. Identification of a novel Orthohepadnavirus in pomona roundleaf bats in China. Arch Virol 160:335–337. doi: 10.1007/s00705-014-2222-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abe A, Inoue K, Tanaka T, Kato J, Kajiyama N, Kawaguchi R, Tanaka S, Yoshiba M, Kohara M. 1999. Quantitation of hepatitis B virus genomic DNA by real-time detection PCR. J Clin Microbiol 37:2899–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Batts W, Yun S, Hedrick R, Winton J. 2011. A novel member of the family Hepeviridae from cutthroat trout (Oncorhynchus clarkii). Virus Res 158:116–123. doi: 10.1016/j.virusres.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 46.Anders K, Yoshimizu M. 1994. Role of viruses in the induction of skin tumours and tumour-like proliferations of fish. Dis Aquat Org 19:215–232. doi: 10.3354/dao019215. [DOI] [Google Scholar]
- 47.Quackenbush SL, Casey JM, Bowser PR, Rovnak J. 2010. Cancers induced by piscine retroviruses, p 191–218. In Dudley J. (ed), Retroviruses and insights into cancer. Springer, New York, NY. [Google Scholar]
- 48.Getchell RG, Casey JW, Bowser PR. 1998. Seasonal occurrence of virally induced skin tumors in wild fish. J Aquat Anim Health 10:191–201. doi: 10.1577/1548-8667(1998)010<0191:SOOVIS>2.0.CO;2. [DOI] [Google Scholar]
- 49.Kew MC. 2011. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J Gastroenterol Hepatol 26:144–152. doi: 10.1111/j.1440-1746.2010.06546.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.