

Abstract
The American Heart Association supports an association between periodontal diseases and atherosclerosis but not a causal association. This study explores the use of the integrin β6−/− mouse model to study the causality. We investigated the ability of a polymicrobial consortium of Porphyromonas gingivalis, Treponema denticola, Tannerella forsythia, and Fusobacterium nucleatum to colonize the periodontium and induce local and systemic inflammatory responses. Polymicrobially infected Itgβ6−/− mice demonstrate greater susceptibility to gingival colonization/infection, with severe gingival inflammation, apical migration of the junctional epithelium, periodontal pocket formation, alveolar bone resorption, osteoclast activation, bacterial invasion of the gingiva, a greater propensity for the bacteria to disseminate hematogenously, and a strong splenic T cell cytokine response. Levels of atherosclerosis risk factors, including serum nitric oxide, oxidized low-density lipoprotein, serum amyloid A, and lipid peroxidation, were significantly altered by polybacterial infection, demonstrating an enhanced potential for atherosclerotic plaque progression. Aortic gene expression revealed significant alterations in specific Toll-like receptor (TLR) and nucleotide-binding domain- and leucine-rich-repeat-containing receptor (NLR) pathway genes in response to periodontal bacterial infection. Histomorphometry of the aorta demonstrated larger atherosclerotic plaques in Itgβ6−/− mice than in wild-type (WT) mice but no significant difference in atherosclerotic plaque size between mice with polybacterial infection and mice with sham infection. Fluorescence in situ hybridization demonstrated active invasion of the aortic adventitial layer by P. gingivalis. Our observations suggest that polybacterial infection elicits distinct aortic TLR and inflammasome signaling and significantly increases local aortic oxidative stress. These results are the first to demonstrate the mechanism of the host aortic inflammatory response induced by polymicrobial infection with well-characterized periodontal pathogens.
INTRODUCTION
Chronic periodontal disease (PD) is an inflammatory condition affecting the periodontal tissues (gingiva, periodontal ligament, and alveolar bone) attributed to a complex polymicrobial biofilm that forms on tooth surfaces in subjects with poor oral hygiene. Interest in studying the systemic effects of PD is growing as evidence linking PD with numerous systemic diseases, especially atherosclerotic vascular disease (ASVD), accumulates (1). The PD-ASVD link is a well-established association for which the mechanism of association is poorly understood. Rodent models of PD are commonly used for these studies, yet current models are limited by the fact that laboratory rodents do not naturally develop PD (2). There are several common models of induced periodontal disease in rodents, including the ligature-induced model, which is falling out of use in favor of more physiologically relevant acute (3) and chronic (4) oral infections with human periodontal pathogenic bacteria. A novel model of PD is the integrin β6−/− (Itgβ6−/−) mouse, which spontaneously develops severe PD characterized by apical migration of the junctional epithelium, gingival epithelial hyperplasia, epithelial edema, infiltration of neutrophils, periodontal pocket formation, and periodontal ligament destruction (5).
Integrin αvβ6 is an epithelial cell-specific integrin, which is expressed postnatally only in the hair follicles, intestinal and junctional epithelia, ameloblasts, and kidney (6, 7). It is able to activate transforming growth factor β1 (TGF-β1) (8), and interestingly, mice deficient in β6 integrin have a normal life span but develop periodontal disease characterized by apical migration of the junctional epithelium on the tooth, periodontal inflammation, and alveolar bone resorption (5, 9). These characteristics are suggested to be linked to poor TGF-β1 activation, as TGF-β1 is considered an anti-inflammatory cytokine in human PD (10). In human PD, the expression of αvβ6 integrin is strongly downregulated in the periodontal pocket epithelium (5), and therefore, it has been proposed that human PD represents “acquired β6 integrin deficiency” (7).
One characteristic of PD found in Itgβ6−/− mice and not in current models is the formation of deep periodontal pockets. Since presently known human periodontal pathogenic bacteria are anaerobic (11), we predicted that the deep periodontal pockets formed in the gingival cavity of these mice would provide a better niche for surface colonization by the bacteria used in our periodontal infection model. Thus, in Itgβ6−/− mice, the bacteria could colonize and proliferate to a greater abundance, as more permissive anaerobic conditions are found in mice with these pockets than in mice lacking these pockets. Furthermore, the higher bacterial burden in these pockets is expected to result in greater tissue destruction, as these bacteria produce a variety of virulence factors that directly damage tissues as well as incite strong inflammatory responses, which in turn would result in more severe bacterium-induced symptoms of PD. Other mouse models used to study this association, including the ApoE−/− mouse model, poorly develop the full range of periodontal disease symptoms because these mice develop oral bone loss but do not exhibit gingival tissue inflammation that characterizes human periodontal disease. Therefore, we sought to investigate the usefulness of this mouse model for studying the mechanistic link between periodontal disease and atherosclerosis. We hypothesized that better development of PD in infected Itgβ6−/− mice will enable better systemic dissemination of bacteria and thus might alter physiological homeostasis sufficiently to develop atherosclerotic plaque without the necessity of a hyperlipidemic background. We investigated our hypothesis using four well-characterized pathogenic, anaerobic periodontal bacterial species, Porphyromonas gingivalis, Treponema denticola, Tannerella forsythia, and Fusobacterium nucleatum, as a polymicrobial periodontal inoculum over a chronic infection period. In this study, we provide evidence to support our hypothesis that the Itgβ6−/− mouse model is a superior model for studies of the causal relationship between PD and inflammatory atherosclerotic vascular disease.
MATERIALS AND METHODS
Microbial strains and inocula.
Strains used were as follows: P. gingivalis FDC 381, T. denticola ATCC 35404, T. forsythia ATCC 43037, and F. nucleatum ATCC 49256. Bacteria were grown as previously described (12–15). The cultures were then harvested by centrifugation at 9,000 × g for 15 min at 4°C, and the pellets were resuspended in reduced transport fluid (RTF). Bacterial concentrations were determined by counting in a Petroff-Hausser bacterial counting chamber, and 2.5 × 108 P. gingivalis, 2.5 × 108 T. denticola, 2.5 × 108 T. forsythia, 2.5 × 108 F. nucleatum bacteria were mixed in equal proportions to attain a final inoculum with a concentration of 109 total bacteria (16).
Ethics statement.
The University of Florida has an assurance with the Office of Laboratory Animal Welfare (OLAW) and follows U.S. Public Health Services (PHS) policy, the Animal Welfare Act and Animal Welfare Regulations, and the Guide for the Care and Use of Laboratory Animals (17). The University of Florida is also AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care) accredited. All animal experimentation procedures were approved by the University of Florida Institutional Animal Care and Use Committee (IACUC) under protocol number 201004539.
Mouse infection, gingival bacterial plaque sampling, and organ collection.
Integrin β6 knockout (Itgβ6−/−) mice on an FVB background were bred at the University of British Columbia, and the corresponding wild-type (WT) FVB NHan/Hsd controls were ordered from Harlan Laboratories (Indianapolis, IN). Twenty-six-week-old mice were randomly divided into infection (n = 12) and control (n = 12) groups for both the Itgβ6−/− and wild-type groups. Mice were acclimated and treated with antibiotics to reduce normal oral flora, as described previously (16). Mice were then inoculated with 109 total bacteria on four consecutive days per week every third week for 24 weeks (8 infections, while sham-infected mice received sterile vehicle). Mouse gingival cavities were swabbed with a sterile cotton swab 1 week after each infection to monitor bacterial colonization.
Upon sacrifice, five right mandibles from each group were stored in 10% neutral buffered formalin (NBF), five right maxillae from each group were stored in liquid nitrogen, and the remaining jaws were autoclaved. Heart, aorta, liver, spleen, kidney, and lung were divided and collected in RNAlater (Life Technologies, Grand Island, NY), RTF, and 10% NBF. Spleens of six mice in each group were collected in RPMI 1640. Blood was collected, and sera were separated and stored at −80°C.
Detection of genomic DNA (gDNA) in gingival bacterial plaque and systemic organs.
Gingival bacterial plaque samples collected after infection were used directly to perform a colony PCR with bacterial species-specific primers to monitor gingival colonization. Total DNA was extracted from the heart, liver, kidney, spleen, and lungs (n = 6 of each organ in each group) and from the aorta (n = 5 in each group), as previously described (13). PCR was run by using a Bio-Rad thermal cycler with the following 16S rRNA bacterial species-specific oligonucleotide primers: 5′-TGTAGATGACTGATGGTGAAAACC-3′ (forward) and 5′-ACGTCATCCCCACCTTCCTC-3′ (reverse) for P. gingivalis, 5′-TAATACCGAATGTGCTCATTTACAT-3′ (forward) and 5′-CTGCCATATCTCTATGTCATTGCTCTT-3′ (reverse) for T. denticola, 5′-AAAACAGGGGTTCCGCATGG-3′ (forward) and 5′-TTCACCGCGGACTTAACAGC-3′ (reverse) for T. forsythia, and 5′-TAAAGCGCGTCTAGGTGGTT-3′ (forward) and 5′-ACAGCTTTGCGACTCTCTGT-3′ (reverse) for F. nucleatum (14, 16). Cycling was performed for each species as previously described (4). These primers were confirmed by NCBI PrimerBLAST to specifically amplify only the bacterial species that they were designed to detect, despite the fact that either individual forward or reverse primer may individually detect numerous cultivable and uncultivable bacterial species.
Serum antibody analysis.
Serum antibody titers against each of the four bacterial species from six mice in each group were determined by an enzyme-linked immunosorbent assay (ELISA) as previously described, using whole-cell formalin-fixed bacteria as the antigen (14, 18). Mouse sera were diluted 1:100 for both IgG and IgM assessments. Mouse serum antibody concentrations were determined by using a gravimetric standard curve that consisted of eight mouse IgG and IgM concentrations (Sigma-Aldrich) (16), mean values for each group were calculated from the six mice, and the statistical significance between the mean values for each group was determined. The fold change in antibody titers between infected and control mice was determined by dividing the mean antibody titer of infected mice by the mean antibody titer of sham-infected mice for both the Itgβ6−/− and WT groups. The quotient, which represents the mean fold change in specific antibody titer due to infection, was graphed.
Morphometric analysis of periodontal alveolar bone resorption.
Mouse maxilla and mandibles collected upon sacrifice were processed and imaged as previously described (12). Images were used to measure the area of alveolar bone resorption between the cementoenamel junction (CEJ) and alveolar bone crest (ABC) to determine horizontal alveolar bone resorption and intrabony defects (16). The percentages of intrabony defects were calculated for all tooth surfaces, where percent values indicate the number of sites found to contain intrabony defects per total number of sites analyzed per group. Broken jaws or jaws missing teeth were excluded from the total.
Histology of gingival inflammation.
Five right mandibles were decalcified in phosphate-buffered saline (PBS) containing 0.4 M EDTA and 2% formaldehyde, embedded in paraffin, and sectioned (4 μm) along the mesiodistal plane. Sections were stained with hematoxylin and eosin (H&E) for histological analysis and scanned with a ScanScope CS system (Aperio, Vista, CA). The scanned slides were viewed at a ×200 magnification with ImageScope viewing software (Aperio). Evidence of inflammation was determined as previously described (18).
Morphometric analysis of aortic sections.
For histological analysis, six aortas were collected from each group and processed as previously described (12). Morphometric analysis of the atherosclerotic plaque area and intimal as well as medial thicknesses was then performed by using the Olympus microscopy analysis system. Measurements were taken twice by a reviewer in a blind manner, and average values for each tissue section were reported. Ratios of the thickness of the intimal layer to that of the medial aortic layer were calculated to reduce variation dependent upon differing aortic sizes from the ascending to descending and abdominal regions (12).
Fluorescence in situ hybridization.
Fluorescence in situ hybridization (FISH) was performed on gingival and aortic sections, as described previously (16), and sections were probed for P. gingivalis and T. denticola together or T. forsythia and F. nucleatum together because of probe hybridization temperature compatibility. The following individual oligonucleotide probes (Invitrogen, Carlsbad, CA) were used to detect 16S rRNA for each species: POGI (5′-CAATACTCGTATCGCCCGTTATTC-3′) for P. gingivalis, TDEN (5′-CATGACTACCGTCATCAAAGAAGC-3′) for T. denticola, B(T)AFO (5′-CGTATCTCATTTTATTCCCCTGTA-3′) for T. forsythia, and FUSO (5′-CTAATGGGACGCAAAGCTCTC-3′) for F. nucleatum (19, 20). Probes POGI and B(T)AFO were Alexa Fluor 488 labeled, and probes TDEN and FUSO were Alexa Fluor 568 labeled. Samples were incubated for 3 h at 46°C for POGI/TDEN and at 48°C for B(T)AFO/FUSO. Tissue sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and mounted with Mowiol. Stained slides were dried overnight before being viewed under a fluorescence microscope. Images were acquired at a ×63 magnification on a Leica upright microscope equipped with filters for wavelengths of 405 nm, 488 nm, and 546 nm for DAPI, Cy3, and fluorescein isothiocyanate (FITC), respectively, by using Zeiss AxioVision v4.8 software.
Aortic inflammatory cell infiltration.
Immunohistochemical staining was performed on aortic cross sections from infected (n = 6) and sham-infected (n = 6) mice, as previously described (12). Positive cells were counted in three high-power fields (magnification, ×100) for each section from each mouse by a reviewer who was blind to the groups, using Image Pro software (12). F4/80-specific primary antibody (catalog number ab100790; Abcam, Cambridge, MA) and CD3-specific antibody (catalog number ab5690; Abcam, Cambridge, MA) were used to detect macrophages and T cells, respectively.
Lipid profile analysis and measurements of oxLDL, serum amyloid A, endogenous nitric oxide, and malondialdehyde levels.
Lipid profile analysis and measurements of oxidized low-density lipoprotein (LDL) (oxLDL), serum amyloid A (SAA), and nitric oxide (NO) levels were performed on sera collected at sacrifice (n = 6 for all analyses). Lipid profiles were analyzed by using a gel filtration high-performance liquid chromatography (HPLC) analysis system for lipid profiles at Skylight Biotech Inc. (Akita, Japan). Serum oxLDL titers were evaluated by using the mouse serum oxidized low-density lipoprotein ELISA kit (Tsz ELISA; Waltham, MA). Serum levels of SAA were estimated by using a colorimetric ELISA kit according to the manufacturer's instructions (Kamiya Biomedical, Seattle, WA). Serum NO concentrations were determined by using a BioVision (Milpitas, CA) NO fluorometric assay kit (13). Malondialdehyde concentrations were measured in livers, gingival tissue extracts, and aortas from 6 mice in each group by using the Lipid Peroxidation Assay kit from BioVision, according to the manufacturer's directions.
Th1/Th2/Th17 splenic cytokine assessment.
Upon sacrifice, spleens from four mice in each group were homogenized in RPMI 1640 with l-glutamine supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% penicillin-streptomycin, 1% HEPES buffer, 1% sodium pyruvate, and 0.27% glucose (RPMI-complete); stored on ice for 1 h; pelleted at 1,200 rpm for 10 min; resuspended in 1.5 ml RPMI-complete–10% dimethyl sulfoxide (DMSO); and stored at −80°C for further analysis. To perform the recall, thawed cells were normalized to 105 cells/ml, and 104 cells were added to wells of a 96-well plate for each mouse in triplicate. The plate was incubated at 37°C in 5% CO2 for 72 h, cells were then pelleted at 1,200 rpm for 10 min, and the supernatant was collected and stored at −80°C for cytokine analysis (12).
Supernatant samples from splenic cell incubations for each mouse in each group of infected and sham-infected mice were analyzed for the presence of select Th1, Th2, and Th17 cytokines by using RayBio Quantibody Mouse TH17 Array 1 (RayBio, Norcross, GA), according to the manufacturer's instructions. Signals were scanned on a GenePix 4400 scanner, using GenePix Pro 7.2.29.002 software. Results were analyzed by using the RayBio Analysis Tool Excel sheet. The cytokines interleukin-13 (IL-13), IL-17F, IL-21, IL-22, and macrophage inflammatory protein 3a (MIP-3a) were excluded from the analysis because their signals were too low to be distinguished from the background. The concentration of each cytokine was determined by using a standard curve included in the array (12).
RT2 Profiler PCR array.
The expression of 84 genes involved in the immune response to bacteria was examined by reverse transcription-quantitative PCR (qRT-PCR) with the RT2 Profiler Mouse Antibacterial Response PCR array (SABiosciences, Valencia, CA). RNA was extracted from aortas (n = 3), converted to cDNA, and run and analyzed as previously described (12). Changes in gene expression levels of >2.5-fold were considered significant (13).
Statistical analysis.
Statistical analyses of horizontal alveolar bone loss, serum antibody responses, and cytokine array data were performed by using analysis of variance (ANOVA) with Bonferroni's multiple-comparison posttest, intrabony defects were analyzed by the χ2 test with GraphPad Prism v. 5 software, and P values of <0.05 were considered statistically significant. Data in graphs are represented as means with standard deviations (SD). Aortic histology measurements were analyzed by ANOVA with Fisher's post hoc least significant difference analysis using the Statview program, P values of <0.05 were considered statistically significant, and data in graphs are represented as means with standard errors.
RESULTS
Infected Itgβ6−/− mice develop severe periodontal disease.
Detection of bacterial DNA in the gingival cavity of Itgβ6−/− mice by PCR revealed a transient gingival colonization pattern (Table 1), suggesting that the bacteria could be residing within the deep periodontal pockets and, hence, inaccessible by our sampling method. Although we did not perform viability assays on our oral plaque samples, Hajishengallis et al. (21) demonstrated that P. gingivalis could be detected in the oral cavity of mice by quantitative PCR (qPCR) up to 6 weeks after final oral infection with P. gingivalis, which suggests that the organisms may be viable in our mice 1 week after infection, when we collected oral samples for PCR. However, serum antibodies against periodontal pathogens suggest bacterial gingival colonization. Serum levels of IgG against P. gingivalis T. denticola, and F. nucleatum in infected Itgβ6−/− mice were significantly elevated (Fig. 1A, top left), while WT mice had significantly elevated IgG levels against all four bacterial species (Fig. 1A, bottom left). Serum IgM levels against F. nucleatum in Itgβ6−/− mice (Fig. 1A, top right) and against T. forsythia in WT mice (Fig. 1A, bottom right) were statistically significantly elevated.
TABLE 1.
Itgβ6−/− and WT mouse gingival plaque samples positive for bacterial gDNA by PCRa
| Group (total no. of samples) | No. of gingival plaque samples positive for P. gingivalis/T. denticola/T. forsythia/F. nucleatum on day: |
||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| Itgβ6−/− mice | |||||||
| Infection (11) | 0/0/0/0 | 0/0/0/0 | 1/0/0/5 | NC | 3/1/2/11 | 0/0/0/0 | 0/0/0/0 |
| Control (9) | 0/0/0/0 | NC | NC | NC | 0/0/0/0 | NC | NC |
| WT mice | |||||||
| Infection (13) | 0/0/0/0 | NC | 0/0/0/0 | NC | 9/8/8/8 | NC | NC |
| Control (13) | 0/0/0/0 | NC | NC | NC | 0/0/0/0 | NC | NC |
NC, not collected to allow the bacterial biofilm to adhere to the gingival surface, invade epithelial cells, and multiply.
FIG 1.

Periodontally infected Itgβ6−/− mice develop periodontal disease with severe periodontal pathology. (A) Serum IgG and IgM responses to orally infected pathogens expressed as fold increases over values for sham-infected controls. Data are representative of results from triplicate experiments. (B) 2-D horizontal alveolar bone resorption. Data are representative of results from triplicate experiments. (C) Representative images showing the area of horizontal bone resorption, outlined from ABC to CEJ. (D) Apical junctional epithelial migration distance (micrometers). (E) Number of leukocytes per 50-μm2 area of gingival tissue. (F) Representative H&E-stained histological jaw tissue sections demonstrating epithelial apical migration. Brackets indicate locations of gingival junctional epithelial migration. Arrowheads indicate the cementoenamel junction, where the gingival junctional epithelium naturally ends. Bar = 100 μm. (G) Number of osteoclasts per 50-μm2 area of gingival tissue. (H) Representative tartrate-resistant acid phosphatase (TRAP)-stained histological jaw tissue sections. Arrowheads indicate TRAP-stained osteoclasts. Bar = 100 μm. (I) Representative images of infected Itgβ6−/− mouse jaws demonstrating positive FISH staining (white arrowheads) for T. denticola (red) (i) and P. gingivalis (green) (ii). Insets show higher magnifications of the boxed area. All the tests were run in triplicates. Data points and error bars represent means ± SD for infected (n = 6) and control (n = 6) mice in each group unless stated otherwise (for panels A and I, n = 6; for panels B to H, n = 5). *, P < 0.05; **, P < 0.01; ***, P < 0.001 (determined by ANOVA with a Bonferroni posttest).
Alveolar bone resorption is the hallmark feature of PD and must occur for disease to be considered periodontitis rather than gingivitis. We found that total bone resorption measured by two-dimensional (2-D) morphometry was significantly (P < 0.001) greater in infected Itgβ6−/− mice than in uninfected Itgβ6−/− mice and infected WT mice (Fig. 1B and C). Sham-infected Itgβ6−/− control mice had greater bone resorption than did uninfected WT control mice, which was expected given the disruption of oral homeostasis due to their inability to activate TGF-β (Fig. 1C). Intrabony defects, which in humans indicate more localized patterns of bone destruction on the tooth surface (Table 2), were significantly more prevalent in infected Itgβ6−/− mice (30%) than in sham-infected Itgβ6−/− mice (10%) and were significantly more prevalent in infected WT mice (12%) than in sham-infected WT mice (3%). Together, the alveolar bone resorption data indicate that infected Itgβ6−/− mice experienced the most severe form of PD of the four mouse groups.
TABLE 2.
Intrabony defect in infected and sham-infected Itgβ6−/− mice and WT mice
| Mouse group | Infection with P. gingivalis, T. denticola, T. forsythia, and F. nucleatum |
Control |
||
|---|---|---|---|---|
| No. of positive sites/total no. of sites | % positive sites | No. of positive sites/total no. of sites | % positive sites | |
| Itgβ6−/− | 36/120 | 30a | 9/84 | 10c |
| WT | 22/180 | 12b | 5/150 | 3 |
P < 0.001 compared to Itgβ6−/− sham-infected mice; P < 0.001 compared to infected WT mice.
P < 0.01 compared to WT sham-infected mice.
P < 0.05 compared to WT sham-infected mice.
Histological examination of gingival inflammation in all four groups revealed the greatest inflammation in infected Itgβ6−/− mice. The apical migration of the junctional/pocket epithelium was significantly (P < 0.001) greater (Fig. 1D and Fi) and leukocyte counts were higher (Fig. 1E) in infected Itgβ6−/− mice. Sham-infected Itgβ6−/− mice had greater numbers of invading leukocytes than did sham-infected WT mice (Fig. 1E), which is expected given their genetic mutation predisposing them to increased periodontal inflammation. The number of activated osteoclasts, the bone-resorbing cells, was significantly (P < 0.001) greater in infected Itgβ6−/− mice (Fig. 1G and Hi) than in sham-infected Itgβ6−/− mice (Fig. 1G and Hii) and infected WT mice (Fig. 1G and Hiii), indicating more active alveolar bone resorption.
Finally, we performed fluorescence in situ hybridization (FISH) to determine if periodontal bacteria had invaded gingival tissues. Numerous T. denticola bacteria were detected within the gingival connective tissue of infected Itgβ6−/− mice (Fig. 1Ii), and P. gingivalis was detected in the gingival epithelium of infected Itgβ6−/− mice (Fig. 1Iii), confirming bacterial invasion of the periodontal tissues; however, T. forsythia and F. nucleatum were not detected. No bacteria were detected in the gingival tissues of infected WT mice.
Bacterial spread induces a peripheral T cell response.
Genomic DNA (gDNA) of all four bacterial species was detected in the hearts and aortas of Itgβ6−/− mice, while fewer WT mouse aortas were positive for bacterial gDNA, although all four species were detected (Tables 3 and 4). Bacterial gDNA was sparse, and the distribution was uneven in the remaining organs examined in both Itgβ6−/− and WT mice. To determine the nature of the systemic immune response to periodontal bacteria, we performed a cytokine array on media from cultures of unstimulated splenocytes to determine the concentrations of select Th1-, Th2-, and Th17-type cytokines in all four groups of mice (Table 5). The levels of all cytokines analyzed were significantly (P < 0.05) elevated in infected Itgβ6−/− mice relative to those in uninfected Itgβ6−/− mice, indicating a large population of stimulated T cells. However, cytokine levels were not significantly different between infected and sham-infected WT mice, indicating that the T cell population in infected WT mice remained largely unexposed to the bacterial antigen. It is likely that infected Itgβ6−/− mice disseminate bacteria into systemic circulation more frequently and at higher levels due to their severe periodontal disease than do infected WT mice, resulting in a larger population of stimulated T cells. We are cautious in drawing conclusions about cytokine responses due to the small number of mice assessed. The levels of many cytokines have a very large SD due to a wide spread or due to a single outlier. For example, even when the outlier for IL-1β is removed, significance is still maintained, indicating that the average value for the other mice is sufficiently high and hence can be considered a true biological phenomenon. This is not true for gamma interferon (IFN-γ), for which there is a wider spread. It may be that since FVB mice are more prone to produce a Th2-type cytokine response upon infection, there is wide variation in the production of Th1-type cytokines, although further studies including more mice will be necessary to address this issue.
TABLE 3.
Distribution of gDNA of P. gingivalis, T. denticola, T. forsythia, and F. nucleatum in systemic organs of Itgβ6−/− mice
| Itgβ6−/− mouse group | No. of systemic tissue samples positive for P. gingivalis/T. denticola/T. forsythia/F. nucleatum |
||||||
|---|---|---|---|---|---|---|---|
| Heart (n = 10) | Aorta (n = 5) | Liver (n = 10) | Spleen (n = 5) | Kidney (n = 10) | Lung (n = 10) | Brain (n = 7) | |
| Bacterial infection | 8/3/7/6 | 4/3/4/4 | 1/0/1/1 | 1/1/1/1 | 6/0/0/3 | 2/0/0/0 | 0/0/0/0 |
| Control | 0/0/0/0 | 0/0/0/0 | 0/0/0/0 | 0/0/0/0 | 0/0/0/0 | 0/0/0/0 | 0/0/0/0 |
TABLE 4.
Distribution of gDNA of P. gingivalis, T. denticola, T. forsythia, and F. nucleatum in systemic organs of WT mice
| WT mouse group | No. of systemic tissue samples positive for P. gingivalis/T. denticola/T. forsythia/F. nucleatum |
||||||
|---|---|---|---|---|---|---|---|
| Heart (n = 13) | Aorta (n = 8) | Liver (n = 13) | Spleen (n = 7) | Kidney (n = 13) | Lung (n = 13) | Brain (n = 7) | |
| Bacterial infection | 6/7/0/0 | 2/5/1/3 | 0/0/2/0 | 2/0/0/1 | 2/0/0/0 | 3/1/0/6 | 1/0/0/0 |
| Control | 0/0/0/0 | 0/0/0/0 | 0/0/0/0 | 0/0/0/0 | 0/0/0/0 | 0/0/0/0 | 0/0/0/0 |
TABLE 5.
Splenocyte cytokine concentrations in infected and sham-infected mice
| Cytokine type | Cytokinee | Mean concn (pg/ml) (×103) ± SD (n = 4) |
|||
|---|---|---|---|---|---|
|
Itgβ6−/− mice |
WT mice |
||||
| Infected | Uninfected | Infected | Uninfected | ||
| Th1 | IFN-γ | 0.384 ± 0.37b | 0.124 ± 0.068 | 0. 922 ± 0.028 | 0.0516 ± 0.021 |
| IL-1β | 4.3 ± 3.3c,d | 0.753 ± 0.44 | 0.451 ± 0.12 | 0.429 ± 0.085 | |
| IL-6 | 10.7 ± 1.9c,d | 3.22 ± 1.9 | 2.15 ± 0.57 | 3.16 ± 0.29 | |
| IL-12p70 | 5.11 ± 2.0c,d | 2.03 ± 1.1 | 1.12 ± 0.32 | 1.01 ± 0.24 | |
| IL-28 | 4.08 ± 0.54c,d | 1.03 ± 0.55 | 0.561 ± 0.098 | 0.555 ± 0.12 | |
| TNF-α | 10.0 ± 0.23c,d | 2.20 ± 1.2 | 2.01 ± 0.79 | 1.07 ± 0.34 | |
| Th2 | IL-2 | 14.5 ± 0.26c,d | 4.09 ± 2.3 | 2.12 ± 0.43 | 2.12 ± 0.47 |
| IL-4 | 30.9 ± 1.2a,d | 16.1 ± 8.5 | 8.09 ± 2.2 | 6.99 ± 1.5 | |
| IL-5 | 12.6 ± 0.11c,d | 3.35 ± 1.9 | 1.87 ± 0.39 | 1.84 ± 0.32 | |
| IL-10 | 19.3 ± 0.62c,d | 5.79 ± 3.2 | 3.26 ± 0.71 | 2.95 ± 0.65 | |
| TGF-β | 1.36 ± 0.29c,d | 0.478 ± 0.26 | 0.258 ± 0.058 | 0.261 ± 0.060 | |
| Th17 | IL-17 | 2.53 ± 0.17c,d | 0.692 ± 0.38 | 0.375 ± 0.087 | 0.359 ± 0.074 |
| IL-23 | 0.403 ± 0.05c,d | 0.132 ± 0.068 | 0.0744 ± 0.022 | 0.0643 ± 0.013 | |
P ≤ 0.05 compared to the Itgβ6−/− sham-infected group.
P ≤ 0.05 compared to the infected WT group.
P ≤ 0.001 compared to the Itgβ6−/− sham-infected group.
P ≤ 0.0001 compared to the infected WT group.
TNF-α, tumor necrosis factor alpha.
Bacterial dissemination elevates levels of systemic risk factors.
Chronic periodontal infection is known to enhance hyperlipidemia of ApoE−/− mice to a more proatherogenic state, but the effect on serum lipids in nonhyperlipidemic mice of the FVB strain had not yet been determined; therefore, we assessed the serum lipid profiles of all four mouse groups. This analysis revealed significantly (P < 0.05) elevated levels of total cholesterol (Fig. 2A) in infected Itgβ6−/− mice compared to sham-infected mice, indicating an increased risk of development of atherosclerosis. Infected WT mice did not exhibit any differences in total cholesterol, triglycerides, or lipoprotein particle fractions between infected and sham-infected mice (Fig. 2B). Unexpectedly, WT mice had significantly higher levels of total cholesterol and total triglycerides than did Itgβ6−/− mice, indicating that the inflammatory state of Itgβ6−/− mice alters the lipid profile yet does not enhance atherogenic potential.
FIG 2.

Periodontal infection enhances systemic risk factors for atherosclerosis. (A) Total serum cholesterol and total triglyceride concentrations; (B) serum lipid fraction concentrations; (C) serum oxLDL concentrations; (D) aortic tissue MDA concentrations; (E) serum NO concentrations; (F) serum SAA concentrations. Chol, cholesterol; Trigly, triglycerides; CM, chylomicrons; VLDL, very-low-density lipoprotein; LDL, low-density lipoprotein; HDL, high-density lipoprotein; i, infected; c, control. All the tests were run in triplicates. Data points and error bars represent means ± SD for infected (n = 6) and control (n = 6) mice in each group unless stated otherwise. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (determined by ANOVA with a Bonferroni posttest).
The level of serum oxLDL (Fig. 2C), a significant risk factor for atherosclerosis development, was significantly elevated in infected Itgβ6−/− mice relative to those in sham-infected mice and infected WT mice. To assess the source of the elevated oxLDL levels, we tested levels of malondialdehyde, a by-product of lipid peroxidation, in gingival tissue extract, liver tissue, and aortic tissue. Levels of malondialdehyde, and therefore lipid peroxidation, were elevated significantly (P < 0.001) in the aortas of infected Itgβ6−/− mice relative to those in sham-infected mice and infected WT mice (Fig. 2D), but no significant differences were detected between groups for gingival tissue extract and liver samples, indicating that oxidizing conditions were localized to the aorta. Additionally, the level of serum nitric oxide, a marker of endothelial function, was decreased in Itgβ6−/− mice relative to those in the sham-infected controls (Fig. 2E), supporting a trend of altered homeostasis in the aorta. The level of serum amyloid A, a systemic inflammatory marker, was significantly elevated in infected Itgβ6−/− mice relative to the levels in sham-infected mice, while infection in WT mice did not alter SAA levels (Fig. 2F). Together, these data suggest that infection promotes inflammatory conditions in the aorta specifically by cytokine production and lipid peroxidation, which may predispose mice to developing atherosclerosis.
To determine if bacterial interaction with the aorta promoted local inflammation that could drive atherosclerotic plaque development, we examined changes in expression levels of genes involved in the antibacterial response (Table 6). We found that between infected Itgβ6−/− mice and sham-infected Itgβ6−/− mice, the levels of 34 out of 84 genes examined were elevated >2.5-fold, and none were downregulated. Between infected Itgβ6−/− mice and infected WT mice, 24 genes were upregulated >2.5-fold. There were 20 genes shared between these two comparisons (Fig. 3), and these 20 genes, which are upregulated under conditions of severe periodontal disease and systemic inflammation, regardless of periodontal infection, are suggested to be key genes for PD-induced atherosclerosis. Unaltered genes in Itgβ6−/− or WT mice included Akt1, Apcs, Bpi, Ccl5, Cd14, Chuk, Crp, Cxcl1, Cxcl3, Fadd, Hsp90aa1, Ifna9, Ifnb1, Ikbkb, Il6, Irak1, Irak3, Jun, Map2k1, Map2k3, Map2k4, Map3k7, Mapk1, Mapk14, Mapk3, Mapk8, Myd88, Nlrc4, Nod1, Nod2, Pik3ca, Prtn3, Rac1, Rela, Ripk2, Slc11a1, Sugt1, Ticam1, Tirap, Tlr4, Tlr5, Tlr6, Tnfrsf1a, Tollip, Traf6, and Xiap. These data indicate that the upregulation of a distinct set of genes is responsible for elevated local aortic inflammation, which could drive plaque development. Further investigation of the role of these genes is warranted to understand the mechanisms involved in aortic inflammation.
TABLE 6.
Antimicrobial response-related gene expression changes in aortad
| Gene group | Gene | Fold change (n = 3) |
|||
|---|---|---|---|---|---|
| Itgβ6−/−i/Itgβ6−/−c | Itgβ6−/−i/WTi | WTi/WTc | Itgβ6−/−c/WTc | ||
| Antimicrobial peptide | Camp | 5.54 | |||
| Ctsg | 4.02 | ||||
| Lcn2 | 7.09 | 6.19 | |||
| Ltf | 27.35 | 9.89 | |||
| Lyz2 | 9.40a | 4.02 | |||
| Slpi | 24.31 | 22.78 | |||
| Apoptosis | Casp1 | 4.03 | |||
| Casp8 | 3.14 | 3.73 | |||
| Il12α | 8.88 | ||||
| Il12β | 8.07 | 3.19 | |||
| Mpo | 5.57 | ||||
| Nfkbiα | 4.20 | ||||
| Inflammatory response | Ccl3 | 10.48 | 4.55 | ||
| Ccl4 | 11.47 | 4.36 | |||
| Il1b | 3.02 | 3.78 | |||
| Tnf | 3.79 | 5.02 | |||
| NOD-like receptor signaling | Birc3 | 4.30 | 4.88 | ||
| Card6 | 3.87 | ||||
| Card9 | 4.38 | 11.42 | 5.25 | ||
| Il18 | 5.33 | 9.36 | 4.41 | ||
| Mefv | 6.35a | ||||
| Naip1 | 4.66 | 3.97 | |||
| Nlrc4 | 5.86 | 4.02 | |||
| Nlrp1a | 4.41a | ||||
| Nlrp3 | 4.85a | ||||
| Pstpip1 | 15.35 | 12.15 | |||
| Pycard | 12.64 | 5.97 | 3.17 | ||
| Toll-like receptor signaling | Irf5 | 6.09 | |||
| Irf7 | 3.43 | 3.69 | |||
| Lpb | 3.59 | 7.85 | |||
| Ly96b | 3.15 | ||||
| Ripk1 | 3.18 | ||||
| Ticam2c | 6.42a | 3.23 | |||
| Tlr1 | 16.11 | 7.11 | |||
| Tlr2 | 3.85 | ||||
| Tlr9 | 11.26 | 6.19 | |||
| Other PRRs | Dmbt1 | 3.93 | |||
| Zbp1 | 10.78 | 5.55 | |||
P ≤ 0.05.
Also known as Md2.
Also known as TRAM.
Itgβ6−/−i, infected Itgβ6−/− mouse group; Itgβ6−/−c, sham-infected control Itgβ6−/− mouse group; WTi, infected WT mouse group; WTc, sham-infected control WT mouse group; PRRs, pattern recognition receptors.
FIG 3.
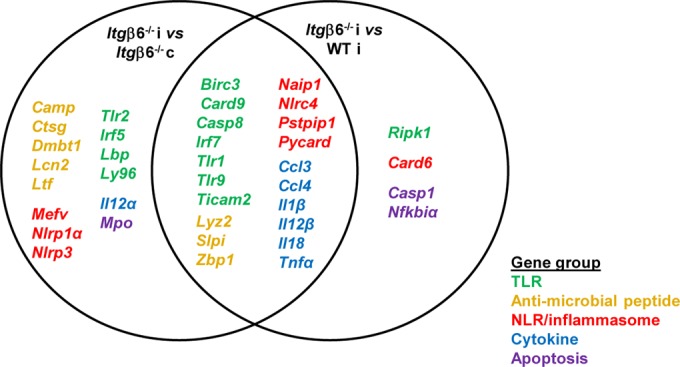
Key genes for periodontal pathogen-induced aortic inflammation. Comparisons of genes upregulated in infected versus uninfected Itgβ6−/− mouse aortas and genes upregulated in infected Itgβ6−/− mouse versus infected WT mouse aortas (Table 6) reveal overlap of 20 genes, all expressed in the context of severe periodontal disease and increased systemic inflammation. These genes are upregulated in infection, may be important for aortic inflammatory responses regardless of periodontal disease status, and warrant further investigation for understanding the mechanisms of aortic inflammation. Genes involved in the same inflammation pathways are the same color. i, infected; c, control.
Aortic plaque localization and histology.
Whether chronic periodontal infection contributes to aortic plaque growth in the FVB mouse strain has never before been determined. Minimal plaque was detected in the aortic arch of mice from all groups (Fig. 4A), although a larger plaque area was detected in both Itgβ6−/− mouse groups than in WT mouse groups. The only significant difference in atherosclerotic plaque area was between the sham-infected mouse groups. The aortic intimal layer thickness of plaques in infected and sham-infected Itgβ6−/− mice was significantly greater than that in sham-infected WT mice (Fig. 4B). The aortic intimal layer thickness/medial layer thickness ratio, which corrects for differences in vessel wall size, indicated that infected Itgβ6−/− mice developed thicker plaques than did infected WT and sham-infected WT mice (Fig. 4C). Arrows in representative images of the aortic root from each group demonstrate small atherosclerotic plaques (Fig. 4D). Together, these data strongly suggest that Itgβ6−/− mice are more prone to plaque development than WT mice, likely because they experience more systemic inflammation.
FIG 4.
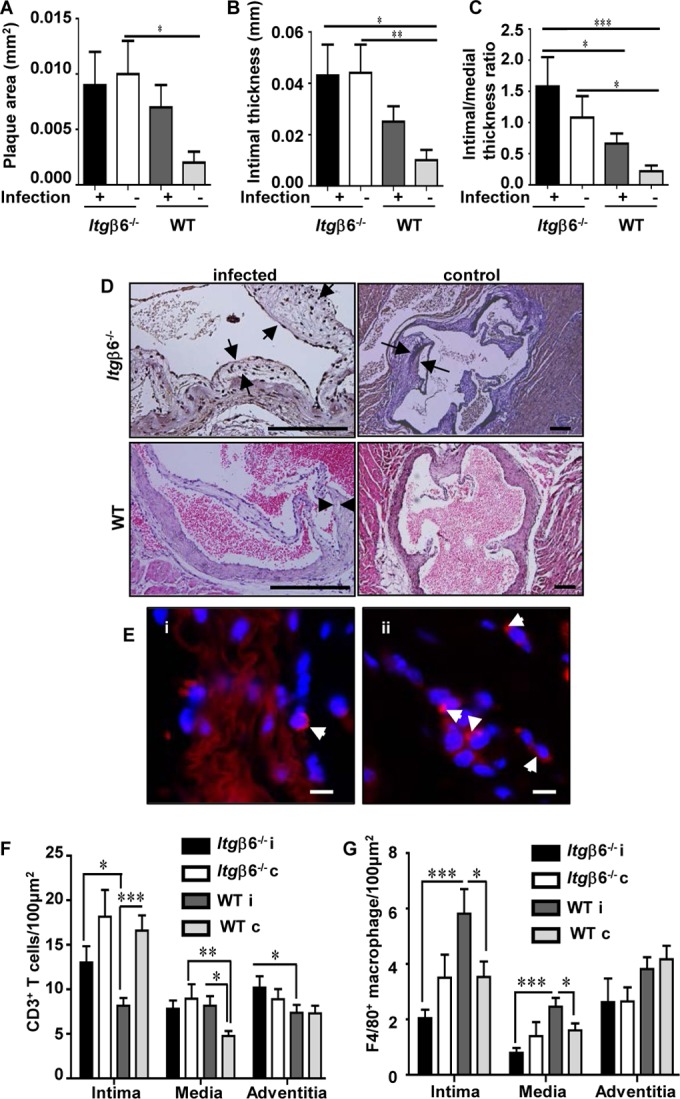
Itgβ6−/− mice exhibit increased aortic plaque accumulation regardless of periodontal infection. (A) Aortic plaque areas. (B) Aortic intimal thicknesses. (C) Aortic intimal layer/medial layer thickness ratios. (D) Representative images of H&E-stained aortic roots from each group. Bar = 50 μm. (E) Representative images of P. gingivalis-positive (red) (white arrowheads) FISH-stained aorta sections within adventitial tissue of infected Itgβ6−/− mice (i) and WT mice (ii). Bar = 10 μm. (F) Aortic CD3+ T cells. (G) Aortic F4/80+ macrophages. All tests were run in triplicates. Data points and error bars represent means ± standard errors of the means for infected (n = 6) and control (n = 6) mice in each group unless stated otherwise (n = 6). i, infected; c, control. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (determined by ANOVA with a Bonferroni posttest). Multiple cross sections were stained and evaluated for measurement and analysis of aortic plaques (2 to 3 sections per aortic area per mouse) by two individuals blind to the treatment groups of the study.
Although minimal aortic plaque was detected in mice, FISH was performed to demonstrate bacterial invasion of the aorta, which may be responsible for aortic gene expression level changes. P. gingivalis was detected in the aortic adventitial layer of four infected Itgβ6−/− mice and two infected WT mice (Fig. 4Ei and ii), indicating vascular dissemination and aortic invasion; however, T. denticola, T. forsythia, and F. nucleatum were not detected. Our attempts to culture live organisms from mouse aortic tissues according to a protocol modified from the one described previously by Rafferty et al. (22) were unsuccessful. Immunohistochemical staining was performed to determine the extent of CD3+ T cell and F4/80+ macrophage infiltration, as both cell types contribute to inflammation and atherosclerotic plaque progression. Infected Itgβ6−/− mice had significantly (P < 0.05) elevated numbers of CD3+ T cells in intimal and adventitial layers of the aorta relative to those in infected WT mice (Fig. 4F). Unexpectedly, sham-infected Itgβ6−/− and WT mice had elevated numbers of T cells in the intimal layer relative to those in their infected counterparts, suggesting that infection probably reduces inflammatory cell recruitment. Infected WT mice had significantly (P < 0.001) elevated numbers of F4/80+ macrophages relative to those in infected Itgβ6−/− mice and WT controls in both the aortic intimal and medial layers (Fig. 4G). Notably, the F4/80+ cell counts were lower than the CD3+ cell counts throughout the vessel wall in all mouse groups, which may explain why minimal atherosclerotic plaque was detected in all mouse types, as macrophages are the major inflammatory cell type involved in atherosclerotic plaque formation.
DISCUSSION
Several observational studies support an association between PD and ASVD, but the causal relationship is unclear (1). A causal relationship between PD and ASVD is difficult to demonstrate without a reproducible animal model of PD; in this study, we provide direct evidence for a causal relationship by using a novel Itgβ6−/− mouse model. The Itgβ6−/− mouse is the only model that mimics severe human-like PD. As mice are naturally resistant to atherosclerosis development, common mouse models to study atherosclerosis rely on genetic mutations that alter physiology and create an environment permissive to fatty atherosclerotic plaque deposition. Our observations presented here revealed that infection of Itgβ6−/− mice induced significantly greater alveolar bone resorption and gingival pathology, with clear histological differences in gingival junctional epithelial migration, numbers of infiltrating lymphocytes, and numbers of activated osteoclasts.
Based on our results, it appears that in mice, tissue disruption (i.e., gingival pocket formation) must be necessary to permit sufficient bacterial colonization and proliferation to elicit an immune response and soft and hard tissue destruction. Itgβ6−/− mice then provide a more holistic model in which to study tissue responses to infectious oral disease. Our decision to use a polymicrobial model of infection was based on the polymicrobial nature of PD (11) and of atherosclerotic vascular disease-affected tissues (1). While specific periodontal bacteria are associated with PD, including P. gingivalis, T. denticola, T. forsythia, and F. nucleatum (11, 23), no single species has been identified as the sole causative agent of PD. Recent studies on the pathogenesis of P. gingivalis demonstrated that shifts in the entire oral community are responsible for alveolar bone resorption (21), a phenomenon further explained by the polymicrobial synergy and dysbiosis (PSD) model of disease (24), supporting the use of our polymicrobial community model of disease induction.
As we expected, significant gingival tissue destruction in infected Itgβ6−/− mice is correlated with bacterial systemic dissemination. We observed significantly increased splenic cytokine responses in Itgβ6−/− mice but not in infected WT mice, demonstrating that sufficient bacterial seeding occurred in Itgβ6−/− mice to induce a peripheral T cell response. Additionally, our observation of localized malondialdehyde, a by-product of lipid peroxidation, only in the aortic tissue further substantiates our hypothesis that local oxidative stress is responsible for the changes in serum oxLDL levels. We demonstrated viable P. gingivalis and T. denticola invasion of aortic tissues by FISH, which were detected exclusively in the adventitial layer. This finding was unexpected given that our previous studies detected bacteria only within the intimal layer (12, 13). It is suggested that immune cells trafficking to the intimal layer from the adventitial layer may be carrying the bacteria into the vessel, rather than bacteria invading the intima from the lumen. In support of this, peripheral blood dendritic cells were shown to carry bacteria, and P. gingivalis may be carried into atherosclerotic plaques by these peripheral blood cells (25). This phenomenon may explain why bacteria were observed only in aortic adventitial tissues in our model.
The FVB mouse strain is considered an atherosclerosis-resistant strain, as shown in previously studies by Dansky et al. which demonstrated that ApoE−/− FVB mice accumulate minimal atherosclerotic plaque despite total serum cholesterol levels being significantly elevated over those in ApoE−/− C57BL/6 mice (26). This surprising result was attributed to differences in genetic susceptibility and was later suggested to be due to differences in the A20 (Tnfaip3) gene (27), which is involved in immune responses. Our results demonstrate that even with active aortic infection, Itgβ6−/− FVB mice remain atherosclerosis resistant, although the Itgβ6 gene deletion results in a minor but increased propensity to form atherosclerotic plaques. Likewise, infection did not significantly increase the number of inflammatory cells in the adventitial layers of either Itgβ6−/− or WT mice relative to those in controls. In fact, fewer macrophages than T cells were detected in all vessel layers in all mouse groups, corresponding to previously reported reduced macrophage recruitment in the FVB mouse model (28). It will be interesting to determine if there are differences in the subsets of T cells and macrophages recruited to the aortic vessel in infected and control Itgβ6−/− and WT mice, as FVB mice tend to mount an M2 macrophage response (29), which is considered part of the anti-inflammatory Th2 response. Also, differences in the recruitment or polarization of immune cell subpopulations may explain our unexpected inflammatory cell counts.
Our aortic gene expression array found a distinct set of 20 genes upregulated in response to PD that promote inflammatory signaling and an antibacterial response. These genes are key candidate genes responsible for promoting atherosclerotic lesions in mice, and further investigation of the pathways involved may provide in-depth mechanistic insights into a causal relationship between PD and atherosclerosis. Our study implicates Toll-like receptor 1 (TLR1) and TLR9 in atherosclerosis development, neither of which is typically considered important in either PD or atherosclerosis, while we observed that TLR2 and TLR4 were not affected, although both are implicated in PD and ASVD. As TLR1 is known to form heterodimers with TLR2, the atherosclerosis-protective effects of TLR2 gene deletion (1, 30) may in fact be because TLR1 is no longer able to bind ligands (31). On the other hand, upregulation of TLR9, NLR, and inflammasome genes, which detect intracellular bacterial components, including DNA, RNA, and peptidoglycan units, indicates that bacterial invasion of aortic vessel cells is occurring. P. gingivalis is known to invade human coronary artery endothelial cells (32), and our finding corroborates this phenomenon in vivo, as intracellular inflammatory response genes are upregulated, including TLR9 and NLR pathway components. Sporadic but chronic exposure to periodontal pathogens through hematogenous spread from the oral cavity may constantly incite inflammation in the aorta, even if the exposure is short and the bacteria are rapidly removed from circulation. Thus, while pathogens may be rapidly cleared, repeated activation of inflammatory signaling in aortic tissues may be occurring and promoting atherosclerotic plaque development.
Toll-like receptor signaling, lipid peroxidation, and decreased NO levels are implicated in atherosclerosis development (33–35). In addition to peroxidizing lipids, reactive oxygen species (ROS) may interact with NO and reduce NO bioavailability, resulting in endothelial dysfunction (35, 36), and we found significantly decreased serum NO levels in infected Itgβ6−/− mice. Thus, our data suggest that local vascular infection induces ROS production via TLR signaling, leading to lipid peroxidation and elevation of serum oxLDL levels, and concomitantly reduces NO concentrations and promotes endothelial dysfunction. Therefore, the conditions in the aortas of infected Itgβ6−/− mice suggest an environment primed for lesion initiation. These data suggest that this may be the major mechanism by which periodontal bacteria causally induce atherosclerosis. In support of this finding, it was recently reported that intravenous injection of live Aggregatibacter actinomycetemcomitans bacteria into spontaneously hyperlipidemic mice (KOR-ApoEshl) promotes the upregulation of TLR gene expression and lipid peroxidation (37). The authors of that study found that increases in the expression levels of ROS-producing enzymes were significant for the response to live bacterial infection but not to heat-killed bacteria, further supporting the notion that viable bacteria are responsible for actively promoting atherosclerotic lesion formation.
Based on our physiological assessment and inflammatory gene expression array, we propose a model by which periodontal infection of Itgβ6−/− mice primes the vasculature for atherosclerotic lesion development (Fig. 5). Itgβ6−/− mice spontaneously develop junctional epithelial migration and form anaerobic periodontal pockets. The anaerobic bacteria used in oral infection are able to colonize these periodontal pockets (Fig. 5A) and establish a severe infection, resulting in significant gingival inflammation, alveolar bone resorption, and, ultimately, systemic dissemination of periodontal bacterial pathogens (Fig. 5B). Bacteria or their components (Fig. 5C) in the bloodstream interact with and actively invade aortic vessel tissue, stimulating innate immune responses, including TLR signaling and inflammasome activation. Signaling from these pathways stimulates the production of inflammatory cytokines that promote local vascular inflammation (Fig. 5D). Additionally, infection increases the local production of reactive oxygen species through inflammatory TLR signaling (Fig. 5E), which can cause oxidation of LDL and reduce NO bioavailability, promoting endothelial dysfunction. We therefore argue that local vascular inflammation under conditions of elevated serum cholesterol levels, oxidative stress with lipid peroxidation, and reduced NO levels primes the vessel for atherosclerotic lesion development (Fig. 5F). Although the mechanisms behind various systemic inflammatory disorders may seem identical, the variations in pathology result from different causative events. These events include factors like the nature of infection, susceptibility of the host, and the host response. This is especially true in the case of our proposed relationship between PD and ASVD. Periodontal disease is caused by dysbiosis of a polymicrobial biofilm that always results in chronic infection of the oral cavity that can, over many years, seed bacteria into systemic circulation, where they can incite a continuous inflammatory response in vascular tissues. This is in contrast to various acute infections of single-bacterium origin that last only several days to a few weeks and could contribute to vascular inflammation only for the short period of infection. Hence, the association between PD and ASVD is unique among inflammatory bacterial infections.
FIG 5.

Proposed model of infection-induced atherosclerosis development in infected Itgβ6−/− mice. Itgβ6−/− mice spontaneously develop PD symptoms and periodontal pockets. (A) Anaerobic bacteria used in oral infection colonize the anaerobic periodontal pocket and establish a severe infection, resulting in significant gingival pathology, alveolar bone resorption, and systemic dissemination of periodontal bacterial pathogens. (B and C) Bacteria or their components interact with and actively invade aortic vessel tissue (B) and stimulate innate immune responses, including TLR signaling and inflammasome activation (C). (D and E) Signaling from these pathways stimulates the production of inflammatory cytokines and promotes local vascular inflammation (D) as well as increased local production of reactive oxygen species (E), which can cause oxidation of LDL and reduce NO bioavailability. (F) Local vascular inflammation under conditions of reduced NO concentrations and elevated serum oxLDL and cholesterol concentrations primes the vessel for atherosclerosis development. Pg, P. gingivalis; Td, T. denticola; Tf, T. forsythia; Fn, F. nucleatum; JNK, Jun N-terminal protein kinase; LBP, lipopolysaccharide-binding protein.
We hypothesized that, with strong induction of PD in the infected Itgβ6−/− mouse model, we would obtain enhanced systemic seeding of bacteria and that this would alter physiological homeostasis sufficiently to progress atherosclerotic plaque development without the need for a hyperlipidemic background. Based on our findings, it can be construed while Itgβ6−/− mice are not prone to infection-induced atheroma formation, they have a greater propensity to form atherosclerotic plaque than do WT mice, which is likely due to their increased inflammatory burden. However, on top of this inflammatory burden, periodontal infection was not sufficient to induce significant atherosclerotic plaque development, although it did promote atherosclerosis-permissive conditions by altering the lipid profile and creating local inflammatory, oxidizing conditions in the aorta. These findings suggests that PD and systemic spread of bacteria that can invade vascular tissues prime the aorta for plaque development but that an additional factor is necessary to initiate atherosclerotic plaque development. Factors such as genetic susceptibility of the host, endothelial cell damage, aging, a longer infection period, or a high-fat diet may be necessary to incite lesion formation, all of which can be tested by using the superior Itgβ6−/− mouse model. We predict that introducing this as-yet-unknown factor into the orally infected Itgβ6−/− mouse model will result in exaggerated aortic plaque development, due to infection-enhanced risk factors that are known to contribute to atherosclerotic lesion initiation. In conclusion, this is the first study to demonstrate that chronic periodontal infection with a polymicrobial consortium can induce local vascular TLR signaling concomitant with local lipid peroxidation and reduction of NO levels, independent of hyperlipidemia, using a holistic model of periodontal disease.
ACKNOWLEDGMENTS
We thank Dean Sheppard (University of California, San Francisco) for providing the Itgβ6−/− mice through Hannu Larjava, University of British Columbia, Vancouver, BC, Canada.
This study was supported by the National Institutes of Health National Institute for Dental and Craniofacial Research (grants R01DE020820 to L.K. and F31DE023492 to I.M.V.) and Canadian Institutes of Health Research (H.L.).
We declare that we have no conflicts of interest.
REFERENCES
- 1.Lockhart PB, Bolger AF, Papapanou PN, Osinbowale O, Trevisan M, Levison ME, Taubert KA, Newburger JW, Gornik HL, Gewitz MH, Wilson WR, Smith SC, Baddour LM, American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young, Council on Epidemiology and Prevention, Council on Peripheral Vascular Disease, Council on Clinical Cardiology. 2012. Periodontal disease and atherosclerotic vascular disease: does the evidence support an independent association? A scientific statement from the American Heart Association. Circulation 125:2520–2544. doi: 10.1161/CIR.0b013e31825719f3. [DOI] [PubMed] [Google Scholar]
- 2.Graves DT, Fine D, Teng Y-TA, Van Dyke TE, Hajishengallis G. 2008. The use of rodent models to investigate host-bacteria interactions related to periodontal diseases. J Clin Periodontol 35:89–105. doi: 10.1111/j.1600-051X.2007.01172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gibson FC, Hong C, Chou H-H, Yumoto H, Chen J, Lien E, Wong J, Genco CA. 2004. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation 109:2801–2806. doi: 10.1161/01.CIR.0000129769.17895.F0. [DOI] [PubMed] [Google Scholar]
- 4.Kesavalu L, Sathishkumar S, Bakthavatchalu V, Matthews C, Dawson D, Steffen M, Ebersole JL. 2007. Rat model of polymicrobial infection, immunity, and alveolar bone resorption in periodontal disease. Infect Immun 75:1704–1712. doi: 10.1128/IAI.00733-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghannad F, Nica D, Fulle MIG, Grenier D, Putnins EE, Johnston S, Eslami A, Koivisto L, Jiang G, McKee MD, Häkkinen L, Larjava H. 2008. Absence of αvβ6 integrin is linked to initiation and progression of periodontal disease. Am J Pathol 172:1271–1286. doi: 10.2353/ajpath.2008.071068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larjava H, Koivisto L, Hakkinen L, Heino J. 2011. Epithelial integrins with special reference to oral epithelia. J Dent Res 90:1367–1376. doi: 10.1177/0022034511402207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larjava H, Koivisto L, Heino J, Häkkinen L. 2014. Integrins in periodontal disease. Exp Cell Res 325:104–110. doi: 10.1016/j.yexcr.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 8.Annes JP, Chen Y, Munger JS, Rifkin DB. 2004. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J Cell Biol 165:723–734. doi: 10.1083/jcb.200312172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohazab L, Koivisto L, Jiang G, Kytömäki L, Haapasalo M, Owen GR, Wiebe C, Xie Y, Heikinheimo K, Yoshida T, Smith CE, Heino J, Häkkinen L, McKee MD, Larjava H. 2013. Critical role for αvβ6 integrin in enamel biomineralization. J Cell Sci 126:732–744. doi: 10.1242/jcs.112599. [DOI] [PubMed] [Google Scholar]
- 10.Dutzan N, Vernal R, Hernandez M, Dezerega A, Rivera O, Silva N, Aguillon JC, Puente J, Pozo P, Gamonal J. 2009. Levels of interferon-gamma and transcription factor T-Bet in progressive periodontal lesions in patients with chronic periodontitis. J Periodontol 80:290–296. doi: 10.1902/jop.2009.080287. [DOI] [PubMed] [Google Scholar]
- 11.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL Jr. 1998. Microbial complexes in subgingival plaque. J Clin Periodontol 25:134–144. doi: 10.1111/j.1600-051X.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 12.Velsko IM, Chukkapalli SS, Rivera MF, Lee J-Y, Chen H, Zheng D, Bhattacharyya I, Gangula PR, Lucas AR, Kesavalu L. 2014. Active invasion of oral and aortic tissues by Porphyromonas gingivalis in mice causally links periodontitis and atherosclerosis. PLoS One 9:e97811. doi: 10.1371/journal.pone.0097811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chukkapalli SS, Rivera MF, Velsko IM, Lee J-Y, Chen H, Zheng D, Bhattacharyya I, Gangula PR, Lucas AR, Kesavalu L. 2014. Invasion of oral and aortic tissues by oral spirochete Treponema denticola in ApoE−/− mice causally links periodontal disease and atherosclerosis. Infect Immun 82:1959–1967. doi: 10.1128/IAI.01511-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Velsko IM, Chukkapalli SS, Rivera-Kweh MF, Chen H, Zheng D, Bhattacharyya I, Gangula PR, Lucas AR, Kesavalu L. 2015. Fusobacterium nucleatum alters atherosclerosis risk factors and enhances inflammatory markers with an atheroprotective immune response in ApoE(null) mice. PLoS One 10:e0129795. doi: 10.1371/journal.pone.0129795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chukkapalli SS, Rivera-Kweh MF, Velsko IM, Chen H, Zheng D, Bhattacharyya I, Gangula PR, Lucas AR, Kesavalu L. 6 February 2015 Chronic oral infection with major periodontal bacteria Tannerella forsythia modulates systemic atherosclerosis risk factors and inflammatory markers. Pathog Dis doi: 10.1093/femspd/ftv009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rivera MF, Lee J-Y, Aneja M, Goswami V, Liu L, Velsko IM, Chukkapalli SS, Bhattacharyya I, Chen H, Lucas AR, Kesavalu LN. 2013. Polymicrobial infection with major periodontal pathogens induced periodontal disease and aortic atherosclerosis in hyperlipidemic ApoE(null) mice. PLoS One 8:e57178. doi: 10.1371/journal.pone.0057178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 18.Bainbridge B, Verma RK, Eastman C, Yehia B, Rivera M, Moffatt C, Bhattacharyya I, Lamont RJ, Kesavalu L. 2010. Role of Porphyromonas gingivalis phosphoserine phosphatase enzyme SerB in inflammation, immune response, and induction of alveolar bone resorption in rats. Infect Immun 78:4560–4569. doi: 10.1128/IAI.00703-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sunde PT, Olsen I, Göbel UB, Theegarten D, Winter S, Debelian GJ, Tronstad L, Moter A. 2003. Fluorescence in situ hybridization (FISH) for direct visualization of bacteria in periapical lesions of asymptomatic root-filled teeth. Microbiology 149:1095–1102. doi: 10.1099/mic.0.26077-0. [DOI] [PubMed] [Google Scholar]
- 20.Moter A, Leist G, Rudolph R, Schrank K, Choi B-K, Wagner M, Göbel UB. 1998. Fluorescence in situ hybridization shows spatial distribution of as yet uncultured treponemes in biopsies from digital dermatitis lesions. Microbiology 144:2459–2467. doi: 10.1099/00221287-144-9-2459. [DOI] [PubMed] [Google Scholar]
- 21.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA. 2011. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rafferty B, Jönsson D, Kalachikov S, Demmer RT, Nowygrod R, Elkind MSV, Bush H Jr, Kozarov E. 2011. Impact of monocytic cells on recovery of uncultivable bacteria from atherosclerotic lesions. J Intern Med 270:273–280. doi: 10.1111/j.1365-2796.2011.02373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fardini Y, Wang X, Témoin S, Nithianantham S, Lee D, Shoham M, Han YW. 2011. Fusobacterium nucleatum adhesin FadA binds vascular endothelial cadherin and alters endothelial integrity. Mol Microbiol 82:1468–1480. doi: 10.1111/j.1365-2958.2011.07905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hajishengallis G, Lamont RJ. 2012. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol 27:409–419. doi: 10.1111/j.2041-1014.2012.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carrion J, Scisci E, Miles B, Sabino GJ, Zeituni AE, Gu Y, Bear A, Genco CA, Brown DL, Cutler CW. 2012. Microbial carriage state of peripheral blood dendritic cells (DCs) in chronic periodontitis influences DC differentiation, atherogenic potential. J Immunol 189:3178–3187. doi: 10.4049/jimmunol.1201053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dansky HM, Charlton SA, Sikes JL, Heath SC, Simantov R, Levin LF, Shu P, Moore KJ, Breslow JL, Smith JD. 1999. Genetic background determines the extent of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 19:1960–1968. doi: 10.1161/01.ATV.19.8.1960. [DOI] [PubMed] [Google Scholar]
- 27.Wolfrum S, Teupser D, Tan M, Chen KY, Breslow JL. 2007. The protective effect of A20 on atherosclerosis in apolipoprotein E-deficient mice is associated with reduced expression of NF-kappaB target genes. Proc Natl Acad Sci U S A 104:18601–18606. doi: 10.1073/pnas.0709011104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stein O, Dabach Y, Ben-Naim M, Halperin G, Stein Y. 2006. Lower macrophage recruitment and atherosclerosis resistance in FVB mice. Atherosclerosis 189:336–341. doi: 10.1016/j.atherosclerosis.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 29.Chaudhuri A, Wilson NS, Yang B, Martinez AP, Liu J, Zhu C, Bricker N, Couto S, Modrusan Z, French D, Cupp J, Ashkenazi A. 2013. Host genetic background impacts modulation of the TLR4 pathway by RON in tissue-associated macrophages. Immunol Cell Biol 91:451–460. doi: 10.1038/icb.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mullick AE. 2005. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest 115:3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 32.Reyes L, Herrera D, Kozarov E, Roldán S, Progulske-Fox A. 2013. Periodontal bacterial invasion and infection: contribution to atherosclerotic pathology. J Clin Periodontol 40(Suppl 14):S30–S50. doi: 10.1111/jcpe.12079. [DOI] [PubMed] [Google Scholar]
- 33.Fogelman AM, Shechter I, Seager J, Hokom M, Child JS, Edwards PA. 1980. Malondialdehyde alteration of low-density lipoproteins leads to cholesteryl ester accumulation in human monocyte-macrophages. Proc Natl Acad Sci U S A 77:2214–2218. doi: 10.1073/pnas.77.4.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiao Y, Darzi Y, Tawaratsumida K, Marchesan JT, Hasegawa M, Moon H, Chen GY, Núñez G, Giannobile WV, Raes J, Inohara N. 2013. Induction of bone loss by pathobiont-mediated Nod1 signaling in the oral cavity. Cell Host Microbe 13:595–601. doi: 10.1016/j.chom.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyrelles SS, Peotta VA, Pereira TM, Vasquez EC. 2011. Endothelial dysfunction in the apolipoprotein E-deficient mouse: insights into the influence of diet, gender and aging. Lipids Health Dis 10:211. doi: 10.1186/1476-511X-10-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tousoulis D, Kampoli A-M, Tentolouris C, Papageorgiou N, Stefanadis C. 2012. The role of nitric oxide on endothelial function. Curr Vasc Pharmacol 10:4–18. doi: 10.2174/157016112798829760. [DOI] [PubMed] [Google Scholar]
- 37.Jia R, Kurita-Ochiai T, Oguchi S, Yamamoto M. 2013. Periodontal pathogen accelerates lipid peroxidation and atherosclerosis. J Dent Res 92:247–252. doi: 10.1177/0022034513475625. [DOI] [PubMed] [Google Scholar]