

Abstract
Many pathogens usurp the host hemostatic system during infection to promote pathogenesis. Yersinia pestis, the causative agent of plague, expresses the plasminogen activator protease Pla, which has been shown in vitro to target and cleave multiple proteins within the fibrinolytic pathway, including the plasmin inhibitor α2-antiplasmin (A2AP). It is not known, however, if Pla inactivates A2AP in vivo; the role of A2AP during respiratory Y. pestis infection is not known either. Here, we show that Y. pestis does not appreciably cleave A2AP in a Pla-dependent manner in the lungs during experimental pneumonic plague. Furthermore, following intranasal infection with Y. pestis, A2AP-deficient mice exhibit no difference in survival time, bacterial burden in the lungs, or dissemination from wild-type mice. Instead, we found that in the absence of Pla, A2AP contributes to the control of the pulmonary inflammatory response during infection by reducing neutrophil recruitment and cytokine production, resulting in altered immunopathology of the lungs compared to A2AP-deficient mice. Thus, our data demonstrate that A2AP is not significantly affected by the Pla protease during pneumonic plague, and although A2AP participates in immune modulation in the lungs, it has limited impact on the course or ultimate outcome of the infection.
INTRODUCTION
The activation of coagulation, resulting in fibrin deposition and fibrin cross-linking, is a host mechanism to stop bleeding and to physically contain invading microorganisms (1, 2). Subsequently, mechanisms to induce fibrinolysis are activated via tightly regulated proteolytic cascades (3). During this phase, tissue-bound and secreted activators mediate the conversion of the circulating zymogen plasminogen into its active serine protease form, plasmin. In addition, the host innate immune system is tightly linked to the hemostatic system. By coordinating host platelet activation and aggregation, coagulation, fibrinolysis, and inflammation, the innate immune response can synchronize the sensing of invading pathogens, immune cell recruitment, and pathogen elimination, as well as subsequent tissue repair (3–5).
As a part of the fibrinolytic system, plasmin degrades fibrin clots with additional biological activities related to tissue remodeling, cell migration, and inflammation (6). In addition, there exist endogenous inhibitors of both active plasmin and plasmin generation to maintain homeostasis. To control the generation of plasmin, plasminogen activator inhibitor 1 (PAI-1) inactivates the plasminogen activators urokinase plasminogen activator (uPA) and tissue plasminogen activator (tPA) (7, 8). On the other hand, α2-antiplasmin (A2AP) inhibits plasmin directly. A2AP is synthesized in the liver and maintained in the circulation to rapidly inhibit freely circulating active plasmin (9). Currently, the only known biological function of A2AP is to inhibit active plasmin; this occurs via a set of interaction steps, which culminates in the formation of an enzymatically inactive 1:1 stoichiometric complex of plasmin-A2AP (10). First, a lysine binding motif within active plasmin interacts with a complementary site in the C-terminal end of A2AP (11). Subsequently, A2AP interacts with the substrate binding pocket within the active site of plasmin, followed by peptide bond cleavage (10). This reaction culminates with the formation of an ester bond between plasmin and A2AP, leaving plasmin inactive (10). Although A2AP can rapidly inhibit active plasmin, both receptor-bound plasmin and fibrin-bound plasmin are protected from A2AP inhibition due to the requirement for the same lysine binding sites for both receptor and A2AP binding (10, 11). Additionally, both infectious and noninfectious disease states are associated with increased circulating A2AP levels, including stroke (12), myocardial infarction (12), and sepsis (13, 14), thus implying a role for inhibition of fibrinolysis via A2AP during many diseases.
Not surprisingly, numerous Gram-positive and Gram-negative bacterial pathogens have independently evolved mechanisms to exploit or usurp host fibrinolytic proteins, including but not limited to Pseudomonas aeruginosa (15), Salmonella enterica serovar Enteritidis (16), Borrelia burgdorferi (17), Staphylococcus aureus (18), group A streptococci (GAS) (19), and Yersinia pestis (20). Among these, the Y. pestis protease Pla is a potent activator of host plasminogen. Pla is an outer membrane protease of the omptin family and cleaves host plasminogen into its active form of plasmin at the same residues recognized by endogenous activators (20–22). Not only has Pla been shown to activate plasminogen, but multiple additional host substrates of Pla associated with the fibrinolytic system have been identified and characterized in vitro, including A2AP and PAI-1 (23–25). Thus, it has been hypothesized that through the activities of Pla, Y. pestis enhances fibrinolysis by potentially targeting multiple regulatory points of the host hemostatic system to aid in virulence (23, 24).
Infections with Y. pestis, the causative agent of plague, manifest in three distinct forms: bubonic, pneumonic, and septicemic (26). Pneumonic plague, the respiratory form of the disease, is both highly infectious and transmissible by aerosols or respiratory droplets. Primary pneumonic plague results in a rapidly progressing, purulent, multifocal severe exudative bronchopneumonia (27, 28). Previous characterizations of pneumonic plague in a mouse model show a biphasic host response, which consists of an initial 24 to 36 h of an anti-inflammatory phase that transitions to a highly proinflammatory phase by 48 h, marked by extensive immune cell influx, inflammatory cytokine production, and tissue damage (27, 29). By 72 h, a severe bronchopneumonia overwhelms the lungs and results in close to 100% mortality if untreated (27).
Pla enhances virulence in mouse models of both pneumonic and bubonic plague, although once the bacterium has reached the blood, the activity of Pla is unnecessary for death via primary or secondary septicemia (22, 30, 31). Secondly, during bubonic plague, the requirement of Pla is also dependent on the presence of host plasminogen, as plasminogen-deficient mice are highly resistant to infection via the subcutaneous route (5). Although Pla has been demonstrated to cleave a number of substrates in vitro, it is unclear what role these molecules play—if any—during infection. Indeed, to date only one in vitro-described Pla substrate, the apoptotic molecule Fas ligand (FasL), has been reported to contribute to disease during pneumonic plague (32).
A2AP has been characterized as a Pla substrate in vitro (24), and it has been hypothesized that cleavage of A2AP could serve as a mechanism by which Y. pestis abrogates the inhibitory effects of A2AP on plasmin in vivo to enhance disease, thus facilitating bacterial outgrowth and dissemination (4, 5, 24, 25, 33, 34). However, the role of A2AP has not yet been investigated during plague. Therefore, we sought to determine if A2AP is cleaved by Pla in the lung airspace and if this contributes to the pathogenesis of Y. pestis during pneumonic plague. Here, we report that while Y. pestis infection results in increased abundance of A2AP in the lungs, the presence of Pla has no impact on the total levels of A2AP in the airspace during pneumonic plague. Indeed, we are unable to detect evidence of significant A2AP cleavage by Pla in the pulmonary compartment, possibly due to the binding of all available free A2AP within plasmin-antiplasmin complexes. Furthermore, we demonstrate that following infection with Y. pestis, A2AP-deficient mice exhibit no significant difference in survival or bacterial burdens from wild-type mice. Instead, we show that in the absence of Pla, A2AP deficiency impacts cytokine production and immune cell infiltration to the lungs, and these changes in the immune response correspond with increased levels of plasminogen and active plasmin. These results suggest that, although Pla may inactivate A2AP in vitro, the cleavage of A2AP by Pla during pneumonic plague is minimal, and while A2AP may contribute to modulating immune responses during infection, this ultimately has limited impact on the outcome of disease.
MATERIALS AND METHODS
Reagents, bacterial strains, and culture conditions.
All reagents used in this study were purchased from Sigma-Aldrich or VWR unless otherwise stated. Brain heart infusion (BHI) broth or agar (Difco) was used to maintain Y. pestis strain CO92 and derivatives thereof. For animal infections, Y. pestis strains were cultured in BHI with the addition of 2.5 mM CaCl2 at 37°C to prime for intranasal inoculations as previously described (31). All experiments using select agent strains of Y. pestis were conducted in a Centers for Disease Control and Prevention-approved biosafety level 3 (BSL-3)/animal BSL-3 (ABSL-3) facility at Northwestern University.
Animal infections.
All procedures involving animals were carried out in compliance with protocols approved by the Institutional Animal Care and Use Committee of Northwestern University. C57BL/6 mice, aged 6 to 8 weeks, were obtained from The Jackson Laboratories or bred at Northwestern University. A2AP-deficient mice were obtained from Roger Lijnen (KU Leuven) on a genetic background of 75% C57BL/6 and 25% 129/SvJ and backcrossed onto the C57BL/6 background for 7 generations (35). Homozygous A2AP deficiency was confirmed by PCR. For infections, mice were anesthetized with ketamine and xylazine and infected by the intranasal route (104 or 108 CFU as indicated) with Y. pestis strains diluted in phosphate-buffered saline (PBS) as previously described (31, 32). The inoculating dose was confirmed by plating on BHI agar. For survival experiments, mice were inoculated intranasally with Y. pestis strains and monitored every 12 h for up to 14 days; mice were allowed to succumb to the disease (death as an endpoint). For all other experiments, mice that were severely moribund (unable to right themselves or to respond to a pinch on the foot or tail) were euthanized by intraperitoneal injection of pentobarbital sodium followed by cervical dislocation and excluded from the experiment. To determine bacterial burden, mice were inoculated intranasally with Y. pestis strains and euthanized at various times postinfection. Subsequently, the lungs and spleens were removed, weighed, homogenized in sterile PBS, serially diluted, and plated onto BHI agar. Following incubation at 26°C for 2 to 3 days, the CFU per organ were enumerated. All animal infections were performed at least twice, and the data were combined.
Histopathology.
Mice were inoculated intranasally with PBS or Y. pestis strains, and at 48 h postinfection, mice were sacrificed and lungs were inflated with 1 ml of 10% neutral buffered formalin via tracheal cannulation. Lungs were removed, fixed in 10% formalin, and embedded in paraffin. Two 4-micrometer sections 200 μm apart per lung were stained with hematoxylin and eosin (H&E) for examination. Tissue embedding, sectioning, and staining with H&E were performed by the Northwestern University Mouse Histology and Phenotyping Laboratory. Slides were imaged using a Zeiss Axioskop/Nuance camera at ×2.5 magnification for lesion size and ×63 magnification for cell recruitment. The numbers of phagocytes present were manually counted within 3 fields of 2 lung sections from 3 independent infections through a single blind analysis. Data represent the mean number of cells per field from each lesion between infections. Additionally, lung sections showing inflamed lesions were analyzed using ImageJ software to calculate the area of inflammation. Data represent the lesion area (square millimeters) per field in 2 sections from 3 mice each.
Innate immune cell quantification.
Mice were infected intranasally with Y. pestis strains or mock infected with PBS, and at 48 h postinfection, mice were sacrificed and bronchoalveolar lavage (BAL) was performed as described previously (32). Bronchoalveolar lavage fluid (BALF) was collected by pooling five 1-ml lavage fluid samples with PBS per animal; the first 1 ml of the BALF was reserved for analyses of cytokines or specific hemostatic proteins (see below). Samples were centrifuged at 300 × g for 5 min to separate cells and cell debris. Cells were washed once each in PBS and twice in flow buffer (2% fetal bovine serum in PBS). For ex vivo cell surface marker detection, cells were stained with antibodies for CD45 (BioLegend; clone 30-F11), CD11b (BioLegend; clone M1/70), CD11c (BD Biosciences; clone HL3), Ly6G (BioLegend; clone IA8), F4/80 (eBioscience; clone BM8), and aqua Live/Dead fixable stain (Invitrogen) for 30 min at 4°C. All antibodies were used at 1:100 dilutions in flow buffer, while Live/Dead cell stain was used at 1:1,000. An anti-CD16/32 FcBlock antibody was included to minimize nonspecific staining (eBioscience). Cells were washed with flow buffer and fixed with 2% paraformaldehyde. Samples were analyzed using a BD FACSCanto II flow cytometer and FlowJo software (TreeStar). Identification of specific cell populations was determined based on the following markers: neutrophils, CD45+ F4/80− Ly6G+ CD11b+; alveolar macrophages, CD45+ F4/80+ Ly6G− CD11b− CD11c+; CD11b-high macrophages, CD45+ F4/80+ Ly6G− CD11bhi; monocytes, CD45+ Ly6G− F4/80− CD11b+ CD11c−; and both CD11b-high and -low dendritic cells, CD45+ Ly6G− F4/80− CD11c+.
Cytokine analysis.
At 48 h after inoculation with PBS or the indicated Y. pestis strains, the levels of tumor necrosis factor alpha (TNF-α), gamma interferon (IFN-γ), monocyte chemoattractant protein 1 (MCP-1), interleukin-12 p70 (IL-12p70), and IL-6 were quantitatively established from the supernatant of the collected BALF using the cytometric bead array technique (BD cytometric bead array mouse inflammation kit; BD Biosciences) as specified by the manufacturer. Data were analyzed using BD cytometric bead array software.
Quantification of total A2AP levels and cleavage.
A2AP levels in BALF from each of the indicated infections were assessed using a total A2AP enzyme-linked immunosorbent assay (ELISA) (Molecular Innovations) per the manufacturer's instructions. Duplicate 100-μl volumes were analyzed from each BALF sample and averaged to yield a nanogram-per-milliliter value for each mouse. The absorbance at 450 nm was measured in a Molecular Devices SpectraMax M5 microplate reader. The data represent 10 mice per group from 2 independent experiments of 5 mice per experiment. For immunoblot analysis, purified recombinant murine A2AP (Abcam) or BALF from each of the indicated infections was separated by SDS-PAGE under reducing or nonreducing conditions, transferred to nitrocellulose, and probed using an anti-mouse A2AP antibody (Santa Cruz; clone SJ-19). A representative blot is shown from 3 independent experiments.
Quantification of plasminogen cleavage and plasmin activity.
Plasmin activity in BALF was assessed using a modified version of the plasminogen activation assay as described previously (31, 36). Briefly, purified murine active plasmin (Hematologic Technologies; 0.3 μg) or 100 μl of BALF from the indicated infections was incubated with the chromogenic substrate D-AFK-ANSNH-iC4H9-2HBr (SN5; Hematologic Technologies; 50 μM) in a total volume of 200 μl PBS at 37°C for 2 h, and the absorbance at 460 nm was measured in a Molecular Devices SpectraMax M5 fluorescence microplate reader. Data represent the relative fluorescence and are combined from 2 independent experiments performed in triplicate. For immunoblot analysis, purified murine plasminogen (Hematologic Technologies), active murine plasmin (Hematologic Technologies), or BALF from each of the indicated infections was separated by SDS-PAGE, transferred to nitrocellulose, and probed using an anti-mouse plasmin(ogen) antibody (Hematologic Technologies). Where noted, immunoblots were stripped in buffer containing 62.5 mM Tris-HCl (pH 6.8), 2% SDS, and 0.07% beta-mercaptoethanol for 1 h with shaking every 15 min at 56°C. Representative blots are shown from 2 or 3 independent experiments as indicated.
Statistics.
In all cases, statistical means are graphed and error bars represent standard errors of the means (SEMs) from pooled experiments. For bacterial load comparisons, the Mann-Whitney U test was performed; for comparison of survival curves, the Mantel-Cox log rank test was used; and all other experiments were analyzed by a Student two-tailed t test or one-way analysis of variance (ANOVA) as indicated using GraphPad Prism 5.
RESULTS
A2AP in the lungs is increased during pneumonic plague but not cleaved by Pla.
Previous studies have demonstrated that the Pla protease of Y. pestis rapidly cleaves A2AP in vitro (24, 36). As Pla is required for the full virulence of Y. pestis during pneumonic plague, we set out to determine if the total levels of A2AP protein are altered in the lungs of mice and if Pla cleaves A2AP in vivo. Therefore, we intranasally inoculated wild-type C57BL/6 mice with PBS (mock) or with equivalent doses of Y. pestis strain CO92 or an isogenic mutant of Y. pestis lacking Pla (31). In addition, as the absence of Pla results in reduced bacterial burden in the lungs compared to that with wild-type Y. pestis (31), we increased the inoculum (indicated as “input CFU,” Fig. 1A) of the Δpla strain so as to match the CFU of the wild-type strain in the lungs at 48 h postinfection (indicated as “output CFU,” Fig. 1A) as we have previously done, to compare the effects of Pla when bacterial loads are equal (32). After 48 h, we performed BALs and analyzed the BALF for the total levels of A2AP present in the lung airspace by ELISA. We found that A2AP levels are significantly increased in the lungs in response to Y. pestis infection compared to mock-infected animals (Fig. 1A). Total A2AP levels are significantly lower in the lung airspace during a Y. pestis Δpla strain infection compared to wild-type Y. pestis infection when the inocula are equivalent; however, this is likely due to the 4- to 5-log difference in bacterial load in the lungs between these infections by 48 h (output CFU). However, when the inoculum of the Δpla strain is adjusted so that the output CFU is equivalent to that of wild-type Y. pestis at 48 h, we found that the abundance of A2AP in the lungs of dose-matched Δpla strain-infected mice is 73.8% ± 10.3% of that of the wild-type infection (not significantly different) (Fig. 1A). These data indicate that infection with Y. pestis stimulates a robust increase in A2AP levels within the lung airspace; however, the presence of Pla does not significantly impact the overall abundance of A2AP in the lungs when bacterial loads are equivalent.
FIG 1.
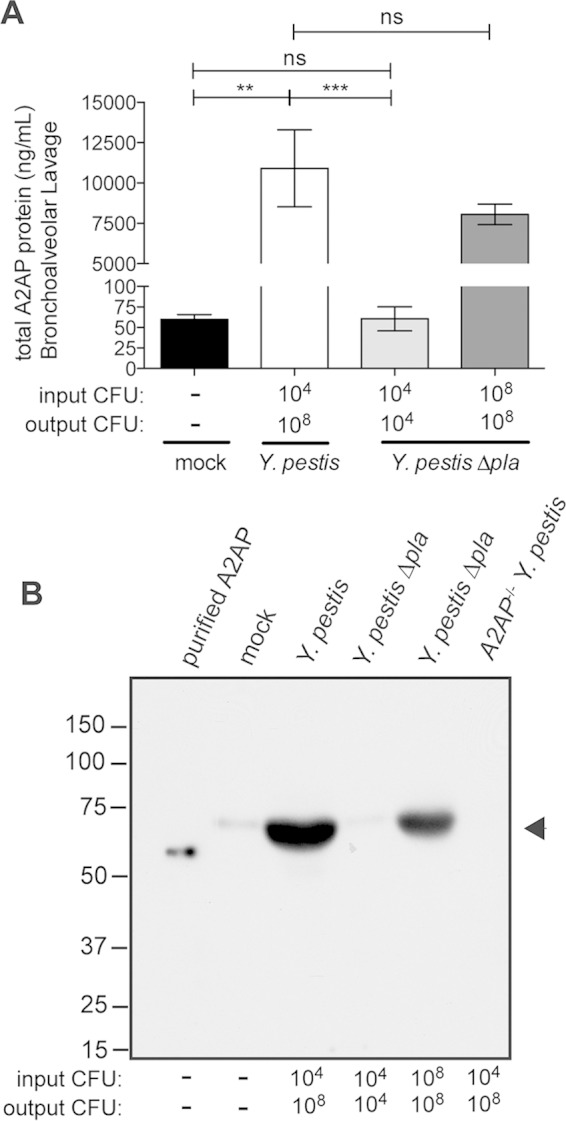
A2AP levels increase in response to Y. pestis but A2AP is not cleaved by Pla. (A) Quantification by ELISA of total A2AP recovered by BAL of C57BL/6 mouse lungs 48 h after inoculation with PBS (mock), 104 CFU of wild-type Y. pestis, 104 CFU of Y. pestis Δpla strain, or 108 CFU of Y. pestis Δpla strain. The input CFU and output CFU are included below the graph to denote the number of bacteria inoculated and the number at the time of assessment, respectively. Data are presented as nanograms per milliliter and are combined from 2 independent experiments (n = 10 for each group); error bars represent SEMs (Student's t test, **, P ≤ 0.01; ***, P ≤ 0.001; ns, not significant). (B) Reducing SDS-PAGE analysis of A2AP recovered by BAL of C57BL/6 mouse lungs 48 h postinoculation. Lanes include recombinant purified murine A2AP (purified A2AP) and BALF from wild-type mice inoculated with PBS (mock), 104 CFU of wild-type Y. pestis, 104 CFU of Y. pestis Δpla strain, or 108 CFU of Y. pestis Δpla strain and from A2AP-deficient mice infected with 104 CFU of wild-type Y. pestis. The arrowhead indicates A2AP protein; numbers to the left of the blot indicate molecular masses in kilodaltons. The blot is representative of 3 independent experiments.
As Pla has been shown to cleave A2AP in vitro, to determine if cleavage of A2AP by Pla could also be observed in vivo, we analyzed BALF from uninfected and infected mice by immunoblotting. We were able to detect full-length A2AP in the lung airspace of mice, which migrates at a slightly higher molecular weight on an SDS-PAGE gel than does recombinant, purified murine A2AP (most likely due to the presence of carbohydrate moieties on endogenous A2AP) (37). Consistent with our ELISA data, we observed an increased abundance of A2AP protein in the lungs following wild-type Y. pestis infection compared to both mock infection and Y. pestis Δpla strain infections; however, we were unable to detect any cleavage products of A2AP by immunoblotting (Fig. 1B). In addition, when output bacterial loads between Y. pestis wild-type and Y. pestis Δpla strains are matched, ImageJ quantification of 3 different exposures of 2 independent immunoblots of the band corresponding to A2AP in the dose-matched Δpla strain infection is 68.3% ± 10.3% of that of wild-type Y. pestis (Fig. 1B), consistent with our ELISA data. Therefore, from the data presented in Fig. 1, we conclude that wild-type Y. pestis infection increases overall A2AP levels in the lungs and that A2AP is not a detectable substrate of Pla during pneumonic plague.
A2AP deficiency does not alter bacterial burden, dissemination, or mouse survival during primary pneumonic plague.
Pla contributes to the outgrowth of Y. pestis in the lungs during pneumonic plague. Although we were unable to detect cleavage of A2AP by Pla within the lungs, it is possible that A2AP contributes to the restriction of bacterial outgrowth in the lungs of mice through the course of pneumonic plague, particularly in the absence of Pla. Wild-type or A2AP-deficient mice were infected with equivalent doses of Y. pestis wild-type or Y. pestis Δpla strains, and CFU in the lungs and spleens were determined at 24, 48, and 72 h postinfection. We found no significant difference in the number of CFU in the lungs at any time between wild-type and A2AP-deficient mice when infected with either wild-type or Δpla Y. pestis (Fig. 2A). As manipulation of the host fibrinolytic system and activation of plasminogen are hypothesized to aid in the invasion by Y. pestis into deeper tissues (38), we also assessed bacterial dissemination to the spleen during pneumonic plague. As observed with the lungs, there was no difference in bacterial load in the spleens at any time point assessed (Fig. 2B). These data demonstrate that A2AP deficiency exhibits no effect on bacterial outgrowth in the lungs or bacterial dissemination to the spleen during primary pneumonic plague.
FIG 2.

A2AP deficiency does not alter bacterial burden, dissemination, or survival of mice infected with Y. pestis. (A and B) Bacterial burden within the lungs (A) and spleens (B) of C57BL/6 mice infected with 104 CFU of wild-type Y. pestis (black squares) or Y. pestis Δpla strain (black circles) or C57BL/6 A2AP−/− mice infected with 104 CFU of wild-type Y. pestis (white squares) or Y. pestis Δpla strain (white circles) over time. Each point represents the numbers of bacteria recovered from a single mouse. An “X” indicates a mouse that succumbed to the infection, the median CFU is denoted by a solid line, and the dashed line indicates the limit of detection. Data are combined from 2 to 3 independent experiments (n = 10 to 15 for each group). (C) Survival over time of C57BL/6 mice infected with 104 CFU of wild-type Y. pestis (black squares) or Y. pestis Δpla strain (black circles) or C57BL/6 A2AP−/− mice infected with 104 CFU of wild-type Y. pestis (white squares) or Y. pestis Δpla strain (white circles). Data are combined from 2 independent experiments (n = 20 for each group). The Mantel-Cox log rank test was used for comparison of survival curves, and the Mann-Whitney U test was used for analysis of bacterial burden measurements (**, P ≤ 0.01; ***, P ≤ 0.001; ns, not significant).
While the 50% lethal dose (LD50) of wild-type Y. pestis following intranasal infection of C57BL/6 mice is 200 to 300 CFU, the LD50 of the Δpla mutant is approximately 104 CFU (27, 31). With this in mind, we next tested the impact of A2AP deficiency on the survival of mice following intranasal infection with Y. pestis. Wild-type or A2AP-deficient mice were infected with equivalent doses of wild-type or Δpla bacteria and monitored for survival. We observed no difference in the survival of A2AP-deficient mice from wild-type controls when infected with Y. pestis wild type or the Δpla strain (Fig. 2C). In total, these data demonstrate that the absence of A2AP does not impact the outgrowth or dissemination of Y. pestis, either in the presence or in the absence of Pla, and does not affect the survival of mice with either bacterial strain following respiratory infection.
A2AP deficiency alters pulmonary pathology and neutrophil recruitment during Y. pestis Δpla strain infection.
Although we observed no impact of A2AP on bacterial burden or the survival of mice, the coordinated links between the hemostatic system and innate immunity prompted investigation into the effect of A2AP deficiency on the histopathology of the lungs following infection with wild-type or Δpla Y. pestis. Wild-type or A2AP-deficient mice were mock infected with PBS or infected with equivalent doses of wild-type or Δpla Y. pestis, and after 48 h, the lungs were inflated and fixed with formalin, and sections were stained with hematoxylin and eosin (H&E) (Fig. 3; also see Fig. S1A in the supplemental material). Examination of pulmonary histopathology revealed no difference in overall lesion size as measured by ImageJ analysis between A2AP-deficient and wild-type mice following infection with wild-type Y. pestis (see Fig. S1B). In contrast, we observed a small but significant increase in the lesion size in A2AP-deficient mice compared to wild-type mice infected with the Y. pestis Δpla mutant. As the overall lesion size differed between wild-type and A2AP−/− mice during a Δpla strain infection, we then assessed the number of cells present within the lesions per field of view. We found that the number of cells per field did not differ significantly between any of the infections, suggesting that the absence of A2AP does not affect the general architecture of the lesion (see Fig. S1C).
FIG 3.

Infection of A2AP-deficient mice with Y. pestis Δpla strain results in altered pulmonary pathology. Sections of formalin-fixed lungs stained with H&E from C57BL/6 or C57BL/6 A2AP−/− mice inoculated with PBS (mock), 104 CFU of wild-type Y. pestis, or 104 CFU of Y. pestis Δpla strain. Representative images of inflammatory lesions are shown (arrows; n = 3). Bars, 100 μm (inset images); 50 μm (larger images).
Therefore, to better quantify both the total and specific immune cells recruited to the lungs in the presence or absence of A2AP, we infected wild-type and A2AP-deficient mice as described above, and after 48 h, we isolated the cells present within the airspace by BAL and analyzed the populations by flow cytometry. A2AP-deficient and wild-type mice infected with wild-type Y. pestis exhibit similar and significant recruitment of immune cells to the lungs during pneumonic plague compared to mock and Y. pestis Δpla strain infections (Fig. 4A). Using the percentages (data not shown) and the total number of immune cells recruited, the absolute numbers of neutrophils, alveolar macrophages, CD11b-high macrophages, monocytes, and both CD11b-high and -low dendritic cells were determined. The absolute numbers of neutrophils, alveolar macrophages, CD11b-high macrophages, monocytes, and both CD11b-high and -low dendritic cells did not differ significantly between wild-type and A2AP-deficient mice when infected with wild-type Y. pestis (Fig. 4B to G). However, we measured a significant increase in the absolute number of neutrophils in the lungs of A2AP-deficient mice compared to wild-type mice infected with the Y. pestis Δpla strain, although no difference in any other cell population was observed (Fig. 4B). These results demonstrate that A2AP deficiency does not alter immune cell numbers during pulmonary infection with wild-type Y. pestis; however, A2AP deficiency specifically increases neutrophil recruitment to the lungs when infected with the Y. pestis Δpla strain. Thus, in the absence of Pla, A2AP participates in the control of the innate immune response in part by restricting the influx of neutrophils into the airspace.
FIG 4.

Increased neutrophil recruitment to the lungs of A2AP-deficient mice following infection with Y. pestis Δpla strain. C57BL/6 or C57BL/6 A2AP−/− mice were inoculated with PBS (mock), 104 CFU of wild-type Y. pestis, or 104 CFU of Y. pestis Δpla strain, and after 48 h, BAL was performed to recover immune cells. (A) The total numbers of cells recruited to the lungs were determined by counting on a hemacytometer. (B to G) Flow cytometry was performed to determine the percentages and absolute numbers of neutrophils (B), alveolar macrophages (C), CD11b-high macrophages (D), monocytes (E), CD11b-high dendritic cells (DCs) (F), and CD11b-low dendritic cells (G) recruited to the lungs. Data are combined from 2 to 3 independent experiments (n = 10 to 15 for each group); error bars represent SEMs (one-way ANOVA with Bonferroni's multiple comparison test; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ns, not significant).
A2AP deficiency alters cytokine production during pneumonic plague.
We next investigated the effects of A2AP deficiency on the production of proinflammatory cytokines in response to pulmonary Y. pestis infection. Wild-type and A2AP-deficient mice were mock infected or infected with Y. pestis strains as described above, and after 48 h, BALF was isolated and assessed for cytokine levels in the airspace via cytometric bead array. We found that the abundance of both IL-12p70 and IFN-γ in the lungs is significantly increased in the absence of A2AP in a Pla-independent manner (Fig. 5A and B). While there are no significant differences in the levels of MCP-1, TNF-α, and IL-6 between wild-type and A2AP-deficient mice during infection with wild-type Y. pestis, the levels of these cytokines are all significantly increased in the absence of A2AP during infection with the Y. pestis Δpla strain (Fig. 5C to E). These results indicate that A2AP suppresses inflammatory cytokine production during infection and that the presence of Pla partially, but not completely, overcomes this effect.
FIG 5.

A2AP deficiency alters the production of pulmonary proinflammatory cytokines during infection with Y. pestis. Forty-eight hours after inoculation with PBS (mock), 104 CFU of wild-type Y. pestis, or 104 CFU of Y. pestis Δpla strain, the abundance of the indicated cytokines present in the BALF of C57BL/6 or C57BL/6 A2AP−/− mice was assessed by cytometric bead array. Data are combined from 2 to 3 independent experiments (n = 10 to 15 for each group); error bars represent SEMs (one-way ANOVA with Bonferroni's multiple comparison test; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ns, not significant).
A2AP deficiency enhances active plasmin levels during infection with Y. pestis Δpla.
A2AP regulates fibrinolysis and inflammation via inhibiting active plasmin. Thus, the effects of A2AP deficiency on neutrophil recruitment and cytokine production in a Y. pestis Δpla strain infection led us to hypothesize that A2AP deficiency increases the levels of active plasmin in the absence of Pla. To test this, wild-type and A2AP-deficient mice were mock infected or infected with Y. pestis strains at equivalent doses; in addition, the Δpla strain was also given at an inoculum to match the dose of wild-type Y. pestis in the lungs at 48 h. BAL was performed at this time point, and the total levels of both plasmin(ogen) and plasmin protein present in BALF were assessed by reducing SDS-PAGE followed by immunoblotting. We found that the amount of plasmin protein is increased in the lungs of mice infected with wild-type Y. pestis compared to both mock-infected and Y. pestis Δpla strain-infected mice (Fig. 6A). However, we also observed a modest increase in the abundance of plasmin in the wild-type mice infected with the Y. pestis Δpla strain compared to mock-infected mice, suggesting that the presence of bacteria in the lungs results in endogenous activation of plasmin(ogen), independent of Pla. Furthermore, when the output CFU of the wild-type and Δpla strains in the lungs are matched in wild-type mice, we observed an increase in total plasmin(ogen) that is dependent on the number of bacteria. However, the presence of Pla results in a vast increase of plasmin compared to that in Pla− bacteria at the same CFU, indicating that Pla does indeed convert plasminogen to plasmin in the lungs during pneumonic plague (Fig. 6A). Additionally, in A2AP-deficient mice, immunoblot analysis revealed an increased abundance of plasmin during infection with wild-type Y. pestis compared to infection with the Δpla strain (Fig. 6A). However, we also observed an increase in the amount of total plasminogen in the lungs of A2AP-deficient mice compared to wild-type mice during infection with the Y. pestis Δpla strain, suggesting that the loss of A2AP dysregulates plasminogen production or recruitment to the lungs during this infection (Fig. 6A).
FIG 6.

A2AP deficiency alters plasmin(ogen) levels and activation in the lungs of Y. pestis Δpla strain-infected mice. (A) Immunoblot analysis of purified murine plasminogen, purified murine plasmin, and plasmin(ogen) recovered by BAL after 48 h from C57BL/6 or C57BL/6 A2AP−/− mice inoculated with PBS (mock), 104 CFU of wild-type Y. pestis, 104 CFU of Y. pestis Δpla strain, or 108 CFU of Y. pestis Δpla strain. Numbers to the left of the blot indicate molecular masses in kilodaltons, the arrow indicates plasminogen, and the arrowhead indicates plasmin. (B) Quantification of active plasmin present in the BALF collected 48 h postinoculation from wild-type (WT) C57BL/6 or A2AP−/− mice infected with PBS (mock), 104 CFU of wild-type Y. pestis, or 104 CFU of Y. pestis Δpla strain or wild-type mice infected with 108 CFU of Y. pestis Δpla strain or purified active murine plasmin. The input CFU and output CFU are included below the graph and blot to denote the number of bacteria inoculated and the number of bacteria at the time of assessment, respectively. Data are presented as the relative fluorescent units of the cleaved product at 120 min and are combined from 2 independent experiments from 3 mice each; error bars represent SEMs (one-way ANOVA with Bonferroni's multiple comparison test; *, P ≤ 0.05; ***, P ≤ 0.001; ns, not significant).
The immunoblot analysis gives an overall summary of protein levels but cannot differentiate between active and inactive plasmin. Therefore, to determine if A2AP deficiency alters the levels of active plasmin within the lungs of mice during pneumonic plague infection, we assessed the levels of active plasmin within BALF using a chromogenic substrate of plasmin. We found no significant difference in plasmin activity between wild-type and A2AP-deficient mice infected with Y. pestis (Fig. 6B). However, we observed slightly but significantly higher plasmin activity in the BALF of A2AP-deficient mice than in that of wild-type mice infected with the Y. pestis Δpla strain (Fig. 6B), suggesting that Y. pestis infection results in the formation of both active and inactive species of plasmin that migrate at the same molecular weight on an SDS-PAGE gel. Furthermore, plasmin activity in BALF from wild-type mice infected with 108 CFU of the Y. pestis Δpla strain exists at an intermediate level between those of wild-type and A2AP-deficient mice infected with 104 CFU of the Y. pestis Δpla strain (Fig. 6B). These results indicate that A2AP deficiency can increase the levels of active plasmin in the lungs, but only during Y. pestis Δpla strain infection, and suggest that A2AP is unable to restrict plasmin activity during pneumonic plague.
Total A2AP in the airspace comigrates with plasmin(ogen) during wild-type, but not Δpla, Y. pestis infection.
Based on the data presented in Fig. 1 and 6, we hypothesized that rather than being cleaved by Pla, all available A2AP in the airspace may instead be complexed with plasmin, thus preventing cleavage of A2AP by Pla during a wild-type Y. pestis infection. To test this, we separated the same BALF samples used in Fig. 1 by nonreducing SDS-PAGE and assessed A2AP by immunoblotting in order to detect supershifts in A2AP, which would suggest complex formation with other factors also present in the BALF. We found that A2AP in the airspace of wild-type mice infected with Y. pestis is contained within a complex(es) that migrates at approximately 100 to 150 kDa (Fig. 7A, top and bottom double arrowheads). Notably, we were unable to detect uncomplexed A2AP migrating at its native molecular mass of 67 kDa. Additionally, A2AP in the airspace of mice given 108 CFU of the Y. pestis Δpla strain primarily migrates at its native, uncomplexed molecular mass of 67 kDa (single arrowhead), with a small but detectable band visible at 150 kDa (double arrowhead) (Fig. 7A).
FIG 7.

Total A2AP comigrates with plasmin(ogen) during wild-type Y. pestis infection. Nonreducing SDS-PAGE analysis of BALF from C57BL/6 or C57BL/6 A2AP−/− mice 48 h postinoculation. Lanes include purified murine plasminogen, purified murine plasmin, recombinant purified murine A2AP (purified A2AP), and BALF from wild-type mice inoculated with PBS (mock), 104 CFU of wild-type Y. pestis, 104 CFU of Y. pestis Δpla strain, or 108 CFU of Y. pestis Δpla strain and A2AP-deficient mice inoculated with 104 CFU of wild-type Y. pestis. The immunoblot was probed with anti-A2AP antibody (A) and stripped and then reprobed with an antiplasminogen antibody (B). The input CFU and output CFU are included below the blots to denote the number of bacteria inoculated and the number of bacteria at the time of assessment, respectively. The double arrowhead and double pound sign indicate supershifted A2AP and plasmin(ogen), respectively; the single arrowhead indicates unshifted (native) A2AP; the single pound sign indicates shifted plasmin(ogen); the closed circle indicates plasmin beta chain; nonspecific bands are denoted by an asterisk. Numbers to the left of the blot indicate molecular masses in kilodaltons, and the panels shown are representative of 2 independent experiments.
As A2AP is known to bind only plasmin, we subsequently stripped and reprobed the blot to determine if supershifted A2AP comigrates with pulmonary plasmin(ogen). We found that the 100- to 150-kDa species present in BALF also reacts with an antibody to plasmin(ogen) (Fig. 7B), suggesting that the supershifted A2AP species may indeed be contained within a plasmin-antiplasmin complex (double pound sign) generated in response to the abundant levels of plasmin produced by Pla. In contrast, the majority of plasmin(ogen) present in the BALF of mice infected with 108 CFU of the Y. pestis Δpla strain migrates at the same molecular weight as does purified plasminogen, supporting the notion that the majority of A2AP is not complexed with plasmin(ogen) in the absence of Pla. Notably, we found that when samples were analyzed under nonreducing conditions, plasmin migrates at a higher molecular weight (pound sign) than under the reducing conditions presented in Fig. 6A, suggesting that plasmin still contains a disulfide-bonded fragment or is complexed with another protein(s) or possibly with itself (Fig. 7B). These data together suggest that the excess plasmin generated during pneumonic plague in a Pla-dependent manner likely binds almost all A2AP present in the lungs.
DISCUSSION
The hemostatic system is tightly regulated in the body, with key zymogens kept in inactive states until specific proteolytic cascades are activated. Endogenous inhibitors prevent excessive fibrinolysis by directly or indirectly controlling the activation of plasmin and its consequent downstream effects. A2AP is considered the major inhibitor of active plasmin, a regulator of fibrinolysis, in the host (9, 39, 40). Thus, in the absence of A2AP it is expected that increased levels of active plasmin would result in enhanced fibrinolysis when the hemostatic system is stimulated. Consistent with its inhibitory role during fibrinolysis, A2AP-deficient mice display normal physiology with no overt bleeding disorders but exhibit increased fibrinolytic potential (35).
In this study, we investigated the potential contribution of A2AP to the development of primary pneumonic plague and interrogated its potential cleavage by Pla in the lungs of infected mice. While Pla has been shown to cleave A2AP in vitro (24, 34, 36), we were unable to detect a significant Pla-dependent change in total A2AP levels or cleavage events in vivo. These data increase our understanding of the multifactorial effects of Pla during pneumonic plague; as Pla is described as both a specific and promiscuous protease, the contribution of each in vitro-described substrate to disease must be reconciled in vivo. The prevailing hypothesis in the field proposes that the cleavage and inactivation of A2AP are significant contributors to Y. pestis disease as an auxiliary means to enhance active plasmin generation and thus promote the outgrowth and dissemination of the bacteria (4, 5, 24, 25, 33, 34). In contrast, our findings indicate that the impact of A2AP on the infection is minor at best and suggest that the extensive levels of plasmin generated by Pla are sufficient to overcome any inhibitory effects of A2AP.
These conclusions are based on the direct comparison of wild-type mice infected with 104 CFU of wild-type Y. pestis and 108 CFU of the Y. pestis Δpla strain, which results in equivalent bacterial CFU of approximately 108 in the lungs at 48 h postinfection (32). We note that, although the courses of colonization and subsequent immune responses between the two infections might be different due to differences in bacterial input, the equivalent numbers of bacteria in the lungs at 48 h allow for investigation of the effects of Pla on A2AP in the lungs. While it remains possible that Pla may cleave A2AP at low levels beyond the limits of our detection, our data suggest that the vast majority of A2AP in the lungs during infection is complexed with plasmin. This, then, suggests a possible explanation for why Pla may be unable to cleave A2AP, as A2AP bound to plasmin may be inaccessible or resistant to cleavage by Pla.
While the amino acid sequence of Pla is 100% conserved among all modern branched strains of Y. pestis, comparative genomics have determined that ancestral strains of Y. pestis contain a single amino acid substitution at position 259. This I259T substitution from ancestral to modern Pla has enhanced virulence during plague (51). Furthermore, the modern variant of Pla is more efficient at activating plasminogen but less efficient at inhibiting A2AP antiprotease activity than its ancestral counterpart (24, 34). This suggests that during its evolution as a respiratory pathogen, the selective pressure on Y. pestis was for enhanced plasmin generation at the expense of A2AP antiprotease activity and may additionally help explain the lack of A2AP cleavage in the lungs during pneumonic plague.
Although we did not observe any evidence that Pla affects the cleavage of A2AP in the pulmonary compartment, we employed A2AP-deficient mice to assess its impact on the outcome of infection. We note that, although the 129/SvJ/C57BL/6 A2AP−/− mice provided by Roger Lijnen were backcrossed onto the C57BL/6 background for 7 generations, we cannot exclude the possibility that additional linked genetic loci may still be present in the mice (41); however, the fact that there were few differences between wild-type and A2AP−/− mice suggests that if this is the case, any linked loci have limited impact on the results of our study. Indeed, we found that A2AP-deficient mice exhibited no difference in bacterial loads in the lungs, bacterial dissemination to the spleen, or survival. These results demonstrate that A2AP is not a restrictive factor for Y. pestis during pneumonic plague. However, we did find that A2AP deficiency impacted the immune response to Y. pestis Δpla strain infection by both enhancing neutrophil recruitment and altering cytokine production. In the absence of Pla, Y. pestis is unable to directly activate plasminogen, and consequently, limited plasmin activity is observed in the pulmonary compartment. Indeed, it appears that the modest level of plasmin generation present in the lungs of A2AP-deficient mice is insufficient to overcome the loss of Pla, suggesting a threshold requirement for plasmin by Y. pestis.
Our results are consistent with the contribution of A2AP to infection with Burkholderia pseudomallei, a respiratory pathogen that is not known to possess any direct plasminogen activator proteins. During B. pseudomallei lung infection, A2AP-deficient mice present with more severe lung pathology, increased neutrophil recruitment, and increased cytokine levels in the lungs (42). In addition, the breakdown of fibrin clots, a known plasmin-mediated process, is increased in A2AP-deficient mice during B. pseudomallei infection (42). Although we detected an increase in neutrophil recruitment and cytokine production when A2AP-deficient mice were infected with the Y. pestis Δpla strain, we did not observe a corresponding effect on bacterial load. This observation suggests that the Y. pestis Δpla strain is able to resist the increased host response in the absence of A2AP and is consistent with findings that Y. pestis is able to evade elimination by neutrophils (43, 44). Our data add to these conclusions by demonstrating that polymorphonuclear leukocyte (PMN) resistance likely occurs in a Pla-independent manner.
Results from our and other studies demonstrate the tight links between the regulation of the fibrinolytic system and the inflammatory response (5, 45). We show here that in the absence of A2AP, the inflammatory response is enhanced during Y. pestis Δpla strain infection, suggesting a role for A2AP in regulating inflammation during Y. pestis infection, most likely through activities associated with plasmin(ogen). A number of studies have found that active plasmin or the activation of plasminogen on the surface of macrophages or monocytes can alter their susceptibility to cell death and the production of proinflammatory cytokines, including IL-6 and TNF-α (46, 47). Pla is absolutely required for the transition of pneumonic plague infection to the proinflammatory phase of disease (31), and our data suggest that in the absence of Pla, A2AP deficiency can restore part of this transition (i.e., immune cell recruitment and proinflammatory cytokine production). Thus, it will be of interest to investigate the contribution of plasminogen activation to the proinflammatory phase of pneumonic plague, as we hypothesize that the fibrinolysis induced by Pla may serve as a mediator of the host inflammatory response.
Although we could find no evidence for A2AP cleavage or inactivation by Pla during pneumonic plague, it is possible that A2AP cleavage may occur during the bubonic form of the disease. The infection sites for bubonic versus pneumonic plague—the dermis and lymph nodes versus lungs, respectively—are vastly different niches. Furthermore, the absence of Pla results in different effects on bacterial outgrowth and the immune response between these infection sites. Investigations on whether A2AP cleavage by Pla contributes to bubonic plague are likely to not only distinguish the substrates of Pla in different tissues but also define the role of plasmin inhibition by A2AP at different infection sites. For instance, during S. aureus skin infection A2AP deficiency increases both lesion size and bacterial loads at the site of infection, the dermis, thus highlighting the varied contributions of A2AP-mediated plasmin inhibition at different infection sites (48).
Our data demonstrate that the Pla-mediated generation of plasmin occurs to an extent that A2AP cannot overcome. Plasmin binds to numerous host and bacterial proteins and once bound is in many instances protected from A2AP inhibition, with significantly lower kinetics of inactivation than that of free plasmin (49, 50). Furthermore, while our data suggest that the majority of A2AP may be complexed with plasmin during pneumonic plague, sufficient free and/or uninhibited plasmin is present to enable the rapid outgrowth of Y. pestis in the lungs and to enhance dissemination to deeper tissues. In summary, this study refutes a long-standing belief that A2AP is a critical substrate of Pla that enhances the virulence of Y. pestis during pneumonic plague and provides a potential explanation for why this may be so. Indeed, our data highlight the need to test the contribution of in vitro-characterized targets of Pla in vivo and indicate that the role(s) of the multifactorial Pla protease during infection is more complex than in vitro studies would suggest.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from Public Health grants R01 AI093727 and R21 AI103658 to W.W.L. This work was supported by the Northwestern University Interdepartmental Immunobiology Flow Cytometry Core Facility. Histopathology was performed by the Northwestern University Mouse Histology and Phenotyping Laboratory, supported by NCI CA060553. Imaging was performed at the Northwestern University Cell Imaging Facility, supported by NCI CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center.
We are grateful to Roger Lijnen, Katholieke Universiteit Leuven, for the kind gift of the A2AP−/− mice and members of the Lathem lab for technical support.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01086-15.
REFERENCES
- 1.Opal SM. 2000. Phylogenetic and functional relationships between coagulation and the innate immune response. Crit Care Med 28:S77–S80. doi: 10.1097/00003246-200009001-00017. [DOI] [PubMed] [Google Scholar]
- 2.Levi M, van der Poll T. 2010. Inflammation and coagulation. Crit Care Med 38:S26–S34. doi: 10.1097/CCM.0b013e3181c98d21. [DOI] [PubMed] [Google Scholar]
- 3.van Gorp EC, Suharti C, ten Cate H, Dolmans WM, van der Meer JW, ten Cate JW, Brandjes DP. 1999. Review: infectious diseases and coagulation disorders. J Infect Dis 180:176–186. doi: 10.1086/314829. [DOI] [PubMed] [Google Scholar]
- 4.Korhonen TK, Haiko J, Laakkonen L, Jarvinen HM, Westerlund-Wikstrom B. 2013. Fibrinolytic and coagulative activities of Yersinia pestis. Front Cell Infect Microbiol 3:35. doi: 10.3389/fcimb.2013.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Degen JL, Bugge TH, Goguen JD. 2007. Fibrin and fibrinolysis in infection and host defense. J Thromb Haemost 5(Suppl 1):S24–S31. [DOI] [PubMed] [Google Scholar]
- 6.Ploplis VA, Castellino FJ. 2002. Gene targeting of components of the fibrinolytic system. Thromb Haemost 87:22–31. [PubMed] [Google Scholar]
- 7.Cubellis MV, Andreasen P, Ragno P, Mayer M, Dano K, Blasi F. 1989. Accessibility of receptor-bound urokinase to type-1 plasminogen activator inhibitor. Proc Natl Acad Sci U S A 86:4828–4832. doi: 10.1073/pnas.86.13.4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Mourik JA, Lawrence DA, Loskutoff DJ. 1984. Purification of an inhibitor of plasminogen activator (antiactivator) synthesized by endothelial cells. J Biol Chem 259:14914–14921. [PubMed] [Google Scholar]
- 9.Collen D. 1976. Identification and some properties of a new fast-reacting plasmin inhibitor in human plasma. Eur J Biochem 69:209–216. doi: 10.1111/j.1432-1033.1976.tb10875.x. [DOI] [PubMed] [Google Scholar]
- 10.Wiman B, Collen D. 1979. On the mechanism of the reaction between human alpha 2-antiplasmin and plasmin. J Biol Chem 254:9291–9297. [PubMed] [Google Scholar]
- 11.Wiman B, Lijnen HR, Collen D. 1979. On the specific interaction between the lysine-binding sites in plasmin and complementary sites in alpha2-antiplasmin and in fibrinogen. Biochim Biophys Acta 579:142–154. doi: 10.1016/0005-2795(79)90094-1. [DOI] [PubMed] [Google Scholar]
- 12.Matsuno H, Kozawa O, Yoshimi N, Akamatsu S, Hara A, Mori H, Okada K, Ueshima S, Matsuo O, Uematsu T. 2002. Lack of alpha2-antiplasmin promotes pulmonary heart failure via overrelease of VEGF after acute myocardial infarction. Blood 100:2487–2493. doi: 10.1182/blood-2001-12-0251. [DOI] [PubMed] [Google Scholar]
- 13.Bagge L, Haglund O, Wallin R, Borg T, Modig J. 1989. Differences in coagulation and fibrinolysis after traumatic and septic shock in man. Scand J Clin Lab Invest 49:63–72. doi: 10.3109/00365518909089079. [DOI] [PubMed] [Google Scholar]
- 14.Lorente JA, Garcia-Frade LJ, Landin L, de Pablo R, Torrado C, Renes E, Garcia-Avello A. 1993. Time course of hemostatic abnormalities in sepsis and its relation to outcome. Chest 103:1536–1542. doi: 10.1378/chest.103.5.1536. [DOI] [PubMed] [Google Scholar]
- 15.Kunert A, Losse J, Gruszin C, Huhn M, Kaendler K, Mikkat S, Volke D, Hoffmann R, Jokiranta TS, Seeberger H, Moellmann U, Hellwage J, Zipfel PF. 2007. Immune evasion of the human pathogen Pseudomonas aeruginosa: elongation factor Tuf is a factor H and plasminogen binding protein. J Immunol 179:2979–2988. doi: 10.4049/jimmunol.179.5.2979. [DOI] [PubMed] [Google Scholar]
- 16.Sjobring U, Pohl G, Olsen A. 1994. Plasminogen, absorbed by Escherichia coli expressing curli or by Salmonella enteritidis expressing thin aggregative fimbriae, can be activated by simultaneously captured tissue-type plasminogen activator (t-PA). Mol Microbiol 14:443–452. doi: 10.1111/j.1365-2958.1994.tb02179.x. [DOI] [PubMed] [Google Scholar]
- 17.Coleman JL, Gebbia JA, Piesman J, Degen JL, Bugge TH, Benach JL. 1997. Plasminogen is required for efficient dissemination of B. burgdorferi in ticks and for enhancement of spirochetemia in mice. Cell 89:1111–1119. doi: 10.1016/S0092-8674(00)80298-6. [DOI] [PubMed] [Google Scholar]
- 18.Collen D. 1998. Staphylokinase: a potent, uniquely fibrin-selective thrombolytic agent. Nat Med 4:279–284. doi: 10.1038/nm0398-279. [DOI] [PubMed] [Google Scholar]
- 19.Sun H, Ringdahl U, Homeister JW, Fay WP, Engleberg NC, Yang AY, Rozek LS, Wang X, Sjobring U, Ginsburg D. 2004. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science 305:1283–1286. doi: 10.1126/science.1101245. [DOI] [PubMed] [Google Scholar]
- 20.Beesley ED, Brubaker RR, Janssen WA, Surgalla MJ. 1967. Pesticins. 3. Expression of coagulase and mechanism of fibrinolysis. J Bacteriol 94:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robbins KC, Summaria L, Hsieh B, Shah RJ. 1967. The peptide chains of human plasmin. Mechanism of activation of human plasminogen to plasmin. J Biol Chem 242:2333–2342. [PubMed] [Google Scholar]
- 22.Sodeinde OA, Subrahmanyam YV, Stark K, Quan T, Bao Y, Goguen JD. 1992. A surface protease and the invasive character of plague. Science 258:1004–1007. doi: 10.1126/science.1439793. [DOI] [PubMed] [Google Scholar]
- 23.Haiko J, Laakkonen L, Juuti K, Kalkkinen N, Korhonen TK. 2010. The omptins of Yersinia pestis and Salmonella enterica cleave the reactive center loop of plasminogen activator inhibitor 1. J Bacteriol 192:4553–4561. doi: 10.1128/JB.00458-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kukkonen M, Lahteenmaki K, Suomalainen M, Kalkkinen N, Emody L, Lang H, Korhonen TK. 2001. Protein regions important for plasminogen activation and inactivation of alpha2-antiplasmin in the surface protease Pla of Yersinia pestis. Mol Microbiol 40:1097–1111. doi: 10.1046/j.1365-2958.2001.02451.x. [DOI] [PubMed] [Google Scholar]
- 25.Suomalainen M, Haiko J, Ramu P, Lobo L, Kukkonen M, Westerlund-Wikstrom B, Virkola R, Lahteenmaki K, Korhonen TK. 2007. Using every trick in the book: the Pla surface protease of Yersinia pestis. Adv Exp Med Biol 603:268–278. doi: 10.1007/978-0-387-72124-8_24. [DOI] [PubMed] [Google Scholar]
- 26.Huang XZ, Nikolich MP, Lindler LE. 2006. Current trends in plague research: from genomics to virulence. Clin Med Res 4:189–199. doi: 10.3121/cmr.4.3.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lathem WW, Crosby SD, Miller VL, Goldman WE. 2005. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc Natl Acad Sci U S A 102:17786–17791. doi: 10.1073/pnas.0506840102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perry RD, Fetherston JD. 1997. Yersinia pestis—etiologic agent of plague. Clin Microbiol Rev 10:35–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price PA, Jin J, Goldman WE. 2012. Pulmonary infection by Yersinia pestis rapidly establishes a permissive environment for microbial proliferation. Proc Natl Acad Sci U S A 109:3083–3088. doi: 10.1073/pnas.1112729109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sebbane F, Jarrett CO, Gardner D, Long D, Hinnebusch BJ. 2006. Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc Natl Acad Sci U S A 103:5526–5530. doi: 10.1073/pnas.0509544103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lathem WW, Price PA, Miller VL, Goldman WE. 2007. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315:509–513. doi: 10.1126/science.1137195. [DOI] [PubMed] [Google Scholar]
- 32.Caulfield AJ, Walker ME, Gielda LM, Lathem WW. 2014. The Pla protease of Yersinia pestis degrades fas ligand to manipulate host cell death and inflammation. Cell Host Microbe 15:424–434. doi: 10.1016/j.chom.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caulfield AJ, Lathem WW. 2012. Substrates of the plasminogen activator protease of Yersinia pestis. Adv Exp Med Biol 954:253–260. doi: 10.1007/978-1-4614-3561-7_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haiko J, Kukkonen M, Ravantti JJ, Westerlund-Wikstrom B, Korhonen TK. 2009. The single substitution I259T, conserved in the plasminogen activator Pla of pandemic Yersinia pestis branches, enhances fibrinolytic activity. J Bacteriol 191:4758–4766. doi: 10.1128/JB.00489-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lijnen HR, Okada K, Matsuo O, Collen D, Dewerchin M. 1999. Alpha2-antiplasmin gene deficiency in mice is associated with enhanced fibrinolytic potential without overt bleeding. Blood 93:2274–2281. [PubMed] [Google Scholar]
- 36.Eddy JL, Gielda LM, Caulfield AJ, Rangel SM, Lathem WW. 2014. Production of outer membrane vesicles by the plague pathogen Yersinia pestis. PLoS One 9:e107002. doi: 10.1371/journal.pone.0107002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clemmensen I, Thorsen S, Mullertz S, Petersen LC. 1981. Properties of three different molecular forms of the alpha 2 plasmin inhibitor. Eur J Biochem 120:105–112. doi: 10.1111/j.1432-1033.1981.tb05675.x. [DOI] [PubMed] [Google Scholar]
- 38.Lahteenmaki K, Edelman S, Korhonen TK. 2005. Bacterial metastasis: the host plasminogen system in bacterial invasion. Trends Microbiol 13:79–85. doi: 10.1016/j.tim.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 39.Moroi M, Aoki N. 1976. Isolation and characterization of alpha2-plasmin inhibitor from human plasma. A novel proteinase inhibitor which inhibits activator-induced clot lysis. J Biol Chem 251:5956–5965. [PubMed] [Google Scholar]
- 40.Mullertz S, Clemmensen I. 1976. The primary inhibitor of plasmin in human plasma. Biochem J 159:545–553. doi: 10.1042/bj1590545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holmdahl R, Malissen B. 2012. The need for littermate controls. Eur J Immunol 42:45–47. doi: 10.1002/eji.201142048. [DOI] [PubMed] [Google Scholar]
- 42.Kager LM, Weehuizen TA, Wiersinga WJ, Roelofs JJ, Meijers JC, Dondorp AM, van 't Veer C, van der Poll T. 2013. Endogenous alpha2-antiplasmin is protective during severe gram-negative sepsis (melioidosis). Am J Respir Crit Care Med 188:967–975. doi: 10.1164/rccm.201307-1344OC. [DOI] [PubMed] [Google Scholar]
- 43.Vagima Y, Zauberman A, Levy Y, Gur D, Tidhar A, Aftalion M, Shafferman A, Mamroud E. 2015. Circumventing Y. pestis virulence by early recruitment of neutrophils to the lungs during pneumonic plague. PLoS Pathog 11:e1004893. doi: 10.1371/journal.ppat.1004893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pechous RD, Sivaraman V, Price PA, Stasulli NM, Goldman WE. 2013. Early host cell targets of Yersinia pestis during primary pneumonic plague. PLoS Pathog 9:e1003679. doi: 10.1371/journal.ppat.1003679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Brien M. 2012. The reciprocal relationship between inflammation and coagulation. Top Companion Anim Med 27:46–52. doi: 10.1053/j.tcam.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Li Q, Laumonnier Y, Syrovets T, Simmet T. 2007. Plasmin triggers cytokine induction in human monocyte-derived macrophages. Arterioscler Thromb Vasc Biol 27:1383–1389. doi: 10.1161/ATVBAHA.107.142901. [DOI] [PubMed] [Google Scholar]
- 47.Mitchell JW, Baik N, Castellino FJ, Miles LA. 2006. Plasminogen inhibits TNFalpha-induced apoptosis in monocytes. Blood 107:4383–4390. doi: 10.1182/blood-2005-07-2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peetermans M, Vanassche T, Liesenborghs L, Claes J, Vande Velde G, Kwiecinksi J, Jin T, De Geest B, Hoylaerts MF, Lijnen RH, Verhamme P. 2014. Plasminogen activation by staphylokinase enhances local spreading of S. aureus in skin infections. BMC Microbiol 14:310. doi: 10.1186/s12866-014-0310-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ellis V, Behrendt N, Dano K. 1991. Plasminogen activation by receptor-bound urokinase. A kinetic study with both cell-associated and isolated receptor. J Biol Chem 266:12752–12758. [PubMed] [Google Scholar]
- 50.Plow EF, Freaney DE, Plescia J, Miles LA. 1986. The plasminogen system and cell surfaces: evidence for plasminogen and urokinase receptors on the same cell type. J Cell Biol 103:2411–2420. doi: 10.1083/jcb.103.6.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zimbler DL, Schroeder JA, Eddy JL, Lathem WW. 2015. Early emergence of Yersinia pestis as a severe respiratory pathogen. Nat Commun 6:7487. doi: 10.1038/ncomms8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.