

Abstract
Group A streptococcus (GAS), the causative agent of pharyngitis and necrotizing fasciitis, secretes the potent cysteine protease SpeB. Several lines of evidence suggest that SpeB is an important virulence factor. SpeB is expressed in human infections, protects mice from lethal challenge when used as a vaccine, and contributes significantly to tissue destruction and dissemination in animal models. However, recent descriptions of mutations in genes implicated in SpeB production have led to the idea that GAS may be under selective pressure to decrease secreted SpeB protease activity during infection. Thus, two divergent hypotheses have been proposed. One postulates that SpeB is a key contributor to pathogenesis; the other, that GAS is under selection to decrease SpeB during infection. In order to distinguish between these alternative hypotheses, we performed casein hydrolysis assays to measure the SpeB protease activity secreted by 6,775 GAS strains recovered from infected humans. The results demonstrated that 84.3% of the strains have a wild-type SpeB protease phenotype. The availability of whole-genome sequence data allowed us to determine the relative frequencies of mutations in genes implicated in SpeB production. The most abundantly mutated genes were direct transcription regulators. We also sequenced the genomes of 2,954 GAS isolates recovered from nonhuman primates with experimental necrotizing fasciitis. No mutations that would result in a SpeB-deficient phenotype were identified. Taken together, these data unambiguously demonstrate that the great majority of GAS strains recovered from infected humans secrete wild-type levels of SpeB protease activity. Our data confirm the important role of SpeB in GAS pathogenesis and help end a long-standing controversy.
INTRODUCTION
Bacterial pathogens often secrete proteases that are important virulence factors (1). Proteases participate in many aspects of pathogen-host interaction, including nutrient acquisition, modification of other bacterial proteins, inactivation of host immune molecules, and tissue destruction (1). Thus, protease virulence factors are essential for the pathogen to colonize mucosal surfaces, evade the host immune response, invade deep soft tissue, disseminate to distant anatomic sites, and spread to new hosts (1). These features make proteases relevant targets for basic research and translational projects geared to creating new diagnostics and vaccines.
Group A streptococcus (GAS), a human-specific pathogen, is an important cause of morbidity and mortality worldwide (2). The pathogen is estimated to cause 600 million infections annually, including 10,000 to 15,000 severe invasive infections in the United States (2). Infections range in severity from self-limiting pharyngitis (“strep throat”) to life-threatening necrotizing fasciitis (“flesh-eating disease”) (3). GAS pathogenesis is mediated by many secreted and cell wall-associated toxins, superantigens, and virulence factors (3). Streptococcal pyrogenic exotoxin B (SpeB), a potent broad-spectrum cysteine protease secreted by GAS, is among the more extensively studied bacterial proteases (4, 5). The chromosomally encoded speB gene is present in virtually all GAS strains (6). Several lines of study have provided strong evidence that SpeB is an important GAS virulence factor (4). SpeB protease can modify many GAS proteins, such as the antiphagocytic M protein, the SmeZ superantigen, and the cytolytic toxin streptolysin O (SLO) (7, 8), cleave the host immune molecules pre-IL-1β (pre-interleukin-1β) and complement component C3b (9), induce the apoptosis of host macrophages and epithelial cells (10), degrade the extracellular matrix proteins fibronectin and vitronectin (11), and activate host matrix metalloproteases responsible for tissue repair (12).
The generation of enzymatically active SpeB by GAS is a very complex multistage process that involves at least 21 gene products (4) (Table 1). speB is transcribed from two promoters and is directly regulated by three transcription factors (13). Transcription and posttranscriptional processing are regulated by various environmental signals, such as changes in pH or electrolytes and expression of other GAS-derived molecules (14, 15). The SpeB zymogen is secreted through an organelle known as the GAS ExPortal (16) before undergoing a series of intra- and intermolecular processing steps to generate the enzymatically active mature SpeB protease (17, 18). Each stage is regulated by multiple GAS accessory factors (Table 1). Disruption of any of these components may render a GAS strain deficient in secreted SpeB protease activity (19–24).
TABLE 1.
Genes implicated in altered secreted SpeB protease activity
| Category and gene | Function | Effect of polymorphisms |
|---|---|---|
| Direct transcriptional regulators | ||
| ropB | Transcription factor | Decreased transcription of speB |
| covRS | Two-component system | Decreased transcription of speB |
| ccpA | Transcription factor | Decreased transcription of speB |
| Indirect regulators and posttranscriptional processing | ||
| clpX | ATP-dependent subunit of Clp protease | Decreased transcription and processing |
| fabT | Transcription factor | Decreased transcription and processing |
| luxS | Transcription factor | Decreased transcription and processing |
| pel | Unknown | Decreased transcription and processing |
| sagP | Transcription factor | Decreased transcription and processing |
| Secretion and posttranslational processing | ||
| ftsH | Membrane-associated protease | Decreased maturation of SpeBz to SpeBm |
| gdpP | c-di-AMP phosphodiesterase | Decreased maturation of SpeBz to SpeBm |
| htrA | Periplasm-associated protease | Decreased maturation of SpeBz to SpeBm |
| mtsR | Transcription factor, regulator of prsA | Decreased maturation of SpeBz to SpeBm |
| prsA | Peptidyl-prolyl cis-trans isomerase | Decreased maturation of SpeBz to SpeBm |
| ropA | Peptidyl-prolyl cis-trans isomerase | Decreased maturation of SpeBz to SpeBm |
| Growth phase and environmental signaling | ||
| codY | Transcription factor, growth phase regulation | Increased transcription of speB |
| lacD.1 | Transcription factor, environmental stimuli | Increased transcription of speB |
| nra | Transcription factor, indirect regulator of speB | Increased transcription of speB |
| svr | Transcription factor, growth phase regulation | Increased transcription of speB |
| vfr | Signal peptide for ropB activation | Increased transcription of speB |
An epidemic serotype emm1 GAS clone has been described and extensively studied because it is a major cause of severe invasive human infections worldwide (25–30). As a consequence of several studies, the idea has arisen that GAS may be under selective pressure to decrease secreted SpeB protease activity during infection (31–33). This decrease has been linked to the initiation of invasive infection, increased disease severity, and a poor outcome on the basis of analysis of convenience samples rather than comprehensive population-based strain samples (23). However, other studies performed by multiple investigators have reported that the secreted SpeB protease is a key virulence factor contributing to tissue destruction, dissemination, and mortality in invertebrate, mouse, and nonhuman primate models (9, 22, 34–41). Moreover, SpeB is expressed in humans with invasive disease (42–44); infected humans generate anti-SpeB antibodies (43–45); and immunization of mice with SpeB or SpeB-derived protein fragments protects against lethal challenge (36, 46–48). Mice treated with a protease inhibitor that inactivates SpeB are similarly protected against invasive disease (49). In this regard, Eriksson et al. reported that acute-phase sera obtained from patients with streptococcal toxic shock syndrome (a severe life-threatening infection) had a significantly lower capacity to neutralize SpeB activity than sera from patients with uncomplicated bacteremia or erysipelas (43). Similarly, acute-phase sera from patients with GAS bacteremia had a significantly lower SpeB-neutralizing ability than sera from patients with uncomplicated tonsillitis (44).
Thus, two divergent ideas have emerged about the role of SpeB in GAS pathogenesis. One idea postulates that SpeB protease is a key contributor to pathogen-host interaction in several phases of pathogenesis. If so, the great majority of strains cultured from diseased humans would be expected to retain the capacity to produce wild-type levels of secreted SpeB protease. Alternatively, if the other idea, that decreased SpeB production is a key driver of invasive infections (31–33), is correct, then the majority of strains cultured from infected humans should lack SpeB protease activity as a consequence of chromosomal mutations in one or more of the 21 genes implicated in SpeB production (Table 1). It is important to resolve this matter, because it has implications for our understanding of GAS pathogenesis and may influence the direction of translational research efforts such as vaccine development.
We recently sequenced the genomes of many thousands of GAS strains recovered from infected humans, focusing on organisms recovered in prospective comprehensive population-based studies conducted in many different parts of the world (27, 50–52). These studies permit us to contribute information that may help in evaluating the two ideas bearing on SpeB production and human infections. We measured the secreted SpeB protease activities made by 6,775 emm1, emm89, emm59, and emm28 GAS strains. These serotypes were selected because they are among the most common causes of severe invasive disease worldwide (2, 53). The results demonstrated that the great majority (84.3%) of GAS strains retain wild-type secreted SpeB protease activity, regardless of the disease type or strain serotype. Similarly, whole-genome sequence analysis of 2,975 emm1 GAS strains recovered in a nonhuman primate model of necrotizing fasciitis found that none had a mutation that would lead to a SpeB-deficient phenotype. We interpret these data as strong evidence for an important role of SpeB in GAS pathogenesis, consistent with findings from many human clinical studies and animal infection models using isogenic mutant strains.
MATERIALS AND METHODS
Serotype emm1 strains.
We studied 3,615 emm1 GAS strains collected over 4 decades from patients in nine distinct geographic locations in North America and Europe (27). This strain sample was recently described in detail, including whole-genome sequence analysis (27). Most strains were collected as part of comprehensive prospective population-based surveillance studies. Invasive strains included 346 from Ontario, Canada (1997 to 2009), 436 from Denmark (1973 to 2013), 155 from East Germany (1969 to 1991), 509 from Finland (1988 to 2011), 50 from Iceland (1988 to 2011), 215 from Norway (1997 to 2009), 482 from Sweden (1996 to 2012), 340 from Georgia, United States (1995 to 2010), and 474 from Minnesota, United States (1995 to 2010). In addition, 11 strains of historic interest from other countries and times were included (27). Strains from Georgia (Atlanta metropolitan area) and Minnesota (statewide) were collected as part of the Active Bacterial Core Surveillance Program administered by the U.S. Centers for Disease Control and Prevention (53). Pharyngitis strains included 597 GAS strains from Finland (1988 to 1997), most of which were recovered in a population-based surveillance study during years overlapping with those of the Finnish invasive-strain study (54).
Serotype emm89 strains.
The 1,181 emm89 strains were collected as part of comprehensive prospective population-based surveillance studies conducted in three countries. These strains were recovered from patients with invasive infections in Finland (n = 286; years, 2003 to 2014), Iceland (n = 24; years, 1997 to 2008) and 10 states in the United States (n = 870; years, 1995 to 2013), and have recently been described in detail (52). The emm89 reference strains MGAS11027 and MGAS23530 have been sequenced to closure (52).
Serotype emm59 strains.
The 704 GAS emm59 invasive strains were collected as part of comprehensive prospective population-based surveillance studies conducted in the United States (n = 50) and Canada (n = 638) or had historic interest (n = 16) (50, 51, 53, 55–57).
Serotype emm28 strains.
The 1,275 serotype emm28 invasive strains were collected as part of comprehensive prospective population-based surveillance studies conducted in Finland (n = 48), Iceland (n = 27), Ontario, Canada (n = 204), and the United States (n = 996).
SpeB secreted protease activity assay.
SpeB protease activity secreted by the 5,775 GAS strains was evaluated with a standard casein hydrolysis (milk plate) assay (34). Briefly, strains were grown from cryopreserved stocks for 12 to 24 h on tryptic soy agar (TSA) supplemented with 5% sheep blood (Becton Dickinson) at 37°C under 5% CO2. Each strain was then subcultured for 16 h to the early-stationary growth phase in Todd-Hewitt medium supplemented with 0.2% yeast extract (Difco) and was inoculated in triplicate by stabbing TSA plates supplemented with 5% skim milk (Teknova) with a solid-bore plastic needle (Globe Scientific). The plates were incubated for 24 h at 37°C in an anaerobic chamber (Mitsubishi Gas Chemical Co.). Secreted SpeB protease activity was determined by measuring the zone of caseinolysis around each inoculation site using a digital caliper (Fisher Scientific). The mean zone size was interpreted relative to those of the SpeB wild-type emm1 strain MGAS2221 (58), the SpeB-deficient emm1 strain MGAS5005, which has an inactivating mutation in the gene encoding CovS (59), and the isogenic mutant SpeB-negative strain MGAS5005ΔspeB, which has the gene encoding SpeB deleted.
Gene sequencing and polymorphism discovery.
The whole-genome sequence data and the bioinformatic analysis strategy used for the 3,615 emm1 strains have been reported previously (27). Genomewide polymorphisms (single nucleotide polymorphisms [SNPs] and insertions and deletions [indels]) were first identified relative to the very high quality genome of the emm1 reference strain MGAS5005 (GenBank accession number CP000017) with VAAL software (60). The nature of SNPs (coding/noncoding, synonymous/nonsynonymous, etc.) was determined with the Perl script SNPeffect-0.2.pl. An R script was used to export a comma-separated values (CSV) file containing all polymorphisms identified in the 21 genes implicated in altered secreted SpeB protease activity (27). The complete upstream noncoding regions and coding regions of the 21 genes were evaluated for every strain identified as having a SpeB-deficient phenotype (Table 1). Synonymous single nucleotide polymorphisms (coding for the same amino acid) and polymorphisms identified in multiple strains with a wild-type SpeB phenotype (not SpeB altering) were excluded from the analysis. For SpeB-deficient strains initially found by this analysis strategy to lack a gene polymorphism that could account for the protease-negative phenotype, the whole-genome sequence data were reanalyzed with a second bioinformatics strategy using Trimmomatic, Musket, SMALT, and FreeBayes as described previously (56). FreeBayes (61) is better able than VAAL to identify minority subpopulations that could contribute to an altered SpeB protease phenotype. All polymorphisms contributing to a SpeB-deficient phenotype were confirmed by visualization of the mapped reads using Tablet (62). Previously published whole-genome sequence data for SpeB-deficient emm89, emm59, and emm28 strains were analyzed using the same bioinformatics process.
Experimental animal infections.
A nonhuman primate model of necrotizing fasciitis was used as described previously (27, 39, 63). Four adult cynomolgus macaques (Macaca fascicularis) (Charles River BRF) were sedated with ketamine, shaved over the upper posterior thorax, and premedicated with a 25-μg/h fentanyl patch that was sutured in place over the shaved area (Sandoz, Inc.). A mesh jacket (Lomir Biomedical, Inc.) was fitted to each animal to protect the fentanyl patch. Each animal was inoculated intramuscularly in the anterior thigh at a uniform depth with 1 × 108 CFU/kg of serotype emm1 strain MGAS2221 in 200 μl phosphate-buffered saline (PBS). Strain MGAS2221 was selected because it has a wild-type allele (i.e., the most common allele) for all major GAS transcriptional regulators, including covRS and ropB, is genetically representative of contemporary epidemic serotype M1 strains, and has been used in numerous animal experiments (27). The inoculation site was marked with a tattoo. The animals were observed continuously, sacrificed at 24 h postinoculation, and necropsied. At necropsy, the quadriceps muscle was removed en bloc, serially sectioned in 0.5-cm slices, and inspected visually. Biopsy specimens of grossly purulent tissue (approximately 0.5 g each) were collected from the inoculation site, 1 cm from the caudal margin, and 1 cm from the cephalic margin. Each muscle sample was homogenized (Omni International), serially diluted in sterile PBS, and plated in quadruplicate on Trypticase soy agar supplemented with 5% sheep blood (Becton Dickinson and Company). Following incubation at 37°C under 5% CO2 for 18 h, the plates were removed and were visually inspected in order to enumerate colonies and confirm the absence of contaminating organisms. In total, 2,954 GAS colonies (range, 698 to 760 per animal; approximately one-third each from the inoculation site, caudal margin, and cephalic margin) were selected for whole-genome sequencing as described previously (27). To avoid introducing bias due to the colony phenotype, every colony was taken from plates prepared from the dilution that produced approximately 100 to 200 colonies/plate. The study protocol was approved by the Institutional Animal Care and Use Committee at the Houston Methodist Research Institute.
RESULTS
Secreted SpeB protease activity among 3,615 emm1 strains.
A casein milk plate hydrolysis assay was used to test the hypothesis that most emm1 GAS strains have a wild-type SpeB proteolytic phenotype. Serotype emm1 strains were selected for study because they are a frequent source of human pharyngeal and sterile-site infections (2), display epidemic behavior (2), and have caused marked increases in the frequency and severity of invasive infections beginning in the mid- to late 1980s (25–30). The 3,615 strains studied were from comprehensive population-based studies conducted in 9 disparate geographic locations over approximately 40 years (27). The majority of these strains were cultured from patients with invasive infections. In agreement with our hypothesis, 3,061 strains (84.6%) had wild-type secreted SpeB protease activity (Fig. 1A).
FIG 1.
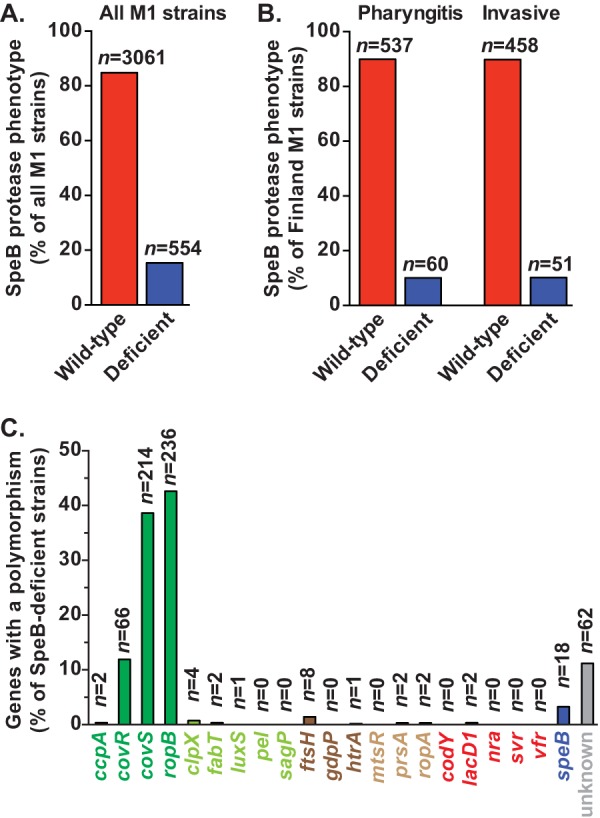
A majority of emm1 GAS strains have a wild-type secreted SpeB protease phenotype. (A) Casein milk plate hydrolysis assays were performed on 3,615 emm1 GAS strains collected in comprehensive population-based studies conducted at 9 geographically disparate sites in North America and Europe. The number of strains with a SpeB-wild-type (red bar) or deficient (blue bar) phenotype is shown. (B) Comparison of secreted SpeB protease activities in strains recovered within overlapping time intervals from patients in Finland with pharyngeal or invasive infections. (C) Whole-genome sequence data were analyzed in order to identify polymorphisms in 21 genes encoding products implicated in secreted SpeB protease activity, including those involved in direct transcriptional regulation (dark green), indirect transcriptional regulation or posttranscriptional processing (light green), secretion (dark brown), posttranslational processing (light brown), or growth phase or environmental signaling (red), as well as speB and its upstream regulatory region (blue). In some SpeB-deficient strains, no likely causative polymorphisms were identified (gray).
A small case series had previously suggested that severe invasive infections were associated with a SpeB-deficient phenotype (23). We compared the SpeB phenotype results for GAS strains recovered from patients with invasive or pharyngeal infections in Finland within overlapping time intervals (27). No significant difference in secreted SpeB protease activity was found between these two patient groups (Fig. 1B) (P, not significant by the chi-square test).
Gene polymorphisms associated with decreased secreted SpeB protease activity among emm1 strains.
Twenty-one genes have been implicated in a complicated process that ultimately generates enzymatically active SpeB (Table 1) (4). To determine the relative frequency of mutations associated with decreased secreted SpeB protease activity, we analyzed whole-genome sequence data for the 554 SpeB-deficient emm1 GAS strains we identified (27). The three most frequently mutated genes encode proteins that are known to directly regulate speB transcription (Fig. 1C). These genes encode the two-component system CovRS (control of virulence regulator/sensor) and RopB (regulator of protease B). A total of 66 (11.9%) SpeB-deficient emm1 strains have mutations in covR, 214 (38.6%) have mutations in covS, and 236 (42.6%) have mutations in ropB. Polymorphisms in the other genes implicated were infrequently identified (range, 0 to 8 polymorphisms per gene [0 to 1.4%]). Only 18 strains (3.2% of SpeB-deficient strains) had a polymorphism in the speB coding region or the upstream promoter region. Of note, no polymorphism that likely explains the SpeB-deficient phenotype was identified for 62 strains (11.2% of SpeB-deficient emm1 strains) (Fig. 1C).
Secreted SpeB protease activity among 1,181 emm89 strains.
To further test our hypothesis that most GAS strains recovered from infected humans with invasive infections have a wild-type SpeB production level, we examined the secreted protease activity in 1,181 emm89 strains for which we also have full-genome sequence data (52). Serotype emm89 strains were analyzed because they are a frequent cause of human infections (2, 53, 64–66). The emm89 strains analyzed were collected in comprehensive population-based studies conducted in Finland, Iceland, and the United States. Like our findings for the emm1 strains, the milk plate hydrolysis data showed that the great majority of emm89 GAS strains (n = 936 [79.3%]) had a wild-type secreted SpeB protease phenotype (Fig. 2A). Among the 245 emm89 strains deficient in SpeB activity, frequently mutated genes included covR (mutated in 37 strains [15.1% of SpeB-deficient emm89 strains]), covS (100 strains [40.8%]), and ropB (96 strains [39.2%]) (Fig. 2B).
FIG 2.
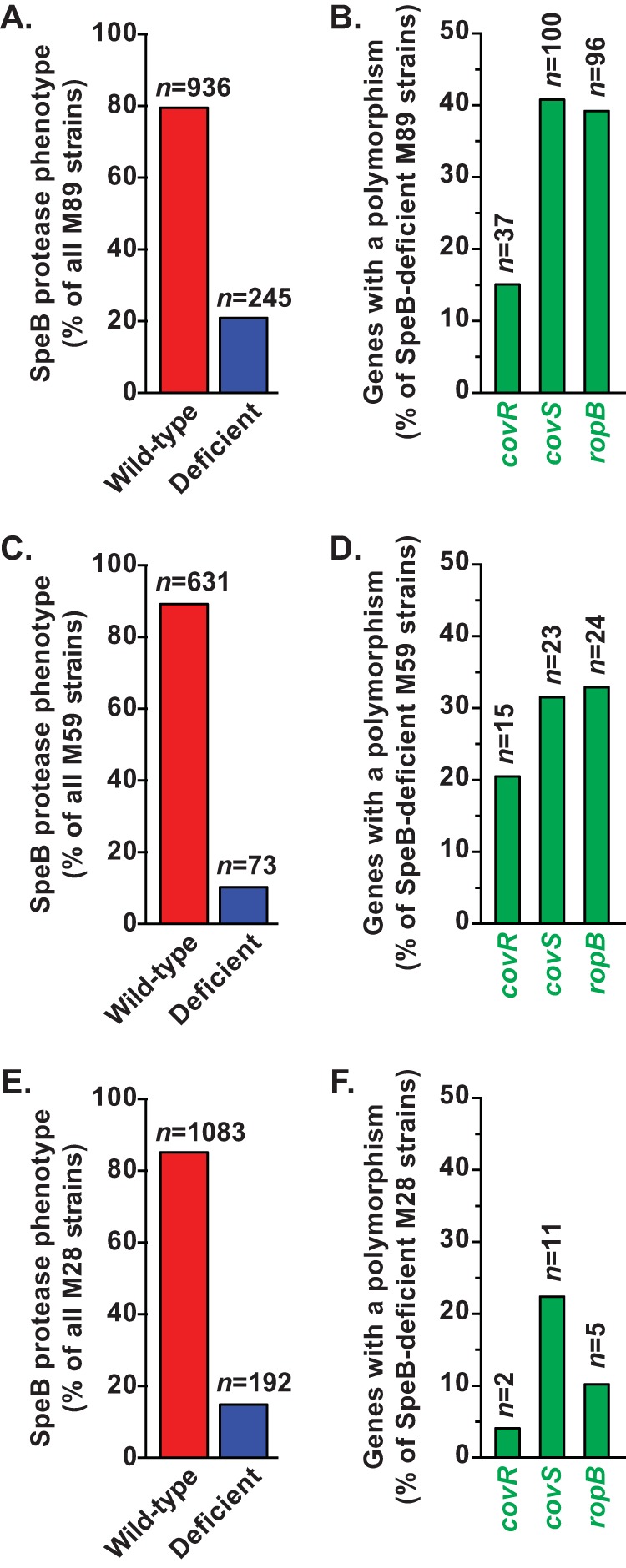
The majority of emm89, emm59, and emm28 strains have a wild-type secreted SpeB protease phenotype. Casein milk plate hydrolysis assays were performed on 1,181 serotype emm89 (A), 704 emm59 (C), and 1,275 emm28 (E) GAS strains collected in comprehensive population-based studies conducted at geographically distinct sites in North America and Europe. The numbers of strains with a SpeB-wild-type (red bars) or deficient (blue bars) phenotype are shown. Whole-genome sequence data were analyzed to identify polymorphisms in covR, covS, and ropB in SpeB-deficient emm89 (B), emm59 (D), and emm28 (F) strains.
Secreted SpeB protease activity among 704 emm59 strains.
We next assessed secreted SpeB protease activity among 704 emm59 strains that have been characterized by whole-genome sequencing (50, 51, 55, 56, 67). Most of these emm59 organisms were recovered as part of comprehensive population-based epidemiologic studies conducted during a countrywide epidemic of severe invasive infections in Canada (57) or were cultured from patients with invasive infections in the United States (53). The results were closely similar to our findings for the emm1 and emm89 strains: the great majority of the emm59 GAS strains (n = 631 [89.6%]) produced wild-type levels of secreted SpeB protease activity (Fig. 2C). The most frequently mutated genes among the protease-deficient emm59 organisms were covR (mutated in 15 strains [20.5% of SpeB-deficient emm59 strains]), covS (23 strains [31.5%]), and ropB (24 strains [32.9%]) (Fig. 2D).
Secreted SpeB protease activity among 1,275 emm28 GAS strains.
We next measured SpeB protease activity secreted by 1,275 emm28 strains. Most of these emm28 GAS organisms were recovered as part of comprehensive population-based epidemiologic studies conducted in Finland, Iceland, Ontario (Canada), and the United States. Serotype emm28 GAS strains are a numerically prominent cause of human infections, particularly life-threatening puerperal sepsis (53, 68). As observed for strains of the other GAS emm types studied, the great majority of emm28 strains (n = 1,083 [84.9%]) had wild-type secreted SpeB protease activity (Fig. 2E). Although not all the genomes of the emm28 GAS strains in these collections have been sequenced yet, data were available for 49 SpeB-deficient organisms. Mutations in covR (mutated in 2 strains [4.1% of SpeB-deficient emm28 strains for which whole-genome sequence data were available]), covS (11 strains [22.4%]), and ropB (5 strains [10.2%]) occurred at rates similar to those found in the emm1, emm89, and emm59 strains (Fig. 2F).
Whole-genome sequence analysis of 2,954 serotype emm1 GAS strains recovered in a nonhuman primate model of necrotizing fasciitis.
When mice are experimentally infected with wild-type GAS, SpeB-deficient mutants (most commonly due to mutations in covRS) arise rapidly and may increase to numerical prominence (19, 33, 58). However, our data clearly demonstrate that the great majority of GAS strains recovered from humans with pharyngitis or invasive infections retain a wild-type SpeB phenotype (Fig. 1 and 2). To further test the hypothesis that most GAS strains recovered in invasive infections secrete SpeB protease, we sequenced the genomes of 2,954 strains recovered from 4 nonhuman primates (the most human-relevant animal model possible) with experimental necrotizing fasciitis caused by a wild-type emm1 GAS strain. In agreement with our hypothesis, none of these strains acquired a mutation in any of the 21 genes known to be needed for GAS to secrete wild-type SpeB protease activity.
DISCUSSION
The progeny of a globally disseminated serotype emm1 GAS clone that arose in the early 1980s are responsible for an ongoing epidemic of severe invasive infections (25, 26). Due to its human health importance, the emm1 epidemic clone has been extensively studied by many investigators (25–30). The identification of polymorphisms in several genes that confer a SpeB-deficient phenotype has led, in part, to the hypothesis that epidemic emm1 GAS strains are under intense selective pressure to decrease secreted SpeB protease activity during infection (19–24). Reinforcing this idea, Kansal et al., using a small sample of strains, reported an inverse relationship between SpeB proteolytic activity and disease severity (23). This report has been repeatedly cited to support the idea that SpeB-deficient strains are driving the emm1 epidemic. However, as shown in the present study, data from 3,615 emm1 GAS strains recovered in comprehensive population-based studies at nine geographically distinct sites on two continents do not support this hypothesis. Our data clearly demonstrate that a SpeB-deficient phenotype is very uncommon among epidemic emm1 organisms (Fig. 1). Moreover, the SpeB-deficient phenotype occurs at the same frequency among strains causing invasive infections and those causing pharyngitis (Fig. 1). Thus, the only reasonable conclusion is that strains with genetic changes resulting in decreased secreted SpeB protease activity are not major contributors to the global epidemic of emm1 GAS infections. Analysis of 1,181 emm89, 704 emm59, and 1,275 emm28 strains produced essentially identical findings (Fig. 2). That is, collectively among 6,775 strains taken from comprehensive population-based collections of 4 different GAS emm types, only 15.7% of strains have a SpeB-deficient phenotype.
Our data have important implications for understanding the emergence of SpeB-deficient strains of emm1 GAS and presumably other emm types of GAS. We and other investigators have described polymorphisms in the two-component signal transduction system covRS that markedly alter the transcriptome profile of GAS, including decreasing speB expression (31, 58, 69). Mutations in covRS are readily generated by passage of wild-type strains through mice to create progeny with an “invasive phenotype” (58). One school of thought has been that elimination of secreted SpeB protease activity, which may degrade other GAS virulence factors expressed in the SpeB wild-type condition, acts as the molecular switch to increased virulence of the epidemic clone during invasive infection (31, 32, 58). However, although loss of SpeB proteolytic activity occurs in experimentally infected mice and some humans, our casein hydrolysis and genomic data rule out the speculative idea that this phenotype is the primary driver of the epidemic GAS disease. If GAS were under selection to develop a SpeB-deficient phenotype in infected humans, then mutations that eliminate secreted SpeB proteolytic activity would be identified at a far higher frequency. Moreover, mutations in speB itself, rather than in transcription factors that regulate many other GAS virulence factors, would be expected to occur more frequently. Our data show that only genes encoding direct regulators of speB expression (covRS and ropB) accumulated more polymorphisms than would be expected to occur by chance alone (Fig. 1 and 2) (27). Furthermore, 28 SpeB-deficient emm1 strains had polymorphisms in both covRS and a second gene that would confer decreased secreted SpeB protease activity. We also identified 19 emm89 and 6 emm59 GAS strains with polymorphisms in both covRS and ropB. That is, covRS and ropB mutations may be occurring in strains that already have a SpeB-deficient phenotype, or vice versa. Two independent studies have similarly described covRS mutations arising in SpeB-deficient strains with preexisting ropB mutations (70, 71). In addition, we identified 8 covRS-mutated SpeB-deficient emm1 strains that were recovered in cases of human pharyngitis, demonstrating that these mutations are not limited to invasive infections. Taken together, our data strongly support an alternative model in which the great majority of emm1 (and emm89, emm59, and emm28) GAS strains causing human infections retain a wild-type SpeB phenotype (Fig. 3). Less commonly, GAS may acquire polymorphisms in major transcription regulators, such as covRS or ropB, that influence the expression of a large number of virulence genes. Thus, selection pressure to eliminate secreted SpeB protease activity may not be a primary factor; rather, GAS may be under selection to increase the expression of genes encoding other potent virulence factors regulated by covRS (58), including the antiphagocytic hyaluronic acid capsule (63), Sda1 DNase (72), and the cytolytic toxins streptolysin O (SLO) and NAD+ glycohydrolase (SPN) (52). In this regard, we recently demonstrated that upregulation of SLO and SPN exotoxin production by recombinational evolution was the critical molecular trigger igniting intercontinental epidemics of emm1 and emm89 GAS that have affected tens of millions of people (27, 52).
FIG 3.
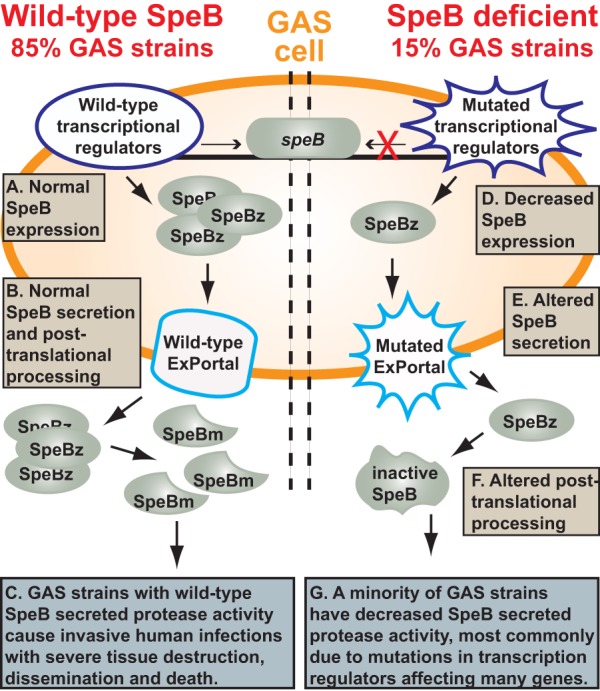
Model summarizing secreted SpeB protease activity. (Left) In wild-type strains (approximately 85% of GAS strains cultured from infected humans), normal amounts of SpeB transcript are made (A), the enzymatically inactive zymogen (SpeBz) is translated and secreted through the ExPortal (B), and SpeBz is processed through multiple intra- and intermolecular steps to yield the potent broad-spectrum mature cysteine protease (SpeBm), which is a key virulence factor for human disease (C). (Right) A minority of GAS strains (15%) have a SpeB-deficient phenotype caused by mutations in genes implicated in transcription or posttranscriptional regulation (D), secretion (E), or posttranslational processing (F). SpeB-deficient strains constitute a minority among organisms causing either human invasive infections or pharyngitis (G).
The relationship between SpeB protease production and disease pathogenesis is a complicated area of investigation that has been studied for decades, with conflicting conclusions reached. Wheeler et al. reported high levels of SpeB protease production among 73% of GAS strains cultured from pediatric patients with bacteremia (73). Holm et al. and Norrby et al. reported high levels of SpeB production among emm1 strains from patients in Scandinavian countries with invasive GAS infections (45, 74). Norgren et al. studied 38 serotype emm1 strains cultured from the blood of patients with serious GAS infections and also reported high levels of SpeB production (75). Talkington et al. analyzed 62 strains cultured from patients with invasive infections, including 32 with streptococcal toxic shock syndrome, and found that emm1 strains were significantly associated with protease production (76). Moreover, protease production was significantly associated with soft tissue necrosis, including myositis and fasciitis (76). Chaussee et al. reported a lack of significant association between SpeB protease production and the severity of human infection based on analysis of strains of various emm serotypes cultured from 112 patients (77). Chatellier et al. analyzed 35 patients with invasive disease with different levels of severity caused by clonally related serotype emm1 strains and failed to identify a significant relationship between the level of SpeB protease production and disease severity (78). In contrast, based on the level of SpeB protease activity made in vitro by serotype emm1 strains, Kansal et al. reported an inverse relationship between disease severity and SpeB expression (23). No data bearing on host factors, such as the level of anti-SpeB antibody present in acute-phase sera from these patients, were reported (23). Similarly, SpeB-negative mutants can be selected rapidly and rise to numerical prominence in experimentally infected mice (33). However, pathogen-host interactions in mice do not necessarily translate to the common human condition (79, 80). In support of a possible host (mouse compared to human) species effect on covRS polymorphism acquisition, we sequenced the genomes of 2,954 emm1 GAS strains recovered from 4 nonhuman primates (the most human-relevant animal model possible) with experimentally induced necrotizing fasciitis. None developed mutations in any gene needed for GAS to produce wild-type SpeB, including covRS. Taken together, our data show that SpeB-deficient organisms have not become predominate members of globally circulating GAS populations, including strains of multiple serotypes and strains recovered in either pharyngitis or invasive infections (Fig. 1 and 2). We interpret our data, and those presented by many other investigators based on divergent lines of research (9, 22, 34–41, 43–45, 48, 73–78), as confirming the important role of SpeB in pathogen-host interaction and GAS pathogenesis (Fig. 3).
ACKNOWLEDGMENTS
We thank Carmen Ardanuy, Dominique A. Caugant, Jessica Darenberg, Magnus Gottfredsson, Birgitta Henriques-Normark, Steen Hoffman, Karl G. Kristinsson, Josefina Linares, Marguerite Lovgren, Donald E. Low, Allison McGeer, Kati Raisanen, Martin Steinbakk, Gregory J. Tyrrell, Chris A. Van Beneden, and Jaana Vuopio for providing strains used in this study.
REFERENCES
- 1.Kaman WE, Hays JP, Endtz HP, Bikker FJ. 2014. Bacterial proteases: targets for diagnostics and therapy. Eur J Clin Microbiol Infect Dis 33:1081–1087. doi: 10.1007/s10096-014-2075-1. [DOI] [PubMed] [Google Scholar]
- 2.Carapetis JR, Steer AC, Mulholland EK, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect Dis 5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 3.Olsen RJ, Musser JM. 2010. Molecular pathogenesis of necrotizing fasciitis. Annu Rev Pathol 5:1–31. doi: 10.1146/annurev-pathol-121808-102135. [DOI] [PubMed] [Google Scholar]
- 4.Carroll RK, Musser JM. 2011. From transcription to activation: how group A streptococcus, the flesh-eating pathogen, regulates SpeB cysteine protease production. Mol Microbiol 81:588–601. doi: 10.1111/j.1365-2958.2011.07709.x. [DOI] [PubMed] [Google Scholar]
- 5.Bohach GA, Hauser AR, Schlievert PM. 1988. Cloning of the gene, speB, for streptococcal pyrogenic exotoxin type B in Escherichia coli. Infect Immun 56:1665–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu CE, Ferretti JJ. 1991. Frequency of the erythrogenic toxin B and C genes (speB and speC) among clinical isolates of group A streptococci. Infect Immun 59:211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nooh MM, Aziz RK, Kotb M, Eroshkin A, Chuang WJ, Proft T, Kansal R. 2006. Streptococcal mitogenic exotoxin, SmeZ, is the most susceptible M1T1 streptococcal superantigen to degradation by the streptococcal cysteine protease, SpeB. J Biol Chem 281:35281–35288. doi: 10.1074/jbc.M605544200. [DOI] [PubMed] [Google Scholar]
- 8.Raeder R, Woischnik M, Podbielski A, Boyle MD. 1998. A secreted streptococcal cysteine protease can cleave a surface-expressed M1 protein and alter the immunoglobulin binding properties. Res Microbiol 149:539–548. doi: 10.1016/S0923-2508(99)80001-1. [DOI] [PubMed] [Google Scholar]
- 9.Terao Y, Mori Y, Yamaguchi M, Shimizu Y, Ooe K, Hamada S, Kawabata S. 2008. Group A streptococcal cysteine protease degrades C3 (C3b) and contributes to evasion of innate immunity. J Biol Chem 283:6253–6260. doi: 10.1074/jbc.M704821200. [DOI] [PubMed] [Google Scholar]
- 10.Tamura F, Nakagawa R, Akuta T, Okamoto S, Hamada S, Maeda H, Kawabata S, Akaike T. 2004. Proapoptotic effect of proteolytic activation of matrix metalloproteinases by Streptococcus pyogenes thiol proteinase (Streptococcus pyrogenic exotoxin B). Infect Immun 72:4836–4847. doi: 10.1128/IAI.72.8.4836-4847.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bryant AE, Bayer CR, Huntington JD, Stevens DL. 2006. Group A streptococcal myonecrosis: increased vimentin expression after skeletal-muscle injury mediates the binding of Streptococcus pyogenes. J Infect Dis 193:1685–1692. doi: 10.1086/504261. [DOI] [PubMed] [Google Scholar]
- 12.Burns EH Jr, Marciel AM, Musser JM. 1996. Activation of a 66-kilodalton human endothelial cell matrix metalloprotease by Streptococcus pyogenes extracellular cysteine protease. Infect Immun 64:4744–4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma Y, Bryant AE, Salmi DB, Hayes-Schroer SM, McIndoo E, Aldape MJ, Stevens DL. 2006. Identification and characterization of bicistronic speB and prsA gene expression in the group A streptococcus. J Bacteriol 188:7626–7634. doi: 10.1128/JB.01059-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loughman JA, Caparon M. 2006. Regulation of SpeB in Streptococcus pyogenes by pH and NaCl: a model for in vivo gene expression. J Bacteriol 188:399–408. doi: 10.1128/JB.188.2.399-408.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Z, Sledjeski DD, Kreikemeyer B, Podbielski A, Boyle MD. 1999. Identification of pel, a Streptococcus pyogenes locus that affects both surface and secreted proteins. J Bacteriol 181:6019–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lyon WR, Caparon MG. 2004. Role for serine protease HtrA (DegP) of Streptococcus pyogenes in the biogenesis of virulence factors SpeB and the hemolysin streptolysin S. Infect Immun 72:1618–1625. doi: 10.1128/IAI.72.3.1618-1625.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doran JD, Nomizu M, Takebe S, Menard R, Griffith D, Ziomek E. 1999. Autocatalytic processing of the streptococcal cysteine protease zymogen: processing mechanism and characterization of the autoproteolytic cleavage sites. Eur J Biochem 263:145–151. doi: 10.1046/j.1432-1327.1999.00473.x. [DOI] [PubMed] [Google Scholar]
- 18.Hauser AR, Schlievert PM. 1990. Nucleotide sequence of the streptococcal pyrogenic exotoxin type B gene and relationship between the toxin and the streptococcal proteinase precursor. J Bacteriol 172:4536–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aziz RK, Pabst MJ, Jeng A, Kansal R, Low DE, Nizet V, Kotb M. 2004. Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. Mol Microbiol 51:123–134. doi: 10.1046/j.1365-2958.2003.03797.x. [DOI] [PubMed] [Google Scholar]
- 20.Chella Krishnan K, Mukundan S, Landero Figueroa JA, Caruso JA, Kotb M. 2014. Metal-mediated modulation of streptococcal cysteine protease activity and its biological implications. Infect Immun 82:2992–3001. doi: 10.1128/IAI.01770-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ender M, Andreoni F, Zinkernagel AS, Schuepbach RA. 2013. Streptococcal SpeB cleaved PAR-1 suppresses ERK phosphorylation and blunts thrombin-induced platelet aggregation. PLoS One 8:e81298. doi: 10.1371/journal.pone.0081298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hollands A, Aziz RK, Kansal R, Kotb M, Nizet V, Walker MJ. 2008. A naturally occurring mutation in ropB suppresses SpeB expression and reduces M1T1 group A streptococcal systemic virulence. PLoS One 3:e4102. doi: 10.1371/journal.pone.0004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kansal RG, McGeer A, Low DE, Norrby-Teglund A, Kotb M. 2000. Inverse relation between disease severity and expression of the streptococcal cysteine protease, SpeB, among clonal M1T1 isolates recovered from invasive group A streptococcal infection cases. Infect Immun 68:6362–6369. doi: 10.1128/IAI.68.11.6362-6369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cole JN, Aquilina JA, Hains PG, Henningham A, Sriprakash KS, Caparon MG, Nizet V, Kotb M, Cordwell SJ, Djordjevic SP, Walker MJ. 2007. Role of group A Streptococcus HtrA in the maturation of SpeB protease. Proteomics 7:4488–4498. doi: 10.1002/pmic.200700626. [DOI] [PubMed] [Google Scholar]
- 25.Musser JM, Krause R. 1998. The revival of group A streptococcal diseases, with a commentary on staphylococcal toxic shock syndrome. Biomed Res Rep 1:185–218. doi: 10.1016/S1874-5326(07)80030-5. [DOI] [Google Scholar]
- 26.Cole JN, Barnett TC, Nizet V, Walker MJ. 2011. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol 9:724–736. doi: 10.1038/nrmicro2648. [DOI] [PubMed] [Google Scholar]
- 27.Nasser W, Beres SB, Olsen RJ, Dean MA, Rice KA, Long SW, Kristinsson KG, Gottfredsson M, Vuopio J, Raisanen K, Caugant DA, Steinbakk M, Low DE, McGeer A, Darenberg J, Henriques-Normark B, Van Beneden CA, Hoffmann S, Musser JM. 2014. Evolutionary pathway to increased virulence and epidemic group A Streptococcus disease derived from 3,615 genome sequences. Proc Natl Acad Sci U S A 111:E1768–E1776. doi: 10.1073/pnas.1403138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Musser JM, Hauser AR, Kim MH, Schlievert PM, Nelson K, Selander RK. 1991. Streptococcus pyogenes causing toxic-shock-like syndrome and other invasive diseases: clonal diversity and pyrogenic exotoxin expression. Proc Natl Acad Sci U S A 88:2668–2672. doi: 10.1073/pnas.88.7.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cleary PP, Kaplan EL, Handley JP, Wlazlo A, Kim MH, Hauser AR, Schlievert PM. 1992. Clonal basis for resurgence of serious Streptococcus pyogenes disease in the 1980s. Lancet 339:518–521. doi: 10.1016/0140-6736(92)90339-5. [DOI] [PubMed] [Google Scholar]
- 30.Musser JM, Kapur V, Szeto J, Pan X, Swanson DS, Martin DR. 1995. Genetic diversity and relationships among Streptococcus pyogenes strains expressing serotype M1 protein: recent intercontinental spread of a subclone causing episodes of invasive disease. Infect Immun 63:994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, McArthur JD, Dinkla K, Aziz RK, Kansal RG, Simpson AJ, Buchanan JT, Chhatwal GS, Kotb M, Nizet V. 2007. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med 13:981–985. doi: 10.1038/nm1612. [DOI] [PubMed] [Google Scholar]
- 32.Cole JN, McArthur JD, McKay FC, Sanderson-Smith ML, Cork AJ, Ranson M, Rohde M, Itzek A, Sun H, Ginsburg D, Kotb M, Nizet V, Chhatwal GS, Walker M. 2006. Trigger for group A streptococcal M1T1 invasive disease. FASEB J 20:1745–1747. doi: 10.1096/fj.06-5804fje. [DOI] [PubMed] [Google Scholar]
- 33.Li J, Liu G, Feng W, Zhou Y, Liu M, Wiley JA, Lei B. 2014. Neutrophils select hypervirulent CovRS mutants of M1T1 group A Streptococcus during subcutaneous infection of mice. Infect Immun 82:1579–1590. doi: 10.1128/IAI.01458-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olsen RJ, Laucirica DR, Watkins ME, Feske ML, Garcia-Bustillos JR, Vu C, Cantu C, Shelburne SA III, Fittipaldi N, Kumaraswami M, Shea PR, Flores AR, Beres SB, Lovgren M, Tyrrell GJ, Efstratiou A, Low DE, Van Beneden CA, Musser JM. 2012. Polymorphisms in regulator of protease B (RopB) alter disease phenotype and strain virulence of serotype M3 group A Streptococcus. J Infect Dis 205:1719–1729. doi: 10.1093/infdis/jir825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lukomski S, Sreevatsan S, Amberg C, Reichardt W, Woischnik M, Podbielski A, Musser JM. 1997. Inactivation of Streptococcus pyogenes extracellular cysteine protease significantly decreases mouse lethality of serotype M3 and M49 strains. J Clin Invest 99:2574–2580. doi: 10.1172/JCI119445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuo CF, Wu JJ, Lin KY, Tsai PJ, Lee SC, Jin YT, Lei HY, Lin YS. 1998. Role of streptococcal pyrogenic exotoxin B in the mouse model of group A streptococcal infection. Infect Immun 66:3931–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Connolly KL, Roberts AL, Holder RC, Reid SD. 2011. Dispersal of group A streptococcal biofilms by the cysteine protease SpeB leads to increased disease severity in a murine model. PLoS One 6:e18984. doi: 10.1371/journal.pone.0018984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lukomski S, Montgomery CA, Rurangirwa J, Geske RS, Barrish JP, Adams GJ, Musser JM. 1999. Extracellular cysteine protease produced by Streptococcus pyogenes participates in the pathogenesis of invasive skin infection and dissemination in mice. Infect Immun 67:1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olsen RJ, Sitkiewicz I, Ayeras AA, Gonulal VE, Cantu C, Beres SB, Green NM, Lei B, Humbird T, Greaver J, Chang E, Ragasa WP, Montgomery CA, Cartwright J, McGeer A, Low DE, Whitney AR, Cagle PT, Blasdel TL, DeLeo FR, Musser JM. 2010. Decreased necrotizing fasciitis capacity caused by a single nucleotide mutation that alters a multiple gene virulence axis. Proc Natl Acad Sci U S A 107:888–893. doi: 10.1073/pnas.0911811107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olsen RJ, Watkins ME, Cantu CC, Beres SB, Musser JM. 2011. Virulence of serotype M3 group A Streptococcus strains in wax worms (Galleria mellonella larvae). Virulence 2:111–119. doi: 10.4161/viru.2.2.14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carroll RK, Shelburne SA III, Olsen RJ, Suber B, Sahasrabhojane P, Kumaraswami M, Beres SB, Shea PR, Flores AR, Musser JM. 2011. Naturally occurring single amino acid replacements in a regulatory protein alter streptococcal gene expression and virulence in mice. J Clin Invest 121:1956–1968. doi: 10.1172/JCI45169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johansson L, Thulin P, Sendi P, Hertzen E, Linder A, Akesson P, Low DE, Agerberth B, Norrby-Teglund A. 2008. Cathelicidin LL-37 in severe Streptococcus pyogenes soft tissue infections in humans. Infect Immun 76:3399–3404. doi: 10.1128/IAI.01392-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eriksson BK, Andersson J, Holm SE, Norgren M. 1999. Invasive group A streptococcal infections: T1M1 isolates expressing pyrogenic exotoxins A and B in combination with selective lack of toxin-neutralizing antibodies are associated with increased risk of streptococcal toxic shock syndrome. J Infect Dis 180:410–418. doi: 10.1086/314872. [DOI] [PubMed] [Google Scholar]
- 44.Norrby-Teglund A, Pauksens K, Holm SE, Norgren M. 1994. Relation between low capacity of human sera to inhibit streptococcal mitogens and serious manifestation of disease. J Infect Dis 170:585–591. doi: 10.1093/infdis/170.3.585. [DOI] [PubMed] [Google Scholar]
- 45.Holm SE, Norrby A, Bergholm AM, Norgren M. 1992. Aspects of pathogenesis of serious group A streptococcal infections in Sweden, 1988–1989. J Infect Dis 166:31–37. doi: 10.1093/infdis/166.1.31. [DOI] [PubMed] [Google Scholar]
- 46.Ulrich RG. 2008. Vaccine based on a ubiquitous cysteinyl protease and streptococcal pyrogenic exotoxin A protects against Streptococcus pyogenes sepsis and toxic shock. J Immune Based Ther Vaccines 6:8. doi: 10.1186/1476-8518-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morefield G, Touhey G, Lu F, Dunham A, HogenEsch H. 2014. Development of a recombinant fusion protein vaccine formulation to protect against Streptococcus pyogenes. Vaccine 32:3810–3815. doi: 10.1016/j.vaccine.2014.04.092. [DOI] [PubMed] [Google Scholar]
- 48.Kapur V, Maffei JT, Greer RS, Li LL, Adams GJ, Musser JM. 1994. Vaccination with streptococcal extracellular cysteine protease (interleukin-1β convertase) protects mice against challenge with heterologous group A streptococci. Microb Pathog 16:443–450. doi: 10.1006/mpat.1994.1044. [DOI] [PubMed] [Google Scholar]
- 49.Björck L, Akesson P, Bohus M, Trojnar J, Abrahamson M, Olafsson I, Grubb A. 1989. Bacterial growth blocked by a synthetic peptide based on the structure of a human proteinase inhibitor. Nature 337:385–386. doi: 10.1038/337385a0. [DOI] [PubMed] [Google Scholar]
- 50.Fittipaldi N, Beres SB, Olsen RJ, Kapur V, Shea PR, Watkins ME, Cantu CC, Laucirica DR, Jenkins L, Flores AR, Lovgren M, Ardanuy C, Linares J, Low DE, Tyrrell GJ, Musser JM. 2012. Full-genome dissection of an epidemic of severe invasive disease caused by a hypervirulent, recently emerged clone of group A Streptococcus. Am J Pathol 180:1522–1534. doi: 10.1016/j.ajpath.2011.12.037. [DOI] [PubMed] [Google Scholar]
- 51.Fittipaldi N, Olsen RJ, Beres SB, Van Beneden C, Musser JM. 2012. Genomic analysis of emm59 group A Streptococcus invasive strains, United States. Emerg Infect Dis 18:650–652. doi: 10.3201/eid1804.111803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu L, Olsen RJ, Nasser W, Beres SB, Vuopio J, Kristinsson KG, Gottfredsson M, Porter AR, DeLeo FR, Musser JM. 1 September 2015. A molecular trigger for intercontinental epidemics of group A Streptococcus. J Clin Invest doi: 10.1172/JCI82478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Centers for Disease Control and Prevention. 2013. Active Bacterial Core Surveillance (ABCs) report, Emerging Infections Program Network, group A Streptococcus, 2012. Centers for Disease Control and Prevention, Atlanta, GA: http://www.cdc.gov/abcs/reports-findings/survreports/gas12.pdf. [Google Scholar]
- 54.Hoe NP, Nakashima K, Lukomski S, Grigsby D, Liu M, Kordari P, Dou SJ, Pan X, Vuopio-Varkila J, Salmelinna S, McGeer A, Low DE, Schwartz B, Schuchat A, Naidich S, De Lorenzo D, Fu YX, Musser JM. 1999. Rapid selection of complement-inhibiting protein variants in group A Streptococcus epidemic waves. Nat Med 5:924–929. doi: 10.1038/11369. [DOI] [PubMed] [Google Scholar]
- 55.Fittipaldi N, Tyrrell GJ, Low DE, Martin I, Lin D, Hari KL, Musser JM. 2013. Integrated whole-genome sequencing and temporospatial analysis of a continuing group A Streptococcus epidemic. Emerg Microbes Infect 2:e13. doi: 10.1038/emi.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olsen RJ, Fittipaldi N, Kachroo P, Sanson MA, Long SW, Como-Sabetti KJ, Valson C, Cantu C, Lynfield R, Van Beneden C, Beres SB, Musser JM. 2014. Clinical laboratory response to a mock outbreak of invasive bacterial infections: a preparedness study. J Clin Microbiol 52:4210–4216. doi: 10.1128/JCM.02164-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tyrrell GJ, Lovgren M, Ibrahim Q, Garg S, Chui L, Boone TJ, Mangan C, Patrick DM, Hoang L, Horsman GB, Van Caeseele P, Marrie TJ. 2012. Epidemic of invasive pneumococcal disease, western Canada, 2005–2009. Emerg Infect Dis 18:733–740. doi: 10.3201/eid1805.110235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog 2:e5. doi: 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sumby P, Porcella SF, Madrigal AG, Barbian KD, Virtaneva K, Ricklefs SM, Sturdevant DE, Graham MR, Vuopio-Varkila J, Hoe NP, Musser JM. 2005. Evolutionary origin and emergence of a highly successful clone of serotype M1 group A Streptococcus involved multiple horizontal gene transfer events. J Infect Dis 192:771–782. doi: 10.1086/432514. [DOI] [PubMed] [Google Scholar]
- 60.Nusbaum C, Ohsumi TK, Gomez J, Aquadro J, Victor TC, Warren RM, Hung DT, Birren BW, Lander ES, Jaffe DB. 2009. Sensitive, specific polymorphism discovery in bacteria using massively parallel sequencing. Nat Methods 6:67–69. doi: 10.1038/nmeth.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garrison E, Marth G. 20 July 2012, revision date. Haplotype-based variant detection from short-read sequencing. arXiv:1207.3907v2 [q-bio.GN]. http://arxiv.org/pdf/1207.3907v2.pdf.
- 62.Milne I, Stephen G, Bayer M, Cock PJ, Pritchard L, Cardle L, Shaw PD, Marshall D. 2013. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform 14:193–202. doi: 10.1093/bib/bbs012. [DOI] [PubMed] [Google Scholar]
- 63.Flores AR, Jewell BE, Olsen RJ, Shelburne SA III, Fittipaldi N, Beres SB, Musser JM. 2014. Asymptomatic carriage of group A Streptococcus is associated with elimination of capsule production. Infect Immun 82:3958–3967. doi: 10.1128/IAI.01788-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karaky NM, Araj GF, Tokajian ST. 2014. Molecular characterization of Streptococcus pyogenes group A isolates from a tertiary hospital in Lebanon. J Med Microbiol 63:1197–1204. doi: 10.1099/jmm.0.063412-0. [DOI] [PubMed] [Google Scholar]
- 65.Shea PR, Ewbank AL, Gonzalez-Lugo JH, Martagon-Rosado AJ, Martinez-Gutierrez JC, Rehman HA, Serrano-Gonzalez M, Fittipaldi N, Beres SB, Flores AR, Low DE, Willey BM, Musser JM. 2011. Group A Streptococcus emm gene types in pharyngeal isolates, Ontario, Canada, 2002–2010. Emerg Infect Dis 17:2010–2017. doi: 10.3201/eid1711.110159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Williamson DA, Moreland NJ, Carter P, Upton A, Morgan J, Proft T, Lennon D, Baker MG, Dunbar R, Fraser JD. 2014. Molecular epidemiology of group A streptococcus from pharyngeal isolates in Auckland, New Zealand, 2013. N Z Med J 127(1388):55–60. [PubMed] [Google Scholar]
- 67.Brown CC, Olsen RJ, Fittipaldi N, Morman ML, Fort PL, Neuwirth R, Majeed M, Woodward WB, Musser JM. 2014. Spread of virulent group A Streptococcus type emm59 from Montana to Wyoming, USA. Emerg Infect Dis 20:679–681. doi: 10.3201/eid2004.130564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Green NM, Zhang S, Porcella SF, Nagiec MJ, Barbian KD, Beres SB, LeFebvre RB, Musser JM. 2005. Genome sequence of a serotype M28 strain of group A Streptococcus: potential new insights into puerperal sepsis and bacterial disease specificity. J Infect Dis 192:760–770. doi: 10.1086/430618. [DOI] [PubMed] [Google Scholar]
- 69.Hollands A, Pence MA, Timmer AM, Osvath SR, Turnbull L, Whitchurch CB, Walker MJ, Nizet V. 2010. Genetic switch to hypervirulence reduces colonization phenotypes of the globally disseminated group A Streptococcus M1T1 clone. J Infect Dis 202:11–19. doi: 10.1086/653124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Flores AR, Sahasrabhojane P, Saldana M, Galloway-Pena J, Olsen RJ, Musser JM, Shelburne SA. 2014. Molecular characterization of an invasive phenotype of group A Streptococcus arising during human infection using whole genome sequencing of multiple isolates from the same patient. J Infect Dis 209:1520–1523. doi: 10.1093/infdis/jit674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ikebe T, Ato M, Matsumura T, Hasegawa H, Sata T, Kobayashi K, Watanabe H. 2010. Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog 6:e1000832. doi: 10.1371/journal.ppat.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sumby P, Barbian KD, Gardner DJ, Whitney AR, Welty DM, Long RD, Bailey JR, Parnell MJ, Hoe NP, Adams GG, DeLeo FR, Musser JM. 2005. Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc Natl Acad Sci U S A 102:1679–1684. doi: 10.1073/pnas.0406641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wheeler MC, Roe MH, Kaplan EL, Schlievert PM, Todd JK. 1991. Outbreak of group A streptococcus septicemia in children. Clinical, epidemiologic, and microbiological correlates. JAMA 266:533–537. [PubMed] [Google Scholar]
- 74.Norrby A, Eriksson B, Norgren M, Ronstrom CJ, Sjoblom AC, Karkkonen K, Holm SE. 1992. Virulence properties of erysipelas-associated group A streptococci. Eur J Clin Microbiol Infect Dis 11:1136–1143. doi: 10.1007/BF01961132. [DOI] [PubMed] [Google Scholar]
- 75.Norgren M, Norrby A, Holm SE. 1992. Genetic diversity in T1M1 group A streptococci in relation to clinical outcome of infection. J Infect Dis 166:1014–1020. doi: 10.1093/infdis/166.5.1014. [DOI] [PubMed] [Google Scholar]
- 76.Talkington DF, Schwartz B, Black CM, Todd JK, Elliott J, Breiman RF, Facklam RR. 1993. Association of phenotypic and genotypic characteristics of invasive Streptococcus pyogenes isolates with clinical components of streptococcal toxic shock syndrome. Infect Immun 61:3369–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chaussee MS, Liu J, Stevens DL, Ferretti JJ. 1996. Genetic and phenotypic diversity among isolates of Streptococcus pyogenes from invasive infections. J Infect Dis 173:901–908. doi: 10.1093/infdis/173.4.901. [DOI] [PubMed] [Google Scholar]
- 78.Chatellier S, Ihendyane N, Kansal RG, Khambaty F, Basma H, Norrby-Teglund A, Low DE, McGeer A, Kotb M. 2000. Genetic relatedness and superantigen expression in group A Streptococcus serotype M1 isolates from patients with severe and nonsevere invasive diseases. Infect Immun 68:3523–3534. doi: 10.1128/IAI.68.6.3523-3534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herdon DN, Davis RW, Xiao W, Tompkins RG. 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun H, Ringdahl U, Homeister JW, Fay WP, Engleberg NC, Yang AY, Rozek LS, Wang X, Sjobring U, Ginsburg D. 2004. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science 305:1283–1286. doi: 10.1126/science.1101245. [DOI] [PubMed] [Google Scholar]