

Abstract
Chlamydia trachomatis infection in the lower genital tract can ascend to and cause pathologies in the upper genital tract, potentially leading to severe complications, such as tubal infertility. However, chlamydial organisms depleted of plasmid or deficient in the plasmid-encoded Pgp3 are attenuated in ascending infection and no longer are able to induce the upper genital tract pathologies, indicating a significant role of Pgp3 in chlamydial pathogenesis. We now report that C. trachomatis Pgp3 can neutralize the antichlamydial activity of human cathelicidin LL-37, a host antimicrobial peptide secreted by both genital tract epithelial cells and infiltrating neutrophils. Pgp3 bound to and formed stable complexes with LL-37. We further showed that the middle region of Pgp3 (Pgp3m) was responsible for both the binding to and neutralization of LL-37, suggesting that Pgp3m can be targeted for attenuating chlamydial pathogenicity or developed for blocking LL-37-involved non-genital-tract pathologies, such as rosacea and psoriasis. Thus, the current study has provided significant information for both understanding the mechanisms of chlamydial pathogenesis and developing novel therapeutic agents.
INTRODUCTION
Chlamydia trachomatis infection in women's lower genital tract can ascend to and cause inflammatory pathologies in the upper genital tract, resulting in complications such as ectopic pregnancy and tubal infertility (1, 2). However, the mechanisms of how the C. trachomatis organisms ascend to the upper genital tract remain unknown. It is thought that the organisms' ability to complete intracellular replication and to spread from cell to cell significantly contributes to chlamydial pathogenicity (3–6). Chlamydial intracellular infection starts with the entry of an infectious elementary body (EB) into an epithelial cell via pathogen-induced endocytosis (7, 8). The endocytosed EB differentiates to a noninfectious but metabolically active reticulate body (RB). After replication, the progeny RBs differentiate back to EBs that exit the infected cells to invade adjacent cells (6). The spreading from cell to cell inevitably exposes the progeny EBs to the extracellular mucosal environment, in which numerous host defense molecules, such as antimicrobial peptides (AMPs), are available for attacking the extracellular EBs. It is unclear how the C. trachomatis EB organisms manage to overcome these host defense mechanisms to ascend to the upper genital tract.
Cationic peptides that possess antimicrobial activities are defined as AMPs, which include human alpha-defensins (HADs) or human neutrophil peptides (HNPs), human beta-defensins (HBDs), and cathelicidin LL-37 (9–12). LL-37 is a C-terminal 37-amino-acid peptide starting with double leucines (LL) processed from human cationic antimicrobial protein 18 (hCAP; 18 kDa) that contains a highly conserved amino-terminal cathelin-like domain and a variable carboxy-terminal domain (9, 11). The mouse orthologue is called cathelin-related antimicrobial peptide (CRAMP) (13). HNPs are produced mainly by neutrophils, while HBDs are produced by epithelial cells. Interestingly, both epithelial cells and neutrophils can produce LL-37 or CRAMP. These extracellular AMPs possess a broad spectrum of antimicrobial activity by inducing pore formation in the bacterial membrane and represent a potent first line of host defense (11, 14). The strong antibacterial activity of AMPs has selected many bacterial species to evolve countermeasures for evading the same defense mechanism (15–17). We have recently shown that the chlamydial chromosome-encoded serine protease CPAF, which is secreted out of the chlamydial inclusion (18), can degrade LL-37 and neutralize its antichlamydial activity (19).
The C. trachomatis organisms share a highly conserved cryptic plasmid that encodes 8 open reading frames (ORFs), designated pORF1-8 or Pgp1-8 (20, 21). Although the plasmid may not be required for chlamydial infection and replication in cell cultures, plasmid-free chlamydial organisms no longer are able to induce pathologies in the upper genital tract or ocular tissues (22–24), suggesting that the plasmid must encode virulence factors that significantly contribute to chlamydial ascending infection and pathogenicity. The plasmid-dependent chlamydial pathogenicity was mapped to Pgp3, since chlamydial organisms deficient in Pgp3 were attenuated and almost phenocopied the plasmid-free organisms (25, 26). Among the 8 ORFs encoded by the cryptic plasmid, only Pgp3 is secreted out of the chlamydial organisms and into chlamydial inclusion and host cell cytosol (20). Pgp3 forms stable trimers (27, 28) and is accumulated in the infected cell cytosol at late stages of infection (20, 27). The lysis of the infected cells may release the cytosolic Pgp3 into the extracellular environment, which is consistent with the observation that Pgp3 is immunogenic during chlamydial infection in humans and animals (29, 30). The above-described analyses have led us to hypothesize that Pgp3 released from the cytosol of the infected cell is able to confront the extracellular AMPs, including LL-37, and neutralize their antichlamydial activity so that the progeny EBs subsequently released from a cytoplasmic inclusion of the same infected cell can have a safe passage for finding the next target cells.
In the current study, we evaluated the roles of the plasmid-encoded Pgp3 in chlamydial evasion of host AMPs. We found that Pgp3 neutralized the antichlamydial activity of LL-37 by binding to and forming stable complexes with LL-37. The middle region of Pgp3 (Pgp3m) was responsible for both the binding and neutralization. Thus, we have identified LL-37 as a host target for the chlamydial plasmid-encoded virulence factor Pgp3, which should further promote our understanding of the chlamydial pathogenic mechanisms. However, these in vitro observations still need validation in animal models.
MATERIALS AND METHODS
Expression and purification of chlamydial proteins using E. coli expression systems.
The pgp3 and pgp4 genes, carried by the pCHL1 plasmid of C. trachomatis serovar D organisms, were cloned into a pGEX-6P2 vector (Amersham Pharmacia Biotech, Inc., Piscataway, NJ) or pET30a vector (Novagen, Madison, WI) and expressed as fusion proteins with glutathione S-transferase (GST) fused to the N terminus or His tag fused to the C terminus as described in references 20 and 31, respectively. To map the binding domain of Pgp3 to host antimicrobial peptide LL-37, various fragments of Pgp3 were expressed as GST- or His-tagged fusion proteins, including the N-terminal fragment, covering the amino acid sequence from the first residue, methionine (M), to serine (S) at position of 66 (Pgp3n), the middle fragment, covering isoleucine (I) 67 to asparagine (N) 132 (Pgp3m), and the C-terminal fragment, covering glycine (G) 133 to the last amino acid, alanine (A) 264 (Pgp3c). The GST fusion proteins bound to glutathione-conjugated agarose beads (Pharmacia) were used in both a GST pulldown assay (as described below) and for further purifying chlamydial proteins or protein fragments. For purification, a precision protease in the form of GST fusion protein (Pharmacia) was used to cleave off the chlamydial proteins from the bead-bound GST fusion proteins. Thus, the cleaved chlamydial proteins or protein fragments were released into solution while the GST-precision enzyme fusion protein was absorbed onto the glutathione beads. The eluents containing the cleaved chlamydial proteins or protein fragments were collected. The His-tagged chlamydial fusion proteins or protein fragments were purified using nickel agarose beads (32050; Qiagen, Valencia, CA) and further washed of excessive imidazole using Centricon units (Millipore, Billerica, MA). All purified proteins were concentrated using Centricon units with the appropriate pore sizes. The concentrated pure proteins were quantitated using a Bio-Rad protein assay dye reagent (500-0006; Bio-Rad, Hercules, CA), and the purity was checked in polyacrylamide gels using Coomassie blue and/or silver staining.
Chlamydial infection in cell cultures.
The C. trachomatis serovar D (UW-3/Cx strain) and L2 (434/Bu strain; data not shown) organisms were propagated in HeLa229 cells (human cervical carcinoma epithelial cells; ATCC CCL2), and the stocks were prepared as described previously (32). For chlamydial infection, host cells grown in 24-well plates with or without coverslips or in tissue culture dishes or flasks containing growth medium, such as Dulbecco's modified Eagle's medium (DMEM; GIBCO BRL, Rockville, MD) with 10% fetal calf serum (FCS; GIBCO BRL), at 37°C in an incubator supplied with 5% CO2 were inoculated with the chlamydial stock organisms. At different time points postinfection as indicated in individual experiments, the infected cultures were processed for immunofluorescence assay or harvested for quantitating live organism recovery or Western blot analyses as described below.
For measuring the antichlamydial activity of host antimicrobial peptides (AMPs), the following AMPs were used at the concentrations indicated in individual experiments to treat chlamydial organisms in 200 μl of sucrose-phosphate-glutamic acid (SPG) medium at room temperature for 2 h. The incubation mixtures then were inoculated onto HeLa cell monolayers. The AMPs used in the current study included HNP1 (human neutrophil peptide 1 or human alpha-defensin 1; 60743), HNP2 (60744; both from AnaSpec, Fremont, CA), HNP3 (PDF-4416-s), HBD1 (human beta-defensin 1; PDF-4337-s), HBD2 (PDF-4338-s), HBD3 (PDF-4382-s), HBD4 (PDF-4406-s) (all 5 were from Peptides International, Louisville, KY), and LL-37 (with a sequence of LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES; 61302; AnaSpec or Peptide 2.0, Chantilly, VA).
To detect the chlamydial Pgp3-mediated neutralization of host AMP antichlamydial activity, the AMPs at the concentrations indicated in individual experiments were preincubated with Pgp3 or its fragments at the appropriate concentrations in SPG buffer for 30 min at 37°C. The preincubation mixtures then were used to treat chlamydial organisms followed by titrating the remaining organisms as described above.
Immunofluorescence assay.
HeLa cells grown on coverslips were fixed with 2% (vol/vol) paraformaldehyde (Sigma) dissolved in phosphate-buffered saline (PBS) for 30 min at room temperature, followed by permeabilization with 2% (wt/vol) saponin (Sigma) for an additional 30 min. After washing and blocking, the cell samples were subjected to antibody and chemical staining. Hoechst (blue; Sigma) was used to visualize DNA. A rabbit antichlamydial organism antibody plus a goat anti-rabbit IgG secondary antibody conjugated with Cy2 (green; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) was used to visualize chlamydial organism-containing inclusions. The immunofluorescence images were acquired using an Olympus AX-70 fluorescence microscope equipped with multiple filter sets and Simple PCI imaging software (Olympus) as described previously (32). The images were processed using the Adobe Photoshop program (Adobe Systems, San Jose, CA). At the same time, both the inclusions and host cells were counted using a 60× subjective oil lens for 5 random views per coverslip, and the results were expressed as infection rates (number of inclusions per 100 cells) in the form of means ± standard deviations calculated from at least 3 independent experiments. For titrating the live organisms recovered from a given sample, the number of inclusions per view was derived from counting 5 random views. Based on the number of inclusions per view, the number of views per coverslip under a given magnification, and sample dilution factors, the total number of live organisms recovered from a given sample was calculated and expressed as log10 inclusion-forming units (IFUs) per sample.
SDS-denatured or native polyacrylamide electrophoresis gels and Western blotting.
Polyacrylamide gels used in this study were prepared by following the procedure provided by the manufacturer (Bio-Rad, Hercules, CA). The running buffer (pH 8.3) used for the native gels contains 24.8 mM Tris base and 191.8 mM glycine. Protein samples run on denaturing gels were heat denatured by boiling for 5 min in a 2× SDS sample buffer (63 mM Tris-HCl, pH 6.8, 10% glycerol, 0.0025% bromophenol blue, 2% [wt/vol] SDS, and 1% [vol/vol] 2-mercaptoethanol). The native gel sample buffer was the same as the denaturing gel sample buffer except without SDS and 2-mercaptoethanol. For detecting chlamydial proteins from chlamydial cultures, the whole cells from a well of a 24-well plate were harvested in 50 μl of 2× SDS buffer. After a brief sonication to shear the genomic DNA and boiling for 5 min, 20 μl was loaded into each lane. For monitoring the GST pulldown assay, GST alone or GST fusion proteins immobilized on glutathione-conjugated agarose beads (each bead in 20 μl) were mixed with a given amount of LL-37 for 30 min at 37°C. After spinning to pellet the agarose beads, both the supernatant and pellet fractions were resuspended in an SDS sample buffer (3 μl 8× for the 20 μl supernatant and 20 μl of 2× for the bead). After boiling, the samples were loaded onto corresponding lanes. The Pgp3/LL-37 complexes were formed by coincubating Pgp3 with LL-37 at the desired concentrations, as indicated in individual experiments, in a total volume of 20 μl for 30 min at 37°C and detected using native gel by loading the entire coincubation mixture into the corresponding lanes.
After electrophoresis, proteins resolved in the polyacrylamide gels were either visualized by staining with Coomassie blue (Sigma) or transferred onto nitrocellulose membranes for Western blot detection with the following primary antibodies: rabbit anti-MOMP (major outer membrane protein) polyclonal antibody (PAb; raised with C. trachomatis serovar D MOMP-GST fusion protein) for detecting MOMP (33), a mouse monoclonal antibody (MAb; clone BC7.1) for detecting C. trachomatis heat shock protein 60 (ctHSP60) (33), a mouse MAb (clone W27; Sigma) for detecting human HSP70, a mouse MAb (clone 3D11; Yacolt Biotech, Plymouth Meeting, PA) for detecting human cathelicidin peptide LL-37, and a mouse MAb (clone 8H6, raised with Pgp3 fusion protein) for detecting Pgp3 (30). These primary antibody bindings were probed with the HRP (horseradish peroxidase)-conjugated goat anti-mouse or -rabbit IgG secondary antibodies (Jackson ImmunoResearch Laboratories, Inc.) and visualized using an enhanced chemiluminescence (ECL) kit (Santa Cruz Biotech, Santa Cruz, CA).
Binding affinity measurement.
A streptavidin-coated biosensor (18-5019; ForteBio, Inc., Menlo Park, CA) was used to immobilize bio-LL-37. LL-37 with the first amino acid conjugated with a biotin (biotin-LC-LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES, where LC is a 6-aminocaprioic acid sequence serving as a long-chain linker) was synthesized by AnaSpec (63693; Fremont, CA). The bio-LL-37-bound biosensor first was immersed in a binding solution (0.1% bovine serum albumin [BSA], 0.02% Tween 20 in PBS, pH 7.4) that contains different concentrations of Pgp3 or other control proteins for 300 s (for measuring association constant or on-rate) and then moved to fresh binding solutions for another 300 s (for measuring dissociation constant or off-rate). During the entire process, the molecules bound to or fallen off from the biosensors were monitored in real-time using a portable BLItz system (45-5000; ForteBio, Inc.). The BLI (biolayer interferometry) technology used in the BLItz system emits white light down the biosensor and then collects any light reflected back. The wavelength shift in the reflected light is affected by the thickness of the coating on the optical layer of the biosensor and captured by a spectrometer as a unique spectral signature, and it is reported in relative intensity units (nanometers). Any change in the number of molecules bound to the biosensor causes a shift in the interference pattern that is measurable in real time.
Statistics.
For quantitative data, analysis of variance (ANOVA) and Student's t test were used. For qualitative data, Fisher's exact test was used. For semiquantitative data, the Wilcoxon signed-rank test was used.
RESULTS
Human cathelicidin LL-37 possesses a robust antichlamydial activity that is neutralized by the chlamydial plasmid-encoded Pgp3.
In the female genital tract, AMPs are both constitutively produced along the menstrual cycle and rapidly induced within a short period of time upon infection (34). Thus, we assessed the antichlamydial activity of the AMPs (Fig. 1). Among the 9 AMPs evaluated, the cathelicidin LL-37 or the mouse homologue CRAMP displayed the highest antichlamydial activity, followed by human beta defensin 3 (HBD3) and human alpha defensin 2, also called human neutrophil peptide 2 (HNP2). At a final concentration of 100 μg/ml, LL-37, CRAMP, HDB3, and HNP2 reduced the chlamydial infection rates from 57.8% ± 2.21% (means ± standard deviations; untreated control) to 6.6% ± 2.41%, 7.4% ± 1.14%, 14.0% ± 2.74%, and 21.6% ± 4.93%, respectively (P < 0.01 for all). Since counting the infection rate is somewhat subjective, we also used more objective methods for quantitating the AMP-reduced chlamydial infection. In a Western blot assay (Fig. 1B), both the chlamydial MOMP and HSP60 proteins were significantly reduced in the cultures infected with the chlamydial organisms pretreated with LL-37 or CRAMP, while the amount of the host cell protein HSP70 was similar among different culture samples, confirming the LL-37- or CRAMP-reduced infection rates observed above. We also titrated the number of live organisms recovered from parallel cultures using fresh HeLa cell monolayers (Fig. 1C). We found that the number of live organisms recovered from the cultures infected with the LL-37- or CRAMP-treated chlamydial organisms was reduced by 10-fold (compared to that from the untreated chlamydial organism-infected culture). Thus, these independent measurements have consistently demonstrated that human cathelicidin LL-37 or its mouse homologue CRAMP possesses strong antichlamydial activity.
FIG 1.

Antichlamydial activity of host antimicrobial peptides. (A) The C. trachomatis serovar D organisms treated with (images b to l) or without (image a) antimicrobial peptides, as indicated at the top of each image, were used to infect HeLa cells grown on coverslips in 24-well plates for ∼50 h, and the cultures were processed for immunofluorescence assay to visualize chlamydial inclusions (green) and host cell nuclei (blue). Both the inclusions and host cells were counted using a 60× subjective oil lens for 5 random views per coverslip, and the results were expressed as infection rates (number of inclusions per 100 cells). The means and standard deviations listed in each image were calculated from 3 independent experiments. The antimicrobial peptide treatment conditions that resulted in a statistically significant reduction in infection rates were marked with white stars. (B) The samples from parallel cultures as described above were harvested for Western blot detection of chlamydial proteins, including the major outer membrane protein (MOMP; row a) and heat shock protein 60 (HSP60; row b). The host HSP70 was detected as a loading control (row c). (C) In parallel experiments, the cultures infected as described above were harvested as cell lysates from each well for quantitating live infectious organisms by titrating the cell lysates on fresh HeLa cell monolayers. The results shown along the y axis were expressed as log10 IFUs recovered per well as means plus standard deviations (calculated from 3 independent experiments). The antimicrobial peptide treatment conditions that resulted in statistically significant reduction of IFU recoveries were marked with asterisks. Asterisks denote statistical significance (*, P < 0.01; **, P < 0.01).
The next question is how the chlamydial organisms overcome the antichlamydial activity of LL-37 during infection in women. The fact that the chlamydial organisms depleted of plasmid or Pgp3 are highly attenuated in ascending infection in mice suggests that the wild-type organisms use the plasmid-encoded virulence factor Pgp3 to neutralize the antichlamydial activity of LL-37. Preincubation of LL-37 with Pgp3 reversed the LL-37-mediated inhibition of chlamydial growth (Fig. 2A). The chlamydial infection rate was reduced by >90% by LL-37 (from 56.2% ± 3.83% to 4.8% ± 0.84%) when LL-37 was used at a final concentration of 30 μg/ml. However, preincubation of LL-37 with Pgp3 restored the chlamydial infection rates in a Pgp3 dose-dependent manner, while Pgp3 alone did not affect the chlamydial infectivity. The reversal of the LL-37 antichlamydial activity by Pgp3 was confirmed by both monitoring the chlamydial protein synthesis (Fig. 2B) and titrating the live chlamydial organism recovery (Fig. 2C) in parallel cultures. The chlamydial MOMP and HSP60 proteins and the number of live organisms were reduced in cultures infected with the chlamydial organisms treated with LL-37 compared to those of cultures infected with the untreated chlamydial organisms. However, both the LL-37-reduced chlamydial proteins and live organisms were reversed by the preincubation of LL-37 with increasing amounts of Pgp3. These observations together have demonstrated that the chlamydial plasmid-encoded Pgp3 can efficiently neutralize the antichlamydial activity of LL-37.
FIG 2.

Pgp3 neutralizes the antichlamydial activity of the human cathelicidin LL-37. (A) LL-37 with (images c to e) or without (b) preincubation with various concentrations of Pgp3 were used to treat C. trachomatis serovar D organisms before the organisms were applied to HeLa cell monolayers. The same serovar D organisms treated without (a) or with Pgp3 alone (f) were used to infect the HeLa cell monolayers as controls. As described in the legend to Fig. 1A, the inclusions were counted and the infection rates were calculated from 3 independent experiments and are shown in the corresponding images. (B) In a parallel culture as described above, the samples were harvested for Western blot detection of chlamydial proteins as described in the legend to Fig. 1B. (C) In experiments parallel to those described for panel A, the cultures were harvested for quantitating live organisms from each and are shown along the y axis in log10 IFUs per well. Asterisks denote statistical significance (*, P < 0.01; **, P < 0.01).
Pgp3 binds to and forms stable complexes with LL-37.
To understand how Pgp3 neutralizes the antichlamydial activity of LL-37, we measured the interactions of Pgp3 with LL-37. In a GST pulldown assay (Fig. 3A), incubation of LL-37 with GST-Pgp3 fusion protein allowed the GST-Pgp3 fusion protein to precipitate most LL-37 into the pellet fraction (lane 6 versus 7), while GST alone failed to do so, leaving most LL-37 in the supernatant (lanes 3 and 4). Interestingly, the GST-Pgp3-LL-37 complexes still were detectable in an SDS denaturing gel, suggesting that the Pgp3-LL-37 complexes are stable. In a native gel assay (Fig. 3B), one copy of His-tagged Pgp3 was found to bind to multiple copies of LL-37, since, when the concentration LL-37 was increased above the equal molar ratio, more LL-37 molecules continued to bind to the His-tagged Pgp3 and increasing amounts of Pgp3-LL-37 complexes migrating above the Pgp3 trimer band were retained in the native gel. Note that free LL-37 ran out of the native gel from the top due to its strong positive charge. Thus, no free LL-37 was detected in the native gel. Pgp3 is known to form stable trimers that can further oligomerize (27). The LL-37 binding appeared to promote the oligomerization of Pgp3, since more Pgp3 aggregates were detected in the native gel when LL-37 reached equal or higher molar concentrations. We further quantitatively measured the Pgp3 interaction with LL-37 in real time using a BLItz instrument (Fig. 3C). We found that the tag-free Pgp3 (purified by cleaving Pgp3 from the GST-Pgp3 fusion proteins) bound to LL-37 immobilized on a biosensor pin with a fast on-rate (steep slope). When the biosensor pin-immobilized Pgp3-LL-37 complexes were subjected to wash in a buffer for 300 s, nothing came off the pin, indicating a slow off-rate. These association and dissociation measurements were specific, since unrelated soluble proteins such as GST and Pgp4 failed to bind to the pin-immobilized LL-37, and Pgp3 was not able to bind to the pin in the absence of LL-37.
FIG 3.

Pgp3 binds to the antimicrobial peptide LL-37 to form stable complexes. (A) Eight micrograms of LL-37 was precipitated alone (lane 1) or with GST agarose beads (lanes 3 and 4) or GST-Pgp3 fusion protein beads (lanes 6 and 7) in a total volume of 200 μl PBS. After spinning down the beads, 10% of each bead pellet (P, lanes 3 and 6) and corresponding remaining supernatant (S, lanes 4 and 7) were loaded onto an SDS-polyacrylamide gel for electrophoresis separation, and the resolved bands were detected with an anti-LL-37 antibody by Western blotting. Note that a portion of LL-37 pulled down by GST-Pgp3 still was complexed with GST-Pgp3 after SDS denaturation. (B) A His-tagged Pgp3 mixed with various concentrations of LL-37, as indicated at the top of the figure, for 30 min at 37°C was loaded onto a native gel with 20 μl/lane for electrophoresis separation, and the resolved bands were probed with an anti-LL-37 (a) or anti-Pgp3 (b) antibody. Due to the strong positive charge of LL-37, the free LL-37 peptide (in the absence of any detergents) ran out of the native gel from the top (lane 1). The anti-LL-37 antibody detected LL-37 complexed with Pgp3, while the anti-Pgp3 antibody detected Pgp3 in trimers or high orders of oligomers, as indicated on the right. (C) Streptavidin-conjugated detection pins, after being coated with biotin-LL-37 at 5,028 nM or without coating (blank), were immersed in PBS containing tag-free Pgp3 (purified by cleaving off from the GST-Pgp3 fusion proteins) at 1,040, 2,604, or 5,208 nM or control protein GST or Pgp4 at 5,208 nM, as indicated on the right of the figure, for 300 s (association period). The pins then were moved to a fresh PBS solution for another 300 s (dissociation period). During the entire process, the molecular mass that bound to the pins was monitored using a portable BLItz system and recorded as response units as shown along the y axis.
The middle region of Pgp3 (Pgp3m) is responsible for both binding to and neutralizing LL-37.
To map the Pgp3 domain responsible for binding to LL-37, we incubated LL-37 with GST alone or the GST fusion proteins containing either the full-length Pgp3 (Pgp3fl), the Pgp3 N-terminal fragment (Pgp3n), the middle fragment (Pgp3m), or the C-terminal fragment (Pgp3c) and precipitated the GST for detection of LL-37 in the pellet fraction using Western blotting (Fig. 4A). The GST-Pgp3fl pulled LL-37 into the pellet fraction while GST alone failed to do so, validating the specificity of the assay. Again, a significant amount of the Pgp3-LL-37 complexes still was detected despite the fact that the samples were subjected to SDS denaturation, indicating the strong interaction of LL-37 with Pgp3. More importantly, the GST-Pgp3m fusion protein, but not the fusion proteins containing Pgp3n or Pgp3c, also pulled down LL-37 into the pellet fraction, indicating that Pgp3m, but not Pgp3n or Pgp3c, is responsible for binding to LL-37. The interaction of Pgp3m with LL-37 also was stable, since the complexes remained intact in SDS denaturing gel, mirroring the stable complexes of the full-length Pgp3 with LL-37. Furthermore, when the purified tag-free Pgp3fl or Pgp3 fragments were used in preincubation with LL-37, both Pgp3fl and Pgp3m but not Pgp3c neutralized the antichlamydial activity of LL-37 (Fig. 4B and C). Pgp3m was as potent as full-length Pgp3fl in reversing the LL-37-mediated inhibition of chlamydial infectivity. Either Pgp3m or Pgp3fl completely blocked the antichlamydial activity of LL-37 at a molar concentration of ∼5-fold lower than that of LL-37, suggesting that one copy of Pgp3m or Pgp3fl was able to bind to at least 5 copies of LL-37. Thus, we have mapped the Pgp3 domain for binding to and neutralizing LL-37 in the Pgp3 middle region (Pgp3m).
FIG 4.
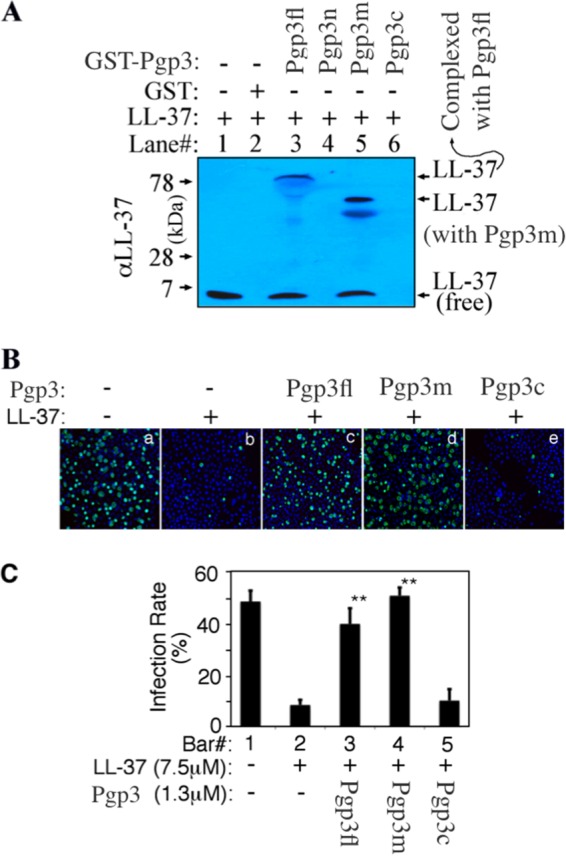
Mapping the binding domain of Pgp3. (A) Eight micrograms of LL-37 was precipitated alone (lane 1) or with GST agarose beads (lane 2) or GST-Pgp3 full length (Pgp3fl; lane 3), GST-Pgp3 N-terminal region (Pgp3n; lane 4), Pgp3 middle region (Pgp3m; lane 5), or Pgp3 C-terminal region (Pgp3c; lane 6) fusion protein bead in a total volume of 200 μl PBS. After centrifugation, the bead pellets were loaded into the corresponding lanes of an SDS-polyacrylamide gel for Western blot detection with an anti-LL-37 antibody. Note that a portion of LL-37 pulled down by either GST-Pgp3fl or GST-Pgp3m still was complexed with GST-Pgp3fl or GST-Pgp3m even after SDS denaturation. (B) LL-37 at 7.5 μM (equivalent to 30 μg/ml) preincubated without (image b) or with 1.3 μM Pgp3fl (image c), Pgp3m (image d), or Pgp3c (image e) was used to treat C. trachomatis serovar D organisms before the organisms were applied to HeLa cell monolayers. The inclusions were counted as described in the legend to Fig. 1A, and the infection rates were calculated from 3 independent experiments and displayed along the y axis of panel C. Double asterisks indicate statistically significant differences (P < 0.01) between chlamydial cultures treated with LL-37 alone (bar 2) and those treated with LL-37/Pgp3fl or LL-37/Pgp3m complexes (bars 3 and 4).
DISCUSSION
Although C. trachomatis infection in the lower genital tract is known to ascend and cause pathology in the upper genital tract, the molecular mechanisms of chlamydial ascension and pathogenicity remain largely unknown. The observation that plasmid-free (22–24) or Pgp3-deficient (25, 26) chlamydial organisms are attenuated in pathogenicity suggests that the plasmid-encoded Pgp3 significantly contributes to chlamydial pathogenicity. This conclusion is consistent with the fact that among all the plasmid-encoded and regulated proteins (35), Pgp3 is the only plasmid-encoded protein that is secreted into host cell cytosol during chlamydial infection (20). In the current study, we have presented evidence for a potential mechanism by which Pgp3 promotes chlamydial pathogenicity. First, Pgp3 neutralized the antichlamydial activity of human cathelicidin LL-37 peptide. The preincubation of LL-37 with Pgp3 blocked the ability of LL-37 to inhibit chlamydial infection. Second, Pgp3 bound to LL-37 with high affinity, suggesting that once Pgp3 is released in the extracellular environment, it can rapidly target the extracellular AMPs for binding. Finally, Pgp3 binding to LL-37 was mapped to the Pgp3 middle region, which is consistent with the structural observation that Pgp3m forms triple helixes that are flexible for interacting with other molecules (28). Once the flexible triple helixes were bound with LL-37, the whole complexes appeared to become more stable, which may explain the resistance of the complexes to SDS denaturation.
The chlamydial induction of pathology in the upper genital tract is dependent on the chlamydial ability to both ascend to and activate pathology-causing inflammation in the upper genital tract. During ascending infection, the progeny chlamydial organisms may inevitably be exposed to extracellular environments where innate immune effectors such as antimicrobial peptides are available for attacking the extracellular organisms (34, 36–38). Antimicrobial peptides are produced both constitutively and in response to infection by epithelial cells and neutrophils in the genital tract (38–40). Many bacterial species have been selected to evolve strategies for evading AMPs via releasing soluble factors to trap (41, 42) or degrade the antimicrobial peptides (15–17, 43). The intrainclusion C. trachomatis organisms are known to secrete various proteins, including CPAF and Pgp3, into host cell cytosol (2, 20, 33, 44–46). Both CPAF and Pgp3 are accumulated in large quantities in the cytosol of the infected cells prior to the lysis of the infected cells (18, 20). Thus, we hypothesize that the prestored CPAF and Pgp3 in the host cell cytosol can be rapidly released to target extracellular antimicrobial peptides upon cell lysis.
Pgp3 has been at the central stage of chlamydial research for decades. Immunization with expression plasmids encoding Pgp3 induced protective immunity in mouse models (47, 48). Pgp3 is an immunodominant antigen during chlamydial infection in humans, and human antibody recognition of Pgp3 is dependent on Pgp3 trimers (27, 29, 30, 49), suggesting that Pgp3 is presented to the immune system in oligomerized forms during C. trachomatis infection in humans. However, the human anti-Pgp3 trimer antibodies may not offer protection against chlamydial infection (27). The question is why humans produce a strong but nonprotective antibody response to Pgp3 trimers. We hypothesize that such a robust anti-Pgp3 trimer antibody response represents another chlamydial strategy for evading adaptive immunity. Thus, the current finding on Pgp3 neutralization of LL-37 may not be the only function of Pgp3 used by chlamydial organisms to benefit chlamydial survival in the infected hosts. Our hunting for new functions of Pgp3 is ongoing.
Apparently, chlamydia has evolved both the chromosome-encoded CPAF and plasmid-encoded Pgp3 for neutralizing the antichlamydial activity of LL-37, suggesting that antimicrobial peptides such as LL-37 are essential for the hosts to control chlamydial ascending infection. Although this hypothesis is consistent with the general concept that antimicrobial peptides play critical roles in controlling mucosal pathogen infections from viruses to fungi (50, 51), a direct test of this hypothesis will require the use of mice deficient in CRAMP, the mouse homologue of LL-37. The combinations of chlamydial genetic mutants with gene knockout mouse models should significantly advance our knowledge on chlamydial pathogenesis. For example, we will be able to address questions such as whether the chlamydial organisms that express Pgp3m are as pathogenic as the organisms that express the full-length Pgp3 or whether the CRAMP deficiency can rescue the pathogenicity of the chlamydial organisms deficient in Pgp3m. At this moment, in the absence of the relevant in vivo data support or validation, we should be cautious in interpreting the in vitro observations presented in the current work. The field learned a painful lesson when in vitro observations were taken as the only proof of chlamydial protein functions (52). It is not known whether a statistically significant reduction in infectivity of chlamydial organisms demonstrated in in vitro assays is sufficient for attenuating the chlamydial pathogenicity in the upper genital tract. However, any reduction in chlamydial infectivity should add up to decrease the risk for the infected host to develop upper genital tract pathology. Most likely, LL-37 only represents one of the many host defense molecules/mechanisms that can impact chlamydial infectivity and pathology. Thus, the antichlamydial activity of LL-37 described in the current study should be interpreted in the context of the overall host defense mechanisms in the genital tract. We also are aware that significant inhibition of chlamydial infection was achieved in the cell culture system only when LL-37 was used at a final concentration of 10 μg/ml or above (Fig. 1). The question is whether this concentration of LL-37 can be reached in the genital tract during chlamydial infection. This question is difficult to address, since the concentration of CRAMP measured in the homogenates of tissues harvested from mice infected with chlamydial organisms may not necessarily represent the physiological levels. This is because when LL-37 is released from either neutrophils or epithelial cells, the local concentrations of LL-37 directly targeting the chlamydial organisms can be very different from the average concentration detected in the tissue. Thus, validating the biological significance of the in vitro observations described in the current manuscript will depend on the experiments using the relevant knockout mouse models.
The role of Pgp3 in chlamydial pathogenicity has been demonstrated using Pgp3-deficient chlamydial organisms in mouse genital tract models (25, 26). Our current in vitro experimental data seem to suggest that CRAMP deficiency in mice is able to significantly restore the pathogenicity of the Pgp3-deficient chlamydial organisms. However, due to potential redundancy, the CRAMP deficiency alone may not be sufficient for a significant restoration. An alterative approach is to use mice with deficiency in a broad spectrum of innate immune effectors. Many innate immune effectors, including the expression of AMPs, are regulated by MyD88-mediated signaling pathways (53). We have previously shown that mice deficient in MyD88 are significantly more susceptible to C. muridarum ascending infection and more vulnerable to develop hydrosalpinx (54), suggesting that MyD88-regulated effector mechanisms are important for mice to control C. muridarum pathogenicity and may represent a selection pressure for the chlamydial organisms to evolve countermeasures for evading the same effector mechanisms. We hypothesize that Pgp3 promotes the chlamydial pathogenicity by aiding in chlamydial evasion of the MyD88-regulated effector mechanisms. Thus, we expect that the MyD88 deficiency is able to significantly restore the ascending infection of the Pgp3-deficient chlamydial organisms. This setup may allow us to further determine which MyD88-regulated effector mechanisms in the genital tract are targeted by Pgp3. For example, mice with cell type-specific knockout of MyD88 (55) can be used to identify the responsible cell types that can further serve as a platform for defining the biologically relevant substrates targeted by Pgp3.
ACKNOWLEDGMENT
This work was supported in part by grants (to G.Z.) from the U.S. National Institutes of Health.
REFERENCES
- 1.Centers for Disease Control and Prevention. 2009. 2008 Sexually transmitted diseases surveillance. Centers for Disease Control and Prevention, Atlanta, GA: http://www.cdc.gov/std/stats08/toc.htm. [Google Scholar]
- 2.Abdelrahman YM, Rose LA, Belland RJ. 2011. Developmental expression of non-coding RNAs in Chlamydia trachomatis during normal and persistent growth. Nucleic Acids Res 39:1843–1854. doi: 10.1093/nar/gkq1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stephens RS. 2003. The cellular paradigm of chlamydial pathogenesis. Trends Microbiol 11:44–51. doi: 10.1016/S0966-842X(02)00011-2. [DOI] [PubMed] [Google Scholar]
- 4.Cheng W, Shivshankar P, Li Z, Chen L, Yeh IT, Zhong G. 2008. Caspase-1 contributes to Chlamydia trachomatis-induced upper urogenital tract inflammatory pathologies without affecting the course of infection. Infect Immun 76:515–522. doi: 10.1128/IAI.01064-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng W, Shivshankar P, Zhong Y, Chen D, Li Z, Zhong G. 2008. Intracellular interleukin-1alpha mediates interleukin-8 production induced by Chlamydia trachomatis infection via a mechanism independent of type I interleukin-1 receptor. Infect Immun 76:942–951. doi: 10.1128/IAI.01313-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hybiske K, Stephens RS. 2007. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci U S A 104:11430–11435. doi: 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clifton DR, Fields KA, Grieshaber SS, Dooley CA, Fischer ER, Mead DJ, Carabeo RA, Hackstadt T. 2004. A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc Natl Acad Sci U S A 101:10166–10171. doi: 10.1073/pnas.0402829101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engel J. 2004. Tarp and Arp: How Chlamydia induces its own entry. Proc Natl Acad Sci U S A 101:9947–9948. doi: 10.1073/pnas.0403633101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kosciuczuk EM, Lisowski P, Jarczak J, Strzalkowska N, Jozwik A, Horbanczuk J, Krzyzewski J, Zwierzchowski L, Bagnicka E. 2012. Cathelicidins: family of antimicrobial peptides. A review. Mol Biol Rep 39:10957–10970. doi: 10.1007/s11033-012-1997-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anaya-Lopez JL, Lopez-Meza JE, Ochoa-Zarzosa A. 2013. Bacterial resistance to cationic antimicrobial peptides. Crit Rev Microbiol 39:180–195. doi: 10.3109/1040841X.2012.699025. [DOI] [PubMed] [Google Scholar]
- 11.Gallo RL, Hooper LV. 2012. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol 12:503–516. doi: 10.1038/nri3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi KY, Chow LN, Mookherjee N. 2012. Cationic host defence peptides: multifaceted role in immune modulation and inflammation. J Innate Immun 4:361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallo RL, Kim KJ, Bernfield M, Kozak CA, Zanetti M, Merluzzi L, Gennaro R. 1997. Identification of CRAMP, a cathelin-related antimicrobial peptide expressed in the embryonic and adult mouse. J Biol Chem 272:13088–13093. doi: 10.1074/jbc.272.20.13088. [DOI] [PubMed] [Google Scholar]
- 14.Korting HC, Schollmann C, Stauss-Grabo M, Schafer-Korting M. 2012. Antimicrobial peptides and skin: a paradigm of translational medicine. Skin Pharmacol Physiol 25:323–334. doi: 10.1159/000341990. [DOI] [PubMed] [Google Scholar]
- 15.Diacovich L, Gorvel JP. 2010. Bacterial manipulation of innate immunity to promote infection. Nat Rev Microbiol 8:117–128. doi: 10.1038/nrmicro2295. [DOI] [PubMed] [Google Scholar]
- 16.Potempa J, Pike RN. 2009. Corruption of innate immunity by bacterial proteases. J Innate Immun 1:70–87. doi: 10.1159/000181144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidtchen A, Frick IM, Andersson E, Tapper H, Bjorck L. 2002. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol Microbiol 46:157–168. doi: 10.1046/j.1365-2958.2002.03146.x. [DOI] [PubMed] [Google Scholar]
- 18.Yang Z, Tang L, Sun X, Chai J, Zhong G. 2015. Characterization of CPAF critical residues and secretion during Chlamydia trachomatis infection. Infect Immun 83:2234–2241. doi: 10.1128/IAI.00275-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang L, Chen J, Zhou Z, Yu P, Yang Z, Zhong G. 2015. Chlamydia-secreted protease CPAF degrades host antimicrobial peptides. Microbes Infect 17:402–408. doi: 10.1016/j.micinf.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Li Z, Chen D, Zhong Y, Wang S, Zhong G. 2008. The chlamydial plasmid-encoded protein pgp3 is secreted into the cytosol of Chlamydia-infected cells. Infect Immun 76:3415–3428. doi: 10.1128/IAI.01377-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas NS, Lusher M, Storey CC, Clarke IN. 1997. Plasmid diversity in Chlamydia. Microbiology 143(Part 6):1847–1854. doi: 10.1099/00221287-143-6-1847. [DOI] [PubMed] [Google Scholar]
- 22.O'Connell CM, Ingalls RR, Andrews CW Jr, Scurlock AM, Darville T. 2007. Plasmid-deficient Chlamydia muridarum fail to induce immune pathology and protect against oviduct disease. J Immunol 179:4027–4034. doi: 10.4049/jimmunol.179.6.4027. [DOI] [PubMed] [Google Scholar]
- 23.Kari L, Whitmire WM, Olivares-Zavaleta N, Goheen MM, Taylor LD, Carlson JH, Sturdevant GL, Lu C, Bakios LE, Randall LB, Parnell MJ, Zhong G, Caldwell HD. 2011. A live-attenuated chlamydial vaccine protects against trachoma in nonhuman primates. J Exp Med 208:2217–2223. doi: 10.1084/jem.20111266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lei L, Chen J, Hou S, Ding Y, Yang Z, Zeng H, Baseman J, Zhong G. 2014. Reduced live organism recovery and lack of hydrosalpinx in mice infected with plasmid-free Chlamydia muridarum. Infect Immun 82:983–992. doi: 10.1128/IAI.01543-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Huang Y, Yang Z, Sun Y, Gong S, Hou S, Chen C, Li Z, Liu Q, Wu Y, Baseman J, Zhong G. 2014. Plasmid-encoded Pgp3 is a major virulence factor for Chlamydia muridarum to induce hydrosalpinx in mice. Infect Immun 82:5327–5335. doi: 10.1128/IAI.02576-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramsey KH, Schripsema JH, Smith BJ, Wang Y, Jham BC, O'Hagan KP, Thomson NR, Murthy AK, Skilton RJ, Chu P, Clarke IN. 2014. Plasmid CDS5 influences infectivity and virulence in a mouse model of Chlamydia trachomatis urogenital infection. Infect Immun 82:3341–3349. doi: 10.1128/IAI.01795-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ang E, Liu Q, Qi M, Liu HG, Yang X, Chen H, Zheng MH, Xu J. 2011. Mangiferin attenuates osteoclastogenesis, bone resorption, and RANKL-induced activation of NF-kappaB and ERK. J Cell Biochem 112:89–97. doi: 10.1002/jcb.22800. [DOI] [PubMed] [Google Scholar]
- 28.Galaleldeen A, Taylor AB, Chen D, Schuermann JP, Holloway SP, Hou S, Gong S, Zhong G, Hart PJ. 2013. Structure of the Chlamydia trachomatis immunodominant antigen Pgp3. J Biol Chem 288:22068–22079. doi: 10.1074/jbc.M113.475012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Zhang Y, Lu C, Lei L, Yu P, Zhong G. 2010. A genome-wide profiling of the humoral immune response to Chlamydia trachomatis infection reveals vaccine candidate antigens expressed in humans. J Immunol 185:1670–1680. doi: 10.4049/jimmunol.1001240. [DOI] [PubMed] [Google Scholar]
- 30.Li Z, Zhong Y, Lei L, Wu Y, Wang S, Zhong G. 2008. Antibodies from women urogenitally infected with C. trachomatis predominantly recognized the plasmid protein pgp3 in a conformation-dependent manner. BMC Microbiol 8:90–95. doi: 10.1186/1471-2180-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Z, Feng Y, Chen D, Wu X, Huang S, Wang X, Xiao X, Li W, Huang N, Gu L, Zhong G, Chai J. 2008. Structural basis for activation and inhibition of the secreted chlamydia protease CPAF. Cell Host Microbe 4:529–542. doi: 10.1016/j.chom.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 32.Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. 1998. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med 187:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong G, Fan P, Ji H, Dong F, Huang Y. 2001. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J Exp Med 193:935–942. doi: 10.1084/jem.193.8.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aubert M, Jerome KR. 2003. Apoptosis prevention as a mechanism of immune evasion. Int Rev Immunol 22:361–371. doi: 10.1080/08830180305213. [DOI] [PubMed] [Google Scholar]
- 35.Carlson JH, Whitmire WM, Crane DD, Wicke L, Virtaneva K, Sturdevant DE, Kupko JJ III, Porcella SF, Martinez-Orengo N, Heinzen RA, Kari L, Caldwell HD. 2008. The Chlamydia trachomatis plasmid is a transcriptional regulator of chromosomal genes and a virulence factor. Infect Immun 76:2273–2283. doi: 10.1128/IAI.00102-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma S, Sethi S, Prasad R, Samanta P, Rajwanshi A, Malhotra S, Sharma M. 2011. Characterization of low molecular weight antimicrobial peptide from human female reproductive tract. Indian J Med Res 134:679–687. doi: 10.4103/0971-5916.90996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valore EV, Park CH, Quayle AJ, Wiles KR, McCray PB Jr, Ganz T. 1998. Human beta-defensin-1: an antimicrobial peptide of urogenital tissues. J Clin Investig 101:1633–1642. doi: 10.1172/JCI1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hickey DK, Patel MV, Fahey JV, Wira CR. 2011. Innate and adaptive immunity at mucosal surfaces of the female reproductive tract: stratification and integration of immune protection against the transmission of sexually transmitted infections. J Reprod Immunol 88:185–194. doi: 10.1016/j.jri.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Com E, Bourgeon F, Evrard B, Ganz T, Colleu D, Jegou B, Pineau C. 2003. Expression of antimicrobial defensins in the male reproductive tract of rats, mice, and humans. Biol Reprod 68:95–104. [DOI] [PubMed] [Google Scholar]
- 40.Doss M, White MR, Tecle T, Hartshorn KL. 2010. Human defensins and LL-37 in mucosal immunity. J Leukoc Biol 87:79–92. doi: 10.1189/jlb.0609382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frick IM, Akesson P, Rasmussen M, Schmidtchen A, Bjorck L. 2003. SIC, a secreted protein of Streptococcus pyogenes that inactivates antibacterial peptides. J Biol Chem 278:16561–16566. doi: 10.1074/jbc.M301995200. [DOI] [PubMed] [Google Scholar]
- 42.Frick IM, Shannon O, Akesson P, Morgelin M, Collin M, Schmidtchen A, Bjorck L. 2011. Antibacterial activity of the contact and complement systems is blocked by SIC, a protein secreted by Streptococcus pyogenes. J Biol Chem 286:1331–1340. doi: 10.1074/jbc.M110.178350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karlsson C, Andersson ML, Collin M, Schmidtchen A, Bjorck L, Frick IM. 2007. SufA–a novel subtilisin-like serine proteinase of Finegoldia magna. Microbiology 153:4208–4218. doi: 10.1099/mic.0.2007/010322-0. [DOI] [PubMed] [Google Scholar]
- 44.Allsworth JE, Peipert JF. 2011. Severity of bacterial vaginosis and the risk of sexually transmitted infection. Am J Obstet Gynecol 205:113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong G, Lei L, Gong S, Lu C, Qi M, Chen D. 2011. Chlamydia-secreted proteins in chlamydial interactions with host cells. Curr Chem Biol 5:29–37. http://benthamscience.com/journals/current-chemical-biology/volume/5/issue/1/page/29/. [Google Scholar]
- 46.Gong S, Lei L, Chang X, Belland R, Zhong G. 2011. Chlamydia trachomatis secretion of hypothetical protein CT622 into host cell cytoplasm via a secretion pathway that can be inhibited by the type III secretion system inhibitor compound 1. Microbiology 157:1134–1144. doi: 10.1099/mic.0.047746-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Z, Wang S, Wu Y, Zhong G, Chen D. 2008. Immunization with chlamydial plasmid protein pORF5 DNA vaccine induces protective immunity against genital chlamydial infection in mice. Sci China C Life Sci 51:973–980. doi: 10.1007/s11427-008-0130-9. [DOI] [PubMed] [Google Scholar]
- 48.Donati M, Sambri V, Comanducci M, Di Leo K, Storni E, Giacani L, Ratti G, Cevenini R. 2003. DNA immunization with pgp3 gene of Chlamydia trachomatis inhibits the spread of chlamydial infection from the lower to the upper genital tract in C3H/HeN mice. Vaccine 21:1089–1093. doi: 10.1016/S0264-410X(02)00631-X. [DOI] [PubMed] [Google Scholar]
- 49.Comanducci M, Cevenini R, Moroni A, Giuliani MM, Ricci S, Scarlato V, Ratti G. 1993. Expression of a plasmid gene of Chlamydia trachomatis encoding a novel 28 kDa antigen. J Gen Microbiol 139:1083–1092. doi: 10.1099/00221287-139-5-1083. [DOI] [PubMed] [Google Scholar]
- 50.Hazrati E, Galen B, Lu W, Wang W, Ouyang Y, Keller MJ, Lehrer RI, Herold BC. 2006. Human alpha- and beta-defensins block multiple steps in herpes simplex virus infection. J Immunol 177:8658–8666. doi: 10.4049/jimmunol.177.12.8658. [DOI] [PubMed] [Google Scholar]
- 51.Tomalka J, Azodi E, Narra HP, Patel K, O'Neill S, Cardwell C, Hall BA, Wilson JM, Hise AG. 2015. β-Defensin 1 plays a role in acute mucosal defense against Candida albicans. J Immunol 194:1788–1795. doi: 10.4049/jimmunol.1203239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Conrad TA, Yang Z, Ojcius D, Zhong G. 2013. A path forward for the chlamydial virulence factor CPAF. Microbes Infect 15:1026–1032. doi: 10.1016/j.micinf.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Warner N, Nunez G. 2013. MyD88: a critical adaptor protein in innate immunity signal transduction. J Immunol 190:3–4. doi: 10.4049/jimmunol.1203103. [DOI] [PubMed] [Google Scholar]
- 54.Chen L, Lei L, Chang X, Li Z, Lu C, Zhang X, Wu Y, Yeh IT, Zhong G. 2010. Mice deficient in MyD88 Develop a Th2-dominant response and severe pathology in the upper genital tract following Chlamydia muridarum infection. J Immunol 184:2602–2610. doi: 10.4049/jimmunol.0901593. [DOI] [PubMed] [Google Scholar]
- 55.Arnold-Schrauf C, Berod L, Sparwasser T. 2015. Dendritic cell specific targeting of MyD88 signalling pathways in vivo. Eur J Immunol 45:32–39. doi: 10.1002/eji.201444747. [DOI] [PubMed] [Google Scholar]