

Background: MicroRNA-155 is a proinflammatory small RNA, but its function in pathological angiogenesis is unknown.
Results: MicroRNA-155 deficiency increases angiogenic protein CCN1 expression, which harnesses retinal neovascularization by reducing both microglia activation and inflammation.
Conclusion: The microRNA-155/CCN1 regulatory axis regulates angiogenic and inflammatory responses in the retina.
Significance: Modulation of microRNA-155 and CCN1 interaction is potentially beneficial in retinal neovascularization therapy.
Keywords: angiogenesis, extracellular matrix protein, inflammation, microRNA (miRNA), retina
Abstract
The response of the retina to ischemic insult typically leads to aberrant retinal neovascularization, a major cause of blindness. The epigenetic regulation of angiogenic gene expression by miRNAs provides new prospects for their therapeutic utility in retinal neovascularization. Here, we focus on miR-155, a microRNA functionally important in inflammation, which is of paramount importance in the pathogenesis of retinal neovascularization. Whereas constitutive miR-155-deficiency in mice results in mild vascular defects, forced expression of miR-155 causes endothelial hyperplasia and increases microglia count and activation. The mouse model of oxygen-induced retinopathy, which recapitulates ischemia-induced aberrant neovessel growth, is characterized by increased expression of miR-155 and localized areas of microglia activation. Interestingly, miR-155 deficiency in mice reduces microglial activation, curtails abnormal vessel growth, and allows for rapid normalization of the retinal vasculature following ischemic insult. miR-155 binds to the 3′-UTR and represses the expression of the CCN1 gene, which encodes an extracellular matrix-associated integrin-binding protein that both promotes physiological angiogenesis and harnesses growth factor-induced abnormal angiogenic responses. Single CCN1 deficiency or double CCN1 and miR-155 knock-out in mice causes retinal vascular malformations typical of faulty maturation, mimicking the vascular alterations of miR-155 gain of function. During development, the miR-155/CCN1 regulatory axis balances the proangiogenic and proinflammatory activities of microglia to allow for their function as guideposts for sprout fusion and anastomosis. Under ischemic conditions, dysregulated miR-155 and CCN1 expression increases the inflammatory load and microglial activation, prompting aberrant angiogenic responses. Thus, miR-155 functions in tandem with CCN1 to modulate inflammation-induced vascular homeostasis and repair.
Introduction
The etiology of numerous vision-threatening ocular diseases, such as retinopathy of prematurity and proliferative diabetic retinopathy, is an aberrant response to injury of the retinal vascular network that palliates the metabolic and oxygen demands of the neural retina (1, 2). Indeed, the vascular beds supplying the retina often sustain injury as a result of diabetes, trauma, hyperoxia, aging, dyslipidemia, or the interaction of genetic predisposition, environmental insults, and age. These injurious stimuli commonly lead to an arrest of vascular development, vaso-obliteration, and/or vascular occlusion. The subsequent pathological response generates disorganized, leaky, and tortuous vessels that leak into the interface between the vitreous and the retinal tissue, attracting fibroglial elements causing severe hemorrhage, retinal detachment, and vision loss.
New therapeutic strategies that are currently receiving increasing attention focus on novel mechanisms regulating normal and abnormal vessel growth by microRNAs (miRNAs),3 a relatively abundant class of gene expression regulators that promote degradation of mRNAs or prevent translation of target genes (3, 4). More than 250 miRNAs are reportedly expressed in the retina, and miRNA gene regulation has been shown to affect retinal development, function, and diseases (5). Dicer and Drosha, the RNase III endonucleases involved in miRNA generation, significantly affect angiogenesis during development, and numerous studies suggest that miRNA function becomes more pronounced in response to pathophysiological stresses (6, 7). Specific functions have recently been attributed to individual endothelial cell-specific miRNAs, although a challenge still remains in validating their relevance in pathological angiogenesis (8, 9). However, normal and abnormal growth of blood vessels occurs through the interactions of vascular and non-vascular cells, including parenchymal, neuronal, and immune cells (10, 11). Therefore, a major focus must be placed on master regulator miRNAs that are able to define how different cell types interact and assemble to form the distinct characteristics of normal and abnormal blood vessel types. This study demonstrates that one such master regulator miRNA is miR-155.
miR-155 is a multifunctional miRNA that has been implicated in various physiological and pathological processes, including hematopoietic lineage differentiation, immunity, inflammation, viral infections, cancer, and cardiovascular diseases (12). miR-155 maps within and is processed from an exon of a noncoding RNA transcribed from the B-cell integration cluster located on chromosome 21 (13). The B-cell integration cluster shows strong sequence homology among humans, mice, and chickens and is highly, although not exclusively, expressed in lymphoid organs, implying an evolutionarily conserved function. Consistent with its widespread expression pattern in myeloid and lymphoid cells, miR-155 was reported to function in hematopoiesis and the immune response. Mice deficient in miR-155 showed clear defects in lymphocyte development and generation of B- and T-cell responses in vivo and an impaired antigen presenting function of dendritic cells (14, 15). Recent studies have shown that miR-155 is also overexpressed in endothelial cells, synovial fibroblasts, and monocytes of rheumatoid joints and during the course of inflammation (16, 17, 18). Thus, miR-155 may play important role(s) in mediating inflammatory and immune responses that are of paramount importance in pathological angiogenesis. A study by Neilsen et al. (19) assembled a comprehensive list of 140 miR-155 mRNA targets that were experimentally confirmed by both the demonstration of endogenous transcript regulation by miR-155 and validation of the miR-155 seed sequence through a reporter assay. This list included regulatory proteins for myelopoiesis and leukemogenesis (AICDA, ETS1, JARID2, SPI1, etc.) and inflammation (BACH1, FADD, IKBKE, INPP5D, MYD88, RIPK1, SPI1, and SOCS) and known tumor suppressors (C/EBPβ, IL17RB, PCCD4, TCF12, ZNF652, etc.). This large number of miR-155 targets probably regulates miR-155 function in numerous biological processes in a context- and tissue type-dependent manner.
Here we provide unprecedented evidence linking miR-155 expression or lack thereof to postnatal retinal vascular development in vivo and abnormal retinal vessel growth in the mouse model of OIR. The antiangiogenic functions of miR-155 correlate with the repression of CCN1, a secreted cysteine-rich and integrin-binding matricellular protein that positively regulates angiogenic signaling either directly by modulating endothelial cell function and behavior through integrin binding or indirectly by fine tuning the activity of growth factor receptors, such as vascular endothelial growth factor (VEGF) receptor (20, 21). We report for the first time how miR-155-dependent suppression of CCN1 regulates both physiological and pathological angiogenesis, at least in part, by modulating the inflammatory response of the retina. The genetic, biochemical, and cell biological data support a model in which negative regulation of CCN1 expression by miR-155 affects retinal vessel patterning and normal growth through alteration of the function and activation of microglia, the resident inflammatory cells of the retina. Concordantly, the miR-155/CCN1 regulatory axis is a key driver for the neuroinflammatory responses associated with physiological and pathological angiogenesis.
Experimental Procedures
Mice
Wild-type mice on a C57BL/6 genetic background (Jackson Laboratory) were bred and housed in the Department of Comparative Medicine at SUNY Downstate Medical Center. Mice deficient in miR-155, miR-155−/−, and the Tg(UBC-cre/ERT2)1Ejb transgenic line have been described previously (22, 23). Generation of CCN1flox/flox mice was described previously (24). All animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the State University of New York Downstate Medical Center.
Generation of CCN1 Conditional Allele and CCN1/miR-155 Double Knock-out Mice
Mice with inducible deletion of CCN1 in all tissue types were generated by cross-breeding CCN1flox/flox with Tg(UBC-cre/ERT2)1Ejb mice to produce CCN1flox/+UBC-Cre−/− and CCN1flox/+UBC-Cre+/− mice. The latter were further crossed among each other or with CCN1flox/flox to produce CCN1flox/+UBC-Cre+/−, CCN1flox/+UBC-Cre−/−, or CCN1flox/floxUBC-Cre+/− mice. A solution of 4-hydroxytamoxifen (4HT) was dissolved in ethanol at 10 mg/ml, and then 4 volumes of sunflower seed oil were added. Samples of 4HT were thawed and diluted in sunflower seed oil prior to intraperitoneal injection of 100 μl to mouse pups. Lactating mothers were alternatively given a single daily injection of 4HT (2 mg) to increase recombination efficiency (25). Genotyping was determined by polymerase chain reaction (PCR) to identify mice with floxed alleles, hemizygous floxed allele and Cre allele (CCN1+/−), and homozygous floxed alleles and one Cre allele (iΔCCN1−/−). Recombination levels in CCN1−/− mice as compared with CCN1+/+ were determined as described previously (24). In addition, CCN1flox/floxUBC-Cre+/− mice were bred with mRNA-155−/− mice to generate heterozygous miR-155+/−/CCN1+/floxUBC-Cre+/− mice. Heterozygous mice were then intercrossed to generate homozygous miR-155−/−/CCN1flox/floxUBC-Cre+/− mice, which were further injected with 4HT to produce miR-155−/−/iΔCCN1−/− double knock-out mice.
Quantification of Junctional Density, Length, and Migration of Retinal Vessels during Development
Mouse eyes were collected at the indicated postnatal days and fixed in 4% paraformaldehyde for 2 h. Retinas were dissected and laid flat on SuperFrost® Plus-coated slides to obtain whole mount preparations. Retinal mounts were then permeabilized in 0.1% Triton X-100 at room temperature for 20 min. Staining was performed with Isolectin B4 (IB4), as described previously (26). Fields of view of the retinal vascular networks from control and mutant mice were captured by using the ×40 objective lens and included regions of capillary-sized vessels directly adjacent to radial arterioles (27). Vascular parameters were measured using the AngioTool software (28). By assessing the variation in foreground and background pixel mass densities across an image, the software determines morphological and spatial parameters, including the overall size of the vascular network, the total and average vessel length, and vessel junctional density. For each parameter, at least four fluorescent images/retina were taken from 4–5 mice. The data are presented as means ± S.E. The statistical significance of differences among mean values was determined by one-way analysis of variance and two-tailed t test.
Intravitreal Injections
In order to assess the effect of Syn-miR-155, mice were given a 1-μl intraocular injection of 5 μmol/μl experimental Syn-miR-155 mimetic (Qiagen) in one eye and 5 μmol/μl control miRNA in the other eye. Mice were sacrificed at the indicated times, and retinas were dissected and analyzed as described above. For gene therapy studies, a lentiviral vector (∼1 μl) was injected into the vitreous in one eye of each animal using a 33-gauge needled syringe. In the contralateral eye, an equal volume of control vector (lnv-luc) was injected. Cells infected with lnv-green fluorescent protein at a multiplicity of infection of 5–10 units achieved transduction efficiency of 70–90%, as determined by counting the number of GFP-positive cells (26). For siRNA-mediated CCN1 suppression studies, siR-CCN1 or scrambled control siRNA was administered at postnatal day 12 (P12) by intravitreal injection behind the ora serrata and directly into the vitreal cavity. At P17, all animals were euthanized and enucleated, and retinas were processed for further analyses. No untoward effects (e.g. redness or opacity) were noted from the intravitreal injections.
Oxygen-induced Retinopathy and Retinal Neovascularization Image Analyses
Ischemic retinopathy was produced in C57BL/6J mice, as described by Smith et al. (29). Neonatal mice and their nursing dams were exposed to 75% oxygen in a PRO-OX 110 chamber oxygen controller from Biospherix Ltd. (Redfield, NY) between P7 and P12, producing vaso-obliteration and cessation of vascular development in the capillary beds of the central retina. On P12, the mice were placed at room air until P17, when the retinas were assessed for maximum neovascular response. For developmental studies, room air mice were raised under normal light and temperature conditions. Mice were sacrificed at the indicated times after birth by CO2 euthanasia and cervical dislocation or by decapitation. Eyes were enucleated and the vascular phenotype of the retinas was analyzed as described above.
Quantification of vascular obliteration, retinal vascularization, and preretinal neovascular tufts was performed at P12 and/or P17. The area of vascular obliteration was measured by delineating the avascular zone in the central retina and calculating the total area using Photoshop CS5 (Adobe) software. Similarly, the area of preretinal neovascularization was calculated by selecting tufts, which appear more brightly stained than normal vasculature, based on pixel intensities. Selected regions were then summed to generate the total area of neovascularization. The avascular areas and zones of neovascularization were expressed as a percentage of the total retinal surface area.
Isolation of Small RNA and MicroRNA Arrays
Small RNAs were isolated using the miRNeasy minikit (Qiagen) in accordance with the manufacturer's instructions. A total of 5 μg of small RNAs pooled from five experimental and five control retinas of age-matched mice reared in room air was used for microarray analysis. RNA samples were diluted to a concentration of 400 ng/μl and reverse transcribed using the MiScript II RT kit (Qiagen). miRNA profiling was performed with an miRNome miScript miRNA PCR array plate in combination with the miScript SYBR Green PCR kit (Qiagen), which contains miScript universal reverse primer and QuantiTect SYBR PCR master mix. The miRNA PCR array plate (Qiagen) includes primers for 84 mature miRNAs and controls. RNU6-2 served as an internal normalizer. Amplification was performed using an ABI 7900 real-time PCR machine (Applied Biosystems). Specificity and identity were verified by dissociation curve analysis. Baseline and threshold were set automatically for all PCR runs. Threshold cycle (Ct) values were exported as an Excel file for further analyses. Data quality control was reviewed to ascertain the PCR reproducibility and reverse transcription efficiency and detect genomic DNA contamination in amplified samples of both groups.
Bromo-2-deoxyuridine (BrdU) Incorporation Assay
BrdU was administered at 10 mg/kg intraperitoneally at P5. For BrdU labeling, retinas were digested with Proteinase K (10 μg/ml), fixed in 4% paraformaldehyde, treated with DNase I (0.1 units/ml) for 2 h at 37 °C, and incubated with anti-BrdU antibody (BD Pharmingen). Endothelial Cells were visualized by staining with isolectin B4 A568 (Molecular Probes), and BrdU was detected using directly conjugated mouse anti-BrdU Alexa 488 (Molecular Probes). The number of BrdU-positive cells per surface unit in equivalent areas of retinas from control and mutant mice was determined.
Immunohistochemistry and Western Immunoblotting
Fixed and permeabilized retinal mounts were stained with Isolectin B4 (IB4) as described above and/or with Iba-1 (Wako), glial fibrillary acidic protein (GFAP) (Invitrogen), and Collagen IV (EMD Millipore). Immunodetection was performed with either rhodamine- or fluorescein-conjugated anti-mouse or anti-rabbit secondary antibody diluted in blocking solution. Retinal mounts were washed several times in phosphate buffer solution between incubations. Images were acquired using a Leica DM5500B fluorescence microscope (Leica).
For retinal protein analysis, mouse eyes were enucleated, and retinas were carefully dissected and homogenized in lysis buffer containing 10 mm NaF, 300 mm NaCl, 50 mm Tris, pH 7.4, 1% Triton X-100, 10% glycerol, and 1 mm EDTA with a 1% volume of phosphatase and protease inhibitor mixture. Protein samples (25 μg) were fractionated in 10% SDS-polyacrylamide gel and transferred to nitrocellulose membrane, and Western blot analysis was performed with each of the following primary antibodies: CCN1 (30), CD68 (Bio-Rad), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Aviva Systems Biology). Immunodetection was performed using enhanced chemiluminescence (Pierce). Protein bands were quantified by ImageJ (National Institutes of Health).
Isolation and Quantitative Analysis of mRNAs
Total RNA was extracted from cells or tissues using the RNeasy column purification protocol (Qiagen). Quantitative analysis by qPCR was performed to determine the levels of a specific mRNA using TaqMan technology (Applied Biosystems). Highly specific primers were designed using Primer3, a Web-based primer design program. The cycling parameters for qPCR amplification reactions were as follows: AmpliTaq activation 95 °C for 10 min, denaturation 95 °C for 15 s, and annealing/extension 60 °C for 1 min (40 cycles). Triplicate Ct values were analyzed with Microsoft Excel using the comparative CT (ΔΔCt) method as described by the manufacturer (Applied Biosystems). The transcript amount (2−ΔΔCt) was obtained by normalizing to the acidic ribosomal phosphoprotein, an endogenous reference that has been shown to be the housekeeper gene least sensitive to oxygen tension in animal models of OIR (31).
Cell Culture
In vitro studies were performed with rat retinal endothelial cells (Cellpro Labs) and maintained in culture according to the manufacturer's instructions. Cells were propagated in 35-mm dishes in predefined endothelial growth medium containing 10% fetal bovine serum (FBS) (Atlanta Biological Inc.). Cells at 80% confluence were treated as described in the text and further processed for various analyses.
Transient Transfection, Mutagenesis, and Reporter Assay
Cultured cells were plated at a density of 1 × 105/cm2 in 35-mm dishes and maintained in serum-containing medium for 18 h. Transfection was then performed using Lipofectamine 2000 transfection reagent in serum-free medium according to the manufacturer's specifications (Invitrogen). Cells were allowed to recover for 16 h in fresh medium containing 10% serum. To assess the functionality of the 3′-UTR of CCN1, full-length CCN1 3′-UTR (U690) was cloned downstream of the Gaussia luciferase reporter in the plasmid pEZX. The plasmid pEZX with no 3′-UTR (MT05) (Genecopeia) was used as a negative control. Additional reporter constructs used include pEZX constructs with serial truncations of the CCN1 3′-UTR, leaving only 548 (U548)- and 229 (U229)-nucleotide-long 3′-UTRs. Additionally, U690 containing mutations in putative miR-155 and/or miR-181 seed sequences were generated by site-directed mutagenesis (Genescript) and used in transfection experiments as well. Constructs were fully sequenced in both directions to confirm successful mutagenesis before use. Cells were co-transfected with pCMV-Cluc2 vector (New England Biolabs) containing the Cypridina luciferase gene to adjust for transfection efficiency. The Lipofectamine 2000:DNA mixture plus serum-free medium was left on cells for 3 h. Cells were then allowed to recover in fresh medium containing 10% serum for 16 h and further incubated in serum-free medium. Medium samples were collected at different time periods and assayed for luciferase activities as both Gaussia and Cypridina luciferases were secreted in the medium. Luciferase activity was measured in a 20/20 Luminometer (Turner Biosystems) using the BioLux assay system (New England Biolabs) as per the manufacturer's instructions. Each experiment was performed at least three times in triplicate, and all experiments included a negative control (MT05). The latter served as a baseline indicator of luciferase activity.
Statistical Analyses
Statistical analyses were performed using Prism for Windows version 4 from GraphPad Inc. (San Diego, CA). To test differences among several means of significance, a one-way analysis of variance with the Newman-Keuls multiple comparison test was used. Where appropriate, a post hoc unpaired t test was used to compare two means/groups, and p values of <0.05 or <0.01 were considered significant. Student's t test with Bonferroni correction was used for pairwise comparisons when an analysis of variance (p) value was statistically significant.
Results
MicroRNA Expression Profile in the Retina of OIR Mice
We used the mouse model of OIR, an ischemia-induced retinal neovascularization model, to identify potential master regulator miRNAs of vascular loss and neovascular growth. First, we performed miRNA array profiling of 84 miRNAs that have been assigned a role in vascular function and/or dysfunction. Overall, microarray results shown in Table 1 were concordant with previous surveys of the miRNA expression pattern in the retina using either microarrays or deep sequencing technologies (5, 32). Several miRNAs with stricter cut-off criteria (log2 >2 or <−2) were differentially expressed at P12 following hyperoxia-induced vaso-obliteration, including miR-103, miR-107, miR-125, miR-126, miR-155, miR-93, miR-16, and miR-195. Real-time PCR-based validation of the expression of several miRNAs confirmed the array result trends (Fig. 1A). Further analyses showed that miR-155 was consistently increased following both hyperoxia at P12, representing the vaso-obliterative phase (phase 1), and ischemia at P17, signifying the neovascularization phase (phase 2), of this model (Fig. 1B). The levels of miR-155 were consistently elevated at every time point during phase 1 and phase 2 (2.4- and 4.2-fold, respectively) as compared with normoxic conditions (Fig. 1B). Thus, miR-155 levels were up-regulated under conditions causing vascular alterations or remodeling.
TABLE 1.
Summary of differentially expressed miRNAs in the retina following hyperoxia
Data were filtered by -fold change >2 or <−2 and p < 0.05.
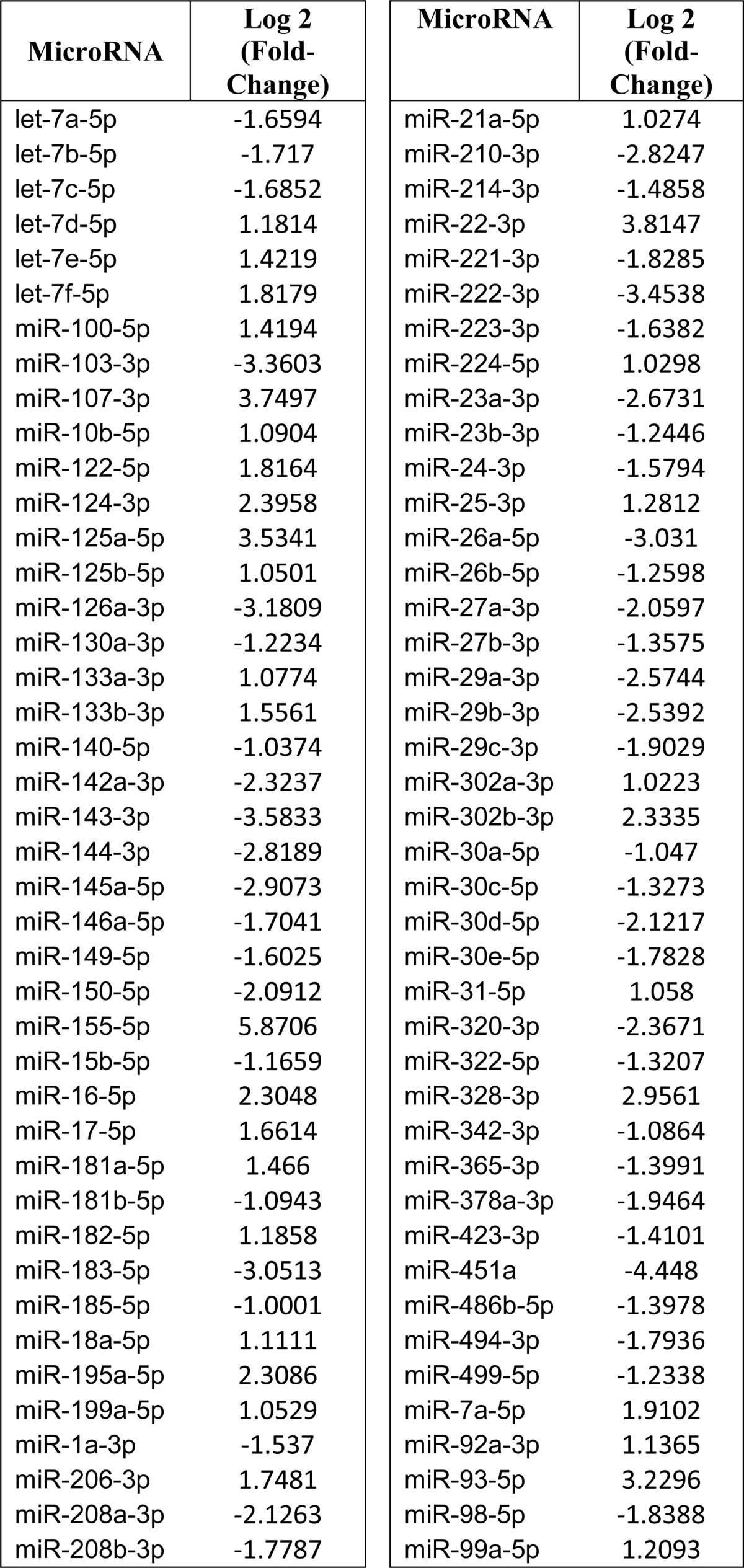
FIGURE 1.
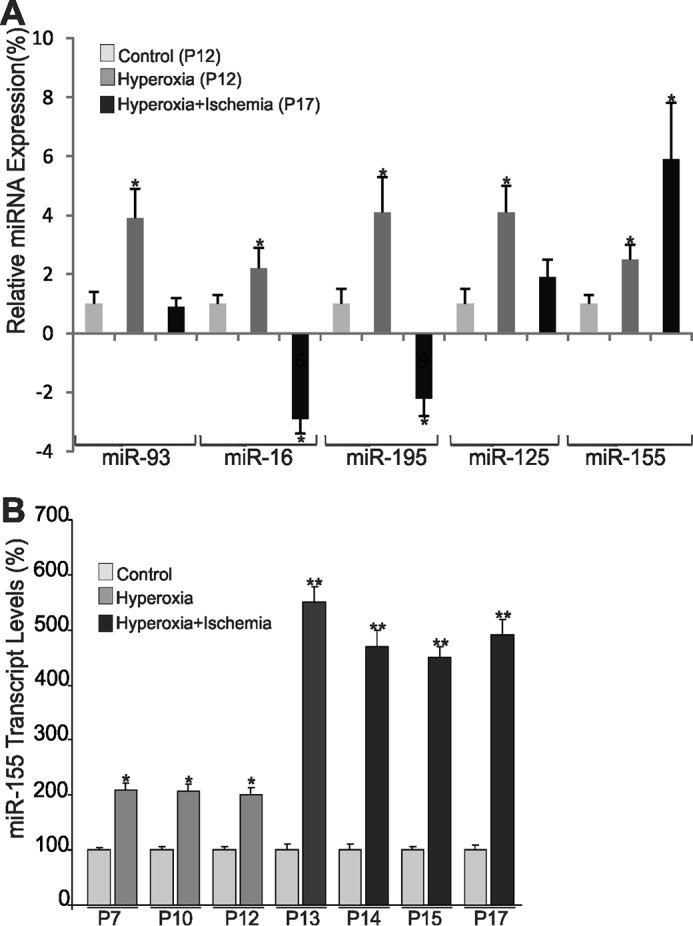
Differential expression of miR-155 in the retina following hyperoxia-induced vasoobliteration and subsequent vascular remodeling. A, C57BL/6 mice were placed in 75% oxygen at P7 and returned to room air at P12. Mice were sacrificed at P12 and P17. Small RNA species were isolated and reverse transcribed into cDNA, and real-time reverse transcription PCR was used to determine the levels of miR-93, miR-16, miR-195, miR-125, and miR-155 relative to the levels of RNU6-2 at P12 and P17. *, p < 0.05 versus control. B, quantitative analysis of miR-155 in the retina throughout the time course of hyperoxia and subsequent ischemia. The mean threshold cycle numbers (ΔCt) for miR-155 were compared with control non-hyperoxic non-ischemic retinas. *, p < 0.05. **, p < 0.001 (n = 5). Error bars, S.E.
Vascular Alterations Associated with miR-155 Expression or Deficiency in the Retina
To examine the role of miR-155 in retinal vascular obliteration and repair, we subjected wild-type and mi-R155−/− mouse pups to OIR and compared their vascular patterns. miR-155-deficient mice did not display any gross anatomical abnormalities and showed normal Mendelian inheritance as reported previously (23). Exposure to hyperoxia (P7 to P12) resulted in vaso-obliteration of the central superficial capillary plexus (Fig. 2A, a–d). In both wild-type and miR-155−/− mice, nearly 32% of the retinal surface area was vaso-obliterated (Fig. 2C). However, miR-155−/− mice exhibited a rapid revascularization of avascular areas during the subsequent normoxic phase (Fig. 2B, a–d). More than 95% of the retinal surface was vascularized in miR-155-deficient mice as compared with only 87% in wild-type mice (Fig. 2D). In miR-155−/− mice, the retinal vasculature exhibited normal morphology and branching in the mid-peripheral and peripheral regions except for sparse areas around the optic nerve. Similarly, preretinal neovascular tufts, which can be seen abundantly in the central and mid-peripheral retina in wild-type mice, were minimally present (<2% of the retinal surface), and in many cases, they were undetectable in miR-155-deficient mouse retinas (Fig. 2E). Thus, although miR-155 deficiency provided no protective effect against hyperoxia-induced vasoobliteration, it allowed rapid revascularization and/or normalization of the retinal vasculature during the ischemic phase of OIR and prevented the development of aberrant neovascularization.
FIGURE 2.
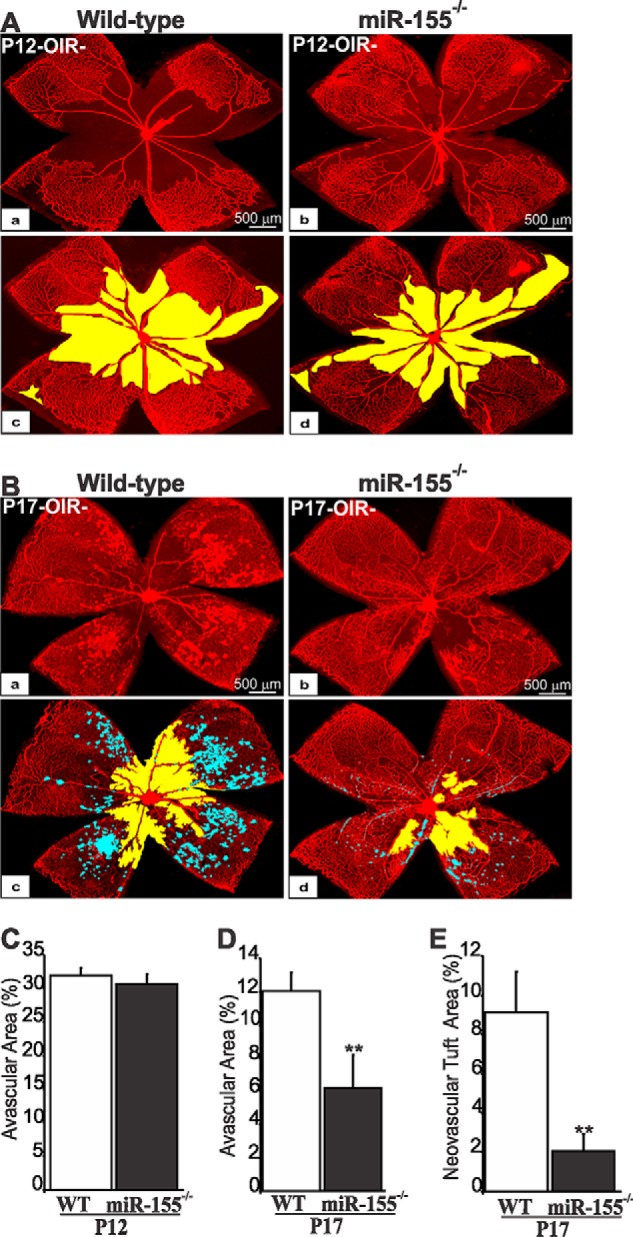
Effects of miR-155 deletion on hyperoxia-induced retinal vaso-obliteration and preretinal neovascularization following OIR. A and B, representative flat mount preparations of IB4-stained retinas eyes at P12 (A) and P17 (B) of wild-type (WT) (a) and miR-155-deficient (b) mice. Areas of vaso-obliteration and preretinal neovascular tuft formation as determined by computer-assisted image analyses are shown in yellow and blue, respectively (c for a and d for b). C–E, compiled data showing percentage of avascular (C and D) and neovascular tuft areas (E) in WT and miR-155−/− mouse retinas. **, p < 0.05 versus WT (n = 6–8). Error bars, S.E.
To determine whether the vascular response to ischemic injury stems from developmental vascular alterations, we examined the net effect of miR-155 deficiency on postnatal retinal vessel development. In wild-type mice, vascularization of the outer retina begins at P0 from the central retinal artery, and endothelial cells sprout to reach the peripheral region at about P8-P9. Retinas from miR-155−/− mice did not show overt defects in vessel growth and specification in arteries, capillaries, and veins (Fig. 3A). However, quantitative assessments of angiogenesis at P3 and P9 by computing several morphometric parameters showed a significant decrease of the junctional density in miR-155−/− retina as compared with wild-type control (Fig. 3B). Conversely, vascular length was increased by more than 40% in miR-155−/− mouse retinas, whereas total vascular area was not significantly affected (Fig. 3, C and D), suggesting that deletion of miR-155 mostly affected vascular density and distribution.
FIGURE 3.

Effects of miR-155 expression and deficiency on retinal vessel growth and development. A, representative immunofluorescence images of IB4-stained whole mount retinas at P3 and P9 of wild-type and miR-155−/− mice. Vascular parameters were analyzed using the AngioTool software. The outline of the vasculature is shown in white, the vasculature skeleton representation is shown in red, and branching points are green (b for a, d for c, f for e, and h for g). Note the strong and dense vascular network in wild-type compared with miR-155−/− mouse retinas. B–D, quantitative analysis of vascular parameters of representative retinas of WT and miR-155−/− mice. Graphical representations of the analysis of junction density (B), vascular length (C), and vascular area (D) are shown. **, p < 0.05 versus same age WT (n = 5). E, effects of miR-155 mimics on retinal vascular development. IB4-labeled vasculature of whole retinal mounts at P6 after Syn-miR-255 treatment at P3. Control mice were injected with scrambled control miRNA. Parallel bars represent distance between the vascular front and the retinal edge. F, quantification of sprouting distance in wild-type mice injected with control or Syn-miR-155. Vascular progression as a function of the retinal radius is shown. *, p < 0.05 versus control (Cntl) (n = 4). G, IB4- and GFAP-labeled whole retinal mounts of wild-type mice injected with scrambled miRNA or Syn-miR-155. Note that endothelial cell sprouting along GFAP-positive astrocytes similarly occurred in control and Syn-miR-155-injected eyes. H, effects of miR-155 gain or loss of function on endothelial cell growth during retinal vessel development. Whole mount retina images of BrdU incorporation (green) together with IB4 staining (red) at P6 of wild-type, Syn-miR-155-injected, and miR-155−/− mice. I, proliferation index at P6 was measured by counting BrdU-positive nuclei. Equivalent areas of retinas of scrambled control miRNA-injected wild-type, Syn-miR-155-injected, and miR-155−/− mice were analyzed. Data are means ± S.E. (error bars). *, p < 0.05; **, p < 0.001 versus wild-type control (Cntl) (n = 5).
Next, the effects of miR-155 gain of function in mice were examined through intravitreous injection of a miR-155 mimic (Syn-miR-155). Syn-miR-155 was injected within the vitreous at P3 in wild-type mice during the active growth of the superficial capillary plexus, and the vascular pattern was analyzed at P6. As shown in Fig. 3E, Syn-miR-155 delayed migration of the radially expanding superficial capillary plexus and hindered advancement toward the retinal edge. Vessels of the Syn-miR-155-injected pups only reached 59% of the distance to the retina edge, compared with 80% in littermate controls injected with a scrambled miRNA (Fig. 3F). In the retina, endothelial cells sprout along GFAP-positive astrocytes that emerge from the optic nerve head. GFAP staining indicated that astrocyte coverage was not affected by miR-155 gain of function (Fig. 3G), suggesting that the retinal endothelial cell sprouting evoked by miR-155 mimic is not secondary to potential astrocyte growth or migration. Interestingly, Syn-miR-155 induced hyperproliferation of endothelial cells at the leading vascular edge with an 65% increase of BrdU-positive cells, whereas miR-155-deficiency resulted in 20% reduction of endothelial cell proliferation as compared with wild-type littermates (Fig. 3, H and I). Growing endothelial cells appeared to incorporate into preformed vessels and no longer directed toward angiogenic sprouts, resulting in thicker and denser vasculature. Taken together, our results indicate that miR-155 modulates retinal vessel growth and remodeling both during development and under ischemic conditions.
miR-155 Represses CCN1 Gene Expression through Direct Interaction with CCN1 3′-UTR
To begin to elucidate the mechanisms whereby miR-155 regulates retinal angiogenesis, we used a bioinformatic approach to identify putative gene targets of miR-155. Among the predicted genes, we focused on CCN1, a matricellular protein best known for its angiogenic potential (11, 21). CCN1 is encoded by an inducible immediate-early gene whose expression substantially increases during postnatal development of the retinal vasculature in mice (33). To determine whether in vivo miR-155 levels are inversely correlated to those of CCN1, we examined CCN1 gene expression in the retina following intravitreal injection at P4 of the Syn-miR-155 mimic in the mouse eye during the active postnatal angiogenic process. As shown in Fig. 4, A and B, Syn-miR-155 reduced both CCN1 mRNA and protein levels below control levels. Conversely, exposure of miR-155−/− mouse pups to OIR increased CCN1 mRNA and protein levels at P12 and markedly at P17 compared with wild-type mice (Fig. 4, C and D). To test whether enhanced expression of the endogenous CCN1 gene in miR-155−/− mouse retina contributed to the vascular phenotype following OIR, mouse pup eyes were injected at P4 prior to OIR with a lentiviral vector encoding a mutant form of the CCN1 protein lacking the C-terminal domain. This mutant variant acts as a dominant negative form of the wild type protein and inhibits CCN1 activity (20). Retinal vessel sensitivity to hyperoxia at P12 was not affected by injection of lnv-CCN1-ΔCT because the degree of vaso-obliteration was similar to that of retinas from eyes injected with the control vector (data not shown). However, OIR-induced neovascularization was significantly increased by 48% in lnv-CCN1-ΔCT-injected eyes as compared with control vector-injected ones (Fig. 4, E and F). In addition, the avascular area in the retina was further increased following the loss of CCN1 activity in miR-155−/− mice (Fig. 4G), suggesting that CCN1 expression contributed to vascular regrowth in miR-155−/− mice subjected to OIR.
FIGURE 4.

MicroRNA-155 directly targets CCN1 gene expression during vessel and neovessel growth. A and B, effects of Syn-miR-155 injection on CCN1 gene expression during retinal vessel development. CCN1 gene expression was determined at the mRNA (A) and protein level (B) in retinal tissue lysates from wild-type mice at P6 after treatment at P3 with either scrambled control miRNA (Cntl) or Syn-miR-155. *, p < 0.05 (n = 3). C and D, expression of the CCN1 gene at the mRNA (C) and protein (D) levels in wild-type and miR-155-deficient mice following OIR. Wild-type and miR-155−/− mice were placed in 75% oxygen at P7 and returned to room air at P12. Retinas were harvested at P12 or P17, and CCN1 expression was analyzed by either qPCR or Western immunoblotting as described under “Experimental Procedures.” CCN1 mRNA levels were normalized to those of acidic ribosomal phosphoprotein. **, p < 0.01 versus wild type. ***, p < 0.001 versus wild type (n = 5). GAPDH was used as a loading control of protein lysate analysis. E–G, effects of CCN1 suppression on the vascularization and neovascularization of the retina of miR-155−/− mice following OIR. Whole mount retinas shown were from eyes injected at P4 with either lnv-luc or lnv-CCN1ΔCT encoding a dominant negative form of CCN1. Mice were subjected to OIR at P7. Areas of vaso-obliteration and preretinal neovascular tufts at P17 as determined by computer-assisted image analyses are shown in yellow and blue, respectively (E). Compiled data showing the percentage of neovascular and avascular areas in eyes injected with lnv-luc and lnv-CCN1ΔCT are shown in F and G, respectively. **, p < 0.01 versus miR-155−/− (n = 5). H, schematic layout of the CCN1 3′-UTR, with the relative location of miR-155, miR-22, miR-181, and miR-221 binding sites. Depiction is not to scale. 3′-UTR sequences of CCN1 of mouse, human, and chimpanzee and predicted interaction with conserved miRNA seeds are shown. The sequence of the CCN1 3′-UTR seed mutant used for reporter assays and predicted disruption of the miR-155 and miR-181 interactions are also shown. I–K, transient transfection of endothelial cells was performed with an empty plasmid (MT05) or pEZX-CCN1 3′-UTR plasmids with and without mutated miRNA sequences depicted in H. Luciferase activity assays were performed on the supernatant media as described under “Experimental Procedures.” Gaussia luciferase values have been normalized to those of Cypridina luciferase, and the percentage of luciferase activity is represented. **, p < 0.05 versus U690. ***, p < 0.001 versus MT05 (n = 4). Error bars, S.E.
To test whether miR-155 represses CCN1 expression through direct interaction with the 3′-UTR of the CCN1 gene, we cloned the 3′-UTR of CCN1 into a reporter plasmid downstream of a luciferase reporter gene driven by the SV40 promoter (Fig. 4, H and I). Cells were transfected with this construct, and a luciferase assay was performed to measure the 3′-UTR activity. As shown in Fig. 4J, the activity of a luciferase reporter fused to the 3′-UTR of CCN1 (U690) was repressed as compared with the empty reporter vector (MT05). According to the TargetScan algorithm, the 3′-UTR of CCN1 contains highly conserved 7–8-mer target “seed” sequences for several miRNAs, including miR-155, miR-22, miR-181, and miR-221 (Fig. 4H). Serial deletion of the CCN1 3′-UTR showed that removal of the distal 150-nucleotide sequence containing miR-221/222 affected the reporter activity similarly as the removal of the full-length 3′-UTR. Deletion of the distal 230-nucleotide sequence containing the highly conserved miR-181 seed sequence reduced the activity by only 15%, suggesting that the proximal 3′-UTR region contains the essential functional repressive elements. Site-directed mutagenesis of the miR-155 seed sequence located in the proximal region reduced the repressive effects of the 3′-UTR by 76% (Fig. 4K). Mutation of both miR-155 and miR-181 seed sequences resulted in similar repressive activity. Thus, miR-155 directly targets the CCN1 3′-UTR, leading to repressed CCN1 gene expression.
Conditional Deletion of CCN1 Causes Retinal Vascular Defects Similar to Those Caused by miR-155 Gain of Function
We hypothesized that if modulation of CCN1 expression by miR-155 plays a non-redundant role in retinal vascular development and repair, loss of CCN1 will result in defective vascular architecture and remodeling. To test this hypothesis, we have generated a conditional null allele of CCN1 because constitutive inactivation of the CCN1 gene leads to early embryonic lethality (34). Mice carrying a loxP-flanked CCN1 gene (CCN1flox/flox) were intercrossed with UBC-Cre-ERT2 transgenics, which ubiquitously express tamoxifen-inducible Cre recombinase (Fig. 5A). UBC-CreERT2-mediated CCN1 deletion effectively eliminated CCN1 gene expression because less than 10% of CCN1 mRNA and less than 20% of residual CCN1 protein were detected in the retina following daily 4HT injection for 3 days (Fig. 5, B and C). At P4, retinas of iΔCCN1−/− mutant mice displayed increased vascularity per surface unit but showed striking defects in the radial expansion of the vascular plexus from the optic nerve head to the periphery (Fig. 5D). At P7, loss of CCN1 resulted in reduced forward progression of the superficial plexus and increased vascular anastomosis at the front of the expanding vascular plexus (Fig. 5E). In addition, iΔCCN1−/− mouse retinas showed widely lumenized vessels with increased diameter, which was most severe close to arteries and the vascular front. Vascular sprouts of wild type mice were generally slender compared with those of iΔCCN1−/− mice, suggesting continued expansion of blood vessels and reduced forward sprouting. This phenotype is strikingly similar to that of endothelium-specific loss of CCN1 that showed reduced forward progression of the superficial plexus but increased speed of growth (35). To test the effects of compound knock-out of CCN1 and miR-155, we generated miR-155−/−/iΔCCN1−/− mice in which miR-155 was constitutively deleted whereas CCN1 alleles were conditional by initially intercrossing miR-155 mice with UBC-Cre-ERT2− CCN1flox/flox mice. Administration of 4HT from P1 to P3 resulted in increased vascularity, widening of blood vessels, and reduced lacunarity of the retinal vasculature of miR-155−/−/iΔCCN1−/− mouse pups (Fig. 5F). These alterations are reminiscent of the retinal vascular phenotype following the loss of CCN1 alone (Fig. 5D). Quantitative analyses of vascular parameters further evidenced increased junctional density but a significantly reduced vessel length average in both iΔCCN1−/− and miR-155−/−/iΔCCN1−/− mouse retinas as compared with wild-type or miR-155−/− mouse retinas (Fig. 5, G and H). Thus, single deletion of CCN1 or double knock-out of both CCN1 and miR-155 phenocopied, at least in part, the vascular phenotype of miR-155 gain of function.
FIGURE 5.

Inducible knock-out of the CCN1 gene recapitulates the retinal vascular abnormalities of miR-155 gain of function. A, schematic diagrams showing the targeted CCN1 genomic locus with the CCN1 flox-neo allele, the CCN1 flox allele with the neomycin resistance cassette deleted by recombination with loxP sites, and the iΔCCN1−/− locus following 4HT-induced Cre-mediated excision. B and C, CCN1 mRNA (B) and protein (C) levels in retinal homogenates of control CCN1+/+ and iΔCCN1−/− mice. Retinas were harvested at P4 following daily injection of 4HT starting at P1 and analyzed for CCN1 expression by qPCR and Western immunoblotting. **, p < 0.001 versus CCN1flox/flox. D–F, representative immunofluorescence images of IB4-stained flat-mount mouse retinas of CCN1+/+, iΔCCN1−/−, miR-155−/−, and miR-155−/−/iΔCCN1−/− mice upon 4HT injection. G and H, analysis and quantification of vascular parameters of representative retinas from CCN1+/+, iΔCCN1−/−, and miR-155−/−/iΔCCN1−/− mice at P5 using the AngioTool software. Fields of view at the sprouting vascular front of the retinal vascular networks from control and mutant mice were captured using a ×40 objective lens and included regions of capillary-sized vessels directly adjacent to radial arterioles. Graphical representations of the analysis of the junctional density and vessel length are shown in G and H, respectively. **, p < 0.01 versus CCN1+/+ (n = 5). Error bars, S.E.
miR-155/CCN1 Regulatory Axis Modulates Microglial Cell Behavior and Activation
Retinal microglial cells, the resident ocular macrophages, are immunocompetent cells that respond to inflammation and infection, phagocytosing debris produced during normal physiological remodeling or degenerative diseases (36). Microglia-deficient mice were found to have reduced numbers of vascular branch points with fewer points of contact between neighboring tip cells in the retina, suggesting that microglia provide scaffolds for sprout fusion and anastomosis (37). Because miR-155 deletion induced the formation of a sparser vascular network with lengthier vessels whereas either gain of miR-155 function or CCN1 deletion abnormally enhanced vessel anastomosis, we hypothesized that miR-155 and CCN1 affect microglia function and behavior. During development, retinal microglia enter the retina from the peripheral margins via the blood vessels of the ciliary body as well as centrally from the embryonic hyaloid artery at the optic nerve head and vitreous (38). Superficial microglial cells are found within the inner plexiform layer in which their density is the highest, the retinal ganglion cell layer, and the nerve fiber layer, whereas deeper microglial cells reside within the outer plexiform layer. In P4 wild-type mouse retinas, Iba-1+ microglia occupy the entire retinal surface, including both vascular and avascular areas (Fig. 6, A and B). However, microglia were sparsely present, and their number was significantly reduced (−15%) in whole retinal mounts of miR-155−/− mice compared with those of wild-type mice (Fig. 6, A and D), suggesting an early loss of microglia as a result of miR-155 deletion. Conversely, loss of either CCN1 alone or both miR-155 and CCN1 was associated with a 35% increase in the number of microglia when compared with wild-type mouse retinas. As sprouting angiogenesis occurs, retinal microglial cells are commonly found at sites of contact between neighboring endothelial tip cells bridging vascular sprouts to add new circuits to the existing vessel network (Fig. 6B). In the largely anastomosed retinal vasculature of iΔCCN1−/− and miR-155−/−/iΔCCN1−/− mice, microglia were frequently positioned at branching points of the excessively anastomosed superficial capillary plexus (Fig. 6C). Thus, single or double knock-out of CCN1 and miR-155 alter both microglial cell number and retinal vascularity.
FIGURE 6.

Altered retinal microglial cell abundance, distribution, and activation in miR-155- and/or CCN1-deficient mice. A and B, representative images of retinal whole mounts stained with Iba-1 of WT, miR-155−/−, iΔCCN1−/−, and miR-155−/−/iΔCCN1−/− mice. B and C, double-staining (Iba-1 in green and IB4 in red) or retinal whole mounts illustrate the relationship between microglia and vasculature in the developing mouse retina at P4. Note the abundance of microglia at sites of contact between neighboring endothelial tip cells at the vascular front of miR-155−/− mice. Microglia are positioned at branching points of the excessively anastomosed superficial capillary plexus of the largely anastomosed vasculature of iΔCCN1−/− and miR-155−/−/iΔCCN1−/− mice. D, microglial cell count per surface unit of retina of WT, miR-155−/−, iΔCCN1−/−, and miR-155−/−/iΔCCN1−/− mice. *, p < 0.05 versus WT (n = 6). Error bars, S.E.
We further examined the effects of ischemia on microglial cells following loss of miR-155 alone or in combination with CCN1. Incidentally, miR-155−/−/iΔCCN1−/− mouse pups abruptly died following exposure to hyperoxia. Mice with compound deletion of miR-155 and CCN1 appeared normal and active during exposure to hyperoxia, and their sudden death occurred within 1 or 2 days under normoxia. It is possible that following hyperoxia, double knock-out mice died of respiratory failure, a common pulmonary complication associated with hyperoxic conditions (39). Pulmonary insufficiency may have occurred as a result of dampened inflammatory response from miR-155 loss together with accelerated lung epithelial cell death previously shown to be associated with loss of CCN1 expression in the lung (40). To circumvent the premature death of miR-155-CCN1 double knock-out mouse pups following hyperoxia, we induced local suppression of CCN1 through intravitreal injection of CCN1-siRNA (siR-CCN1) in miR-155−/− mice. Small inhibitory RNAs have been used successfully to selectively inhibit gene expression in the retina (41, 42). As shown in Fig. 7, A and B, loss of miR-155 promoted a near normalization of the vasculature after OIR. However, miR-155−/−/siR-CCN1 mouse retinas exhibited vascular alterations similar to those of wild-type OIR animals with persistence of avascular areas in the central retina and the appearance of neovascular tufts (Fig. 7, A–C). In miR-155−/−/siR-CCN1 mice, retinal vessels of the superficial capillary plexus appeared highly anastomosed with enlarged lumen, which is reminiscent of developing vessels in CCN1-deficient mouse retinas (Fig. 7C). Microglial cell count was determined following OIR in representative field areas of the peripheral vascularized zone, central avascular zone, and mid-retinal neovascularized zone. In wild-type OIR mice, the numbers and distribution of microglial cells in the central zone were similar to those in the peripheral zone (Fig. 7, D and E). However, the number of microglial cells significantly increased in the neovascular areas in which IB4 and Iba-1+ cells were densely packed within and in close proximity to neovascular tufts. In miR-155-deficient OIR mice, microglial cell counts were relatively uniform in all three zones of whole retinal mounts concomitantly with a visibly normalized retinal vasculature (Fig. 7, F and G). Conversely, loss of both miR-155 and CCN1 recapitulated microglia distribution and count obtained in wild-type OIR mice in zones 1, 2, and 3 (Fig. 7, H and I), suggesting that CCN1 is, at least in part, responsible for microglial cell-dependent alterations of the retinal vasculature during OIR.
FIGURE 7.

Effects of ischemia-induced retinal neovascularization on microglial cell localization and abundance in wild-type (WT) and miR-155−/− mice left untreated or injected with siR-CCN1. A–C, representative retinal whole mounts of mice subjected to 5-day hyperoxia and 5-day normoxia and double-stained with Iba-1 and IB4. Highly magnified areas illustrate the vascular pattern in central areas of retinas and microglial cell distribution. D–I, microglial cell distribution (D, F, and H) and count (E, G, and I) in the central avascular (1), middle neovascular (2), and vascularized peripheral (3) zones of WT and miR-155−/− mice left untreated or injected with siR-CCN1. *, p < 0.05; **, p < 0.001 versus zone 1 (n = 5). Error bars, S.E.
Typical resting Iba-1+ microglial cells commonly exhibit a ramified morphology with a small ovoid or triangular body from which three or four primary processes have sprouted and then divided dichotomously into secondary ones. These cells respond to ischemic stress and injurious stimuli by adopting an activated state characterized by alterations in cellular morphology and function, tissue distribution, migration, proliferation, and expression of various cytokines and growth factors. Microglial cells at the advancing ischemic retinal vascular edge adopted an activated state by retracting their processes and becoming lectin-positive in wild-type mice following injection of either control or Syn-miR-155 (Fig. 8A). Microglia with a similar morphology and activation features were found in retinal mounts of CCN1-deficient mice (data not shown). Loss of miR-155 was associated with a slight decrease of CD68, a single-chain heavily glycosylated protein of 90–110 kDa that is expressed on the lysosomal membrane of active retinal microglial cells (Fig. 8, E and F). However, CCN1 deletion alone or in combination with miR-155 deletion significantly increased CD68 protein levels as compared with those in the corresponding control mouse retinas. In normoxic controls, dendritic-like microglial cells ran parallel to the retinal surface and were located in perivascular areas in the peripheral retina (Fig. 8B). Conversely, active microglial cells were found in higher numbers around the vascular tufts between the central avascular and peripheral vascularized zones in P17 OIR mouse retinas (Fig. 8C, a and b). Morphologically, active cells largely found in the superficial layers retracted their processes and became IB4-positive (Fig. 8C, c), whereas those in the deeper layers exhibited a normal resting morphology (Fig. 8C, d), suggesting that the formation of vascular tufts occurs in the midst of activated neighboring superficial microglia. Conversely, resting ramified microglia were also found around the normalized retina of miR-155−/− mice after OIR (Fig. 8D). As shown in Fig. 8, E–G, CD68 protein levels were reduced in retinal lysates of miR-155−/− OIR mice, but their levels were significantly increased in retinal lysates of miR-155−/−/siR-CCN1 mice as compared with those of wild-type OIR mice. Thus, loss of CCN1 increased microglial cell activation in the retinas both during development and under ischemic conditions.
FIGURE 8.

miR-155 and CCN1 alter microglial cell activation. A, representative images of the retinal vasculature of WT mice pretreated with control miRNA (Cntl miR) or Syn-miR-155. Whole retinal mounts were stained with either IB4 (red) or IB4 and Iba-1 (green) and show localization of IB4- and Iba-1-positive microglia at points of sprout fusion and anastomosis. B and D, microglial cell morphology in WT and miR-155−/− mice under normoxic conditions or following OIR. Resting microglia in WT mouse retinas exhibit a ramified morphology with a small ovoid body from which three or four primary processes have sprouted (B). In WT mice exposed to OIR (C), microglia of the superficial layers of the retina (a) were in higher numbers around the vascular tufts. They become activated by retracting their processes and also appear IB4-positive (b and c). In the deeper layers (d), microglia show no morphological signs of activation. D, microglial cells still adopt a resting state morphology in miR-155−/− mouse retinas after OIR. E–G, Western blot analyses of CD68 protein levels in retinal lysates from WT, miR-155−/−, and iΔCCN1−/− normoxic mice at P6 and in WT and miR-155−/− treated with either scrambled control siRNA (siR-Scrbl) or siR-CCN1 following OIR. CD68 protein band intensities were determined by densitometric measurements and normalized to that of GAPDH to account for total protein loading (F and G). **, p < 0.001 versus control/WT. *, p < 0.001 versus WT-OIR− (n = 3). Error bars, S.E.
Regulation of Microglial Marker and Inflammatory Cytokine Expression via miR-155 and CCN1
We determined the expression of a battery of microglial markers in retinal lysates of miR-155−/− and CCN1−/− mice and examined a potential correlation of their levels with that of a panel of key inflammatory cytokines that have been functionally linked to microglia activation and angiogenesis. As shown in Fig. 9A, the expression of the mitochondrial translocator protein (TSPO), a selective marker of microglia in their highly reactive state (43), and DNAX activation protein of 12 kDa (DAP12), a proinflammatory adaptor protein (44), were down-regulated in miR-155-deficient mouse retina, whereas CD200R, which triggers an inhibitory intracellular signaling cascade that blocks proinflammatory activation in microglial cells (45), was increased by nearly 28% as compared with wild-type mouse retinas. In CCN1−/− mutant mouse retinas, the expression of TSPO was increased, whereas that of CD200R was markedly decreased, suggesting that CCN1 harnesses, in large part, microglial cell activation. Expression of DAP12 was not affected following CCN1 inactivation, which indicates selective CCN1 effects on microglial marker expression. The resting state of microglial cells in miR-155−/− mouse retinas was also associated with reduced IL-1β and IL-10 expression but no significant alterations of TNF-α and IL-6 levels (Fig. 9A). Conversely, IL-1β, IL-6, and IL-10 were remarkably increased in CCN1-deficient mouse retinas, as compared with wild-type ones, suggesting that the expression of CCN1 (or suppression of miR-155) reduced inflammatory cytokines levels in the retina. Furthermore, alterations of inflammatory marker expression were nearly identical in iΔCCN1−/− and miR-155−/−/iΔCCN1−/− mouse retinas (data not shown). Under ischemic conditions (i.e. OIR at P17), miR-155 deficiency alone reduced microglial cell activation markers (e.g. TSPO and DAP12) and inflammatory cytokines (e.g. IL-1β and IL-10) as compared with wild-type mouse OIR retinas (Fig. 9B). The alteration of IL-1β and IL-10 levels were reversed in miR-155−/−/siR-CCN1 mouse retinas, suggesting a specific effect of CCN1 on the expression of these cytokines. Of note, CD200R expression was strongly up-regulated in miR-155−/−/siR-CCN1 retinas, suggesting that its expression is either directly or indirectly regulated by the dual action of CCN1 and miR-155. Functionally, CD200R up-regulation may occur as a compensatory reaction to suppress the inflammatory response associated with loss of CCN1. Meanwhile, miR-155 deficiency did not significantly alter IL-6 expression, whereas dual deletion of miR-155 and CCN1 induced >3-fold increase of IL-6 as compared with wild-type OIR mouse retinas, indicating that IL-6 expression is potentially sensitive to the levels of CCN1 in the tissue.
FIGURE 9.
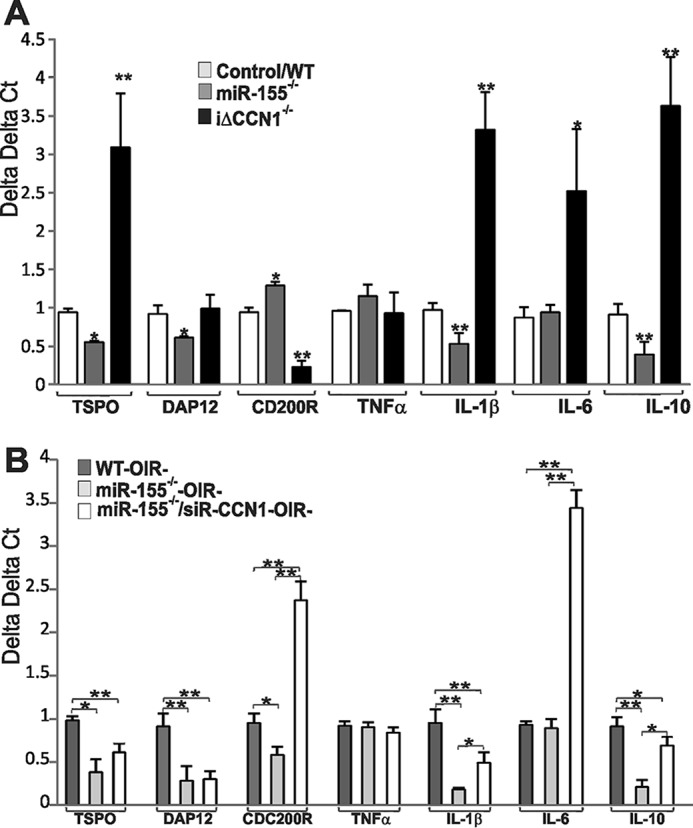
miR-155- and CCN1-dependent regulation of microglial markers and inflammatory cytokine during development and in response to OIR. A, gene expression of microglial markers TSPO; DAP12; CDC200R; and inflammatory cytokines TNF-α, IL-1 β, IL-6, and IL-10 was quantified by real-time PCR in retinal lysates from P5 WT and miR-155−/− and iΔCCN1−/− mutant mice. The data shown are the means ± S.E. (error bars) of three determinations each performed in triplicate. *, p < 0.05 versus control. **, p < 0.01 versus control. B, gene expression of microglial markers and inflammatory cytokines in WT, miR-155−/−, and miR-155−/−/siR-CCN1 mice at P17 following OIR. The transcript levels were determined by qPCR and normalized to those of acidic ribosomal phosphoprotein levels. Transcript levels in miR-155−/− were compared with those in WT mice subjected to OIR and represent the average of three determinations (n = 3). *, p < 0.05; **, p < 0.001.
Discussion
The key findings of this study are that expression of miR-155 or lack thereof has a major impact on the regulation of retinal vascular homeostasis and remodeling. The matricellular CCN1, a potent angiogenic protein that is required for proper vessel development and repair, is identified as a molecular target and downstream effector of miR-155. Analysis of mice with either single or compound deletion for miR-155 and CCN1 evidenced a pivotal function of these molecules in modulating the inflammatory response within the retina by engaging microglial cells to allow for normal vessel morphogenesis and/or normalization of the retinal vasculature under ischemic conditions.
Deregulation of miR-155 has been associated with different kinds of cancers and cardiovascular and inflammatory diseases (12). The large number of miR-155 targets identified in different tissues and organs suggests that miR-155 controls numerous biological processes in a context- and tissue type-dependent manner (46). Thus far, evidence of the involvement of miR-155 in vessel development and pathophysiology has remained scanty. Retinal angiogenesis, an essential component of postnatal development of the retina and a prerequisite for proper retinal function, is a complex process involving miRNA-dependent targeted gene activation and/or repression and modulation of specific cell signaling pathways (3). We found that miR-155 was expressed at low levels in the retina during postnatal development of the vasculature. However, constitutive miR-155 deficiency in mice resulted in retinal vascular alterations, including increased vessel length, reduced vascular branch point number, and decreased microglial cell population in the retina. Along these lines, low expression levels of miR-155 have previously been reported in lymphocytes, macrophages, dendritic cells, and progenitor stem cell populations (23), yet mice deficient in miR-155 showed defects in lymphocyte development and generation of B- and T-cell responses in vivo (15, 47). Thus, even low levels of miR-155 in cells or tissues may be important in tissue function and/or structure, which further underscores the need for miR-155 expression to be stringently regulated.
miR-155 has various roles in different cell types and physiological situations, and individual targets probably make significant contributions to miR-155 function in various tissues. The important role of miR-155 in retinal vessel growth and morphogenesis was further evidenced in our study by the formation of denser and largely anastomosed retinal vessels following exogenous intravitreal injection of miR-155 mimic. miR-155 has been found to be expressed in endothelial cells and was shown to regulate hypoxia by targeting the ELK3 gene (48, 49). Herein, we identified and characterized a direct link between miR-155 and CCN1, an angiogenic factor largely produced by vascular cells, whereby miR-155 can directly repress expression of CCN1 and impede its function as an angiogenic factor orchestrating normal vessel growth and patterning. miR-155 directly interacts with the 3′-UTR of the CCN1 gene and enhanced the repressive effects of the 3′-UTR by more than 70%. The 3′-UTR-mediated post-transcriptional gene regulation is critical for CCN1 gene expression duration and silencing, whereas promoter-mediated transcriptional activation of the CCN1 gene is important for gene induction. Consistent with our data, a study by Nakagawa et al. (50) revealed that regulatory elements that include the miR-155 binding site in the proximal half of the 3′-UTR of the CCN1 gene form a stable secondary stem-loop structure required for posttranscriptional regulation of the CCN1 gene. Although several putative targets of miR-155 have been predicted through bioinformatic and proteomic approaches, our data showed that knockdown of CCN1 can mimic many of the effects of miR-155 gain of function. The delivery of miR-155 mimic, which concomitantly reduced CCN1 levels in the retina, caused the formation of hyperplastic and largely anastomosed vessels as a result of increased growth of endothelial cells. Similarly, deletion of CCN1 caused retinal vessels to coalesce into large flat hyperplastic sinuses with subsequent loss of their hierarchical organization into arteries, capillaries, and veins, which recapitulates endothelium-specific loss of CCN1 (35). A study by Roitbak et al. (48) has demonstrated that interference with miR-155 in endothelial cells significantly enhances capillary tube formation, including the number of tubule branch points and tubular length. Interestingly, the direct effects of recombinant CCN1 on capillary tube formation recapitulate those of miR-155 inhibition (20, 51). The CCN1 protein regulates several aspects of endothelial cell function, including adhesion, migration, proliferation, and survival. Many of these activities derive from CCN1 interaction with integrin receptors localized in each of the CCN1 protein constitutive domains, which contain binding sites bearing structural homologies to αvβ3, αvβ5, and α2β3 integrin binding sequences (52). However, these biological activities are context-specific and depend on the interaction of the CCN1 protein with other growth factors and/or receptors (53, 54, 55). In vivo studies using mouse genetics have demonstrated that CCN1 activity is integrated with VEGF receptor 2 activation and downstream signaling pathways required for tubular network formation (35).
Our study design further demonstrated the importance of microglia in mediating the action of miR-155 and CCN1 on vessel patterning and morphogenesis. Homeostatic microglia function to control developmental apoptosis, phagocytosis of cell debris, shaping of neuronal connections, and guidance of primary vessel growth (56, 57). Loss of CCN1 expression or negative regulation of CCN1 by miR-155 altered resident microglia recruitment, distribution, and reactivity in the retina. miR-155 deletion was associated with 15% reduction of the number of microglia, the majority of which exhibited a non-reactive morphology. Conversely, CCN1 deficiency was associated with a 35% increase of the number of microglia. Microglial cells invade the retina around embryonic day 16 and are first located at the vitreo-retinal surface close to blood vessels (38). Microglial reactivity is characterized by increasing soma swelling, retraction of protrusions, and generalized disturbance of the microglial network (58). The number of amoeboid phagocytic microglia is the highest around P5, the time point when ganglion cell death peaks in the retina (59). This is consistent with their involvement in phagocytosis of dying neurons before they occupy their strategic positions in both plexiform layers of the adult retina. Concomitant with a reduced number of amoeboid microglial cells in the outer plexiform layers, the expression of proinflammatory factors, such as IL-10, IL-1β, TSPO, and DAP12, was significantly decreased in miR-155-deficient retinas. Conversely, ablation of CCN1 reversed the effects of miR-155 deletion on the expression of the same cytokines as well as that of IL-6. Dual deletion of miR-155 and CCN1 recapitulated the vascular phenotype associated with CCN1 deletion during development. Thus, miR-155 knock-out and CCN1 re-expression reduced the inflammatory load and microglial activation in the retina, in agreement with other studies that have shown that miR-155 is one of the most influential miRNA of proinflammatory pathways (60).
During vascular development, retinal microglial cells have a unique spatial relationship with angiogenic tip cells, which appear in close contact with microglial cells at the point of turning and branching. Reduced microglial cell count in miR-155−/− mouse retinas is consistent with the formation of a sparser superficial capillary plexus. Conversely, increased vascular density and anastomosis of the superficial capillary plexus in iΔCCN1−/− mice is consistent with increased microglial cell number and reactivity, which reflects enhanced angiogenesis and/or defective remodeling. How miR-155 expression and/or CCN1 deficiency affect microglial cell-dependent regulation of vascular density and vascular traffic direction remains to be elucidated. A study by Stefater et al. (61) has shown that retinal microglial cells modulates vessel network formation directly by producing VEGF inhibitory receptor Flt1 through non-canonical Wnt ligands. Interestingly, CCN1 regulates both the expression of and cell response to Wnt signaling components (33, 62). Conceptually, CCN1 might reduce the signaling sensitivity threshold for the Wnt-Flt1 pathway in microglial cells as the sprouting process occurs in the retina, although the possibility of more complex pathways involving other cell types as intermediates cannot be excluded. Future studies will use mice with compound gene deletion to determine whether and how paracrine stimulation of microglial cells by CCN1 might modulate autocrine stimulation of myeloid Wnt ligands.
The connection between miR-155 and CCN1 has relevant implications for not only physiological angiogenesis but also retinal vascular pathophysiology, such as retinopathy. OIR is a common non-diabetic model to study VEGF-dependent neovascularization resulting from ischemia (63). Our data showed a good correlation between the neovascular growth caused by OIR and miR-155 expression and down-regulation of CCN1 gene expression. Increased miR-155 levels were associated with the formation of neovascular tufts that grow abnormally into the avascular vitreous with a simultaneous increase of inflammatory factor expression and reactive microglial cells in the avascular and neovascular areas of the retina. The neovascular tufts formed consist of poorly perfused immature vessels lacking NG2-positive pericyte coverage (64). This vascular phenotype, at least in part, phenocopied retinal vasculature features upon CCN1 or CCN1 and miR-155 deletion. An extensive amount of reactive amoeboid microglial cells aggregate in and around neovascular tufts formed midway between the central avascular and peripheral vascularized areas of the retina. High VEGF levels around neovascular tufts can potentially support migration and growth of microglial cells that express VEGF receptors 1 and 2 (65). Although it is unlikely that microglia significantly contribute to the overall VEGF production, their tropism to neovessels could locally affect vessel sprouts and thereby substantially influence neovascular tuft formation (66). Of particular interest is the improved vascular recovery (i.e. reduced vaso-obliteration and neovascular tuft formation) in miR-155-deficient mice exhibiting a concomitant increase of CCN1 expression, whereas deletion of both CCN1 and miR-155 exacerbated neovascular alterations. Inhibition of CCN1 abrogated the retinal vascular phenotype of miR-155 mice subjected to OIR, further linking miR-155 to CCN1 during vascular repair. There are a number of possible explanations that may account for the ability of CCN1 to reduce the formation of neovessels. First, CCN1 dampens VEGF receptor 2 activity through up-regulation and association of Src homology 2 domain-containing protein tyrosine phosphatase-1 (SHP-1) with the VEGF receptor (35). By inducing the recruitment of SHP-1 and rapid dephosphorylation of VEGF receptor 2, CCN1 helps safeguard against an excessive and aberrant neoangiogenic response. SHP-1 has also been reported as a direct target of miR-155, which represses SHP-1 expression through direct 3′-UTR interactions (67), suggesting that the dual molecular action of miR-155 and CCN1 on SHP-1 expression and activity may harmoniously fine tune physiological angiogenesis. Second, the expression of CCN1 reduced the inflammatory load in hypoxic retina, providing a physiological angiogenesis-friendly environment. During OIR, CCN1 expression and miR-155 suppression define the “on” and “off” status in microglial activation, which correlate to “amoeboid” and “ramified” morphologies, respectively. In support of this idea is the observation that expression of IL-1β, which was reported to sustain microglial activation and promote microvascular injury via the release of proapoptotic repulsive semaphoring 3A from neurons (68), was significantly reduced in the miR-155−/− mouse retinas following OIR. Likewise, the levels of IL-10, which reduces endothelial cell proliferation in response to hypoxia (69), were diminished in the mutant mice. IL-10 deficiency in mice has been shown to significantly reduce pathological retinal angiogenesis (69). Not only did the combined deletion of CCN1 and miR-155 reverse these effects on IL-1 and IL-10, but it also strikingly increased the expression of IL-6, which further exacerbates neovascular growth (70).
Moreover, the effects of the miR-155/CCN1 regulatory axis on neovascular growth do not preclude a direct physical and/or functional interaction between CCN1 and other inflammatory cells, such as macrophages. Macrophages can exhibit both pro- and antiangiogenic functions based largely on their polarization by cytokines in the resident tissue microenvironment (71). Of these cytokines, IL-10 may possess the most significant influence on the polarization of macrophages and their ability to regulate angiogenesis in the eye (72). Low levels of IL-10, which is a characteristic feature of the retinal environment of miR-155−/− mice after OIR, are associated with “classically activated” macrophage, or M1 macrophage, that displays an antiangiogenic phenotype. High levels of IL-10 and reduced levels of TNF-α and IL-6, a characteristic feature of the retinal environment of CCN1−/− mice, are associated with “alternatively activated” macrophages or M2 macrophages that are proangiogenic. In contrast to a previous in vitro study that demonstrated that CCN1 regulates a proinflammatory genetic program in murine macrophage in culture through binding to integrin αMβ2 and the heparin sulfate proteoglycan syndecan-4 (73), the presence of CCN1 in the retinal environment rather polarizes inflammatory cells toward an antiangiogenic phenotype. These in vivo biological functions of CCN1 should be dissociated from its effects in in vitro cultured macrophages.
Because both miR-155 deletion and CCN1 expression exhibit overlapping functions and regulate critical aspects of pathological angiogenesis, therapeutic manipulation of their interaction represents a potential strategy in treating retinal neovascularization in patients with retinopathy of prematurity, diabetic retinopathy, and other vascular diseases. The identification of miR-155 and CCN1 as important regulators of microglia activation and deactivation also suggests roles for these molecular effectors in other inflammatory diseases.
Author Contributions
B. C. conceived and oversaw the study, gathered data, and wrote the paper. L. Y. performed miRNA arrays, characterized the vascular phenotype of miR-155-deficient mice, and analyzed miR-155-CCN1 promoter interactions. S. L. generated and characterized the vascular phenotype of CCN1−/− and miR-155−/−/CCN1−/−-deficient mice. M. B. G. contributed to the vascular phenotype analyses and interpretation. J. A. and D. R. L. contributed to the conception, design, and implementation of the study. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We are thankful to Lester Lau for the generous gift of CCN1flox/flox mice. Viral vectors were produced under the auspices of the Gene Therapy Resource Program of NHLBI, National Institutes of Health. We thank the personnel and staff who prepared and amplified the viral vectors in the Preclinical Vector Core Laboratory at the University of Pennsylvania. We also thank Heather Gallagher, Trevor Cerbini, and all of our laboratory members for assistance with the experiments, data analyses, and/or proofreading the manuscript.
This work was supported, in whole or in part, by National Institutes of Health, NEI, Grants EY022091 and EY019387 (to B. C.) and EY007739 (to M. B. G.). This work was also supported by a grant from Research to Prevent Blindness (to D. R. L.). The authors declare that they have no conflicts of interest with the contents of this article.
- miRNA
- microRNA
- CCN1
- cysteine-rich protein 61-connective tissue growth factor-nephroblastoma overexpressed 1
- OIR
- oxygen-induced retinopathy
- IB4
- isolectin B4
- TSPO
- mitochondrial translocator protein
- SHP-1
- Src homology 2 domain-containing protein tyrosine phosphatase-1
- DAP12
- DNAX activation protein of 12 kDa
- Iba-1
- ionized calcium-binding adapter molecule 1
- 4HT
- 4-hydroxytamoxifen
- GFAP
- glial fibrillary acidic protein
- qPCR
- quantitative PCR
- Pn
- postnatal day n.
References
- 1. Tolentino M. J. (2009) Current molecular understanding and future treatment strategies for pathologic ocular neovascularization. Curr. Mol. Med. 9, 973–981 [DOI] [PubMed] [Google Scholar]
- 2. Yan L., Chaqour B. (2013) Cysteine-rich protein 61 (CCN1) and connective tissue growth factor (CCN2) at the crosshairs of ocular neovascular and fibrovascular disease therapy. J. Cell Commun. Signal. 7, 253–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Agrawal S., Chaqour B. (2014) MicroRNA signature and function in retinal neovascularization. World J. Biol. Chem. 5, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roy S., Sen C. K. (2012) miRNA in wound inflammation and angiogenesis. Microcirculation 19, 224–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Karali M., Peluso I., Gennarino V. A., Bilio M., Verde R., Lago G., Dollé P., Banfi S. (2010) miRNeye: a microRNA expression atlas of the mouse eye. BMC Genomics 11, 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leung A. K., Vyas S., Rood J. E., Bhutkar A., Sharp P. A., Chang P. (2011) Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol. Cell 42, 489–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang W. J., Yang D. D., Na S., Sandusky G. E., Zhang Q., Zhao G. (2005) Dicer is required for embryonic angiogenesis during mouse development. J. Biol. Chem. 280, 9330–9335 [DOI] [PubMed] [Google Scholar]
- 8. Small E. M., Sutherland L. B., Rajagopalan K. N., Wang S., Olson E. N. (2010) MicroRNA-218 regulates vascular patterning by modulation of Slit-Robo signaling. Circ. Res. 107, 1336–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Su Z., Si W., Li L., Zhou B., Li X., Xu Y., Xu C., Jia H., Wang Q. K. (2014) MiR-144 regulates hematopoiesis and vascular development by targeting meis1 during zebrafish development. Int. J. Biochem. Cell Biol. 49, 53–63 [DOI] [PubMed] [Google Scholar]
- 10. Liebner S., Plate K. H. (2010) Differentiation of the brain vasculature: the answer came blowing by the Wnt. J. Angiogenes. Res. 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chaqour B. (2013) Molecular control of vascular development by the matricellular proteins (CCN1/Cyr61) and (CTGF/CCN2). Trends Dev. Biol. 7, 59–72 [PMC free article] [PubMed] [Google Scholar]
- 12. Elton T. S., Selemon H., Elton S. M., Parinandi N. L. (2013) Regulation of the MIR155 host gene in physiological and pathological processes. Gene 532, 1–12 [DOI] [PubMed] [Google Scholar]
- 13. Lagos-Quintana M., Rauhut R., Yalcin A., Meyer J., Lendeckel W., Tuschl T. (2002) Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12, 735–739 [DOI] [PubMed] [Google Scholar]
- 14. Vigorito E., Perks K. L., Abreu-Goodger C., Bunting S., Xiang Z., Kohlhaas S., Das P. P., Miska E. A., Rodriguez A., Bradley A., Smith K. G., Rada C., Enright A. J., Toellner K. M., Maclennan I. C., Turner M. (2007) microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity 27, 847–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang L., Zhang H., Rodriguez S., Cao L., Parish J., Mumaw C., Zollman A., Kamoka M. M., Mu J., Chen D. Z., Srour E. F., Chitteti B. R., HogenEsch H., Tu X., Bellido T. M., Boswell H. S., Manshouri T., Verstovsek S., Yoder M. C., Kapur R., Cardoso A. A., Carlesso N. (2014) Notch-dependent repression of miR-155 in the bone marrow niche regulates hematopoiesis in an NF-κB-dependent manner. Cell Stem Cell 15, 51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kumar S., Kim C. W., Simmons R. D., Jo H. (2014) Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitive athero-miRs. Arterioscler. Thromb. Vasc. Biol. 34, 2206–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stanczyk J., Pedrioli D. M., Brentano F., Sanchez-Pernaute O., Kolling C., Gay R. E., Detmar M., Gay S., Kyburz D. (2008) Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 58, 1001–1009 [DOI] [PubMed] [Google Scholar]
- 18. Urbich C., Kuehbacher A., Dimmeler S. (2008) Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 79, 581–588 [DOI] [PubMed] [Google Scholar]
- 19. Neilsen P. M., Noll J. E., Mattiske S., Bracken C. P., Gregory P. A., Schulz R. B., Lim S. P., Kumar R., Suetani R. J., Goodall G. J., Callen D. F. (2013) Mutant p53 drives invasion in breast tumors through up-regulation of miR-155. Oncogene 32, 2992–3000 [DOI] [PubMed] [Google Scholar]
- 20. Choi J., Lin A., Shrier E., Lau L. F., Grant M. B., Chaqour B. (2013) Degradome products of the matricellular protein CCN1 as modulators of pathological angiogenesis in the retina. J. Biol. Chem. 288, 23075–23089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jun J. I., Lau L. F. (2011) Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 10, 945–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ruzankina Y., Pinzon-Guzman C., Asare A., Ong T., Pontano L., Cotsarelis G., Zediak V. P., Velez M., Bhandoola A., Brown E. J. (2007) Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 1, 113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thai T. H., Calado D. P., Casola S., Ansel K. M., Xiao C., Xue Y., Murphy A., Frendewey D., Valenzuela D., Kutok J. L., Schmidt-Supprian M., Rajewsky N., Yancopoulos G., Rao A., Rajewsky K. (2007) Regulation of the germinal center response by microRNA-155. Science 316, 604–608 [DOI] [PubMed] [Google Scholar]
- 24. Kim K. H., Chen C. C., Monzon R. I., Lau L. F. (2013) Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol. Cell Biol. 33, 2078–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weber T., Böhm G., Hermann E., Schütz G., Schönig K., Bartsch D. (2009) Inducible gene manipulations in serotonergic neurons. Front. Mol. Neurosci. 2, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chintala H., Liu H., Parmar R., Kamalska M., Kim Y. J., Lovett D., Grant M. B., Chaqour B. (2012) Connective tissue growth factor regulates retinal neovascularization through p53 protein-dependent transactivation of the matrix metalloproteinase (MMP)-2 gene. J. Biol. Chem. 287, 40570–40585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chang K. H., Chan-Ling T., McFarland E. L., Afzal A., Pan H., Baxter L. C., Shaw L. C., Caballero S., Sengupta N., Li Calzi S., Sullivan S. M., Grant M. B. (2007) IGF binding protein-3 regulates hematopoietic stem cell and endothelial precursor cell function during vascular development. Proc. Natl. Acad. Sci. U.S.A. 104, 10595–10600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zudaire E., Gambardella L., Kurcz C., Vermeren S. (2011) A computational tool for quantitative analysis of vascular networks. PLoS One 6, e27385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith L. E., Wesolowski E., McLellan A., Kostyk S. K., D'Amato R., Sullivan R., D'Amore P. A. (1994) Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis. Sci. 35, 101–111 [PubMed] [Google Scholar]
- 30. Schober J. M., Chen N., Grzeszkiewicz T. M., Jovanovic I., Emeson E. E., Ugarova T. P., Ye R. D., Lau L. F., Lam S. C. (2002) Identification of integrin αMβ2 as an adhesion receptor on peripheral blood monocytes for Cyr61 (CCN1) and connective tissue growth factor (CCN2): immediate-early gene products expressed in atherosclerotic lesions. Blood 99, 4457–4465 [DOI] [PubMed] [Google Scholar]
- 31. van Wijngaarden P., Brereton H. M., Coster D. J., Williams K. A. (2007) Stability of housekeeping gene expression in the rat retina during exposure to cyclic hyperoxia. Mol. Vis. 13, 1508–1515 [PubMed] [Google Scholar]
- 32. Zhao R., Qian L., Jiang L. (2015) Identification of retinopathy of prematurity related miRNAs in hyperoxia-induced neonatal rats by deep sequencing. Int. J. Mol. Sci. 16, 840–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hasan A., Pokeza N., Shaw L., Lee H. S., Lazzaro D., Chintala H., Rosenbaum D., Grant M. B., Chaqour B. (2011) The matricellular protein cysteine-rich protein 61 (CCN1/Cyr61) enhances physiological adaptation of retinal vessels and reduces pathological neovascularization associated with ischemic retinopathy. J. Biol. Chem. 286, 9542–9554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mo F. E., Muntean A. G., Chen C. C., Stolz D. B., Watkins S. C., Lau L. F. (2002) CYR61 (CCN1) is essential for placental development and vascular integrity. Mol. Cell Biol. 22, 8709–8720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chintala H., Krupska I., Yan L., Lau L., Grant M., Chaqour B. (2015) The matricellular protein CCN1 controls retinal angiogenesis by targeting VEGF, Src homology 2 domain phosphatase-1 and Notch signaling. Development 142, 2364–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karlstetter M., Scholz R., Rutar M., Wong W. T., Provis J. M., Langmann T. (2015) Retinal microglia: just bystander or target for therapy? Prog. Retin. Eye Res. 45, 30–57 [DOI] [PubMed] [Google Scholar]
- 37. Rymo S. F., Gerhardt H., Wolfhagen Sand F., Lang R., Uv A., Betsholtz C. (2011) A two-way communication between microglial cells and angiogenic sprouts regulates angiogenesis in aortic ring cultures. PLoS One 6, e15846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perry V. H., Gordon S. (1988) Macrophages and microglia in the nervous system. Trends Neurosci. 11, 273–277 [DOI] [PubMed] [Google Scholar]
- 39. Mizushina Y., Shirasuna K., Usui F., Karasawa T., Kawashima A., Kimura H., Kobayashi M., Komada T., Inoue Y., Mato N., Yamasawa H., Latz E., Iwakura Y., Kasahara T., Bando M., Sugiyama Y., Takahashi M. (2015) NLRP3 protein deficiency exacerbates hyperoxia-induced lethality through Stat3 protein signaling independent of interleukin-1β. J. Biol. Chem. 290, 5065–5077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jin Y., Kim H. P., Ifedigbo E., Lau L. F., Choi A. M. (2005) Cyr61 protects against hyperoxia-induced cell death via Akt pathway in pulmonary epithelial cells. Am. J. Respir. Cell Mol. Biol. 33, 297–302 [DOI] [PubMed] [Google Scholar]
- 41. Marshall B., Mo J., Covar J., Atherton S. S., Zhang M. (2014) Decrease of murine cytomegalovirus-induced retinitis by intravenous delivery of immediate early protein-3-specific siRNA. Invest. Ophthalmol. Vis. Sci. 55, 4151–4157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tien T., Muto T., Barrette K., Challyandra L., Roy S. (2014) Downregulation of Connexin 43 promotes vascular cell loss and excess permeability associated with the development of vascular lesions in the diabetic retina. Mol. Vis. 20, 732–741 [PMC free article] [PubMed] [Google Scholar]
- 43. Liu G. J., Middleton R. J., Hatty C. R., Kam W. W., Chan R., Pham T., Harrison-Brown M., Dodson E., Veale K., Banati R. B. (2014) The 18 kDa translocator protein, microglia and neuroinflammation. Brain Pathol. 24, 631–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weigelt K., Ernst W., Walczak Y., Ebert S., Loenhardt T., Klug M., Rehli M., Weber B. H., Langmann T. (2007) Dap12 expression in activated microglia from retinoschisin-deficient retina and its PU.1-dependent promoter regulation. J. Leukoc. Biol. 82, 1564–1574 [DOI] [PubMed] [Google Scholar]
- 45. Lyons A., Downer E. J., Crotty S., Nolan Y. M., Mills K. H., Lynch M. A. (2007) CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J. Neurosci. 27, 8309–8313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Faraoni I., Antonetti F. R., Cardone J., Bonmassar E. (2009) miR-155 gene: a typical multifunctional microRNA. Biochim. Biophys. Acta 1792, 497–505 [DOI] [PubMed] [Google Scholar]
- 47. Rodríguez A. E., Hernández J. Á., Benito R., Gutiérrez N. C., García J. L., Hernández-Sánchez M., Risueño A., Sarasquete M. E., Fermiñán E., Fisac R., de Coca A. G., Martín-Núñez G., de Las Heras N., Recio I., Gutiérrez O., De Las Rivas J., González M., Hernández-Rivas J. M. (2012) Molecular characterization of chronic lymphocytic leukemia patients with a high number of losses in 13q14. PLoS One 7, e48485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Roitbak T., Bragina O., Padilla J. L., Pickett G. G. (2011) The role of microRNAs in neural stem cell-supported endothelial morphogenesis. Vasc. Cell 3, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Robertson E. D., Wasylyk C., Ye T., Jung A. C., Wasylyk B. (2014) The oncogenic MicroRNA Hsa-miR-155–5p targets the transcription factor ELK3 and links it to the hypoxia response. PloS One 9, e113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nakagawa Y., Minato M., Sumiyoshi K., Maeda A., Hara C., Murase Y., Nishida T., Kubota S., Takigawa M. (2013) Regulation of CCN1 via the 3′-untranslated region. J. Cell Commun. Signal. 7, 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kireeva M. L., Mo F. E., Yang G. P., Lau L. F. (1996) Cyr61, a product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, and adhesion. Mol. Cell Biol. 16, 1326–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lau L. F. (2012) CCN1 and CCN2: blood brothers in angiogenic action. J. Cell Commun. Signal. 6, 121–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen N., Leu S. J., Todorovic V., Lam S. C., Lau L. F. (2004) Identification of a novel integrin αvβ3 binding site in CCN1 (CYR61) critical for pro-angiogenic activities in vascular endothelial cells. J. Biol. Chem. 279, 44166–44176 [DOI] [PubMed] [Google Scholar]
- 54. Jun J. I., Lau L. F. (2010) The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 12, 676–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu H., Yang R., Tinner B., Choudhry A., Schutze N., Chaqour B. (2008) Cysteine-rich protein 61 and connective tissue growth factor induce deadhesion and anoikis of retinal pericytes. Endocrinology 149, 1666–1677 [DOI] [PubMed] [Google Scholar]
- 56. Bessis A., Béchade C., Bernard D., Roumier A. (2007) Microglial control of neuronal death and synaptic properties. Glia 55, 233–238 [DOI] [PubMed] [Google Scholar]
- 57. Schafer D. P., Lehrman E. K., Kautzman A. G., Koyama R., Mardinly A. R., Yamasaki R., Ransohoff R. M., Greenberg M. E., Barres B. A., Stevens B. (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rojas B., Gallego B. I., Ramírez A. I., Salazar J. J., de Hoz R., Valiente-Soriano F. J., Avilés-Trigueros M., Villegas-Perez M. P., Vidal-Sanz M., Triviño A., Ramírez J. M. (2014) Microglia in mouse retina contralateral to experimental glaucoma exhibit multiple signs of activation in all retinal layers. J. Neuroinflammation 11, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bodeutsch N., Thanos S. (2000) Migration of phagocytotic cells and development of the murine intraretinal microglial network: an in vivo study using fluorescent dyes. Glia 32, 91–101 [DOI] [PubMed] [Google Scholar]
- 60. Freilich R. W., Woodbury M. E., Ikezu T. (2013) Integrated expression profiles of mRNA and miRNA in polarized primary murine microglia. PLoS One 8, e79416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stefater J. A. 3rd, Lewkowich I., Rao S., Mariggi G., Carpenter A. C., Burr A. R., Fan J., Ajima R., Molkentin J. D., Williams B. O., Wills-Karp M., Pollard J. W., Yamaguchi T., Ferrara N., Gerhardt H., Lang R. A. (2011) Regulation of angiogenesis by a non-canonical Wnt-Flt1 pathway in myeloid cells. Nature 474, 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Latinkic B. V., Mercurio S., Bennett B., Hirst E. M., Xu Q., Lau L. F., Mohun T. J., Smith J. C. (2003) Xenopus Cyr61 regulates gastrulation movements and modulates Wnt signalling. Development 130, 2429–2441 [DOI] [PubMed] [Google Scholar]
- 63. Grossniklaus H. E., Kang S. J., Berglin L. (2010) Animal models of choroidal and retinal neovascularization. Prog. Retin. Eye Res. 29, 500–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Feng Y., vom Hagen F., Pfister F., Djokic S., Hoffmann S., Back W., Wagner P., Lin J., Deutsch U., Hammes H. P. (2007) Impaired pericyte recruitment and abnormal retinal angiogenesis as a result of angiopoietin-2 overexpression. Thromb. Haemost. 97, 99–108 [PubMed] [Google Scholar]
- 65. Couturier A., Bousquet E., Zhao M., Naud M. C., Klein C., Jonet L., Tadayoni R., de Kozak Y., Behar-Cohen F. (2014) Anti-vascular endothelial growth factor acts on retinal microglia/macrophage activation in a rat model of ocular inflammation. Mol. Vis. 20, 908–920 [PMC free article] [PubMed] [Google Scholar]
- 66. Ryu J. K., Cho T., Choi H. B., Wang Y. T., McLarnon J. G. (2009) Microglial VEGF receptor response is an integral chemotactic component in Alzheimer's disease pathology. J. Neurosci. 29, 3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. O'Connell R. M., Chaudhuri A. A., Rao D. S., Baltimore D. (2009) Inositol phosphatase SHIP1 is a primary target of miR-155. Proc. Natl. Acad. Sci. U.S.A. 106, 7113–7118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rivera J. C., Sitaras N., Noueihed B., Hamel D., Madaan A., Zhou T., Honoré J. C., Quiniou C., Joyal J. S., Hardy P., Sennlaub F., Lubell W., Chemtob S. (2013) Microglia and interleukin-1β in ischemic retinopathy elicit microvascular degeneration through neuronal semaphorin-3A. Arterioscler. Thromb. Vasc. Biol. 33, 1881–1891 [DOI] [PubMed] [Google Scholar]
- 69. Dace D. S., Khan A. A., Kelly J., Apte R. S. (2008) Interleukin-10 promotes pathological angiogenesis by regulating macrophage response to hypoxia during development. PLoS One 3, e3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cai M., Zhang X., Li Y., Xu H. (2015) Toll-like receptor 3 activation drives the inflammatory response in oxygen-induced retinopathy in rats. Br. J. Ophthalmol. 99, 125–132 [DOI] [PubMed] [Google Scholar]
- 71. Apte R. S. (2010) Regulation of angiogenesis by macrophages. Adv. Exp. Med. Biol. 664, 15–19 [DOI] [PubMed] [Google Scholar]
- 72. Kelly J., Ali Khan A., Yin J., Ferguson T. A., Apte R. S. (2007) Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J. Clin. Invest. 117, 3421–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bai T., Chen C. C., Lau L. F. (2010) Matricellular protein CCN1 activates a proinflammatory genetic program in murine macrophages. J. Immunol. 184, 3223–3232 [DOI] [PMC free article] [PubMed] [Google Scholar]