

Background: The recently discovered lytic polysaccharide monooxygenases (LPMOs) are important in enzymatic conversion of lignocellulosic biomass.
Results: We describe structural and functional studies of NcLPMO9C, which cleaves both cellulose and certain hemicelluloses.
Conclusion: NcLPMO9C has structural and functional features that correlate with the enzyme's catalytic capabilities.
Significance: This study shows how LPMO active sites are tailored to varying functionalities and adds to a growing LPMO knowledge base.
Keywords: biodegradation, bioenergy, copper monooxygenase, crystal structure, electron paramagnetic resonance (EPR), isothermal titration calorimetry (ITC)
Abstract
The recently discovered lytic polysaccharide monooxygenases (LPMOs) carry out oxidative cleavage of polysaccharides and are of major importance for efficient processing of biomass. NcLPMO9C from Neurospora crassa acts both on cellulose and on non-cellulose β-glucans, including cellodextrins and xyloglucan. The crystal structure of the catalytic domain of NcLPMO9C revealed an extended, highly polar substrate-binding surface well suited to interact with a variety of sugar substrates. The ability of NcLPMO9C to act on soluble substrates was exploited to study enzyme-substrate interactions. EPR studies demonstrated that the Cu2+ center environment is altered upon substrate binding, whereas isothermal titration calorimetry studies revealed binding affinities in the low micromolar range for polymeric substrates that are due in part to the presence of a carbohydrate-binding module (CBM1). Importantly, the novel structure of NcLPMO9C enabled a comparative study, revealing that the oxidative regioselectivity of LPMO9s (C1, C4, or both) correlates with distinct structural features of the copper coordination sphere. In strictly C1-oxidizing LPMO9s, access to the solvent-facing axial coordination position is restricted by a conserved tyrosine residue, whereas access to this same position seems unrestricted in C4-oxidizing LPMO9s. LPMO9s known to produce a mixture of C1- and C4-oxidized products show an intermediate situation.
Introduction
Naturally abundant polysaccharides such as cellulose and chitin are enticing supplements to fossil resources in the future production of fuels, chemicals, and other materials, but the economical integration of these alternative carbon sources into fuel production pathways is hampered by their recalcitrance to hydrolytic deconstruction. Until recently, the degradation of polysaccharides in plant biomass, such as cellulose and various hemicelluloses, was thought to be entirely driven by hydrolytic enzymes. However, it has been shown recently that oxidative degradation catalyzed by lytic polysaccharide monooxygenases (LPMOs)5 (1) plays a significant role (2–6). LPMOs are copper-dependent biocatalysts that use molecular oxygen and an electron donor to break glycosidic bonds (5–9). Electrons may be provided by a reducing agent such as ascorbic acid, gallate, or reduced glutathione (5, 6) or by a co-secreted enzyme such as cellobiose dehydrogenase (10). The catalytic copper-binding site of LPMOs has unique features that are shared with only one other type of enzyme, methane monooxygenase (5, 11, 12).
Because of their potential use in commercial biomass decomposing enzyme mixtures (3), LPMOs have attracted considerable attention since their discovery in 2010 (6). Initially, LPMO activity was demonstrated for cellulose and chitin. However, it was noted that the abundance, the large sequence variation, and the varying domain composition of LPMOs encoded in the genomes of biomass-degrading microorganisms suggest that some of these enzymes could have other substrates (1). Indeed, three studies published in 2014 have broadened the LPMO paradigm by demonstrating cleavage of soluble cellodextrins (13), hemicelluloses with β(1→4)-glucan backbones (14), and even starch (15). Based on sequence characteristics, LPMOs are currently categorized in auxiliary activity (AA) families 9–11 and 13 of the CAZy database (16). AA families 9, 11, and 13 almost exclusively contain fungal enzymes, whereas bacterial LPMOs occur in family 10. So far, chitin activity has been described for families 10 and 11, cellulose activity for families 9 and 10, hemicellulose activity for family 9, and starch activity for family 13. It has been shown that the action of family 9 and 10 LPMOs on β(1→4)-glycosidic linkages may lead to oxidation of either the C1 or the C4 carbon of the glycan (8, 17). Based on sequence analysis and the known variation in oxidative outcome of the reaction, it has been suggested that AA9 LPMOs may be divided into C1-oxidizing (LPMO1 group), C4-oxidizing (LPMO2 group), and both C1- and C4-oxidizing (LPMO3 group) enzymes (18).
We have recently discovered that NcLPMO9C, a C4-oxidizing AA9 LPMO (hereafter LPMO9) from Neurospora crassa also known as NCU02916 or NcGH61–3, shows activity on soluble cellodextrins and hemicelluloses with β(1→4)-glucan backbones, including xyloglucan, glucomannan, and β-glucan (13, 14). NcLPMO9C is a two-domain protein, containing an N-terminal catalytic AA9 domain and a family 1 carbohydrate-binding module (CBM1), that is connected through a serine- and threonine-rich linker comprising ∼50 amino acid residues. CBM1s are primarily found in multidomain proteins from fungi and are known to bind both crystalline and amorphous cellulose (19, 20). Approximately 20% of all AA9s contain at least one CBM1 domain. Until now, the effect of such binding domains on LPMO activity and substrate specificity has not been thoroughly investigated.
In this study, we set out to explore the structural basis of the unique functional properties of NcLPMO9C. We report the cloning, purification, and structural determination of NcLPMO9C-N, which is a C-terminally truncated variant of NcLPMO9C comprising only the AA9 domain. Further insights into NcLPMO9C properties were obtained from EPR studies in the presence or absence of soluble substrate, studies of metal and substrate binding affinities using ITC, determination of the redox potential, and activity assays using several substrates and NcLPMO9C variants with and without the CBM1. Notably, the unique functional properties of NcLPMO9C allowed for enzyme-substrate interaction studies that have not been possible to date for LPMOs, and the novel structure of the enzyme enabled a structural comparison revealing structural determinants of the oxidative regioselectivity of these enzymes.
Experimental Procedures
Cloning, Expression, and Purification
Full-length LPMO9C from N. crassa (NCU02916; NcLPMO9C) was produced in Pichia pastoris using a codon-optimized gene inserted into the pPICZα-A plasmid (21) and purified as described previously (22). A gene fragment encoding the AA9 domain of NcLPMO9C (nucleotides 1–729, encoding 243 residues) was PCR-amplified from the pPICZαA_NcLPMO9C vector. The amplification primers containing EcoRI and Acc65I restriction sites for cloning (underlined) were F1, TTTCGAAACGGAATTCGAAACGATGAAGACTGGTTCCATCTTGGC, and R2, ATGGCCGGCCGGTACCTCAACAAGTGAACAAAGCTGGACCT. The PCR product was ligated into the vector fragment emerging after cleaving the pPINK_GAP_AfCel12A vector (23) with restriction enzymes EcoRI and Acc65I, using the In-Fusion HD cloning kit (Clontech). This construct leads to constitutive expression of the protein driven by the GAP promoter, and secretion of the protein is driven by its native signal peptide. After the DNA sequence was confirmed by GATC Biotech AG (Germany), the resulting pGAP_NcLPMO9C-N plasmid was linearized with the restriction enzyme AflII and was used to transform competent PichiaPinkTM strain 4 (Invitrogen), according to the supplier's instructions. Positive transformants were selected on Pichia adenine dropout plates (Teknova, Hollister, CA) and were further cultivated in yeast extract/peptone/dextrose (YPD) media for detection of LPMO expression and activity.
For the production of NcLPMO9C-N, 50 ml of YPD medium, containing 2% (w/v) dextrose, was inoculated with a single colony of a positive P. pastoris transformant. After growth at 30 °C and 150 rpm for 16 h, these 50 ml were used as preculture to inoculate 1.5 liters of YPD medium to a starting optical density (OD) of 0.15. Cultivation was carried out for 5 days at 30 °C and 150 rpm in shake flasks with standard baffles until the final OD of 19. Starting YPD media contained 1% (w/v) yeast extract, 2% (w/v) peptone, and 1% (w/v) glucose in 0.1 m potassium phosphate buffer, pH 6.0. Every 24 h, 1% (w/v) of glucose was added to the culture medium, and the NcLPMO9C-N expression level was monitored by SDS-PAGE.
As the first purification step, culture supernatant was recovered by centrifugation at 18,000 rpm for 15 min followed by sequential filtering through 0.45- and 0.2-μm polyethersulfone (PES) filters (Millipore, Billerica, MA). A Vivaflow 200 tangential cross-flow concentrator (molecular mass cutoff of 10,000 Da, Sartorius Stedim Biotech GmbH, Germany) was then used to concentrate the supernatant 10-fold and to exchange buffer to 25 mm Tris-HCl, pH 8.0. The concentrated sample was loaded onto a 150-ml DEAE-Sepharose FF CL-6B column (GE Healthcare) equilibrated in 25 mm Tris, pH 8.0, and bound proteins were eluted by applying a linear gradient up to 0.25 m NaCl in the same buffer. Selected fractions were pooled, concentrated, and applied to a Superdex 75 gel filtration column (GE Healthcare) in 50 mm MES buffer, pH 6.5, supplemented with 150 mm NaCl. Protein purity was assessed by SDS-PAGE, and protein concentrations were determined by measuring absorbance at 280 nm, using theoretical extinction coefficients calculated using the ExPASy server (24) (NcLPMO9C, 46,910 m−1 cm−1; NcLPMO9C-N, 32,680 m−1 cm−1).
Preparation of Crystals and Structural Determination
The search for NcLPMO9C-N crystallization conditions was performed in 96-well sitting drop trays, using a Mosquito crystallization robot (TTP Labtech, UK) and commercially available screens. Diffracting crystals grew at room temperature in a variety of conditions containing PEG 3350 and at a protein concentration of 1.4 mg/ml (see “Results” for more details). Various efforts were made to obtain crystals of NcLPMO9C-N in complex with substrate analogs, such as thio-linked cello- and xylo-oligosaccharides. In attempts to obtain ligand complexes, NcLPMO9C-N was deactivated using EDTA or potassium cyanide prior to co-crystallization or soaking experiments with cellulosic or xyloglucan oligomers. NcLPMO9C-N crystals were soaked in mother liquor supplemented with 20% glycerol as cryoprotectant and then flash-frozen in liquid N2 prior to transportation and x-ray data collection on beamline I911-3 at MAX-lab (Lund, Sweden). The data were integrated with XDS (25) and scaled using the program Aimless in the CCP4 suite (26). The structure was solved by molecular replacement using PHASER (27) with the coordinates of another N. crassa LPMO, NcLPMO9D, also known as NcGH61–4 or NCU01050 (PDB code 4EIR (18)), as a search model. NcLPMO9D is a single domain C4-oxidizing LPMO sharing 47% sequence identity with NcLPMO9C-N.
REFMAC5 (28) was used for structure model refinements, and manual model rebuilding was performed with Coot (29, 30), with maximum likelihood σA-weighted 2Fobs − Fcalc electron density maps (29). For cross-validation and R and Rfree calculations, 5% of the data were excluded from the structure refinement (31). Solvent molecules were automatically added using the automatic water picking function in the ARP/wARP package (32). Picked water molecules were selected or discarded manually by visual inspection of the 2Fobs − Fcalc electron density maps. The copper or the zinc ions bound in the active site of the enzyme in the two different NcLPMO9C-N structures were introduced at a final stage of the structure refinement. The coordinates for the two final structure NcLPMO9C models, and the structure factors, have been deposited in the Protein Data Bank (PDB) with accession codes 4D7U and 4D7V. PyMOL (version 1.5) and UCSF Chimera (version 1.10.1) were used for analysis of the structures and figure preparations.
Substrate Docking Experiments
The docking study evaluated 10 different scenarios (Table 1) in an attempt to identify putative soluble substrate-binding sites for NcLPMO9C-N. Autodock version 4.2 was used to compute and cluster the grid-based free energies of the enzyme·ligand complexes (33). Four different ligands, corresponding to those evaluated in the biochemical assessment, were included in the docking studies as follows: xyloglucan (XXGX), cellotetraose (Glc4), cellopentaose (Glc5), and cellohexaose (Glc6). The XXGX xyloglucan pattern was selected to encompass the glycosidic linkage thought to be the cleavage site in tamarind xyloglucan. The ligand conformational state was either fixed with the pyranose ring “backbone” in a near-linear conformation or allowed to be flexible (Table 1). Oligomers allowed to adopt different conformations throughout the docking calculation are termed “flexible” in this study. This simply means the torsion angles of the molecules were free of constraints. A rigid version of cellulosic oligomers was considered because the backbone of cello-oligomers does not change significantly in solution. Here, “rigid” refers to a molecule in which the torsion angles associated with the glycosidic linkages were restrained.
TABLE 1.
NcLPMO9C-N ligand docking cases considered in this study
The “receptor” column describes the metal ions included in the active site. The “torsion angles” column describes whether torsion angles along the glycosidic linkages were held fixed or not. Search parameters for each case are given in the remaining columns. More details are provided under “Experimental Procedures.”
| Receptor | Ligand | Torsion angles | Grid box | Runs | Population | Minimum survivors |
|---|---|---|---|---|---|---|
| No metals | Glc4 | Rigid | 60*120*120 | 200 | 200 | 3 |
| 1 Cu+, 2 zinc | Glc4 | Rigid | 60*120*120 | 100 | 300 | 3 |
| 2 Cu+, 1 zinc | Glc4 | Rigid | 60*120*120 | 200 | 300 | 3 |
| 1 Cu+, 2 zinc | Glc4 | Flexible | 100*100*100 | 300 | 300 | 1 |
| 2 Cu+, 1 zinc | Glc4 | Flexible | 100*100*100 | 300 | 300 | 1 |
| 2 Cu+, 1 zinc | Glc5 | Rigid | 60*120*120 | 200 | 200 | 3 |
| 2 Cu+, 1 zinc | Glc6 | Rigid | 60*120*120 | 200 | 200 | 3 |
| No metals | XXGX | Flexible | 60*120*120 | 200 | 200 | 3 |
| 1 Cu+, 2 zinc | XXGX | Flexible | 60*120*120 | 200 | 200 | 3 |
| 2 Cu+, 1 zinc | XXGX | Flexible | 60*120*120 | 200 | 200 | 3 |
The participation of NcLPMO9C-N-bound metal ions in substrate binding was considered through variation of both type and number. Three different protein scenarios were considered as follows: 1) NcLPMO9C without any metal ions; 2) NcLPMO9C with one Cu+ in the histidine brace at the active site and two zinc ions in the structurally resolved metal-binding locations; and 3) NcLPMO9C with Cu+ in the histidine brace, one Cu+ in the metal-binding site closest to the active center, and a zinc ion in the distal loop region. The latter scenario was considered based on an early hypothesis that two copper ions were participating in catalysis. Ultimately, as described under “Results,” the EPR studies clearly indicate only a single copper atom is required for NcLPMO9C to catalyze cleavage of the β-1,4-linkage.
Autogrid4 was used to set up the grid maps centered on the NcLPMO9C-N active site metal atoms and the surrounding protein surface. Grid dimensions for each case are given in Table 1; a grid spacing of 0.375 Å was used for each case. Using Lamarckian Genetic Algorithm, the search for the most favorably docked ligand structures was carried out over 200 independent docking runs. The search parameters for the calculations were set as follows: mutation rate of 0.02, crossover rate of 0.80, and a maximum number of energy evaluations per run of 25 million. Initial population and maximum number of top individual survivors are given in Table 1.
Copper is a nonstandard atom type in Autodock 4.2. Thus, parameters for non-hydrogen-bonding copper were incorporated in the parameter file (34). The sum of the van der Waals radii of two copper atoms was set to 3.40 Å. The van der Waals well depth was 0.050 kcal/mol. The atomic solvation volume was set to 20.58 Å3, and the atomic solvation parameter was −0.00110.
Electron Spin Resonance Studies
To prepare apo NcLPMO9C for electron spin resonance (EPR) experiments, purified enzyme was treated with 100 mm EDTA, followed by desalting using a NAP-5 column (GE Healthcare) equilibrated with Chelex-treated 20 mm BisTris/HCl buffer, pH 6.0. To reconstitute the copper-active site, 10 molar eq of Cu2+ were added from an acidic CuCl2 solution. After 10 min of incubation at room temperature, excess Cu2+ was removed by repeating the desalting procedure described above. EPR samples containing 225 μm apo NcLPMO9C, 160 μm Cu2+-loaded NcLPMO9C, 160 μm Cu2+-loaded NcLPMO9C and 20 mg/ml cellohexaose (Megazyme International, Ireland), or 160 μm Cu2+-loaded NcLPMO9C and 15 mg/ml xyloglucan isolated from tamarind seeds (Megazyme International) were frozen and stored in liquid nitrogen.
EPR spectra were recorded using a BRUKER EleXsys 560 SuperX instrument equipped with an ER 4122 SHQE SuperX high sensitivity cavity and a cold finger. The spectra were recorded using 0.05–1 milliwatt microwave power and 2 or 10 G modulation amplitude at a temperature of 77 K. Spin quantification was carried out by comparing double integrated spectra of samples to a standard of 500 μm CuCl2 in 1 m HClO4. The EasySpin toolbox developed for MATLAB (MathWorks) was used to simulate and fit EPR spectra (35). Only the gz and |Az| values were determined accurately, whereas gx, gy, |Ax|, and |Ay| were estimated using the EasySpin automatic fitting procedure.
Isothermal Titration Calorimetry Studies
ITC experiments were performed with a VP-ITC system from Microcal, Inc. (Northampton, MA) (36). Solutions were thoroughly degassed prior to experiments to avoid air bubbles in the calorimeter. Chelex-treated 20 mm MES buffer, pH 5.5, was used for all binding studies. For binding of Cu2+ to apo NcLPMO9C-N (prepared as described above for NcLPMO9C, eluted with 20 mm Chelex-treated MES buffer, pH 5.5), typically 5 μm enzyme was placed in the reaction cell with a volume of 1.42 ml, and 150 μm CuSO4 was placed in the ITC syringe. Aliquots of 4 μl were injected into the reaction cell at 180-s intervals with a stirring speed of 260 rpm and a temperature of 10 °C. The titrations were complete after 40 injections. For binding of Glc6 to NcLPMO9C, 30 μm copper-saturated enzyme (prepared as described above) was placed in the reaction cell and 11 mm ligand in the syringe. Aliquots of 8 μl were injected at 180-s intervals with a stirring speed of 260 rpm and temperature of 25 °C. The titrations were complete after 40 injections. For binding of NcLPMO9C or NcLPMO9C-N to the xyloglucan, 0.9 μm xyloglucan (225 kDa, from tamarind seeds, Megazyme) was placed in the reaction cell, and 500 μm enzyme in the ITC syringe. Aliquots of 6 μl were injected into the reaction cell at 180-s intervals with a stirring speed of 260 rpm and temperature of 10 °C. The titrations were complete after 50 injections. For binding of NcLPMO9C or NcLPMO9C-N to phosphoric acid-swollen cellulose (PASC; prepared from Avicel as described by T. M. Wood (37)), 0.146 mg/ml substrate was placed in the reaction cell. The concentration of PASC was set to be 4.5 μm based on an estimation that the average chain length of Avicel-derived PASC is 200 glucose units (38). 500 μm enzyme was placed in the ITC syringe. Aliquots of 6 μl were injected into the reaction cell at 180-s intervals with a stirring speed of 260 rpm and temperature of 25 °C. The titrations were complete after 50 injections. ITC data were collected automatically using the Microcal Origin version 7.0 software accompanying the VP-ITC system (36). Prior to further analysis, all data were corrected for heat of dilution by subtracting the heat produced by continuing injections of ligand into the reaction cell after completion of the binding reaction. These heats had the same magnitudes as the heats of titrating ligand into buffer alone. The data were fitted using a nonlinear least squares algorithm using a single-site binding model employed by the Origin software that accompanies the VP-ITC system, yielding the stoichiometry (n), the equilibrium binding association constant (Ka), and the enthalpy change (ΔHr0) of the reaction. Errors in ΔHr0, Kd, and ΔGr0 were obtained as standard deviations of at least three experiments. Errors in ΔSr0 and −TΔSr0 were obtained through propagation of error.
Determination of Redox Potential
The cell potential (E0) for the NcLPMO9C-Cu2+/NcLPMO9C-Cu+ redox couple was determined as described previously (7, 39). In short, 50 μl of 70 μm Cu2+-saturated NcLPMO9C was incubated at room temperature with 50 μl of an oxygen-free solution of 200 μm reduced N,N,N′,N′-tetramethyl-1,4-phenylenediamine (TMPred) in Chelex-treated 20 mm MES, pH 5.5. Solutions were made oxygen-free by bubbling of N2 (g) through the buffer for 1 h prior to addition to TMPred and concentrated LPMO solutions. The reactions took place in UVettes (Eppendorf), and the absorbance at 610 nm was monitored using a Hitachi U-1900 spectrophotometer until the signal became stable. From this, the concentration of TMPox was calculated based on its extinction coefficient of 14 mm−1 cm−1 (39) followed by determination of the equilibrium constant for the electron transfer reaction. Finally, from the known cell potential of 273 mV for the TMPox/TMPred redox couple (40), the cell potential of NcLPMO9C-Cu2+/NcLPMO9C-Cu1+ was determined as outlined previously (7, 17).
Substrate Degradation Studies
Prior to activity measurements, pure NcLPMO9C and NcLPMO9C-N were copper-saturated as described above for NcLPMO9C. To analyze activity on polymeric substrates, we employed the fact that the C4-oxidizing NcLPMO9C generates novel reducing ends (14). 5 mg/ml xyloglucan from tamarind seed (Megazyme International) or PASC was incubated with 4 μm copper-saturated NcLPMO9C or NcLPMO9C-N in 40 mm sodium phosphate, pH 6.5, and in the presence or absence of 2 mm ascorbic acid. Reactions were incubated at 50 °C and shaking at 1000 rpm. At appropriate time points, 40-μl samples were taken, and the enzyme reaction was quenched by addition of 60 μl of DNS solution (0.04 m 3,5-dinitrosalicylic acid in 1.0 m potassium-sodium tartrate, 0.4 m NaOH). After mixing, the samples were incubated for 10 min in boiling water. The samples were centrifuged, and 50 μl of the supernatant was transferred to a 384-well plate, and the absorbance at 540 nm was measured using a Multiscan FC microplate photometer (Thermo Scientific, Waltham, MA). Calibration was done with cellobiose (Glc2; Megazyme) for PASC samples and a xyloglucan heptasaccharide called XG7 (a glucose tetramer where all glucose units, except the one at the reducing end, are substituted with xylose, XXXG; Megazyme) for xyloglucan samples. All results shown have been corrected for background absorbance from reactions containing only the substrate or substrate and ascorbic acid.
Reactions for degradation of soluble cello-oligosaccharides contained 0.2 mm cellopentaose (Glc5) and 1.5 μm copper-saturated NcLPMO9C or NcLPMO9C-N in 20 mm MES, pH 6.0, with or without 1 mm ascorbic acid. Reactions were incubated at 50 °C and shaken at 1000 rpm. At appropriate time points, 50-μl samples were taken and mixed with 50 μl of 0.2 m NaOH to quench the enzyme reaction. Products were analyzed by high performance anion exchange chromatography using an ICS3000 system (Dionex, Sunnyvale, CA) as described previously (41). A 2.5-μl sample was injected onto a CarboPac PA1 2×250-mm analytical column (Dionex) coupled to a CarboPac PA1 2×50-mm guard column kept at 30 °C. Cello-oligosaccharides were eluted in 0.1 m NaOH at 0.25 ml/min by increasing the concentration of sodium acetate linearly from 0 to 0.1 m in 10 min and then from 0.1 to 0.18 m in 10 min, followed by a 2-min exponential gradient to 1 m sodium acetate. Between each run, the column was reconditioned by running initial conditions for 14 min. Cello-oligosaccharides with a degree of polymerization of 2–5 were used as standards.
Results
Overall Structural Analysis and Metal Binding
NcLPMO9C-N was crystallized in space group P21 with approximate unit cell dimensions of a = 44 Å, b = 67 Å, c = 66 Å, β = 101° and with two molecules in the asymmetric unit. We present two structures of NcLPMO9C-N. The first one (PDB code 4D7U), obtained at 1.56 Å resolution from a crystal grown in the presence 0.2 m ammonium citrate and 20% PEG3350, has one copper ion bound in the catalytic center, similar to other known AA9 structures. The second structure (PDB code 4D7V), determined at 1.9 Å resolution from a crystal grown in the presence of 0.2 m zinc acetate and 20% PEG3350, contains three zinc ions per protein molecule. Data collection and refinement statistics for both structures are summarized in Table 2; Fig. 1 shows structural features that are discussed further below. The two structures are highly similar, with a root-mean-square deviation of 0.55 Å for all Cα atoms. The biggest variations occur in the loop that includes residues 64–78, which binds one of the zinc ions (Fig. 1, c and d). Similar to other LPMO9s of known structure, NcLPMO9C-N folds into a β-sandwich of two β-sheets consisting of eight β-strands in total. The first β-sheet is formed of β-strands β1 (residues 3–10), β3 (residues 56–63), and β6 (residues 130–136), and the second one is composed of strands β5 (residues 106–114), β4 (residues 86–94), β7 (residues 143–153), and β8 (residues 164–175). In both structures, residues 180–185 display increased temperature factors, and the corresponding electron density was weak or even absent, especially for Ser-183 and Asn-184 in some monomers. Weak electron density is also observed in the region spanning residues 68–74 of monomer B of the three-metal structure. Neither of the two structures showed signs of glycosylation.
TABLE 2.
Diffraction data and refinement statistics for the two NcLPMO9C-N structures
| 1-metal structure | 3-metal structure | |
|---|---|---|
| Space group details | ||
| Cell dimensions (a, b, c) (Å) | a = 44.3, b = 66.8, c = 66.08 | a = 44.3, b = 67.5, c = 65.6 |
| α, β, γ (ο) | 90, 101.3, 90 | 90, 100.9, 90 |
| Space group | P21 | P21 |
| No. of molecules per asymmetric unit | 2 | 2 |
| Data collection and processing statistics | ||
| Station (synchrotron) | I911-3 Lund max lab | I911-3, Lund max lab |
| Wavelength (Å) | 1.000 | 1.000 |
| No. of observations | 166,092 | 198,749 |
| No. of unique reflections | 49,150 | 29,831 |
| Maximum resolution (A) | 1.56 | 1.9 |
| Completeness (outermost shell) (%) | 91.3 (29.2) | 99.3 (98.4) |
| Rmerge (%)a | 4.0 | 31.1 |
| Mean((I)/sd(I)) (outermost shell) | 15.5 (3.1) | 6.8 (2) |
| CC1/2 | 99.9 (94.8) | 98.9 (87.2) |
| Multiplicity | 3.3 (1.8) | 6.7 (6.6) |
| Outermost shell (Å) | 1.59–1.56 | 1.94–1.9 |
| Refinement statistics | ||
| Resolution range (Å) | 64.81–1.56 | 64.41–1.9 |
| Rwork/Rfree (%) | 15.7/18.6 (25/28) | 21.3/26.8 (31/31) |
| R.m.s.d., bond lengths (Å) | 0.005 | 0.01 |
| R.m.s.d., bond angles (°) | 1.1 | 1.44 |
| No. of reflections | 46,509 | 28,300 |
| No. of protein atoms | 3376 | 3362 |
| No. of solvent molecules | 561 | 427 |
| No. of glycerol molecules | 5 (2 in chain A, 3 in chain B) | 2 (chain A) |
| No. of acetate ions | 8 (3 in chain A and 5 in chain B) | |
| No. of metal atoms | 2 (copper) | 6 (zinc) |
| Average B factor (Å2) for protein residues | ||
| Overall | 12.6 | 9.7 |
| Main chain atoms | 12.6 | 9.8 |
| Side chain atoms | 12.6 | 9.7 |
| Average B factor (Å2) for heteroatoms | ||
| Water molecules | 24.14 | 18.6 |
| Metal atoms | 8.04 | 16.4 |
| Glycerol molecules | 16.67 | 23.2 |
| Acetate ions | 16.3 | |
| Ramachandran plot (%) | ||
| Favored region | 97.3 | 97.5 |
| Allowed region | 2.7 | 2.5 |
| PDB entry | 4D7U | 4D7V |
FIGURE 1.
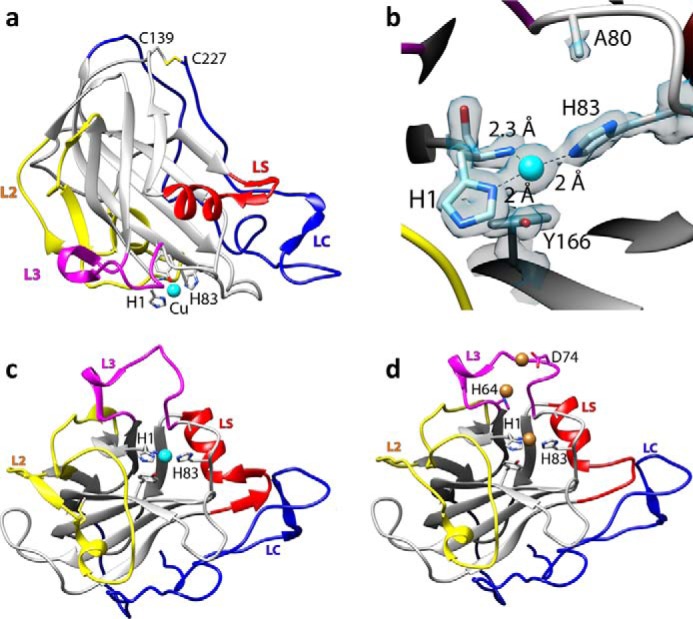
Structural representations of NcLPMO9C-N. a, cartoon representation of the copper-bound structure; copper is depicted as a cyan sphere; b, close up of the copper-binding site with the electron density map around the active site in gray mesh (contoured at 1σ); c, overall structure of the copper-loaded protein rotated by 90° along the horizontal axis compared with the view in a; d, structure of the zinc-loaded protein with the three bound zinc ions depicted as brown spheres; the orientation is similar to that in c. Note the structural variation in the loop coordinating the third zinc ion (residues 70–76 in the L3 loop, colored in pink).
There are two disulfide bridges in the NcLPMO9C-N structure. The first one, formed between Cys-39 and Cys-169 (partially cleaved due to x-ray radiation damage), is common among all known LPMO9 structures and connects loop L2 to strand β8 (Fig. 2). The second disulfide bond, formed between Cys-139 and the C-terminal amino acid, Cys-227, is observed in about half of LPMO9s with known structures (Fig. 2).
FIGURE 2.

Structure-based sequence alignment of LPMO9s with known structures. The proteins included are as follows: NcLPMO9C-N (PDB code 4D7U), NcLPMO9D (PDB code 4EIR), TtLPMO9E (PDB code 3EII), PcLPMO9D (PDB code 4B5Q), TaLPMO9A (PDB code 3ZUD), NcLPMO9M (PDB code 4EIS), and HjLPMO9B (PDB code 2VTC). Fully conserved residues are shown in white on a red background. Blue frames indicate that more than 70% of the residues in the corresponding columns exhibit similar physico-chemical properties (indicated as red residues on a white background). Blue triangles indicate residues coordinating the active site metal, and yellow triangles indicate residues involved in binding of two additional zinc ions. The secondary structure assignment (β-strands indicated as blue arrows and α-helices as red cylinders) refers to NcLPMO9C-N and was determined with the program DSSP (56). The oxidative regio-specificity of the LPMOs, indicated on the left, was assigned based on experimental evidence (4, 5, 13, 57) or, for HjLPMO9B and TtLPMO9E, by inference from the sequence-based categorization (43, 45). The residue numbered 80, which affects the accessibility of the solvent-facing axial copper coordination site, as shown in Fig. 4, c and d, and discussed in detail in the text, is indicated by a black asterisk. The loop regions that contribute to shaping the substrate-binding surface, named L2, LC, L3, and LS (see text), are marked by horizontal bars below the sequence, with color coding as in Fig. 2. The figure was prepared with ESPript.
The copper ion lies in the center of a flat surface that interacts with the substrate (Fig. 1, a and b) (7). The copper site is very similar to that in other LPMOs, resembling an octahedral coordination environment with tetragonal distortion due to the Jahn-Teller effect and with four of six coordination positions occupied by protein ligands (5, 42). The equatorial plane includes the protein's N-terminal amino group (at 2.3 Å), the Nδ of His-1 (2.0 Å), and the Nϵ of His-83 (2.0 Å), forming what has been termed a “histidine-brace” (5). Although His-1 tends to be methylated in LPMO9s produced in fungi (5), His-1 in Pichia-produced NcLPMO9C was not methylated, as observed previously for C1-oxidizing PcLPMO9D (or PcGH61D), which was also expressed in P. pastoris (42). There is no density in the fourth equatorial coordination position that is facing the solvent. Access to the protein-facing axial position is limited by the phenolic oxygen of a buried tyrosine, Tyr-166 (3.0 Å). In the solvent-facing axial position, there is extra density both in the σA-weighted Fo − Fc map contoured at 3σ and the σA-weighted 2Fo − Fc map contoured at 1σ. This density could perhaps reflect a citrate molecule with partial occupancy, because there was 0.2 m citrate in the crystallization solution, but the data did not allow reliable modeling of a citrate molecule or any other compound in the electron density map.
In the structure of the zinc-soaked protein, the histidine brace is occupied by zinc instead of copper. Like the copper ion, the zinc ion is coordinated by the N-terminal amino group and the Nδ of His-1 (with distances of 2.2 and 2.1 Å, respectively) and the Nϵ of His-83 (2.1 Å). An overview over all distances related to metal binding is provided in Table 3. The fourth equatorial position and the solvent-facing axial position are both occupied by acetate ions with distances of 2.3 and 3.3 Å from the copper to the oxygens of the carboxyl group, respectively. The axially interacting acetate is replaced by a water molecule in some of the NcLPMO9C-N molecules and was thus modeled with partial occupancy. The oxygen atom of the Tyr-166 side chain is located at a distance of 3.8 Å from the bound zinc atom. Superposition of the copper and zinc structures indicated that zinc is displaced by ∼0.8 Å relative to the copper, explaining the longer distance to Tyr-166 (3.8 and 3.0 Å, respectively). Relative to the copper structure, the zinc-coordinating histidines show an 8° shift in their ψ dihedral angle (Δψ = −8°).
TABLE 3.
Distances (in Å) between the metal ion bound in the primary metal-binding site (i.e. the copper site) and the closest residues in the active site of NcLPMO9C-N in the copper and the zinc structure
| NcLPMO9C-N structure | PDB 4D7U (copper) |
PDB 4D7V (zinc) |
||
|---|---|---|---|---|
| Chain A | Chain B | Chain A | Chain B | |
| Me-N(His-1) | 2.3 | 2.3 | 2.2 | 2.3 |
| Me-Nδ(His-1) | 2.0 | 2.0 | 2.1 | 2.2 |
| Me-Nϵ(His-83) | 2.0 | 2.0 | 2.1 | 2.1 |
| Me-O (Tyr-166) | 3.0 | 3.0 | 3.8 | 3.5 |
The zinc structure reveals two possible additional metal-binding sites that are also located in the flat planar substrate-binding surface. The second zinc ion is coordinated by His-64 and is located at a distance of ∼7.7 Å from the active site ion, whereas the third zinc ion is coordinated by Asp-74 and is 7.0 Å away from the second zinc atom (Fig. 1d). Both metals are coordinated by amino acids belonging to a sequence insertion that seems unique for strictly C4-oxidizing LPMO9s, also referred to as the LPMO2 subfamily (Figs. 1d and 2) (43). Notably, the EPR and ITC experiments described below strongly indicate that NcLPMO9C binds only one copper ion with high affinity and that this is sufficient for activity.
Because NcLPMO9C has been found to act on soluble substrates, including cellodextrins and xyloglucan fragments (13, 14), attempts were made to obtain crystallographic data for an enzyme·substrate complex. Despite the use of a variety ofsubstrates (thio-linked cello-oligosaccharides with a degree of polymerization of 2–5, thio-linked xylopentaose, cello-oligosaccharides with a degree of polymerization of 5 and 7, xylogluco-oligosaccharides, low molecular lignin, and β-glucan) and a massive number of crystallization trials using both soaking and co-crystallization, such data were not obtained. The protein molecules in the NcLPMO9C-N crystals are very tightly packed, especially close to the catalytic center of the enzyme, with a distance of ∼5 Å between two protein molecules. This distance is relatively small, and there is thus very little room left for a ligand molecule to diffuse into the crystal and bind in the active center of the enzyme.
Comparison with Other LPMO Structures
Using the Dali server (44), the closest structural homolog of NcLPMO9C was found to be another C4-oxidizing LPMO, NcLPMO9D (PDB code 4EIR) (4). The second closest structural homolog is NcLPMO9M (NCU07898, PDB code 4EIS, 39% sequence identity; C1/C4-oxidizing (43)), followed by a Thermoascus aurantiacus LPMO (TaGH61A, PDB code 3ZUD, 39% sequence identity; C1/C4-oxidizing (5)), GH61E from Thielavia terrestris (TtGH61E, PDB code 3EJA, 44% sequence identity; predicted to be C1-oxidizing (18)), and Cel61B from Hypocrea jecorina (HjLPMO9B, PDB code 2VTC, 31% sequence identity; predicted to be C1/C4-oxidizing (18)).
Importantly, although the histidine brace and the tyrosine in the protein-facing axial position are structurally highly conserved, comparison of the seven available LPMO9 structures reveals differences in the accessibility of the solvent-facing axial position that correlate with the regioselectivity of the oxidation reaction. The two C1 oxidizers have a tyrosine in this position, whose hydroxyl group blocks access to the axial position to essentially the same extent as what is achieved by a conserved alanine in C1-oxidizing LPMO10s (11, 17). The mixed C1 and C4 oxidizers have a proline instead of a tyrosine providing better access to the axial position (Fig. 3, c and d). In strict C4 oxidizers, the axial position appears fully accessible due to the tyrosine being replaced by an alanine in NcLPMO9C and an aspartate pointing away from the copper in NcLPMO9D. Another strictly C4-oxidizing N. crassa LPMO9, named NCU02240 (43), whose structure has not yet been determined, also has an aspartate in this position.
FIGURE 3.

Structural comparison of LPMO9s. a, superposition of PcGH61D (green; PDB code 4B5Q) with NcLPMO9C-N (colored as in Fig. 2). b, comparison of protruding surface residues in NcLPMO9C (colored as in a) with residues at equivalent positions in C1-oxidizing PcLPMO9D (green). b is rotated 90° along the horizontal axis compared with a. c and d, superposition of the copper sites of seven LPMO9s with known structure; C1, C1/C4, and C4 oxidizers are colored green, yellow, and magenta, respectively. The orientation shown in c is similar to that in a, whereas the orientation in d resembles that of b. Residue numbers refer to NcLPMO9C, with the exception of green labels in b, which refer to PcLPMO9D. The copper in NcLPMO9C is shown as a cyan sphere in all panels.
As observed in earlier studies (18, 42, 43) and as clearly visible in Figs. 1 and 3, LPMO9s show large variation in their putative substrate-binding surfaces. Three of these regions (Fig. 1) are known as the L2 (residues 10–49), LS (residues 114–128), and LC (residues 176–226) loops (42, 45). In addition, strictly C4-oxidizing AA9 LPMOs contain an insertion, referred to here as the L3 loop (resides 64–78), that potentially also affects the substrate-binding surface (Fig. 2). The variability in these loops is likely to provide a tunable scaffold for substrate specificity. Fig. 3b shows that the variation in these loops indeed results in quite different binding surfaces, when comparing PcLPMO9D (active on cellulose but not on xyloglucan (14)) with NcLPMO9C. Compared with PcLPMO9D, the substrate-binding surface of NcLPMO9C is more extended and crowded with polar residues. It should be noted that nothing is known about possible activities on xyloglucan for the other five LPMOs with known structures.
Further Insight into the Catalytic Center from EPR Spectroscopy, ITC Measurements, and Determination of Redox Potentials
EPR spectroscopy was used to assess whether the unique properties of NcLPMO9C are reflected in the electronic structure of the bound copper ion. The spin Hamiltonian values gz = 2.267 and |Az| = 152 × 10−4 cm−1 obtained for the Cu2+-loaded full-length enzyme are very similar to those obtained previously for cellulose-oxidizing LPMOs in the AA9 and AA10 families analyzed at pH values varying from 5.0 to 6.5 (5, 9, 17, 46) and indicate a type 2 copper active site. Spin quantification of Cu2+ in Cu2+-loaded NcLPMO9C was consistent with one copper-binding site/monomer. Interestingly, the addition of the soluble substrates cellohexaose or tamarind seed xyloglucan to Cu2+-loaded NcLPMO9C in the absence of reductant led to clear and similar changes in the EPR spectra (Fig. 4), indicative of structural changes in the Cu2+ active site and providing the first ever experimental evidence for substrate-binding effects on the copper site. The simulated spin Hamiltonian values changed to gz = 2.226 and |Az| = 175 × 10−4 cm−1 for both substrates upon binding (Fig. 4a) and superhyperfine splittings, reflecting the interaction between the unpaired electron and nitrogen nuclei adjacent to the Cu2+, were greatly enhanced (Fig. 4b). The spectral envelopes for NcLPMO9C interacting with d-cellohexaose or xyloglucan were nearly identical, showing that the two substrates affect the catalytic center of NcLPMO9C in a similar manner.
FIGURE 4.

NcLPMO9C EPR spectra. a, EPR spectra of 160 μm Cu2+-loaded NcLPMO9C in the absence (upper panel) or presence (lower panel) of the soluble substrates cellohexaose (20 mg/ml) or xyloglucan (15 mg/ml). Spectra were recorded at 77 K, 1 milliwatt of microwave power, and 10 gauss modulation amplitude. The simulated spectra (SIM) for each species are shown below the corresponding experimental spectra (simulation parameters for the cellohexaose and xyloglucan spectra were identical). b, effect of substrate on super hyperfine splitting in the high field region. The splitting constants (|A| ∼13 × 10−4 cm) are consistent with coupling to nearby nitrogen nuclei. Spectra were recorded at 77 K, 1 milliwatt of microwave power, and 2 gauss modulation amplitude.
ITC measurements of the binding of Cu2+ to NcLPMO9C at pH 5.5 yielded data that fitted well to a single-site binding model, yielding the stoichiometry (n), the equilibrium binding association constant (Ka), and the enthalpy change (ΔHr0) of the reaction (data not shown). The value of n was found to be between 0.9 and 1.1 per enzyme molecule. The dissociation constant was determined to be 33 ± 10 nm with ΔGr0 = −9.7 ± 0.2 kcal/mol, ΔHr0 =−5.4 ± 0.2 kcal/mol, and −TΔSr0 = −4.3 ± 0.3 kcal/mol.
The redox potential for the NcLPMO9C-Cu2+/NcLPMO9C-Cu+ redox couple was determined as described previously (7, 39), yielding a value of 224 ± 3 mV. Combining the redox potential and dissociation constants for Cu2+ in three thermodynamic relationships (7) allowed estimation of the dissociation constant for reduced copper (Cu+), resulting in a value of 6.1 nm. These values for copper binding and the redox potential are similar to those previously found for LPMO10s at pH 5.5–6.0 (7, 9, 17).
Enzyme Activity and Substrate Specificity
Previous studies have shown that NcLPMO9C is active on both xyloglucan and cellulosic substrates, including soluble β-glucans and cellodextrins (13, 14). To compare the activities on various substrates and to determine the influence of the CBM1 domain, reaction conditions were optimized so as to obtain reasonably linear initial progress curves allowing rate comparisons. This entailed incubating Cu2+-saturated enzyme, with or without the binding module, with tamarind xyloglucan, PASC, or cellopentaose in the presence of ascorbic acid as reducing agent. Samples taken at appropriate time points were analyzed using the DNS assay (for polymeric substrates) or HPLC (for cellopentaose). The results show no effect of the removal of the CBM1 domain on the degradation rate of PASC (Fig. 5a) or cellopentaose, but for xyloglucan, removal of the binding module had a negative effect on the degradation rate (Fig. 5b). The degradation rate for the full-length enzyme on polymeric xyloglucan was ∼0.1 s−1, whereas all other reactions gave rates of ∼0.04 s−1.
FIGURE 5.

Comparison of substrate degradation rates. Degradation of 5 mg/ml PASC (left) or tamarind xyloglucan (right) by 4 μm NcLPMO9C with or without (−N) the CBM domain at 50 °C was monitored by measuring the formation of reducing ends. In the absence of a reductant, the enzyme reactions did not yield reducing ends (not shown).
The binding reaction between Cu2+-saturated NcLPMO9C and different substrates in the absence of reductant was assessed using ITC. The first substrate investigated was polymeric xyloglucan from tamarind seeds with a molecular mass of 225 kDa and estimated degree of polymerization of the main chain of 594 (calculated from the sugar composition). The binding dissociation constant at pH 5.5 and 10 °C was determined to be 2.3 ± 0.5 μm with ΔGr0 = −7.3 ± 0.2 kcal/mol, ΔHr0 =−0.4 ± 0.1 kcal/mol, −TΔSr0 = −6.9 ± 0.2 kcal/mol, and a binding stoichiometry of 30 ± 8 (Fig. 6). The shape of the ITC binding curve is described by the so-called Wiseman c value (36), which can be expressed as follows: c = nKa[M]t (1), where n is the stoichiometry of the reaction; Ka is the equilibrium binding association constant; and [M]t is the protein concentration. It is well established that c values within the range of 10 < c < 1000 are a prerequisite for meaningful calculations of Ka (36). Under the given conditions, the Wiseman c value is 14. When the truncated version (NcLPMO9C-N) was titrated against the same substrate, the binding isotherm changed from being sigmoidal to hyperbolic, indicating weaker binding and a c value below 10 (Fig. 6) (36, 47). It has been shown that binding thermodynamics can be obtained even if c is in the range of 0.01 < c < 10 if a sufficient portion of the binding isotherm is used for analysis (47). This is achieved by ensuring a high molar ratio of ligand versus protein at the end of the titration, accurate knowledge of the concentrations of both ligand and receptor, an adequate level of signal-to-noise in the data, and known stoichiometry. Fitting of the theoretical data to the experimental data suggested a 10-fold lower binding affinity (∼24 μm). Because the stoichiometry of the reaction is not known, this value is uncertain. Still, it is clear that the truncated form binds weaker to the xyloglucan than the full-length form.
FIGURE 6.

Thermograms. a, upper panels, binding isotherms with theoretical fits (lower panels) obtained for the titration of NcLPMO9C (left) and NcLPMO9C-N (right) into 0.9 μm xyloglucan (top) and 0.146 mg/ml PASC (bottom). The concentration of PASC was set to be 4.5 μm based on an estimated degree of polymerization of 200 (38). All experiments were carried out at pH 5.5. The temperature was 25 °C for PASC and 10 °C for xyloglucan; b, upper panel, binding isotherms with theoretical fits (lower panel) obtained for the binding of 11 mm Glc6 to 30 μm NcLPMO9C-Cu2+ at t = 25 °C.
Next, both full-length and truncated forms were titrated with PASC with an estimated average degree of polymerization of 200, at pH 5.5 and t = 25 °C. Interestingly, the full-length form binds in two separate modes (Fig. 6) where one is clearly stronger (Kd = 0.013 ± 0.004 μm with ΔGr0 = −10.8 ± 0.2 kcal/mol, ΔHr0 = −1.1 ± 0.1 kcal/mol, −TΔSr0 = −9.7 ± 0.2 kcal/mol, and n = 2.6 ± 0.6) than the other (Kd = 0.64 ± 0.04 μm, ΔGr0 = −8.4 ± 0.1 kcal/mol, ΔHr0 = −1.1 ± 0.1 kcal/mol, and n = 11.4 ± 0.2). The truncated form only displayed a single binding mode (Kd = 0.54 ± 0.16 μm, ΔGr0 = −8.5 ± 0.2 kcal/mol, ΔHr0 =−1.1 ± 0.1 kcal/mol, −TΔSr0 = −7.4 ± 0.2 kcal/mol, and n = 5.2 ± 0.4). All three fits yielded acceptable c values ranging from 60 to 900. The data suggest that upon removal of the CBM1, the high affinity binding mode is lost.
Binding of the soluble substrate Glc6 to NcLPMO9C (pH 5.5, t = 25 °C) was found to be much weaker, having a Kd of 0.81 ± 0.08 mm, with ΔGr0 = −4.3 ± 0.2 kcal/mol, ΔHr0 = −2.5 ± 0.5 kcal/mol, −TΔSr0Δ = −1.8 ± 0.5 kcal/mol (Fig. 6b). These values were obtained assuming a 1:1 binding stoichiometry between NcLPMO9C and Glc6. Considering the binding surface of NcLPMO9C, it is conceivable that two Glc6 molecules can bind simultaneously. Assuming a 2:1 binding stoichiometry, the fit yielded a virtually identical Kd value within experimental errors (0.85 ± 0.09 mm), albeit with a lower enthalpy change (ΔHr0 = −1.0 ± 0.4 kcal/mol) and a higher entropy change (−TΔSr0 = −2.3 ± 0.5 kcal/mol).
Substrate Docking Experiments
An extensive computational docking study was carried out to identify potential substrate-binding sites on NcLPMO9C-N. Using Autodock version 4.2 to compute and cluster grid-based free energies of enzyme·ligand complexes (33), we evaluated 10 different scenarios (Table 1) involving four known substrates and three different bound metal ion configurations (Fig. 7). For the substrates, both flexible and rigid conformational states were considered to examine the limitations of fixed torsion angles. Clustering analysis of the docked ligand position conformations (with a 0.5 Å root-mean-square deviation) and calculated binding free energies revealed no clearly preferred conformation for any of the considered ligand binding conditions. As in the case of structural studies attempting to capture NcLPMO9C-N·substrate complexes, we were unable to conclusively identify a favorable binding orientation for any of the four ligands under any metal binding conditions. Increasing degrees of freedom through reduced dihedral restrictions were also unsuccessful in enabling the solution of a minimum energy-bound conformation.
FIGURE 7.
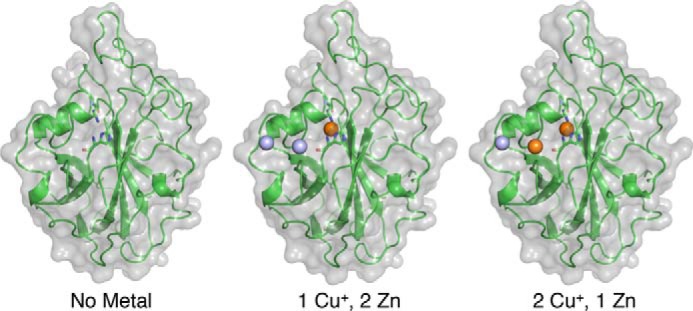
Illustration of the three different bound metal scenarios considered in the computational docking study of NcLPMO9C-N.
Discussion
The primary aim of this study was to analyze the structural and biochemical features of NcLPMO9C with its unprecedented substrate specificity toward soluble cello-oligosaccharides and hemicellulosic substrates. To this end, the crystal structure of the catalytic domain of the enzyme was determined, and several functional analyses were conducted. Considering the ability of NcLPMO9C to bind productively to soluble substrates, one major goal of our work was to produce the first ever crystallographic insight into LPMO-substrate interactions. However, despite multiple efforts using a wide variety of experimental conditions, such insight was not obtained; no enzyme·substrate complexes could be captured through the structural studies, and docking experiments were inconclusive. In contrast, our EPR data clearly reveal effects of substrate binding on the configuration of the copper site, and the ITC data provide the first glimpse of substrate binding affinities involved in LPMO action.
After some initial uncertainty regarding the nature of the active site metal (3, 6), it is now well established that LPMOs are copper-dependent monooxygenases (5, 7, 9). Although binding of multiple bivalent metal ions has been observed in the crystal structures for several LPMOs (5, 17), available experimental and theoretical data consistently indicate that the active site copper centers are mononuclear (11, 12, 17, 49). The additional metal ions observed in the zinc structure of NcLPMO9C are coordinated by amino acids belonging to a sequence insertion unique for strictly C4-oxidizing LPMO9s, but Fig. 2 shows that the residues interacting with zinc ions in NcLPMO9C are not conserved in C4-oxidizing NcLPMO9D. The metal sites do not look like high affinity sites (only one ligand) and are too far away from the copper site to affect the catalytic center. Still, because the additional sites are located in the flat planar substrate-binding surface, it cannot be excluded that they play a role in enzyme-substrate interactions, thus perhaps co-determining the unique specificity of NcLPMO9C.
Detailed functional characterization revealed that NcLPMO9C resembles other well studied LPMOs in many ways. Enzyme rates are in the order of 0.04–0.1/s, i.e. similar to rates observed for other LPMOs (6, 50). Likewise, both the copper affinity and the redox potential of NcLPMO9C resemble those of other LPMOs (7, 9, 17), although higher affinity for copper (<1 nm) has been reported for TaLPMO9A (5). The dissociation constant for Cu2+ determined here closely resembles dissociation constants that were recently determined for a C1-oxidizing (31 nm) and a C1/C4-oxidizing (12 nm) cellulose-active LPMO10 at the same pH (5.5) (17). Notably, these LPMO10 enzymes are also very similar to NcLPMO9C in terms of their spin Hamiltonian values.
The fact that NcLPMO9C is active on soluble substrates allowed us to obtain a first glimpse of substrate binding affinities in LPMOs. We obtained reliable data for binding of full-length NcLPMO9C to (soluble) polymeric xyloglucan, showing a dissociation constant of 2.3 ± 0.5 μm. Binding to cellohexaose was about 350 times weaker. Encouraged by these results, we also obtained ITC data for binding of (insoluble) PASC, revealing two binding modes, one of with a considerably lower Kd (0.013 ± 0.004 μm), relative to binding of xyloglucan. Interestingly, the data suggest that only the lower affinity binding mode for PASC is maintained in the truncated protein. The data for the truncated protein show that the catalytic domain of NcLPMO9C has higher affinity for cellulose (Kd = 0.54 ± 0.16 μm) than for xyloglucan (Kd roughly estimated at 24 μm; Fig. 6A). The CBM1 improves the affinity for both substrates, and the effect seems larger for cellulose, as one could expect considering the known substrate specificities of CBM1s. We note that the affinities for cellulose determined in this study are high compared with literature data (51, 52) and that further work on these affinities is of major interest. Future studies could address the cooperativity between binding of the CBM1 and the catalytic domain and the impact of having different binding modes in the catalytic domain (e.g., LPMOs versus processive glycoside hydrolases).
Low affinities for short soluble substrates offer a convenient explanation as to the failed attempts at obtaining crystals of enzyme·substrate complexes and may also explain why extensive computational docking studies did not reveal NcLPMO9C-specific binding of cellotetraose or xyloglucan fragments at any point along the protein surface. Furthermore, low affinities for soluble substrates may be taken to indicate that the natural substrate of the LPMO is polymeric. The latter intuitively makes sense, because cellulolytic microbes have many other enzymes capable of cleaving soluble dextrins, whereas cleavage of polymeric glucans and their co-polymeric complexes is more challenging.
Measurement of LPMO kinetics is notoriously difficult, and the literature is almost devoid of kinetic data. Under the conditions used here to obtain reasonably linear progress curves, the differences in substrate affinity were hardly reflected in the obtained rates. Apparently, under these substrate concentrations, substrate binding is not the main rate-limiting factor. More detailed kinetic analyses, as well as more extended binding studies, preferably under “natural conditions,” are needed to obtain further insight into the biological function of NcLPMO9C, and other LPMOs, in biomass conversion. The activity measurements depicted in Fig. 5 indicate that the CBM1 domain of NcLPMO9C is of importance for the activity on xyloglucan, possibly because the affinity of the truncated protein is so low that substrate binding becomes a rate-limiting factor.
In the quantum mechanical calculations described by Kjaergaard et al. (12), the closest modeled Cu2+ ligands in the equatorial plane include the three nitrogen atoms of the histidine brace and hydroxide. Kjaergaard et al. (12) also confirmed the presence of this hydroxide/water using extended x-ray absorption fine structure spectroscopy. Although density corresponding to a hydroxide/water molecule in the fourth equatorial position has been observed in some LPMO structures, such density was not observed in the structure of NcLPMO9C, possibly because of photo-reduction of the copper by the x-ray beam (53). The EPR envelope though confirms a d(x2 − y2) ground state (gz > gx, gy) typical for an elongated octahedron. Thus, the singly occupied molecular orbital is centered at the Cu2+ and encompasses the N/O ligands in the equatorial plane.
The weak axial interaction between the copper ion and the hydroxyl group of Tyr-166 observed in the NcLPMO9C crystal structure is in agreement with a tetragonal distortion of the Cu2+ site due to the Jahn-Teller effect. The spectral change observed when binding substrate to NcLPMO9C reveals substrate-induced conformational changes. Such conformational changes may include a general distortion of the amino acid Cu2+ ligands or displacement/reordering of coordinating water molecules. The increased intensity of the superhyperfine splittings indicates that the structural change upon substrate binding enhances the interaction between the coordinating 14N atoms of the histidine brace and the unpaired electron. Binding of cellohexaose and xyloglucan had similar effects on super hyperfine splitting in the gx-gy area (Fig. 4b). This suggests that the copper site only interacts with the (common) β-glucan part of the xyloglucan and that the chemical outcome of the reaction should be the same for both substrates, as is indeed observed (cleavage of a β(1→4) glucose-glucose bond with oxidation of C4 (14)).
All in all, the presented data indicate that the unique functionality of NcLPMO9C is not due to properties of the enzyme's catalytic site as such but rather to features of its substrate-binding surface and, at least to some extent, the presence of a CBM1. The most noticeable feature of the substrate-binding surface is that it is quite extended and highly polar (Fig. 3b), properties that align well with the ability to bind complex and branched polysaccharides such as xyloglucan. The structural determinants of the substrate specificity of LPMOs remain mysterious, and it should be noted that differences in these specificities might not only rely on binding as such but also on binding geometries being sufficiently optimized for binding to become productive.
Most interestingly, accumulating structural data for LPMO9s reveal structural differences in the copper coordination sphere that correlate with the oxidative outcome of the reaction (Fig. 3, c and d). In the strictly C1-oxidizing LPMO9s, access to the solvent-facing axial position is restricted by the hydroxyl group of a conserved tyrosine, whereas access to this same position seems unrestricted in strictly C4-oxidizing LPMO9s. In terms of access to the solvent-facing axial position, LPMO9s that oxidize C1 and C4 show an intermediate situation (Fig. 3). A similar difference has been observed when comparing a strictly C1-oxidizing LPMO10 with an LPMO10 that can oxidize both C1 and C4 (17). It has been shown that end-on binding of dioxygen is energetically favorable (12, 49); however, although Kim et al. (49) model O2 binding at the exposed axial position, Kjaergaard et al. (12) propose a model where the equatorial OH ligand is replaced by O2. This discrepancy is probably a result of different initial structural models and basis sets used in the density functional theory (DFT) calculations. The preferred binding mode (or modes) of O2 has not been determined experimentally. It is tempting to speculate that O2 binding in LPMOs with unrestricted access to the axial site leads to C4 oxidation and that C1 oxidation perhaps requires binding of dioxygen in another position. Alternatively, structural variation at the exposed axial site could affect the geometry of the enzyme·substrate complex, which could cause a different oxidative outcome, even if dioxygen binding to the copper does not vary. From an evolutionary point of view, development of both C1- and C4-oxidizing LPMOs seems logical, because different cellulases, such as cellobiohydrolases moving into opposite directions, will respond differently to the different oxidations (54, 55).
Author Contributions
A. S. B. and M. D. planned and performed all structural experiments, interpreted data, and helped write the manuscript. T. I. planned and performed the activity studies, performed the ITC experiments, interpreted data, and helped write the manuscript. Mo. S. designed, performed, and analyzed the ITC experiments and helped write the manuscript. A. A. K. and C. M. P. designed and carried out ligand docking experiments, interpreted the docking data, and helped write the manuscript. A. S. B., G. M., and A. V. carried out all molecular biology, protein expression and purification, and interpreted data; Å. K. R. planned and performed all EPR experiments, interpreted EPR data, and helped write the manuscript; Ma. S. and V. G. E. designed the study, planned the experiments, interpreted data, and wrote the manuscript. All authors have given approval to the final version of the manuscript.
Acknowledgments
C. M. P. and A. A. K. thank the August T. Larsson Guest Researcher Programme at the Swedish University of Agricultural Sciences for the opportunity to work alongside the Sandgren and Ståhlberg research groups. Computational time for this research was provided in part by the National Science Foundation through Extreme Science and Engineering Discovery Environment, which is supported by National Science Foundation Grant number ACI-1053575 under allocation number TG-MCB090159.
This work was supported by the Faculty for Natural Resources and Agriculture, Swedish University of Agricultural Sciences through the research program MicroDrivE (Microbially Derived Energy), and by the Norwegian Research Council Grants 209335, 214613, and 216162. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 4D7U and 4D7V) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- LPMO
- lytic polysaccharide monooxygenase
- ITC
- isothermal titration calorimetry
- PDB
- Protein Data Bank
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- TMP
- N,N,N′,N′-tetramethyl-1,4-phenylenediamine
- AA
- auxiliary activity
- PASC
- phosphoric acid-swollen cellulose
- YPD
- yeast extract/peptone/dextrose.
References
- 1. Horn S. J., Vaaje-Kolstad G., Westereng B., Eijsink V. G. (2012) Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 5, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Forsberg Z., Vaaje-Kolstad G., Westereng B., Bunæs A. C., Stenstrøm Y., MacKenzie A., Sørlie M., Horn S. J., Eijsink V. G. (2011) Cleavage of cellulose by a CBM33 protein. Protein Sci. 20, 1479–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harris P. V., Welner D., McFarland K. C., Re E., Navarro Poulsen J. C., Brown K., Salbo R., Ding H., Vlasenko E., Merino S., Xu F., Cherry J., Larsen S., Lo Leggio L. (2010) Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry 49, 3305–3316 [DOI] [PubMed] [Google Scholar]
- 4. Phillips C. M., Beeson W. T., Cate J. H., Marletta M. A. (2011) Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 6, 1399–1406 [DOI] [PubMed] [Google Scholar]
- 5. Quinlan R. J., Sweeney M. D., Lo Leggio L., Otten H., Poulsen J. C., Johansen K. S., Krogh K. B., Jørgensen C. I., Tovborg M., Anthonsen A., Tryfona T., Walter C. P., Dupree P., Xu F., Davies G. J., Walton P. H. (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U.S.A. 108, 15079–15084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vaaje-Kolstad G., Westereng B., Horn S. J., Liu Z., Zhai H., Sørlie M., Eijsink V. G. (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222 [DOI] [PubMed] [Google Scholar]
- 7. Aachmann F. L., Sørlie M., Skjåk-Bræk G., Eijsink V. G., Vaaje-Kolstad G. (2012) NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc. Natl. Acad. Sci. U.S.A. 109, 18779–18784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beeson W. T., Phillips C. M., Cate J. H., Marletta M. A. (2012) Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 134, 890–892 [DOI] [PubMed] [Google Scholar]
- 9. Hemsworth G. R., Taylor E. J., Kim R. Q., Gregory R. C., Lewis S. J., Turkenburg J. P., Parkin A., Davies G. J., Walton P. H. (2013) The copper active site of CBM33 polysaccharide oxygenases. J. Am. Chem. Soc. 135, 6069–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Langston J. A., Shaghasi T., Abbate E., Xu F., Vlasenko E., Sweeney M. D. (2011) Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl. Environ. Microbiol. 77, 7007–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hemsworth G. R., Davies G. J., Walton P. H. (2013) Recent insights into copper-containing lytic polysaccharide mono-oxygenases. Curr. Opin. Struct. Biol. 23, 660–668 [DOI] [PubMed] [Google Scholar]
- 12. Kjaergaard C. H., Qayyum M. F., Wong S. D., Xu F., Hemsworth G. R., Walton D. J., Young N. A., Davies G. J., Walton P. H., Johansen K. S., Hodgson K. O., Hedman B., Solomon E. I. (2014) Spectroscopic and computational insight into the activation of O2 by the mononuclear Cu center in polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 111, 8797–8802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Isaksen T., Westereng B., Aachmann F. L., Agger J. W., Kracher D., Kittl R., Ludwig R., Haltrich D., Eijsink V. G., Horn S. J. (2014) A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J. Biol. Chem. 289, 2632–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Agger J. W., Isaksen T., Várnai A., Vidal-Melgosa S., Willats W. G., Ludwig R., Horn S. J., Eijsink V. G., Westereng B. (2014) Discovery of LPMO activity on hemicelluloses shows the importance of oxidative processes in plant cell wall degradation. Proc. Natl. Acad. Sci. U.S.A. 111, 6287–6292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vu V. V., Beeson W. T., Span E. A., Farquhar E. R., Marletta M. A. (2014) A family of starch-active polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 111, 13822–13827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Levasseur A., Drula E., Lombard V., Coutinho P. M., Henrissat B. (2013) Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 6, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Forsberg Z., Mackenzie A. K., Sørlie M., Røhr Å. K., Helland R., Arvai A. S., Vaaje-Kolstad G., Eijsink V. G. (2014) Structural and functional characterization of a conserved pair of bacterial cellulose-oxidizing lytic polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 111, 8446–8451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li X., Beeson W. T. 4th., Phillips C. M., Marletta M. A., Cate J. H. (2012) Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure 20, 1051–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mattinen M. L., Linder M., Drakenberg T., Annila A. (1998) Solution structure of the cellulose-binding domain of endoglucanase I from Trichoderma reesei and its interaction with cello-oligosaccharides. Eur. J. Biochem. 256, 279–286 [DOI] [PubMed] [Google Scholar]
- 20. Reinikainen T., Ruohonen L., Nevanen T., Laaksonen L., Kraulis P., Jones T. A., Knowles J. K., Teeri T. T. (1992) Investigation of the function of mutated cellulose domains of Trichoderma reesei cellobiohydrolase I. Proteins 14, 475–482 [DOI] [PubMed] [Google Scholar]
- 21. Sygmund C., Kracher D., Scheiblbrandner S., Zahma K., Felice A. K., Harreither W., Kittl R., Ludwig R. (2012) Characterization of the two Neurospora crassa cellobiose dehydrogenases and their connection to oxidative cellulose degradation. Appl. Environ. Microbiol. 78, 6161–6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kittl R., Kracher D., Burgstaller D., Haltrich D., Ludwig R. (2012) Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol. Biofuels 5, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Várnai A., Tang C., Bengtsson O., Atterton A., Mathiesen G., Eijsink V. G. (2014) Expression of endoglucanases in Pichia pastoris under control of the GAP promoter. Microb. Cell Fact. 13, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., Bairoch A. (2005) in The Proteomics Protocols Handbook (Walker J. M., ed) pp. 571–601, Humana Press Inc., Totowa, NJ [Google Scholar]
- 25. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Evans P. R. (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 67, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 31. Collaborative Computational Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 32. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Op't Holt B. T., Merz K. M. Jr. (2007) Insights into Cu(I) exchange in HAH1 using quantum mechanical and molecular simulations. Biochemistry 46, 8816–8826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stoll S., Schweiger A. (2006) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55 [DOI] [PubMed] [Google Scholar]
- 36. Wiseman T., Williston S., Brandts J. F., Lin L. N. (1989) Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 179, 131–137 [DOI] [PubMed] [Google Scholar]
- 37. Wood T. M. (1988) Preparation of crystalline, amorphous, and dyed cellulase substrates. Methods Enzymol. 160, 19–25 [Google Scholar]
- 38. Zhang Y. H., Lynd L. R. (2005) Determination of the number-average degree of polymerization of cellodextrins and cellulose with application to enzymatic hydrolysis. Biomacromolecules 6, 1510–1515 [DOI] [PubMed] [Google Scholar]
- 39. Sørlie M., Seefeldt L. C., Parker V. D. (2000) Use of stopped-flow spectrophotometry to establish midpoint potentials for redox proteins. Anal. Biochem. 287, 118–125 [DOI] [PubMed] [Google Scholar]
- 40. Liu Y., Seefeldt L. C., Parker V. D. (1997) Entropies of redox reactions between proteins and mediators: the temperature dependence of reversible electrode potentials in aqueous buffers. Anal. Biochem. 250, 196–202 [DOI] [PubMed] [Google Scholar]
- 41. Westereng B., Agger J. W., Horn S. J., Vaaje-Kolstad G., Aachmann F. L., Stenstrøm Y. H., Eijsink V. G. (2013) Efficient separation of oxidized cello-oligosaccharides generated by cellulose degrading lytic polysaccharide monooxygenases. J. Chromatogr. A 1271, 144–152 [DOI] [PubMed] [Google Scholar]
- 42. Wu M., Beckham G. T., Larsson A. M., Ishida T., Kim S., Payne C. M., Himmel M. E., Crowley M. F., Horn S. J., Westereng B., Igarashi K., Samejima M., Ståhlberg J., Eijsink V. G., Sandgren M. (2013) Crystal structure and computational characterization of the lytic polysaccharide monooxygenase GH61D from the Basidiomycota fungus Phanerochaete chrysosporium. J. Biol. Chem. 288, 12828–12839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vu V. V., Beeson W. T., Phillips C. M., Cate J. H., Marletta M. A. (2014) Determinants of regioselective hydroxylation in the fungal polysaccharide monooxygenases. J. Am. Chem. Soc. 136, 562–565 [DOI] [PubMed] [Google Scholar]
- 44. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Morgenstern I., Powlowski J., Tsang A. (2014) Fungal cellulose degradation by oxidative enzymes: from dysfunctional GH61 family to powerful lytic polysaccharide monooxygenase family. Brief. Funct. Genomics 13, 471–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Forsberg Z., Røhr A. K., Mekasha S., Andersson K. K., Eijsink V. G., Vaaje-Kolstad G., Sørlie M. (2014) Comparative study of two chitin-active and two cellulose-active AA10-type lytic polysaccharide monooxygenases. Biochemistry 53, 1647–1656 [DOI] [PubMed] [Google Scholar]
- 47. Turnbull W. B., Daranas A. H. (2003) On the value of c: can low affinity systems be studied by isothermal titration calorimetry? J. Am. Chem. Soc. 125, 14859–14866 [DOI] [PubMed] [Google Scholar]
- 48. Kleywegt G. J., Jones T. A. (1996) φ/ψ-chology: Ramachandran revisited. Structure 4, 1395–1400 [DOI] [PubMed] [Google Scholar]
- 49. Kim S., Ståhlberg J., Sandgren M., Paton R. S., Beckham G. T. (2014) Quantum mechanical calculations suggest that lytic polysaccharide monooxygenases use a copper-oxyl, oxygen-rebound mechanism. Proc. Natl. Acad. Sci. U.S.A. 111, 149–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Loose J. S., Forsberg Z., Fraaije M. W., Eijsink V. G., Vaaje-Kolstad G. (2014) A rapid quantitative activity assay shows that the Vibrio cholerae colonization factor GbpA is an active lytic polysaccharide monooxygenase. FEBS Lett. 588, 3435–3440 [DOI] [PubMed] [Google Scholar]
- 51. Guo J., Catchmark J. M. (2013) Binding specificity and thermodynamics of cellulose-binding modules from Trichoderma reesei Cel7A and Cel6A. Biomacromolecules 14, 1268–1277 [DOI] [PubMed] [Google Scholar]
- 52. Colussi F., Sørensen T. H., Alasepp K., Kari J., Cruys-Bagger N., Windahl M. S., Olsen J. P., Borch K., Westh P. (2015) Probing substrate interactions in the active tunnel of a catalytically deficient cellobiohydrolase (Cel7). J. Biol. Chem. 290, 2444–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gudmundsson M., Kim S., Wu M., Ishida T., Momeni M. H., Vaaje-Kolstad G., Lundberg D., Royant A., Ståhlberg J., Eijsink V. G., Beckham G. T., Sandgren M. (2014) Structural and electronic snapshots during the transition from a Cu(II) to Cu(I) metal center of a lytic polysaccharide monooxygenase by x-ray photoreduction. J. Biol. Chem. 289, 18782–18792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cannella D., Hsieh C. W., Felby C., Jørgensen H. (2012) Production and effect of aldonic acids during enzymatic hydrolysis of lignocellulose at high dry matter content. Biotechnol. Biofuels 5, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vermaas J. V., Crowley M. F., Beckham G. T., Payne C. M. (2015) Effects of lytic polysaccharide monooxygenase oxidation on cellulose structure and binding of oxidized cellulose oligomers to cellulases. J. Phys. Chem. B 119, 6129–6143 [DOI] [PubMed] [Google Scholar]
- 56. Kabsch W., Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 57. Westereng B., Ishida T., Vaaje-Kolstad G., Wu M., Eijsink V. G., Igarashi K., Samejima M., Ståhlberg J., Horn S. J., Sandgren M. (2011) The putative endoglucanase PcGH61D from Phanerochaete chrysosporium is a metal-dependent oxidative enzyme that cleaves cellulose. PLoS ONE 6, e27807. [DOI] [PMC free article] [PubMed] [Google Scholar]