

Background: Mycobacterium tuberculosis synthesizes ergothioneine, a sulfur-containing molecule with unknown function.
Results: egtD encodes for a histidine methyltransferase that is essential for ergothioneine biosynthesis and is negatively regulated through M. tuberculosis serine/threonine protein kinase D.
Conclusion: M. tuberculosis modulates intracellular ergothioneine levels in response to starvation.
Significance: Mechanisms by which M. tuberculosis senses and adapts to nutrient starvation is essential for understanding persistence and disease latency.
Keywords: bacterial protein kinase, bacterial signal transduction, microbiology, Mycobacterium tuberculosis, thiol, ergothioneine, histidine methylation
Abstract
Ergothioneine (EGT) is synthesized in mycobacteria, but limited knowledge exists regarding its synthesis, physiological role, and regulation. We have identified Rv3701c from Mycobacterium tuberculosis to encode for EgtD, a required histidine methyltransferase that catalyzes first biosynthesis step in EGT biosynthesis. EgtD was found to be phosphorylated by the serine/threonine protein kinase PknD. PknD phosphorylates EgtD both in vitro and in a cell-based system on Thr213. The phosphomimetic (T213E) but not the phosphoablative (T213A) mutant of EgtD failed to restore EGT synthesis in a ΔegtD mutant. The findings together with observed elevated levels of EGT in a pknD transposon mutant during in vitro growth suggests that EgtD phosphorylation by PknD negatively regulates EGT biosynthesis. We further showed that EGT is required in a nutrient-starved model of persistence and is needed for long term infection of murine macrophages.
Introduction
Mycobacterium tuberculosis, the causative agent of tuberculosis, senses and adapts its physiology to ensure survival within the host, enabling it to overcome oxidative and nitrosative challenges associated with intracellular infection. Xenobiotics, including free radicals, produced by the host to cope with infection; antibiotics; and general bacterial respiration are countered by an intricate detoxification system utilized by the bacterium that is composed of (a) enzymes such as catalase, superoxide dismutase, and alkyl hydroperoxidase; (b) truncated hemoglobins; (c) oxidoreductases; and (d) redox coupling systems (1). The most common defense mechanism against reactive oxygen and nitrogen species damage in eukaryotes and Gram-negative bacteria relies on glutathione, a low molecular weight thiol. Mycobacteria, like most Gram-positive bacteria, do not produce glutathione; rather, they synthesize mycothiol (MSH)2 at millimolar levels, making it the most abundant low molecular weight thiol in these species (2). Similarly to glutathione, MSH also serves as an antioxidant that is important in maintaining a highly reducing environment inside the cell (3). Several studies have demonstrated the role of MSH in detoxifying reactive species by either (i) donating a reducing equivalent (4) or (ii) forming an S-conjugate composed of MSH and the respective agent (5). Consistent with these findings, mycobacterial species deficient in MSH are found to have varying but increased sensitivity to H2O2 (3, 4, 6), nitric oxide (NO) (7), and other redox cycling agents (3, 4, 8, 9). Despite the importance of MSH in protecting mycobacteria against oxidative and nitrosative stressors, M. tuberculosis strains lacking MSH remain viable in vivo, suggesting a compensatory mechanism (10).
Fahey and co-workers (11) showed that Mycobacterium smegmatis mutants devoid of MSH displayed a marked elevation in another sulfur-containing low molecular weight compound known as ergothioneine (EGT). EGT exists primarily as a thione under physiological conditions and differs significantly from other cysteine-containing thiols as it possesses a lower redox couple value (E0′ = −0.06 V) (12). Although a poorer reductant, EGT is described as an effective antioxidant through quenching of singlet oxygen, scavenging of hydroxyl radicals, and inhibition of heavy metal-catalyzed reactions (for a review, see Ref. 13), and its depletion in mammalian cells leads to augmented oxidative damage and cell death in the presence of exogenous stressors (14). Despite the presence of EGT in mammalian cells, biosynthesis is limited to a subset of organisms, which includes actinobacteria, cyanobacteria, and specific fungi and yeast (15–18). Mammals obtain EGT from dietary sources and concentrate it in tissues exposed to high levels of oxidative stress via a specific transporter (19).
Among microorganisms, EGT biosynthesis follows a similar pathway, that is the conversion of histidine to EGT through the intermediate hercynine. The genes encoding EGT biosynthesis in M. smegmatis have been identified (20), paving the way to study the role EGT in M. tuberculosis physiology and pathogenesis. Still, it remains to be discovered as to which physiological conditions trigger M. tuberculosis to up-regulate EGT biosynthesis in vivo. As observed in Fig. 1, EGT biosynthesis is an energetically costly process for mycobacteria. The pathway follows five enzymatic steps that consume the amino acids histidine and cysteine and cofactors such as ATP, S-adenosylmethionine (AdoMet), and pyridoxal phosphate. Therefore, EGT production is likely to be tightly regulated to avoid metabolic stress under unfavorable environmental conditions.
FIGURE 1.
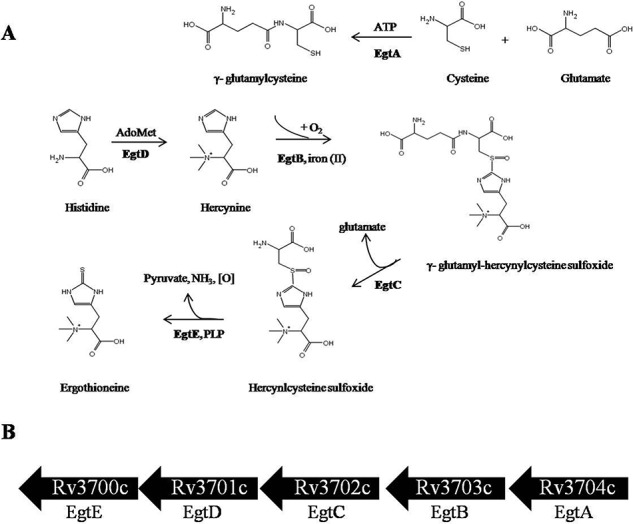
EGT biosynthetic pathway in M. tuberculosis. A, EGT biosynthesis occurs through five enzymatic steps, and the genes encode for a γ-glutamylcysteine synthase (EgtA), a formylglycine-generating enzyme-like protein (EgtB), a glutamine amidotransferase (EgtC), a methyltransferase (EgtD), and a pyridoxal 5-phosphate protein (EgtE). The pathway proceeds from l-histidine through the intermediary precursor hercynine, hercynyl γ-glutamylcysteine sulfoxide, and hercynylcysteine sulfoxide. EGT acquires its sulfur from γ-glutamylcysteine. B, in silico analysis identified the gene cluster Rv3700c–Rv3704c to encode for EGT biosynthesis in M. tuberculosis. PLP, pyridoxal 5-phosphate.
Mycobacterial adaptation to varying conditions is dependent on both the classical bacterial two-component systems comprising histidine kinases and response regulators and the eukaryotic-like Ser/Thr protein kinases (STPKs) (21). Signaling through STPKs has recently emerged as a key regulatory mechanism in M. tuberculosis, playing roles in the transport of metabolites (22), cell division (23), and virulence (24). Several metabolic pathways such as mycolic acid (25–28), glutamine (29), phthiocerol dimycocerosates (30), and glucan (31) biosyntheses have been shown to be regulated by M. tuberculosis STPK phosphorylation.
In the present study, using combined biochemical and genetic analyses, we showed that EGT biosynthesis is dependent on EgtD, a histidine methyltransferase catalyzing the first reaction step in EGT biosynthesis. We further demonstrated that EgtD is under phosphorylation control by the STPK PknD, leading to increased up-regulation of EGT biosynthesis during starvation and enhancing the survival of M. tuberculosis in an in vitro model of persistence.
Experimental Procedures
Bacterial Strains and Growth Conditions
M. tuberculosis H37Rv cultures were grown aerobically at 37 °C on Middlebrook 7H10 agar plates with 10% (v/v) oleic acid/albumin/dextrose/catalase (OADC) enrichment (BD Biosciences) or in Middlebrook 7H9 broth supplemented with 0.05% (v/v) Tween 80, 0.2% (v/v) glycerol, and 10% (v/v) OADC. Hygromycin (50 μg/ml) and kanamycin (25 μg/ml) were added for the selection of the appropriate M. tuberculosis strains. Escherichia coli DH5α and BL21(DE3) were grown at 37 °C in Luria-Bertani (LB) broth or on LB agar and were supplemented with kanamycin (50 μg/ml) or ampicillin (100 μg/ml) when required.
Cloning and Protein Expression and Purification
All genes were amplified by PCR from H37Rv genomic DNA using standard methods for cloning. Recombinant plasmids were further transformed into E. coli BL21(DE3) cells for protein expression. Strains harboring the genes to produce recombinant protein were used to inoculate LB broth from an overnight culture (1:100), and the cells were induced with 1-thio-β-d-galactopyranoside once an A600 of 0.6–0.8 was reached. Protein purification was carried out on either nickel-nitrilotriacetic acid resin (Qiagen) or glutathione-agarose (Sigma-Aldrich) columns according to the manufacturers' supplied guidelines. Site-directed mutagenesis was performed following the Stratagene QuikChange protocol, and parental pMV261-egtD and pET28-egtD were used as a template.
EgtD in Vitro Methylation Activity
A reaction containing 10 mm histidine, 4 mm AdoMet, 1 mm Mg(OAc)2, 5 mm NaCl, 20 μg of EgtD, and 5 μg of S-adenosylhomocysteine hydrolase was prepared as described (20) and incubated overnight at 37 °C. Detection of methylated histidine products in the reaction was carried out by ESI-MS at the University of Victoria Genome British Columbia Proteomics Centre as detailed below. EgtD substrate specificity was analyzed using a continuous enzyme-coupled SAM510TM methyltransferase assay (G-Biosciences) according to the manufacturer's protocol. Reactions were initiated immediately following the addition of a 1 mm concentration of each tested substrate and monitored at 510 nm for 30 min at room temperature.
Construction and Complementation of the M. tuberculosis ΔegtD Mutant
The ΔegtD mutant strain was constructed via allelic exchange using the conditionally replicating mycobacteriophage phAE159 as described previously (32). To construct the egtD knock-out phage, flanking regions comprising 1000-bp upstream and downstream regions of the egtD gene were amplified from H37Rv genomic DNA. The up- and downstream flanking regions of egtD were cloned into the p0004S cosmid prior to ligation of this recombinant cosmid with phAE159. The ligated DNA was packaged into phage λ with Gigapack III Gold packaging extract (Stratagene) and E. coli HB101 cells that were previously grown in MgSO4 and maltose overnight. Colonies were selected for growth on LB plates containing 150 μg/ml hygromycin, and phage DNA was extracted and electroporated into M. smegmatis mc2155. Transformation plates were incubated at 30 °C for 3 days. Plaques were picked to prepare high titer phage stocks (109 pfu/ml) in M. smegmatis. Phages were transduced into M. tuberculosis H37Rv and plated on Middlebrook 7H10 supplemented with OADC and hygromycin (50 μg/ml). After 4 weeks, hygromycin-resistant colonies appeared and were cultured for analysis by PCR and Southern hybridization to identify clones in which allelic exchange had occurred within the Rv3701c gene.
Southern Blot Hybridization
Southern blotting was performed using the digoxigenin hybridization system (Roche Applied Science). Chromosomal DNA (12 μg) from both the H37Rv wild-type and Rv3701c-null mutant strains was digested with AflIII. Digested DNA was resolved on a 1% agarose gel prior to its transfer to a nylon membrane via capillary method overnight. Hybridization was performed at 68 °C overnight with a digoxigenin-11-dUTP-labeled probe. Anti-digoxigenin antibodies were used to detect the probe hybridized to its DNA target.
EGT Extraction from M. tuberculosis
M. tuberculosis strains were grown to their desired A600, and 4 ml of cells were harvested by centrifugation. Cells were washed twice in the same volume of double distilled H2O. Following washing, the cells were resuspended in 2 ml of 70% acetonitrile with 25 ng/ml internal standard 1-methyl-4-phenylpyridinium ion. Cells were disrupted with the MagNAlyser (Roche Applied Science) and 0.1-mm silica beads (BioSpec) at a speed of 7000 rpm for 60-s intervals followed by 2 min of rest at −20 °C (repeated four times). The extract was then filter-sterilized using 0.22-μm nylon polypropylene Spin-X® centrifuge tubes prior to exiting the biosafety containment level 3 laboratory for further analysis by ESI LC-MS/MS.
ESI LC-MS/MS Analysis of EGT
EGT was quantified using an Agilent Technologies 1200 binary HPLC system coupled to an AB Sciex 5500 Q-Trap triple quadrupole mass spectrometer. Separation was performed on a Zorbax HILIC Plus column (Agilent Technologies; 100 × 2.1 mm, 3.5-μm particle size). Acetonitrile (76%) and water (24%), both containing 0.1% formic acid, were used as the mobile phase at a flow rate of 200 μl/min. The peak area of EGT was measured using Analyst 1.5.2 (AB Sciex) and normalized by the weighted contribution of the peak areas of the 1-methyl-4-phenylpyridinium ion internal standard. Identification of EGT was based on its theoretical m/z value, MS/MS fragmentation data, and its retention time, which was verified by analyzing a pure standard (Oxis International Inc.). Calibration curves were generated through a series of EGT standard additions to the sample. Regression coefficients of each calibration curve were all greater than 0.99.
Protein-Protein Interaction Assay
Protein-protein interactions were investigated using the mycobacterial protein fragment complementation assay as described previously (33). M. tuberculosis EgtD and STPKs (PknA, PknB, PknD, and PknK) were amplified by PCR and cloned into pUAB100 (expressing murine dihydrofolate enzyme fragments F1 and F2) and pUAB200 (expressing murine dihydrofolate fragment F3), respectively. EgtD was co-transformed with each of the four kinases into M. smegmatis mc2155, and co-transformants were selected for on 7H11/kanamycin/hygromycin plates. Co-transformants were replated on 7H11/kanamycin/hygromycin plates supplemented with 0 and 10 μg/ml trimethoprim and analyzed for growth over 4–5 days.
In Vitro Kinase Assay
An in vitro phosphorylation screen was performed as described previously (34) using 1 μg of EgtD in 20 μl of the assay buffer (20 mm Tris-HCl, pH 7.4, 5 mm MgCl2, 5 mm MnCl2, 1 mm DTT) and varying concentrations of kinase (0.1–1 μg) to obtain optimal autophosphorylation activity. Kinases used for screening in this assay were PknA, PknB, PknD, PknE, PknF, PknG, PknH, and PknK. Reactions were commenced by the addition of 10 μCi of [γ-32P]ATP (PerkinElmer Life Sciences; 3000 Ci/mmol) and incubated at room temperature (23 °C) for 30 min. Following the incubation period, reactions were arrested using SDS sample loading buffer and heated at 95 °C for proteins bands. EgtD dose-dependent phosphorylation kinetics were performed as described above with minor changes. First, the kinase was left to autophosphorylate for 20 min prior to the addition of EgtD. Cold ATP (100 μm) was spiked into 10 μCi of [γ-32P] ATP prior to the addition of ATP to the reactions. The reaction kinetics were monitored by excising the bands corresponding to EgtD and subjecting them to scintillation counting (Beckman Coulter LS 6500). Kinetic parameters were calculated using Prism Software (GraphPad 6.04). EgtD phosphorylation sites were analyzed using LC-MS/MS (phosphopeptide analysis) as described (24) with 1 mm non-radiolabeled ATP.
Cell-based Phosphorylation
egtD was cloned into pGEX-4T3, and pknD was cloned into pET-28. Both plasmids were co-transformed into E. coli BL21. Co-transformants were selected on LB plates containing ampicillin and kanamycin. Cultures were induced with 1 mm 1-thio-β-d-galactopyranoside and further grown for 16 h at 25 °C. Both proteins were purified as described above and resolved by SDS-PAGE to ensure adequate expression of both proteins in the culture. Approximately 20 μg of recombinant protein was then subjected to phosphopeptide analysis by LC-MS/MS to determine egtD phosphorylation sites in a cell-based system.
TLC Analysis of Phosphorylated EgtD Activity
Phosphorylated EgtD was obtained through the in vitro kinase assay outlined above. The reaction varied slightly in that 8 μm EgtD, 10 μm non-radiolabeled ATP, and 0.4 μm kinase were used. Reactions were incubated at room temperature for 1.5 h to obtain the maximum yield of phosphorylated EgtD. Next, 15 mm histidine, 2.5 mm NaCl, 500 μm Mg(OAc)2, and 10 μm S-adenosylhomocysteine nucleosidase were added to the 8 μm phosphorylated EgtD. The methylation reaction was initiated upon addition of 10 μmol of S-[methyl-14C]adenosyl-l-methionine (PerkinElmer Life Sciences; 60 mCi/mmol). Ten-microliter samples were taken from the reaction at various time points, stopped with 1 μl of 1% trifluoroacetic acid, and stored at −20 °C until use. Samples were subjected to TLC using PEI Cellulose F plates (EMD Millipore, Darmstadt, Germany) and developed in butanol/water/acetic acid (60:25:15, v/v). The separation of radiolabeled AdoMet and methylated histidine was visualized using PhosphorImager SI (GE Healthcare) following 7 days of exposure. Ninhydrin (0.03%) was used to detect histidine on the plate. The spots corresponding to histidine were cut from the plate and subjected to scintillation counting (Beckman Coulter LS 6500) to quantify the formation of methylated histidine over time.
Macrophage Infection
Murine J774A.1 macrophages were purchased from the American Type Culture Collection (ATCC catalogue number TIB-67) and were stored and prepared according to the manufacturer's guidelines. Macrophages were prepared by seeding onto a 24-well plate at a density of 2.5 × 105 cells/well in culture medium (Dulbecco's modified Eagle's medium, high glucose supplemented with 1% glutamine, 10% fetal bovine serum, 1% HEPES, 1% non-essential amino acids). Cells were left overnight. The following day, J774A.1 cells were infected with exponentially growing M. tuberculosis (A600 = 0.5) at a multiplicity of infection of 5:1. J774A.1 cells were incubated with M. tuberculosis for 3 h at 37 °C in 5% CO2. Wells were next washed three times and resuspended in culture medium containing 100 μg/ml gentamicin to remove any remaining extracellular M. tuberculosis. For cfu counting, cells were washed three times with J774A.1 culture medium, and the macrophages were lysed using 0.025% SDS at the selected time points postinfection. Serial dilutions of the lysate were plated onto Middlebrook 7H10 agar medium supplemented with OADC and the appropriate antibiotics. Colonies were counted after incubation for 3 weeks at 37 °C.
Starvation Studies
Mycobacterial cultures were grown with shaking in Middlebrook 7H9, 0.2% (v/v) glycerol, 10% OADC, 0.05% tyloxapol to an A600 of 0.8. Cells were washed twice with PBS and then resuspended in PBS prior to leaving the cultures to stand at 37 °C in sealed bottles (35). M. tuberculosis viability during starvation was determined by counting cfu from triplicate cultures over a 4-week period. Serial dilutions of the cells were performed and followed by plating onto Middlebrook 7H10 agar medium supplemented with OADC and the appropriate antibiotics. Colonies were counted after incubation for 3 weeks at 37 °C. The extraction and quantification of intracellular EGT levels were performed at various time points throughout starvation as described in the methods above.
Results
EGT Biosynthesis Pathway in M. tuberculosis
Previously, the EGT biosynthetic pathway was identified in M. smegmatis and was described to consist of five clustered genes (20). These genes encode for γ-glutamylcysteine ligase (egtA), a formylglycine-like enzyme (egtB), a glutamine amidotransferase (egtC), a histidine methyltransferase (egtD), and lastly a pyridoxal 5-phosphate-binding protein (egtE). Using the NCBI Basic Local Alignment Search Tool, we identified the open reading frames Rv3700c--Rv3704c (Table 1) to encode for EGT biosynthesis in M. tuberculosis.
TABLE 1.
Homology of the M. tuberculosis EGT biosynthetic gene cluster (Rv3700c–Rv3704c) with M. smegmatis (msmeg6246–6250)
| Enzyme | Accession number | Identity | Similarity | E-valuea |
|---|---|---|---|---|
| % | % | |||
| EgtA | NP_218221.1 | 66 | 77 | 0.0 |
| EgtB | NP_218220.1 | 77 | 83 | 0.0 |
| EgtC | NP_218219.1 | 74 | 82 | 4e−116 |
| EgtD | NP_218218.1 | 74 | 81 | 6e−173 |
| EgtE | NP_338356.1 | 66 | 79 | 9e−158 |
a Obtained from BLAST analysis.
Rv3701c and Rv3704c are predicted to encode EgtD and EgtA (Fig. 1B), respectively, and commit the necessary amino acids to the pathway, suggesting an optimal site for post-translational modification. Transposon site hybridization studies identified Rv3701c, but not Rv3704c, to be essential for growth in animal models and murine macrophages (36, 37). Therefore, the requirement for Rv3701c during infection may not only implicate EGT in the pathogenesis of M. tuberculosis but may also represent a critical point in the pathway responsible for orchestrating EGT biosynthesis in response to changes in the environment of the bacilli.
Rv3701c Encodes for egtD in EGT Biosynthesis
We cloned Rv3701c from M. tuberculosis H37Rv for expression in E. coli and assayed the purified recombinant protein. Using the SAM510 assay, the methylation activity of EgtD was assessed in the presence of each of the proteinogenic amino acids with the exception of cysteine due to its interference with the assay (38). The consumption of AdoMet in the reaction was continuously monitored at 1-min intervals over a 30-min time period. No change in absorbance was observed, suggesting that histidine was the only amino acid that could undergo methylation (data not shown). We further explored substrate specificity using the same methods and ruled out methylation activity with histamine, imidazole, and the histidine derivatives 1-methyl-l-histidine, 3-methyl-l-histidine, and α-methyl-dl-histidine. These findings validate EgtD as a methyltransferase with high specificity for the amino acid histidine.
The methyltransferase assay we used for the enzyme characterization does not identify the number of methyl groups added to a substrate but solely indicates whether methylation is in fact occurring. Thus, we performed ESI-MS analysis on a methylation reaction containing EgtD and histidine in an attempt to isolate hercynine (α-N,N,N-trimethylhistidine) variants. Fig. 2A illustrates that EgtD produces three methylation products, mono-, di-, and trimethylated histidine. As observed in Fig. 2B, when the same reaction was prepared in the absence of EgtD, histidine was not transformed into any of the expected methylated products. The dependence of reaction velocity on histidine concentration was measured in the presence of 5.3 μm EgtD using the SAM510 assay at various concentrations of histidine (0–15 mm). Experimental data were best fitted using the Michaelis-Menten equation (R2 = 0.99) and the Vmax (114.5 ± 1.8 μm/min·mg) and Km (422.0 ± 37.4 μm). Based on these values, we further calculated the kcat (1.3 × 10−2 s−1) and kcat/Km (30.8 m−1·s−1) of the enzyme.
FIGURE 2.
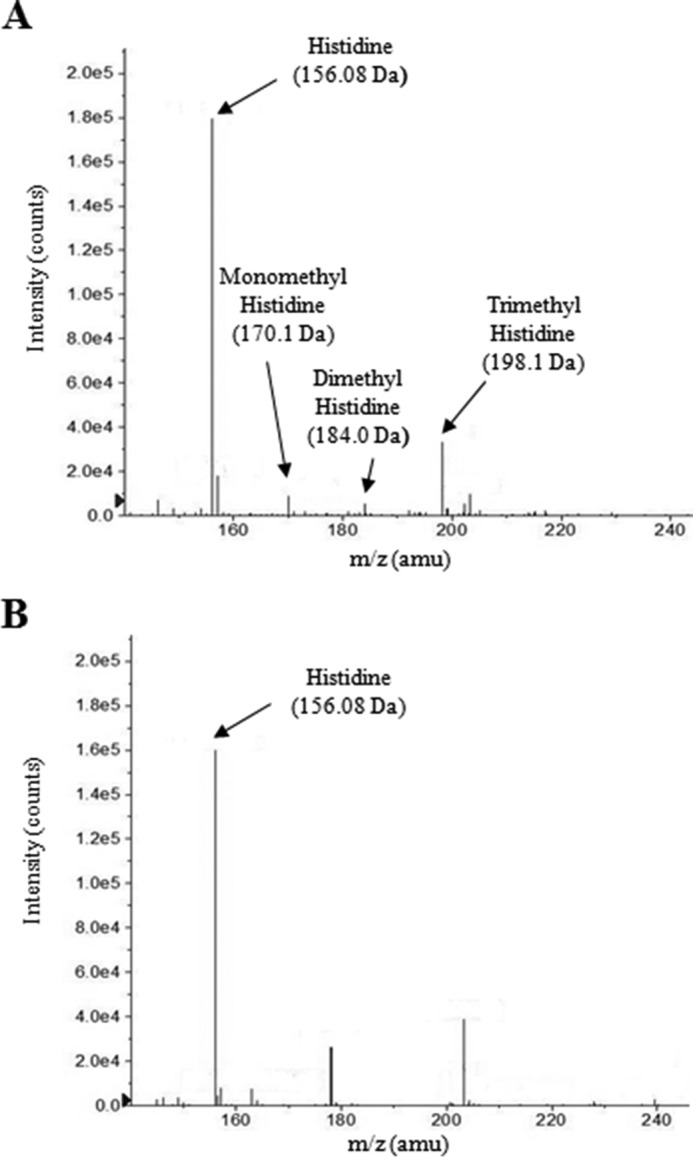
Rv3701c encodes for a histidine methyltransferase. The methylation activity of Rv3701c in the presence of AdoMet and histidine was analyzed by ESI-MS. A, Rv3701c catalyzes the methylation of the α-amino nitrogen atom of histidine to form mono-, di-, and trimethylated histidine. B, reaction in the absence of Rv3701c. No methylated histidine products were observed. amu, atomic mass units.
egtD Is Essential for EGT Biosynthesis but Not Needed for Growth
To study the necessity and role of EgtD in EGT biosynthesis, a M. tuberculosis ΔegtD null mutant was constructed in M. tuberculosis H37Rv. We replaced the genomic Rv3701c open reading frame by specialized transduction with a hygromycin resistance cassette (Fig. 3A) and verified the knock-out strain formation by Southern blotting and PCR (Fig. 3, B and C, respectively). Intracellular EGT levels were quantified in the parental wild-type M. tuberculosis, the Rv3701c-null mutant, and a complemented strain in which the Rv3701c mutant was transformed with a plasmid containing the Rv3701c gene (pMV261:ΔegtD). EGT could not be detected in extracts prepared from the ΔegtD mutant, whereas both the wild-type and complemented strains synthesized considerable amounts of EGT (Fig. 3D). Through this experiment, we demonstrated that EgtD encoded by Rv3701c is required for EGT biosynthesis in M. tuberculosis.
FIGURE 3.

Construction and in vitro characterization of ΔegtD in M. tuberculosis. A, schematic diagram of the Rv3701c region of the chromosome of M. tuberculosis. Genomic DNA was digested with AflIII, and the blot was probed with a digoxigenin-11-dUTP-labeled DNA fragment containing 314 bp of the egtD 3′-flanking sequence. B, confirmation of the ΔegtD mutant through Southern blotting. AflIII-digested genomic DNA gave rise to the expected 1.77-kbp fragment in wild-type M. tuberculosis (lane 1) and 2.74-kbp fragment in the hygromycin-resistant transductant in which egtD was disrupted with the hyg marker (lane 2). C, PCR analysis of the hygromycin-resistant transductant genomic DNA for the ΔegtD. Left panel, PCR amplification of egtD (966 bp). Right panel, PCR amplification of the hygromycin-resistant cassette (∼700 bp), which replaced egtD in the mutants. D, intracellular EGT levels extracted from wild-type M. tuberculosis, ΔegtD M. tuberculosis, and ΔegtD transformed with pMV261:egtD and quantified by ESI LC-MS/MS. Error bars indicate the means ± S.D. of three independent experiments. Col, colony.
EgtD Is a Substrate of M. tuberculosis Ser/Thr Kinases
M. tuberculosis uses a number of signal transduction systems as a means for adaptive gene expression and regulation of metabolic processes in response to external stimuli (39). Screening of a set of M. tuberculosis STPKs for their ability to phosphorylate purified EgtD identified PknA, PknB, PknD, and PknK as kinases capable of phosphorylating EgtD (Fig. 4A). No radioactive bands were observed in the presence of the other STPKs or when EgtD was incubated in the absence of kinase. These findings indicate that EgtD is an in vitro substrate of several of M. tuberculosis STPKs, suggesting that EGT biosynthesis could be under phosphorylation control in mycobacteria.
FIGURE 4.

EgtD is a substrate of multiple M. tuberculosis STPKs. A, in vitro phosphorylation of EgtD by multiple kinases. M. tuberculosis STPKs purified as GST or His fusions were incubated with His-tagged EgtD and [γ-32P]ATP. Samples were separated by SDS-PAGE and stained with Coomassie Blue followed by visualization by autoradiography. Upper bands represent autophosphorylation activity of each kinase (Pkn); lower bands reflect phosphorylated EgtD. B, interaction between EgtD and M. tuberculosis STPKs facilitates the reassembly of complementary fragments F1 and F2 and fragment F3 of murine dihydrofolate reductase and thus confers M. smegmatis resistance to trimethoprim (TMP). Growth was monitored over 4 days on kanamycin/hygromycin plates supplemented with 0 and 10 μg/ml trimethoprim. Control plates without trimethoprim revealed growth of all strains. Positive Control, M. tuberculosis ESAT-6 (F1 and F2) and CFP-10 (F3); Negative Control, EgtD (F1 and F2) with F3 alone. Experiments are shown in duplicates.
To test whether EgtD interacts with the four identified kinases in vivo, a cell-based interaction assay using the mycobacterial protein fragment complementation assay was performed in M. smegmatis (33).The mycobacterial protein fragment complementation assay involves the reassembly of complementary fragments F1 and F2 (expressed by pUAB100) and fragment F3 (expressed by pUAB200) of murine dihydrofolate reductase enzyme, conferring resistance to trimethoprim. As illustrated in Fig. 4B, growth of M. smegmatis co-transformed with pUAB100-egtD and pUAB200 containing pknB, pknD, or pknK was present. No growth was observed in the strain co-expressing EgtD and PknA, suggesting that these two proteins do not interact under in vivo growth conditions. The interaction between EgtD and the kinases was weaker than the positive control, CFP-10 and ESAT-6, which is expected due to the transient nature of kinase-substrate interactions. Therefore, the mycobacterial protein fragment complementation assay provided evidence that EgtD is potentially a substrate for the M. tuberculosis STPKs PknB, PknD, and PknK in mycobacteria.
PknD Preferentially Phosphorylates EgtD in Vitro
It is apparent from Fig. 4A that PknD possesses the greatest phosphorylation capacity toward EgtD in comparison with PknB and PknK. To verify this, we analyzed the phosphorylation kinetics of EgtD. We confirmed that PknD possesses the greatest kcat/Km and lowest Km for the methyltransferase (Table 2). As these findings suggest the PknD interaction to have the greatest relevance, we therefore focused on the effect of PknD phosphorylation of EgtD for the remainder of the study.
TABLE 2.
EgtD phosphorylation kinetics
Various concentrations of EgtD (1–12 μm) were phosphorylated by 0.7–1 nm kinase. The transfer of γ-32P was measured via scintillation counting to determine phosphorylation kinetics. Data are representative of three independent experiments and presented as average values ±S.E.
| Kinase | Vmax | Km | kcat | kcat/Km |
|---|---|---|---|---|
| nmol/min/mg | μm | s−1 | m−1·s−1 | |
| PknB | 27.7 ± 2.3 | 2.3 ± 0.6 | 0.01 | 1.2 × 104 |
| PknD | 15.7 ± 0.8 | 0.2 ± 0.09 | 0.02 | 8.4 × 104 |
| PknK | 10.3 ± 1.1 | 0.6 ± 0.2 | 0.02 | 1.7 × 104 |
Phosphorylation Negatively Regulates EgtD Methyltransferase Activity
Phosphorylation of a protein introduces a negative charge on the targeted amino acid(s) that can ultimately affect protein activity. To investigate the effect of phosphorylation on the methylation activity of EgtD, we developed an assay designed to monitor the transfer of methyl-14C from AdoMet to histidine. Phosphorylated and unphosphorylated EgtD was prepared using the in vitro kinase assay in the presence or absence of PknD. Following 2 h of incubation at room temperature, the kinase assays containing EgtD were added to the methylation assay containing S-[methyl-14C]adenosyl-l-methionine and histidine. Samples were taken at various time points from the reaction and separated by TLC (Fig. 5A). The formation of [methyl-14C]histidine was visualized by autoradiography (upper), whereas total histidine was observed on TLC plates developed with ninhydrin (lower). As expected, the combined kinase and methylation assay lacking EgtD did not form [methyl-14C]histidine after 2 h. However, methylation activity of phosphorylated EgtD was visibly slower than that of the unphosphorylated enzyme. Histidine spots were then excised from the TLC plate, and the cellulose stationary phase was added to scintillation fluid for quantification. Counts per minute (cpm) phosphorylated and non-phosphorylated EgtD were plotted as a function of time for both enzyme sets (Fig. 5B). An approximate 20% reduction in the activity of phosphorylated EgtD was observed compared with non-phosphorylated EgtD. Although ATP concentrations were in excess in these reactions, it remains likely that a mixed population of phosphorylated and non-phosphorylated protein existed, potentially underestimating the effect of phosphorylation on the methylation activity of EgtD. Nonetheless, the rate of formation of [methyl-14C]histidine was significantly slower for phosphorylated EgtD, suggesting that PknD may negatively regulate EgtD in M. tuberculosis.
FIGURE 5.

EgtD methylation activity is negatively regulated by phosphorylation. A, phosphorylated and non-phosphorylated EgtD was obtained from an in vitro kinase assay and added to a methylation assay containing S-[methyl-14C]adenosyl-l-methionine (2 μCi/ml). The transfer of methyl-14C to histidine was monitored over a 2-h period and analyzed by one-dimensional TLC using butanol/acetic acid/water (60:15:25, v/v). Upper, detection of [methyl-14C]histidine was performed by autoradiography, exposing the TLC plate to x-ray cassettes for 1 week. Lower, TLC plate developed with ninhydrin to visualize histidine. B, graphical representation of the effect of phosphorylation on EgtD methylation activity from A. ***, p < 0.0005 for comparison of phosphorylated versus non-phosphorylated EgtD. Error bars indicate the means ± S.E. of three independent experiments. ′, minutes.
Thr213 Is the Major Phosphorylation Site of EgtD
Mass spectrometry was used to identify the nature and location of the phosphorylation site(s) on M. tuberculosis EgtD as performed previously (24). MS/MS analysis of purified recombinant EgtD incubated in the presence of PknD identified phosphorylation on the trypsin-digested 205AYDDPGGVTAQFNR218 peptide that was located on Thr213 in the C terminus of the protein (Fig. 6A). No autophosphorylation was observed in the negative control containing EgtD incubated with ATP in the absence of PknD.
FIGURE 6.

Identification of EgtD phosphorylation by PknD. A, MS/MS spectra at +2 representing peptide positions 205–218 with a monoisotopic mass of 1,510.69 Da from EgtD phosphorylated by PknD in vitro. Phosphorylation at Thr213 was shown by the “y” C-terminal daughter ion series where all y ions identified lose phosphoric acid (−98 Da) after the phosphorylated residue. pT, phosphothreonine; amu, atomic mass units. B, in vitro kinase assay confirmed Thr213 as the major phosphorylation site of EgtD by PknD. EgtD T213A is defective in phosphorylation. Upper, phosphorimage; lower, Coomassie Blue stain. The arrowhead points to EgtD. C, MS/MS spectra m/z 795.83 (+2) representing peptide positions 205–218 from EgtD phosphorylated in a cell-based system with PknD showing phosphorylation of Thr213. Phosphorylation at Thr213 is shown by the y C-terminal daughter ion series where all y ions after Thr213 lose phosphoric acid.
The phosphopeptide identified by mass spectrometry was validated by substituting EgtD Thr213 with alanine through site-directed mutagenesis to prevent phosphorylation. The autoradiogram in Fig. 6B shows that T213A site-directed mutagenesis resulted in abrogation of EgtD phosphorylation compared with intact EgtD. The above findings further illustrate that EgtD Thr213 is the major phosphorylation site for PknD in vitro.
We next wanted to further validate the phosphorylation site of EgtD in a cell-based system. Due to potential interference of other mycobacterial STPKs, we decided to study the interaction between EgtD and PknD using E. coli as a heterologous host because it lacks any known STPKs. The active kinase domain of PknD and full-length recombinant wild-type EgtD were subcloned into pET-28a and pGEX-4T3, respectively, to achieve compatible expression conditions. Purified EgtD expressed in the presence and absence of PknD inside E. coli was subjected to LC-MS/MS analysis to identify the phosphorylation sites. As seen previously in vitro, EgtD co-expressed with PknD was monophosphorylated on peptide 205AYDDPGGVTAQFNR218 (Fig. 6C), but no phosphorylation was observed in the absence of the kinase in E. coli. From these results, we concluded that Thr213 is the major phosphorylation site of EgtD both in vitro and in an in vivo cell-based system.
M. tuberculosis EgtD Phosphomimetic Mutant Synthesizes Lower EGT Levels
Introduction of a negative charge through the substitution of acidic residues such as Asp or Glu has been shown previously to mimic phosphorylation of a protein with regard to functional activity (25, 27, 28, 31). We wanted to further determine the effect of phosphorylation on EGT levels in M. tuberculosis and thus constructed EgtD variant strains with constitutively altered activities. The ΔegtD mutant was transformed with pMV261 derivatives allowing constitutive expression of different egtD alleles under the control of the hsp60 promoter: egtD_WT, phosphomimetic egtD_T213E, and phosphoablative egtD_T213A. As shown in Fig. 7A, EGT levels in the EgtD_T213A-overexpressing strain were comparable with those in EgtD_WT with a minor reduction likely due to the nature of the amino acid substitution. In contrast, overexpression of EgtD_T213E was accompanied by a significant decrease in EGT levels in comparison with EgtD_T213A and EgtD_WT, providing stronger evidence that EgtD phosphorylation negatively regulates EGT biosynthesis in M. tuberculosis.
FIGURE 7.

Phosphorylation of EgtD reduces EGT levels in M. tuberculosis. A, electrocompetent H37Rv M. tuberculosis cells were transformed with pMV261_egtD_WT, pMV261_egtD_T213A, and pMV261_egtD_T213E to allow for the constitutive expression of the egtD alleles under the control of the hsp60 promoter. Bacteria were harvested at mid-log phase, washed with purified water, and lysed in 70% acetonitrile. Bacterial lysates were collected and analyzed for EGT by ESI LC-MS. EGT intracellular levels were normalized to the number of cells. B, CDC1551 wild type and a PknD:Tn (point of insertion at bp 1166) generously provided by The John Hopkins Mutant Library were grown to mid-log phase prior to extraction, and EGT was quantified by ESI LC-MS. The results presented for both A and B are expressed as the mean of three independent experiments ±S.E. (error bars). **, p < 0.005; ***, p < 0.0005.
To further confirm the effect of EgtD phosphorylation on EGT biosynthesis, we investigated EGT levels in the CDC1551 pknD:Tn mutant (Fig. 7B). Because PknD is expressed during mid-log phase (40) and we observed phosphorylation to negatively regulate EgtD methylation activity and EGT biosynthesis, we hypothesized that the pknD mutant would exhibit higher levels of EGT than wild-type M. tuberculosis in culture. Intracellular EGT was extracted from CDC1551 wild type and pknD:Tn mutant at an A600 of 0.5. As expected, the pknD:Tn mutant had significantly higher levels of EGT than wild type, indicating that in the absence of PknD regulation the enzymatic activity of EgtD was enhanced.
Intracellular EGT Levels Increase during Late Logarithmic Phase
We explored a potential role for EGT in M. tuberculosis growth. In vitro, MSH was shown to be essential for M. tuberculosis growth in the absence of catalase (10, 41). As EGT was previously suggested to have overlapping functions with MSH in mycobacteria (42), we tested whether the ΔegtD mutant possesses similar growth defects under in vitro growth conditions. No differences in growth were observed in rolling or standing cultures containing Middlebrook 7H9 supplemented with OADC or albumin, dextrose, and sodium chloride for the wild-type, ΔegtD, and ΔegtD complemented strains after 14 days (data not shown). However, when we quantified intracellular EGT levels during the various stages of H37Rv wild-type growth, we observed EGT to begin accumulating during late logarithmic phase (Fig. 8A). These findings suggest that M. tuberculosis may be involved in conditions involving growth-limiting factors such as nutrient limitation, endogenous waste production, or low pH.
FIGURE 8.
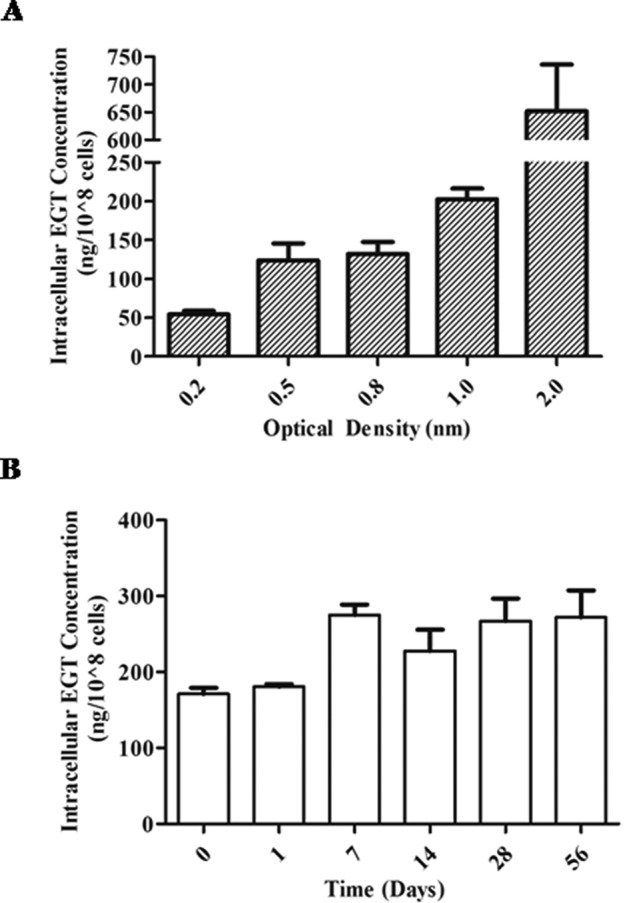
Intracellular EGT levels in H37Rv wild type under nutrient rich and starvation conditions. A, ESI LC-MS/MS quantification of intracellular EGT levels of M. tuberculosis at different stages of growth. Cultures were grown in Middlebrook 7H9 supplemented with 0.2% glycerol, 10% OADC, and 0.05% tyloxapol, and EGT was extracted from each culture at various optical densities. B, monitoring intracellular EGT levels under starved cultures. M. tuberculosis was starved in standing cultures for up to 6 weeks in PBS containing 0.05% tyloxapol. EGT was extracted from M. tuberculosis at weekly intervals for quantification by ESI LC-MS/MS. Both A and B are expressed as mean of three independent experiments ± S.E. (error bars).
EGT Is Required for M. tuberculosis Survival during Starvation
To further understand the relevance of M. tuberculosis regulation of EGT biosynthesis, we further sought out conditions where intracellular EGT levels may be regulated. Microarray studies analyzing the global adaptation of M. tuberculosis to nutrient starvation discovered pknD to be down-regulated after 4 h (43). As we described PknD to be a negative regulator of M. tuberculosis EgtD, we therefore hypothesized that EGT levels will increase during starvation.
First, to identify whether EGT plays a role in later stages of starvation, we grew wild-type H37Rv in PBS containing 0.05% tyloxapol and extracted intracellular EGT from cells at five time points over a 6-week period. After 1 week, we observed an increase in intracellular EGT levels that was maintained over 6 weeks (Fig. 8B). Because intracellular EGT levels are elevated during long term starvation, it was important for us to determine whether EGT is required for the survival of M. tuberculosis under these conditions. We starved our cultures in PBS containing 0.05% tyloxapol and monitored bacterial survival by counting cfu for up to 4 weeks. The H37Rv wild-type and the ΔegtD complemented strains did not show a significant loss in viability over this time period; however, the ΔegtD mutant cfu were reduced by a magnitude greater than one log after 3 weeks (Fig. 9A). From these findings, we conclude that M. tuberculosis requires EGT for its survival during long term starvation.
FIGURE 9.
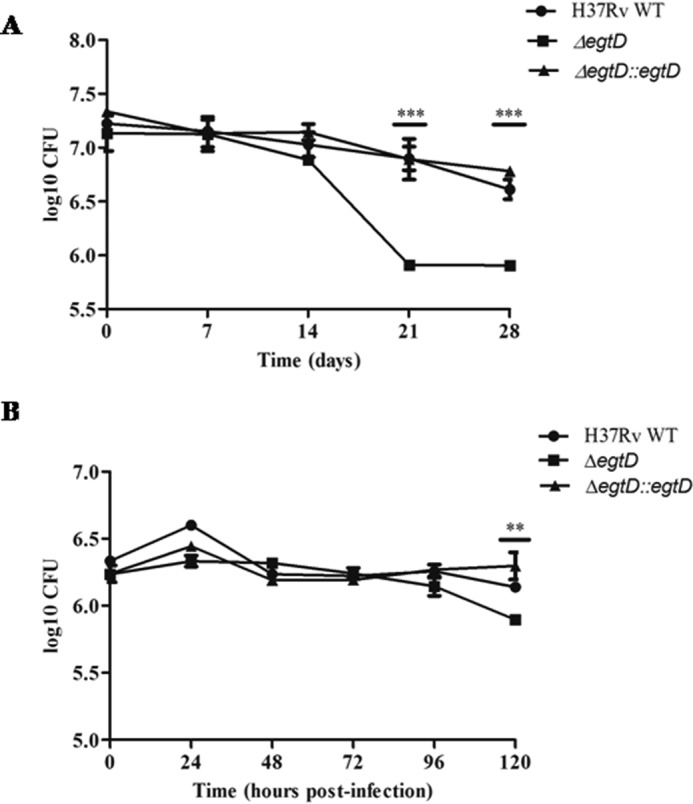
Role of EgtD in the viability of M. tuberculosis under nutrient starvation and during macrophage infection. A, survival of H37Rv WT, ΔegtD, and ΔegtD::egtD strains in 4-week-starved cultures. M. tuberculosis was incubated as standing cultures at 37 °C and starved in PBS with 0.05% tyloxapol. Samples were taken on a weekly basis to assess viability by cfu counts. The results of the three experiments (A–C) are representative of two independent experiments ±S.E. (error bars). ***, p < 0.0001. D, replication and survival of H37Rv WT, ΔegtD, and the corresponding complemented strain (ΔegtD::egtD) in J774A.1 macrophages infected at a multiplicity of infection of 5:1. cfu were calculated at the specified time points. Results are representative of two independent experiments ±S.E. (error bars). **, p < 0.01.
Growth of M. tuberculosis ΔegtD Is Attenuated in Murine J774A.1 Macrophages
As EGT has been shown to scavenge a number of reactive oxygen and nitrogen species in vitro, we wanted to assess whether EGT was able to protect M. tuberculosis from bactericidal micromolecules generated by the macrophage. Controversy still exists as to whether human macrophages produce adequate levels of NO. Because the expression of functional inducible NO synthase in mouse macrophages has been clearly demonstrated, we decided to study the role of EGT in J774A.1 macrophages (44). Independent infection of the J774A.1 cells showed an approximate half-log reduction in cfu at 120 h in the ΔegtD mutant (Fig. 9B). The minor impact EgtD has on the survival of M. tuberculosis inside the macrophage suggests that EGT is probably needed to a greater extent at later stages of infection.
Discussion
The inherent energy cost of synthesizing EGT in mycobacteria led us to believe that biosynthesis is likely regulated as it would not be energetically economic for the cell to continuously canalize numerous metabolites into the pathway. The identification of Rv3701c as part of the EGT biosynthetic cluster in mycobacteria and its implication in pathogenesis prompted us to further characterize its function in M. tuberculosis. Using a combination of genetic and biochemical assays, we confirmed the role of Rv3701c in M. tuberculosis EGT biosynthesis and annotated the enzyme as EgtD because of its activity as a histidine methyltransferase and involvement in the production of hercynine. Partially methylated histidine products were also formed in the EgtD methylation reaction. In earlier work, Reinhold et al. (45) also observed the conversion of histidine to α-N-methyl-l-histidine and α-N,N-dimethyl-l-histidine derivatives and hercynine in the presence of a cell-free extract of Neurospora crassa mycelium. These methylated intermediates are the preferential substrates of EgtD in N. crassa (45) and M. smegmatis (20), indicating that EgtD methylates histidine in a stepwise reaction due to an increased affinity for the methylated intermediates (41). The specificity of EgtD for these methylated derivatives likely acts as a regulatory mechanism, limiting the quantity of histidine taken up for EGT biosynthesis.
We have found that similarly to M. smegmatis EgtD is essential in the production of EGT in M. tuberculosis (42). These findings along with the commitment of histidine to the pathway suggested EgtD to be a likely candidate for regulation in EGT biosynthesis. We identified EgtD to act as a substrate of PknD and negatively regulate EGT biosynthesis in M. tuberculosis. The fact that Thr213 was demonstrated to be the site of phosphorylation in vitro and inside E. coli combined with the negative effect of phosphorylation on EgtD activity and EGT biosynthesis points to Thr213 as a critical residue in catalysis. Through the crystallization of M. smegmatis EgtD, Vit et al. (41) demonstrated Thr213 to be responsible for binding the imidazole ring of histidine through a water-mediated hydrogen bond. Therefore, the introduction of a phosphate at this residue impedes the interaction between the methyltransferase and its substrate, resulting in the observed loss in EgtD activity.
The decline (20%) in the catalytic efficiency observed when EgtD was phosphorylated in vitro (Fig. 5) varied from the levels of EGT synthesized from the genetic experiments (Fig. 7) typical to differences between in vitro and in vivo experiments. Enzymatic reactions proceed with greater efficiency in their natural environments, which likely describes the enhanced suppression of EGT production in the pknD:Tn mutant (35%) in comparison with our in vitro experiment (20%). Furthermore, our in vitro assay monitors the formation of methylated histidine and not the synthesis of ergothioneine (which was measured in the EgtD phosphomutant strains). We suggest that phosphorylation of EgtD inhibits the di- or trimethylation of histidine, preventing EGT biosynthesis from proceeding.
To gain a greater appreciation for the regulation of EGT biosynthesis in M. tuberculosis and to clarify its role in pathogenesis, we assessed the survival of the ΔegtD mutant in murine macrophages. During infection, the mutant was only slightly attenuated (half-log) at 120 h, an unexpected result because of the reputation of EGT as an antioxidant. Perhaps MSH provides a compensatory effect in M. tuberculosis in the absence of EGT ex vivo; however, current work does not show increased levels of MSH in the absence of EGT (11, 42). The pknD:Tn mutant showed no difference in viability when infecting J774A.1 macrophages (13), further supporting the notion that EGT levels have a limited effect on the survival of M. tuberculosis in murine macrophages (43). Additional studies examining the survival of M. tuberculosis ΔegtD in gp91Phox−/− and NOS2−/− macrophages are needed to attribute a clearer role to EGT protecting the bacilli against host-generated reactive oxygen and nitrogen species.
We did not observe any growth defect between the ΔegtD mutant and the wild-type and complemented strains when grown in the presence or absence of catalase. These findings differ from those observed with MSH where catalase is essential when growing M. tuberculosis in vitro (10). As a result, we believe that EGT does not provide protection against peroxides during in vitro growth of M. tuberculosis. Studies examining the survival of M. smegmatis ΔegtD when challenged with cumene hydroperoxide or tert-butyl hydroperoxide found the mutant to be similar to wild type (42), which additionally suggests little to no involvement of EGT in the elimination of peroxides in mycobacteria.
Additional growth studies identified intracellular EGT levels to be correlated with M. tuberculosis growth. Specifically, a substantial rise was recorded when M. tuberculosis reached late logarithmic phase, and an even greater amount of EGT accumulated during stationary phase. Elevated levels of intracellular EGT were also observed during the stationary phase of M. smegmatis by Ta et al. (11), but the reverse was shown by Sao Emani et al. (42).
Stationary phase is characterized by a number of growth-limiting factors, one of which is nutrient deprivation. An earlier study using nutrient starvation in M. tuberculosis identified pknD to be significantly down-regulated 4 h following starvation (35). Because EgtD is negatively regulated by PknD, we expected to observe increased levels of intracellular EGT during starvation. Despite being metabolically costly to the cell, M. tuberculosis maintained elevated intracellular EGT levels over 5 weeks, suggesting that EGT is needed during long term starvation. It is possible that the discrepancy in EGT levels between our starved (weeks 1–6; ∼300 ng/108 cells) and stationary phase (A600 = 2.0; 750 ng/108 cells) cultures were the result of M. tuberculosis sensing the gradual depletion of nutrients under non-chemostatic culture conditions, providing the bacilli with the opportunity to accumulate more EGT before entering stationary phase. We further assessed the contribution of EGT to the survival of M. tuberculosis during nutrient starvation and found the ΔegtD mutant to have over a 1-log reduction in viability at weeks 3 and 4 in comparison with the wild-type strain, confirming the role of EGT in long term starvation.
Earlier EGT was shown to accumulate during nutrient-limited growth of Schizosaccharomyces pombe under a wide range of glucose concentrations (0–111 mm) (46). EGT was also found to accumulate 1 h following nitrogen starvation in S. pombe (47). The doubling time of S. pombe is substantially shorter compared with M. tuberculosis, and thus it is plausible that EGT is required by the cell sooner and is synthesized more quickly than we observed in M. tuberculosis. Interestingly, these low glucose and nitrogen conditions in S. pombe are characterized by the cells transitioning from a dividing to a quiescent state (46, 47).
The fungi Colletotrichum graminicola and N. crassa were found to contain 17 and 5 times the amount of EGT in their conidial fractions than mycelium, respectively (48). The exact function of accumulating EGT in the conidia of ascomycetous fungi is unknown; however, EGT was shown to play a role in conidial longevity and germination (48, 49). Analysis of the ability of endogenous EGT to protect N. crassa against the toxic effects of menadione and cupric sulfate showed no effect on mycelial growth or spore germination (48). EGT also did not offer any protection against DNA damage when conidia were exposed to 254 nm UV light (49). The only antioxidant property EGT exerted in N. crassa was that against exogenous peroxide where the germination of the EGT-minus mutant was significantly more sensitive to tert-butyl hydroperoxide than the wild type (48). However, the quiescent S. pombe deletion and overexpression EGT mutants did not show any sensitivity or resistance to hydrogen peroxide or tert-butyl hydroperoxide (18). Based on the above findings that EGT provides minimal protection in microorganisms against oxidative stress and in conjunction with its chemical properties (poor reducing power and resistance to auto-oxidation), we postulate that the primary function of EGT in M. tuberculosis is as not as an antioxidant.
As seen with other sulfur-containing compounds, the primary function of EGT could be in biosynthesis or energy metabolism. For example, EGT was recently observed to be involved in C8 sugar transfer and activation in the biosynthesis of the antibiotic lincomycin A in Streptomyces lincolnensis (50). In this reaction, EGT served as a carrier during the first glycosylation step to channel the lincosamine unit for further condensation with a methylated derivative of 4-propyl-l-proline. Interestingly, the reaction does not consume but recycles EGT through a thiol exchange with MSH, which may explain the low abundance of this molecule in cells. In addition to glycosylation reactions, EGT was also found to be involved in the biosynthesis of two bohemamine-type pyrrolizidine alkaloids (spithioneines A and B) synthesized by the marine bacterium Streptomyces spinoverrucosus. During spithioneine A and B synthesis, EGT is incorporated directly to form a polyketide, which is thought to result from an atom of EGT carrying out a nucleophilic attack on the epoxide of the bohemamine (51).The association of EGT with glycosylation reactions and polyketide synthesis suggests its involvement in enzymatic reactions. Although unable to synthesize lincomycin A, M. tuberculosis encodes a number of polyketide synthases and performs a number glycosylation reactions that are primarily involved in the synthesis of cell wall lipids. Interestingly, under starvation conditions and in a murine model of chronic infection, sulfolipid synthesis is up-regulated, whereas the majority of cell wall synthesis genes are repressed (35, 52). The direct role sulfolipids play in M. tuberculosis virulence remains unclear; however, these lipids have been identified to negatively regulate the intracellular survival of M. tuberculosis in a species-specific manner and mediate the susceptibility of M. tuberculosis to human cationic antimicrobial peptides (53).
A number of studies have implicated PknD in regulating the adaptation of M. tuberculosis to extracellular stressors through cell wall remodeling. During cell wall synthesis, PknD positively regulates the transport of phthiocerol dimycocerosate (54). Later work also described one of the substrates of PknD, osmosensory protein A (OprA), to be up-regulated in the presence of increasing extracellular osmolarity and to enable adaptation through modifying peptidoglycan thickness (55, 56). Several other enzymes involved in the synthesis of cell wall components are also substrates of PknD, including malonyl-CoA:acyl carrier protein transacylase (FabD) and the β-ketoacyl-acyl carrier protein synthases KasA and KasB (39).
It is evident that EGT plays a role in the long term survival and non-replicating/dormant state of bacteria, fungi, and yeast, but whether its mechanism involves cell wall synthesis or long term energy storage or whether it acts as a cytoprotectant remains to be discovered. In tuberculosis, both the phagosome and granuloma are thought to be sites of nutrient deprivation during M. tuberculosis infection (57, 58). Starvation and hostile conditions in the host are suspected to act as external triggers that terminate growth and render the bacilli phenotypically resistant to drugs (59). This state of non-replicating persistence not only makes latent disease difficult to eradicate, but it is now accepted that this subset of bacteria is responsible for prolonged treatment in active tuberculosis cases. Therefore, it is of relevance to clarify the role of EGT as an antioxidant and further investigate enzymatic reactions related to metabolism that may involve EGT in M. tuberculosis. Studies are currently underway in our laboratory to address such questions and to further understand the contribution of EGT to M. tuberculosis physiology and pathogenesis.
Author Contributions
Y. A.-G. and M. R.-G. conceived and coordinated the study and wrote the paper. M. R.-G., H. B., A. J. C. S., and Y. A.-G. designed experiments. M. R.-G. performed experiments. J. A. designed and performed EGT mass spectrometric analysis. S. P.-D. designed and performed the mouse macrophages infection studies described in Fig. 9B. L. W. cloned PknD. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Peifu Zhou and Dr. Anaximandro Gomez Velasco for providing the clones of various STPKs and Mary Ko, Xingji Zheng, and Dr. Dennis Wong for technical assistance and guidance. We also thank the British Columbia Centre for Disease Control for the use of the containment level 3 facility.
This work was supported by grants from the British Columbia Lung Association (to Y. A.-G.), University of British Columbia's four-year doctoral fellowship (to M. R.-G.), and the Friedman Scholars Program (to M. R.-G.). The authors declare that they have no conflicts of interest with the contents of this article.
- MSH
- mycothiol
- EGT
- ergothioneine
- AdoMet
- S-adenosylmethionine
- STPK
- Ser/Thr protein kinase
- OADC
- oleic acid/albumin/dextrose/catalase
- ESI
- electrospray ionization
- Tn
- transposon.
References
- 1. Kumar A., Farhana A., Guidry L., Saini V., Hondalus M., Steyn A. J. (2011) Redox homeostasis in mycobacteria: the key to tuberculosis control? Expert Rev. Mol. Med. 13, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Newton G. L., Arnold K., Price M. S., Sherrill C., Delcardayre S. B., Aharonowitz Y., Cohen G., Davies J., Fahey R. C., Davis C. (1996) Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes. J. Bacteriol. 178, 1990–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rawat M., Newton G. L., Ko M., Martinez G. J., Fahey R. C., Av-Gay Y. (2002) Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob. Agents Chemother. 46, 3348–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ung K. S., Av-Gay Y. (2006) Mycothiol-dependent mycobacterial response to oxidative stress. FEBS Lett. 580, 2712–2716 [DOI] [PubMed] [Google Scholar]
- 5. Rawat M., Uppal M., Newton G., Steffek M., Fahey R. C., Av-Gay Y. (2004) Targeted mutagenesis of the Mycobacterium smegmatis mca gene, encoding a mycothiol-dependent detoxification protein. J. Bacteriol. 186, 6050–6058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Newton G. L., Unson M. D., Anderberg S. J., Aguilera J. A., Oh N. N., delCardayre S. B., Av-Gay Y., Fahey R. C. (1999) Characterization of Mycobacterium smegmatis mutants defective in 1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside and mycothiol biosynthesis. Biochem. Biophys. Res. Commun. 255, 239–244 [DOI] [PubMed] [Google Scholar]
- 7. Miller C. C., Rawat M., Johnson T., Av-Gay Y. (2007) Innate protection of Mycobacterium smegmatis against the antimicrobial activity of nitric oxide is provided by mycothiol. Antimicrob. Agents Chemother. 51, 3364–3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rawat M., Johnson C., Cadiz V., Av-Gay Y. (2007) Comparative analysis of mutants in the mycothiol biosynthesis pathway in Mycobacterium smegmatis. Biochem. Biophys. Res. Commun. 363, 71–76 [DOI] [PubMed] [Google Scholar]
- 9. Rawat M., Kovacevic S., Billman-Jacobe H., Av-Gay Y. (2003) Inactivation of mshB, a key gene in the mycothiol biosynthesis pathway in Mycobacterium smegmatis. Microbiology 149, 1341–1349 [DOI] [PubMed] [Google Scholar]
- 10. Vilchèze C., Av-Gay Y., Attarian R., Liu Z., Hazbón M. H., Colangeli R., Chen B., Liu W., Alland D., Sacchettini J. C., Jacobs W. R. Jr. (2008) Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol. Microbiol. 69, 1316–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ta P., Buchmeier N., Newton G. L., Rawat M., Fahey R. C. (2011) Organic hydroperoxide resistance protein and ergothioneine compensate for loss of mycothiol in Mycobacterium smegmatis mutants. J. Bacteriol. 193, 1981–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Calvin M. (1954) in Glutathione: Proceedings of the Symposium Held at Ridgefield, Connecticut, November, 1953 (Colowick S., Schwarz D. R., Lazarow A., Stadtman E., Racker E., Waelsch H., eds) pp. 3–30, Academic Press Inc., New York [Google Scholar]
- 13. Cheah I. K., Halliwell B. (2012) Ergothioneine; antioxidant potential, physiological function and role in disease. Biochim. Biophys. Acta 1822, 784–793 [DOI] [PubMed] [Google Scholar]
- 14. Paul B. D., Snyder S. H. (2010) The unusual amino acid L-ergothioneine is a physiologic cytoprotectant. Cell Death Differ. 17, 1134–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pfeiffer C., Bauer T., Surek B., Schömig E., Gründemann D. (2011) Cyanobacteria produce high levels of ergothioneine. Food Chem. 129, 1766–1769 [Google Scholar]
- 16. Genghof D. S., Inamine E., Kovalenko V., Melville D. B. (1956) Ergothioneine in microorganisms. J. Biol. Chem. 223, 9–17 [PubMed] [Google Scholar]
- 17. Genghof D. S., Vandamme O. (1964) Biosynthesis of ergothioneine and hercynine by mycobacteria. J. Bacteriol. 87, 852–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pluskal T., Ueno M., Yanagida M. (2014) Genetic and metabolomic dissection of the ergothioneine and selenoneine biosynthetic pathway in the fission yeast, S. pombe, and construction of an overproduction system. PLoS One 9, e97774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gründemann D., Harlfinger S., Golz S., Geerts A., Lazar A., Berkels R., Jung N., Rubbert A., Schömig E. (2005) Discovery of the ergothioneine transporter. Proc. Natl. Acad. Sci. U.S.A. 102, 5256–5261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seebeck F. P. (2010) In vitro reconstitution of mycobacterial ergothioneine biosynthesis. J. Am. Chem. Soc. 132, 6632–6633 [DOI] [PubMed] [Google Scholar]
- 21. Av-Gay Y., Everett M. (2000) The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol. 8, 238–244 [DOI] [PubMed] [Google Scholar]
- 22. Spivey V. L., Molle V., Whalan R. H., Rodgers A., Leiba J., Stach L., Walker K. B., Smerdon S. J., Buxton R. S. (2011) Forkhead-associated (FHA) domain containing ABC transporter Rv1747 is positively regulated by Ser/Thr phosphorylation in Mycobacterium tuberculosis. J. Biol. Chem. 286, 26198–26209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Molle V., Kremer L. (2010) Division and cell envelope regulation by Ser/Thr phosphorylation: Mycobacterium shows the way. Mol. Microbiol. 75, 1064–1077 [DOI] [PubMed] [Google Scholar]
- 24. Chao J. D., Papavinasasundaram K. G., Zheng X., Chávez-Steenbock A., Wang X., Lee G. Q., Av-Gay Y. (2010) Convergence of Ser/Thr and two-component signaling to coordinate expression of the dormancy regulon in Mycobacterium tuberculosis. J. Biol. Chem. 285, 29239–29246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khan S., Nagarajan S. N., Parikh A., Samantaray S., Singh A., Kumar D., Roy R. P., Bhatt A., Nandicoori V. K. (2010) Phosphorylation of enoyl-acyl carrier protein reductase InhA impacts mycobacterial growth and survival. J. Biol. Chem. 285, 37860–37871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Veyron-Churlet R., Zanella-Cléon I., Cohen-Gonsaud M., Molle V., Kremer L. (2010) Phosphorylation of the Mycobacterium tuberculosis β-ketoacyl-acyl carrier protein reductase MabA regulates mycolic acid biosynthesis. J. Biol. Chem. 285, 12714–12725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Molle V., Gulten G., Vilchèze C., Veyron-Churlet R., Zanella-Cléon I., Sacchettini J. C., Jacobs W. R. Jr., Kremer L. (2010) Phosphorylation of InhA inhibits mycolic acid biosynthesis and growth of Mycobacterium tuberculosis. Mol. Microbiol. 78, 1591–1605 [DOI] [PubMed] [Google Scholar]
- 28. Corrales R. M., Molle V., Leiba J., Mourey L., de Chastellier C., Kremer L. (2012) Phosphorylation of mycobacterial PcaA inhibits mycolic acid cyclopropanation: consequences for intracellular survival and for phagosome maturation block. J. Biol. Chem. 287, 26187–26199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cowley S., Ko M., Pick N., Chow R., Downing K. J., Gordhan B. G., Betts J. C., Mizrahi V., Smith D. A., Stokes R. W., Av-Gay Y. (2004) The Mycobacterium tuberculosis protein serine/threonine kinase PknG is linked to cellular glutamate/glutamine levels and is important for growth in vivo. Mol. Microbiol. 52, 1691–1702 [DOI] [PubMed] [Google Scholar]
- 30. Gómez-Velasco A., Bach H., Rana A. K., Cox L. R., Bhatt A., Besra G. S., Av-Gay Y. (2013) Disruption of the serine/threonine protein kinase H affects phthiocerol dimycocerosates synthesis in Mycobacterium tuberculosis. Microbiology 159, 726–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leiba J., Syson K., Baronian G., Zanella-Cléon I., Kalscheuer R., Kremer L., Bornemann S., Molle V. (2013) Mycobacterium tuberculosis maltosyltransferase GlgE, a genetically validated antituberculosis target, is negatively regulated by Ser/Thr phosphorylation. J. Biol. Chem. 288, 16546–16556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bardarov S., Bardarov S. Jr., Pavelka M. S. Jr., Sambandamurthy V., Larsen M., Tufariello J., Chan J., Hatfull G., Jacobs W. R. Jr. (2002) Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148, 3007–3017 [DOI] [PubMed] [Google Scholar]
- 33. Singh A., Mai D., Kumar A., Steyn A. J. (2006) Dissecting virulence pathways of Mycobacterium tuberculosis through protein-protein association. Proc. Natl. Acad. Sci. U.S.A. 103, 11346–11351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng X., Papavinasasundaram K. G., Av-Gay Y. (2007) Novel substrates of Mycobacterium tuberculosis PknH Ser/Thr kinase. Biochem. Biophys. Res. Commun. 355, 162–168 [DOI] [PubMed] [Google Scholar]
- 35. Betts J. C., Lukey P. T., Robb L. C., McAdam R. A., Duncan K. (2002) Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43, 717–731 [DOI] [PubMed] [Google Scholar]
- 36. Sassetti C. M., Rubin E. J. (2003) Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U.S.A. 100, 12989–12994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rengarajan J., Bloom B. R., Rubin E. J. (2005) Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc. Natl. Acad. Sci. U.S.A. 102, 8327–8332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luo D., Smith S. W., Anderson B. D. (2005) Kinetics and mechanism of the reaction of cysteine and hydrogen peroxide in aqueous solution. J. Pharm. Sci. 94, 304–316 [DOI] [PubMed] [Google Scholar]
- 39. Chao J., Wong D., Zheng X., Poirier V., Bach H., Hmama Z., Av-Gay Y. (2010) Protein kinase and phosphatase signaling in Mycobacterium tuberculosis physiology and pathogenesis. Biochim. Biophys. Acta 1804, 620–627 [DOI] [PubMed] [Google Scholar]
- 40. Vanzembergh F., Peirs P., Lefevre P., Celio N., Mathys V., Content J., Kalai M. (2010) Effect of PstS sub-units or PknD deficiency on the survival of Mycobacterium tuberculosis. Tuberculosis 90, 338–345 [DOI] [PubMed] [Google Scholar]
- 41. Vit A., Misson L., Blankenfeldt W., Seebeck F. P. (2015) Ergothioneine biosynthetic methyltransferase EgtD reveals the structural basis of aromatic amino acid betaine biosynthesis. Chembiochem 16, 119–125 [DOI] [PubMed] [Google Scholar]
- 42. Sao Emani C., Williams M. J., Wiid I. J., Hiten N. F., Viljoen A. J., Pietersen R. D., van Helden P. D., Baker B. (2013) Ergothioneine is a secreted antioxidant in Mycobacterium smegmatis. Antimicrob. Agents Chemother. 57, 3202–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Be N. A., Bishai W. R., Jain S. K. (2012) Role of Mycobacterium tuberculosis pknD in the pathogenesis of central nervous system tuberculosis. BMC Microbiol. 12, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mestas J., Hughes C. C. (2004) Of mice and not men: differences between mouse and human immunology. J. Immunol. 172, 2731–2738 [DOI] [PubMed] [Google Scholar]
- 45. Reinhold V. N., Ishikawa Y., Melville D. B. (1970) Conversion of histidine to hercynine by Neurospora crassa. J. Bacteriol. 101, 881–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pluskal T., Hayashi T., Saitoh S., Fujisawa A., Yanagida M. (2011) Specific biomarkers for stochastic division patterns and starvation-induced quiescence under limited glucose levels in fission yeast. FEBS J. 278, 1299–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sajiki K., Pluskal T., Shimanuki M., Yanagida M. (2013) Metabolomic analysis of fission yeast at the onset of nitrogen starvation. Metabolites 3, 1118–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bello M. H., Barrera-Perez V., Morin D., Epstein L. (2012) The Neurospora crassa mutant NcΔEgt-1 identifies an ergothioneine biosynthetic gene and demonstrates that ergothioneine enhances conidial survival and protects against peroxide toxicity during conidial germination. Fungal Genet. Biol. 49, 160–172 [DOI] [PubMed] [Google Scholar]
- 49. Bello M. H., Mogannam J. C., Morin D., Epstein L. (2014) Endogenous ergothioneine is required for wild type levels of conidiogenesis and conidial survival but does not protect against 254 nm UV-induced mutagenesis or kill. Fungal Genet. Biol. 73, 120–127 [DOI] [PubMed] [Google Scholar]
- 50. Zhao Q., Wang M., Xu D., Zhang Q., Liu W. (2015) Metabolic coupling of two small-molecule thiols programs the biosynthesis of lincomycin A. Nature 518, 115–119 [DOI] [PubMed] [Google Scholar]
- 51. Fu P., MacMillan J. B. (2015) Spithioneines A and B, two new bohemamine derivatives possessing ergothioneine moiety from a marine-derived Streptomyces spinoverrucosus. Org. Lett. 17, 3046–3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rodríguez J. E., Ramírez A. S., Salas L. P., Helguera-Repetto C., Gonzalez-y-Merchand J., Soto C. Y., Hernández-Pando R. (2013) Transcription of genes involved in sulfolipid and polyacyltrehalose biosynthesis of Mycobacterium tuberculosis in experimental latent tuberculosis infection. PLoS One 8, e58378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gilmore S. A., Schelle M. W., Holsclaw C. M., Leigh C. D., Jain M., Cox J. S., Leary J. A., Bertozzi C. R. (2012) Sulfolipid-1 biosynthesis restricts Mycobacterium tuberculosis growth in human macrophages. ACS Chem. Biol. 7, 863–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pérez J., Garcia R., Bach H., de Waard J. H., Jacobs W. R. Jr., Av-Gay Y., Bubis J., Takiff H. E. (2006) Mycobacterium tuberculosis transporter MmpL7 is a potential substrate for kinase PknD. Biochem. Biophys. Res. Commun. 348, 6–12 [DOI] [PubMed] [Google Scholar]
- 55. Greenstein A. E., MacGurn J. A., Baer C. E., Falick A. M., Cox J. S., Alber T. (2007) M. tuberculosis Ser/Thr protein kinase D phosphorylates an anti-anti-σ factor homolog. PLoS Pathog. 3, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hatzios S. K., Baer C. E., Rustad T. R., Siegrist M. S., Pang J. M., Ortega C., Alber T., Grundner C., Sherman D. R., Bertozzi C. R. (2013) Osmosensory signaling in Mycobacterium tuberculosis mediated by a eukaryotic-like Ser/Thr protein kinase. Proc. Natl. Acad. Sci. U.S.A. 110, E5069–E5077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nyka W. (1974) Studies on the effect of starvation on mycobacteria. Infect. Immun. 9, 843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wayne L. G. (1994) Dormancy of Mycobacterium tuberculosis and latency of disease. Eur. J. Clin. Microbiol. Infect. Dis. 13, 908–914 [DOI] [PubMed] [Google Scholar]
- 59. Wayne L. G., Sohaskey C. D. (2001) Nonreplicating persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 55, 139–163 [DOI] [PubMed] [Google Scholar]